Beyond the RNA-dependent function of LncRNA genes
- Institute of Cardiovascular Regeneration, Centre for Molecular Medicine, Goethe University, Germany
- Faculty of Science, Benha University, Egypt
Abstract
While long non-coding RNA (lncRNA) genes have attracted a lot of attention in the last decade, the focus regarding their mechanisms of action has been primarily on the RNA product of these genes. Recent work on several lncRNAs genes demonstrates that not only is the produced RNA species important, but also that transcription of the lncRNA locus alone can have regulatory functions. Like the functions of lncRNA transcripts, the mechanisms that underlie these genome-based functions are varied. Here we highlight some of these examples and provide an outlook on how the functional mechanisms of a lncRNA gene can be determined.
LncRNA genes in the genome
The complex genome of eukaryotes is pervasively transcribed and efforts to comprehensively define all transcripts have led to the idea that about half of the genome can be transcribed into RNA in an individual cell (Djebali et al., 2012). The units that produce RNAs – the genes - can roughly be categorized into the two main biotypes: protein-coding genes (PCGs) and non-protein-coding genes (NCGs). The largest and most coherent category is the PCG, which encodes RNAs that serve as the template for all the peptides and proteins in the cell. The NCG category is a highly heterogenous collection and can be sub-grouped into small ncRNA (non-coding RNA) and long ncRNA (lncRNA) genes, where the term long refers to the arbitrary length of 200 nucleotides or longer. In particular, the lncRNA genes have attracted a lot of attention in recent years due to their wide range of action and mostly unexplored functions. While their number was overestimated after their initial discovery, similar to the overestimation of the number of PCGs at the beginning of the human genome project (Lander et al., 2001), current and careful curation projects, such as the GENCODE and FANTOM projects, list 17,957 and 27,919 lncRNA genes, respectively (Figure 1A), in their most recent data releases of the human genome (Frankish et al., 2019; Hon et al., 2017). Hence, the number of lncRNA genes are in the same range, or even a bit higher, than the number of PCGs (19,954). In the future, this currently very heterogeneous class of NCGs may be sub-categorized further into more specific biotypes.
Figure 1
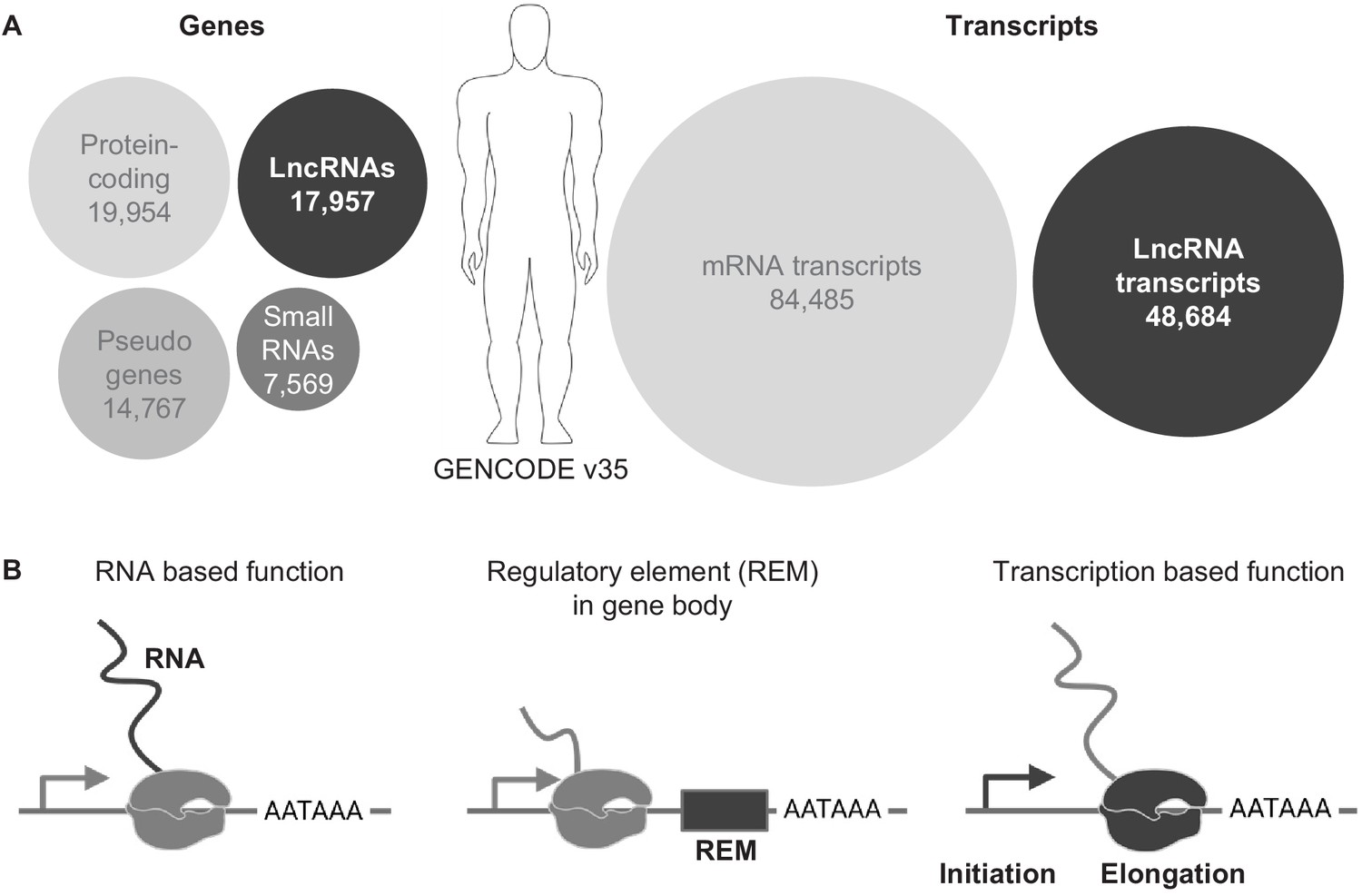
LncRNA genes in the genome.
(A) Overview of genes and transcript numbers in the human genome (GENCODE v35). Circle area represents relative quantities. (B) Schematics of three possible functional properties of lncRNA loci.
Currently, three major functional principles can be assigned to lncRNA loci (Figure 1B): (1) either the RNA is the functional biomolecule and interacts with other components in the cell, for example DNA, proteins or RNAs, (2) a gene regulatory element is embedded in the transcription body of a lncRNA gene and the activity of the lncRNA gene directs the activity of the regulatory element or (3) the process of transcription influences genome and thereby gene activity. A lncRNA locus can haveone of these functions or a mixture of them (Yin et al., 2015). In this review we will focus on the latter two functional lncRNA properties, in which the RNA is, at least partially dispensable for the lncRNA gene function.
The transcription of genes
The generation of RNA using the genome as a template, or the process of transcription, depends on certain functional genomic elements (Figure 2). The core element of a gene that initiates the production of an RNA is the promoter. A GC-rich element that is accessible (open chromatin) will attract the polymerase machinery and general transcription factors (TFs). This minimal core element serves as a core promoter and can be sufficient to initiate transcription (Deaton and Bird, 2011). Transcription of RNA starts at the transcriptional start site (TSS), which is located within the core promoter. Like PCGs, most lncRNAs are transcribed by POL II (RNA polymerase 2, a multiprotein complex), but are more tissue-specific compared to PCGs (for review see Ransohoff et al., 2018). Both biotypes (PCGs and lncRNAs) have conserved core promoter sequences with fewer overlapping TF binding motifs in lncRNA promoters, resulting in an overall lower expression level compared to PCGs (Figure 2; Mattioli et al., 2019). Thus, the architecture of the core promoter is the first player that defines the degree of lncRNA expression (Batut and Gingeras, 2017; Mattioli et al., 2019). The second important element that influences the transcription of genes are enhancers, which are cis-regulatory elements that can either have a positive or a negative (which are then often referred to as repressors) impact on their target genes. Consequently, enhancers are genomic regions that encode binding sites for sequence-specific activator or repressor TFs. These elements often confer specificity in spatiotemporal expression. Many lncRNAs can also be generated from such enhancer elements, which contributes to their overall more tissue-specific expression when compared to PCGs (Mattioli et al., 2019).
Figure 2

Distinguishing features of transcript generation of PCGs and lncRNAs (A) LncRNA and (B) mRNAs: lncRNA genes are lowly expressed as fewer transcription factors (TFs) bind the promoter.
In addition, lncRNA TSS, exon and/or pA site more often associate with transposable elements (TEs), while TEs contribute mostly to UTRs and/or introns of mRNAs. In addition, mRNAs are more efficiently spliced.
The core promoter initiates transcription and thereby the generation of an RNA that may or may not be further diversified by splicing (Figure 2). This depends on whether splice sites are present between the promoter and the transcription termination element, the polyadenylation signal (pA). The mechanism of PCG and lncRNA splicing is similar, although the splicing efficiency of lncRNAs is lower than PCGs, likely due to the loss of proximal RNA POL II phosphorylation over 5’ splice sites (Krchnáková et al., 2019). In addition, lncRNAs show signs of co-transcriptional cleavage and premature termination with Thr4p PolII enriched over the entire lncRNA body (Schlackow et al., 2017). At some point the transcriptional machinery will run into a termination signal, a DNA sequence element consisting of AATAAA and downstream GU (or U)-rich motifs (Eaton et al., 2020). These elements are ubiquitously present in the genome. In humans, one can find 569,005 elements that meet the criterion of a pA signal (301,001 in mouse and 20,931 in C. elegans) (Herrmann et al., 2020). Moreover, this high number likely ensures successful termination of transcription (Eaton and West, 2020).
Another class of genetic elements that play an important role for gene and genome activity are transposable elements (TEs) (for review see Chuong et al., 2017). These mobile genomic elements make up more than 44% of the human genome (Lander et al., 2001) and attracted attention as important regulators of gene and genome activity (Bourque et al., 2018). In this respect, TEs are an important component of lncRNA biology as well (Figure 2A). Approximately, 75% of lncRNA transcripts contain sequence elements from TEs (Kapusta et al., 2013) and some of them represent important sequence elements to direct lncRNA localization (Lubelsky and Ulitsky, 2018). In addition, 25% of TEs are found to overlap with TSS and pA signals of lncRNA genes (Kapusta et al., 2013). Hence, they are an important driving force of lncRNA expression. One recent example is the primate-specific lncRNA XACT (Table 1), which has been shown to protect the active X chromosome from being silenced (antagonizing XIST lncRNA effect) and whose sequence contains elements derived from a TE (Casanova et al., 2019). Interestingly, XACT lncRNA is also regulated by a TE-derived enhancer element that harbors pioneer pluripotency factor binding sites. This exemplifies that TEs containing embedded TF motifs can direct tissue-specific expression when they insert next to a promoter element. Several other TE-derived lncRNAs are described elsewhere (Kapusta et al., 2013).
Table 1
Selection of lncRNA genes with RNA independent function.
| LncRNA | Relative location of respective TSSs target gene | Literature | Mode of action | |
|---|---|---|---|---|
| Regulatory element located within the transcription unit | ||||
| Haunt (Halr1) | 40 kb downstream of HOXA | Yin et al., 2015 | Activation of HOXA | |
| Lockd | 4 kb downstream of Cdkn1b | Paralkar et al., 2016 | Positive regulation of Cdkn1b via loop formation | |
| Meteor | 80 kb upstream of Eomes | Alexanian et al., 2017 | Positive licensing of Eomes expression | |
| ThymoD | 844 kb downstream of Bcl11b | Isoda et al., 2017 | DNA methylation, CTCF-binding | |
| Pcdhα-as | Pcdhα | Canzio et al., 2019 | DNA methylation, CTCF-binding | |
| GAL10-ncRNA | GAL10 antisense transcript | Houseley et al., 2008 | GAL10 promoter acetylation | |
| AIRN | 28 kb Antisense to Igfr2 | Latos et al., 2012 | Promoter methylation | |
| Upperhand (Hand2os1) | 0,1 kb upstream of Hand2 | Anderson et al., 2016; Han et al., 2019 | Promotes enhancer accessibility for Hand2 activation | |
| Activity exerted by transcription initiation or elongation | ||||
| Ftx | 140 kb upstream of Xist | Furlan et al., 2018 | Xist activation independent of Ftx RNA | |
| Chaserr | 16 kb upstream of Chd2 | Rom et al., 2019 | Negative regulation of Chd2 | |
| PVT1 | 52 kb downstream of Myc | Cho et al., 2018 | Enhancer boundary element | |
| Handsdown (Handlr) | 11 kb downstream of Hand2 | George et al., 2019; Ritter et al., 2019 | Transcriptional elongation-based enhancer shielding | |
In summary, the genome stores the information required to generate the RNAs that are necessary for a cell’s proper function, whether the RNA is protein-coding or not. An elaborate machinery is established that controls the specific activation of genes and whole genomic regions via positive or negative mechanisms. These regulatory mechanisms require energy investment from the cell. It is conceivable that sometimes it can be ‘cheaper’ for a cell to let spurious transcription of non-harmful transcripts occur, might they be coding or non-coding, than to invest energy in silencing all of these transcriptionally active sites.
Layers of gene regulation
The expression of genes and whole genomic regions is controlled by several layers of regulation. In addition to the genomic elements described above, DNA is packed with histone proteins into chromatin. These protein components can be modified to act as signaling centers for the transcription machinery (for review see Talbert et al., 2019). In addition, the proteins of the nucleus also regulate the 3D arrangement of genomic DNA in such a way that functionally connected elements of gene regulation come together. In short, each chromosome is composed of sub-megabase units known as topologically associated domains (TADs), the structural and functional unit of the chromosome (for review see Szabo et al., 2019). Such genome arrangements can allow for promoter-enhancer contacts and organize functionally dependent regulatory elements together (Hnisz et al., 2017). The major factors that regulate this organization are CTCF (CCCTC-binding TF) and the cohesin complex (Ali et al., 2016; Rao et al., 2017). CTCF binding frequently co-localizes and interacts with the cohesin complex at TAD borders (Li et al., 2020). Indeed, elimination of cohesin dissolves all chromatin TADs even in the presence of CTCF (Rao et al., 2017). Interestingly, disruption of the TADs either by removal of CTCF or cohesin results in unexpected mild effects on gene expression (Nora et al., 2017; Rao et al., 2017). While it has been accepted that gene expression and 3D genome folding are correlated, their functional relevance is still to be elucidated (Ibrahim and Mundlos, 2020).
All of these enhancers and genome organizing regions must be functionally regulated to accurately control gene and genome activity. As many such regulatory sites are associated with lncRNAs, these lncRNA loci might be important functional support elements. The process of transcription can assist in reorganizing chromatin marks (van Steensel and Furlong, 2019), allowing regions to be accessible for other factors or prevent others by diverting/directing the transcription machinery to nearby genes.
Current annotations in the database are a work-in-progress
Current annotations of genomic databases categorize genes according to various criteria. One that appears, on the surface, to be very simple is the separation of protein-coding genes (PCGs) and non-protein-coding genes (NCGs). It was already found some time ago that RNAs originating from NCGs do actually associate with ribosomes, the machinery that translates mRNAs into proteins (Ingolia et al., 2011; van Heesch et al., 2014). This association is not surprising, as the ribosomes function is to bind RNAs in the cytosol and attempt to translate it into a peptide or protein. However, just because an RNA is bound to a ribosome does not mean it is translated and even if translated, the pure presence of a peptide does not prove a function of this peptide. In more recent in-depth studies, it was found that some lncRNAs do produce peptides and that some of these peptides are even functional (Chen et al., 2020; Ji et al., 2015; van Heesch et al., 2019), including within 5’ and 3’ untranslated regions (UTR) of mRNAs. Hence, until databases are updated with suitable information that incorporates the presence of peptides derived from expressed RNAs, a peptide coding probability always must be taken into consideration when studying lncRNA function. Equally important, many PCG or NCGs have a high number of splice variants, some of which might encode a peptide and others not.
The revolution of high-throughput sequencing of fragmented cDNA libraries revealed the complexity of expression from the genome. Enrichment of lowly expressed transcripts and subsequent sequence analysis identified an even more complex pattern of splice variants (Mercer et al., 2012). However, these analyses relied on the sequencing of fragmented cDNA libraries and subsequent reconstruction of the transcriptome to a reference genome. The most recent generation of long read sequencers, such as the PacBio or the Nanopore systems, allows the direct analysis of RNAs and eliminates the intermediate step of a fragmented cDNA library. Capturing lncRNA genes specifically and resequencing by Long-read platform (known as Capture Long Sequence or CLS) determined the full variety of splice variants of the mammalian transcriptome (Lagarde et al., 2017). The advantage of this technology is the capability to precisely determine 5’ and 3’ ends and, ideally, all splice variants of a transcript. For example, the estimated mean number of exons per lncRNA using CLS was 4.27 compared to 3.59 measured by short-reads RNA-seq method (Lagarde et al., 2017). While this approach doesn’t eliminate the necessity to carefully determine the splice variants from a lncRNA locus entirely, it does provide a very good starting point for detailed analysis. In particular, when CLS data are not available for your locus-of-interest or your tissue-of-interest, one should determine the full transcript length, splice variants and regulatory elements of the lncRNA-of-interest. Only then can a successful strategy to study the lncRNA be initiated.
Gene regulation by lncRNA genes – regulatory elements within the transcription unit
Surveying the chromatin and DNA modification landscape led to the annotation of potential regulatory regions across the genome and sometimes even for specific tissues and cell types. Regulatory elements, whether they are promoters or other regulatory elements, can be found within or far away from the transcription unit of a gene. The occurrence of such a regulatory element within a transcription unit, for example of a lncRNA gene, can indicate that the function of this element might be affected by its activity.
One interesting lncRNA gene example that reflects the duality of lncRNA genes with respect to their RNA-based mechanism on one side, and an enhancer element on the other side, is Haunt. While the RNA of Haunt is thought to be required for negative regulation of HoxA, the Haunt locus contains regulatory elements to activate the HoxA locus during in vitro differentiation of pluripotent stem cells (Yin et al., 2015). While it is shown that these enhancers can interact with HoxA directly, the elements are not further defined nor how their function might depend on Haunt transcriptional activity.
A similar early example of a lncRNA locus that contains a regulatory element within its transcription unit is the Lockd lncRNA locus, which regulates its cis gene Cdkn1b. The deletion of the entire locus of Lockd, including TSS upstream elements, leads to a reduction of Cdkn1b expression (Paralkar et al., 2016). While the 5’ genomic region of Lockd interacts genomically with the promoter of Cdkn1b, this interaction is not altered if the transcription of Lockd is depleted by a pA signal inserted into the first exon of Lockd. Thus, the genomic locus itself is important as an regulatory element rather than its transcriptional activity.
Even if a specific regulatory element cannot be defined, careful analysis and genetic dissection of a lncRNA can point toward such a regulatory principle. The TSS of the Meteor lncRNA locus is important to license its cis-located gene Eomes for activation in the mesendoderm (Alexanian et al., 2017). The lack of Meteor expression by TSS deletion causes the loss of Eomes activation during mesendoderm differentiation of mouse ESCs. Decreasing levels of Meteor RNA during this process did not alter expression of downstream genes, arguing against an RNA-based function of Meteor. Interestingly, endogenous activation of Meteor is not only licensing Eomes gene activation, but other cardiac mesodermal genes as well. Moreover, transcriptional inhibition of Meteor using a polyadenylation element insertion downstream of the Meteor TSS does not cause the Eomes gene to be silenced during mesendoderm differentiation (Alexanian et al., 2017; Engreitz et al., 2016). This finding argues against a transcription-based mechanism of Meteor and suggests that the genomic locus Meteor harbors important regulatory elements to render the cis-located Eomes gene activatable during differentiation.
An excellent example of a lncRNA with a defined regulatory element within transcription unit is the ThymoD lncRNA locus. Its transcription prevents methylation of a CTCF-binding site located within its transcriptional unit (Isoda et al., 2017; Figure 3A). The binding of CTCF allows looping of the Bcl11b transcription unit in the same domain as activating regions of Bcl11b. This activation is lost when the transcription of ThymoD is blocked by insertion of a pA signal after exon two and before the CTCF-binding site and, consequentially, the CTCF-binding site is methylated (Figure 3A). Therefore, the transcriptional activity has an indirect, structural effect on the regulation of Bcl11b while the ThymoD RNA is dispensable.
Figure 3
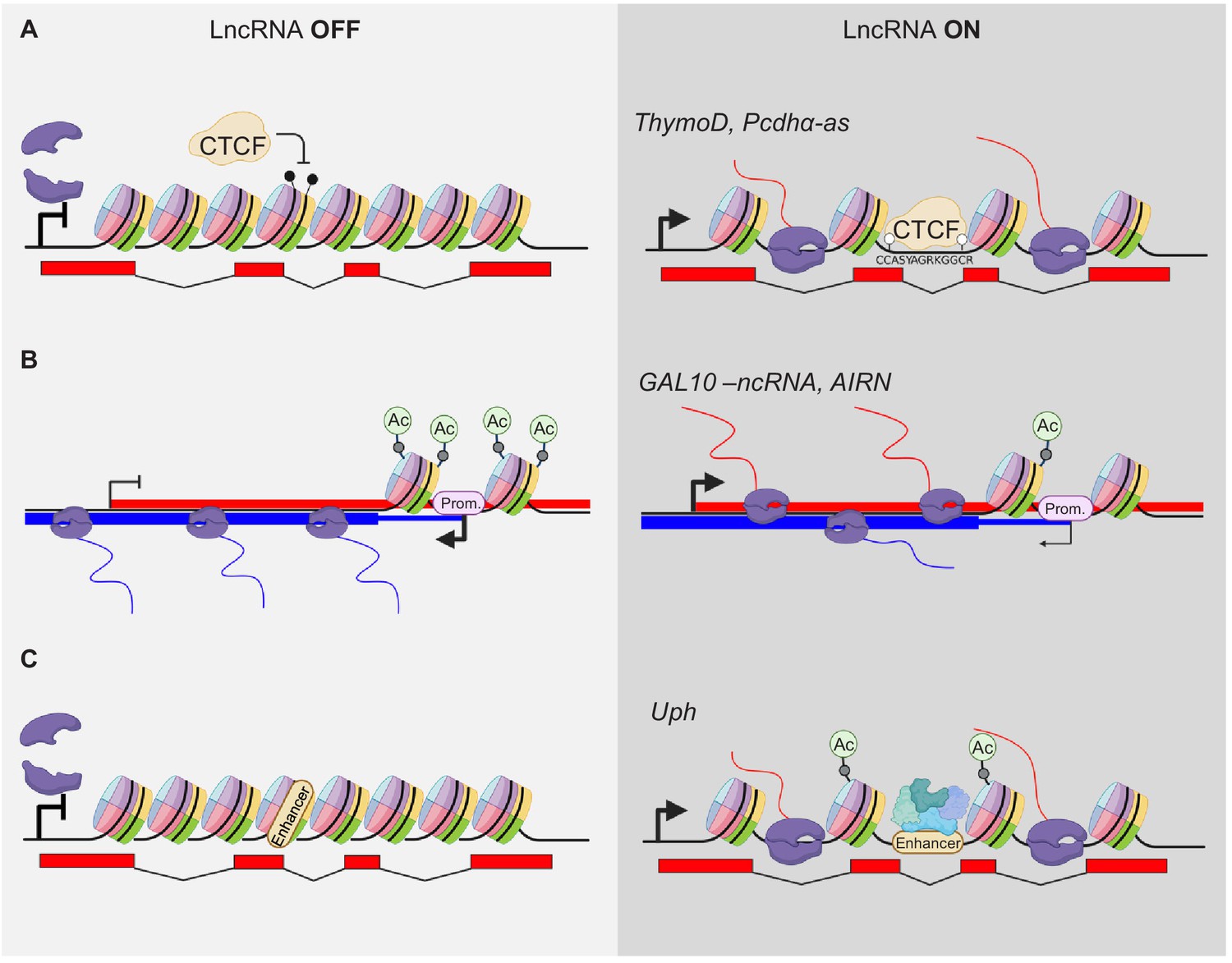
Modulation of gene expression by lncRNA transcription.
(A) Transcriptional activity modulates DNA methylation and thereby alters occupation of DNA binding factors within the gene body, for example CTCF. The POL2 complex is indicated in violet. Black drumsticks indicate methylated CpGs, white drumstick non-methylated CpGs. (B) LncRNA expression alters promoter (Prom.) activity by modifying e.g. acetylation of histones at TSS sites. (C) Transcription elongation can activate poised enhancers within their gene body (only acetylation shown).
A more complex situation of several antisense transcripts regulating their cis gene is the Protocadherin alpha (Pcdhα) cluster. The variable, stochastic expression from several Protocadherin clusters provide cell-surface proteins for cellular identity recognition in the neuronal system to allow dendrites and axons to distinguish from self and other neurons. This stochastic expression is partly regulated by a distal enhancer region. The cluster of Pcdhα produces three distinct variants from three alternative TSSs to achieve stochastic expression of splice variants from this cluster. The first exon of each of these variants contains an antisense lncRNA transcript (Pcdhα-as) (Canzio et al., 2019). The expression of the lncRNAs precedes the expression of the PCGs and positively regulates the most nearby PCG expression. Mechanistically, the Pcdhα lncRNAs act similar to the ThymoD lncRNA (above) (Figure 3A). Expression of the Pcdhα-as variants leads to the demethylation of a CTCF-binding site in the region upstream of the Pcdhα PCG, thereby allowing for a stable loop formation with the distal enhancer region and a positive effect on the PCGs expression.
There are also examples of lncRNA genes that reside within a different transcriptional entity from cis target genes. Here, it is even more conceivable that their activity has an impact on the gene they are embedded in. One of the first examples was a ncRNA within the GAL10 gene cluster in yeast Saccharomyces cerevisiae. Under 0% galactose, the TF Reb1 binds to the promoter region of GAL10-ncRNA antisense to GAL10 and fully activates its expression (Houseley et al., 2008). The transcriptional unit of GAL10-ncRNA overlaps with the TSS of GAL10 and GAL1, leading to inhibition of the GAL10 and GAL1 gene by promoting high levels of H3K36me3 methylation and hypoacetylation at the GAL10 and GAL1 promoters. Addition of galactose to the growth medium blocks GAL10-ncRNA expression and hyperacetylation of the GAL10 and GAL1 promoters, leading to expression of genes that encode galactose fermenting proteins (Figure 3B).
A similar principle was shown in higher eukaryotes at the AIRN (antisense Igf2r RNA non-coding) locus. The TSS of the lncRNA AIRN is located in the second intron of the Igf2r PCG and AIRN is transcribed antisense to Ifg2r. Transcription of AIRN negatively regulates Igfr2 (Santoro et al., 2013). When transcription of AIRN is blocked by a polyA insertion before the promoter of Igf2r, this negative regulation is abolished (Figure 3B). However, if the same pA is inserted after the promotor of Igf2r, this negative regulatory effect on Igf2r is not observed (Latos et al., 2012). These findings support the hypothesis that the transcription of AIRN, and not the RNA product itself, is important for the transcriptional regulation of the Igfr2.
A lncRNA gene transcription that influences an enhancer is Upperhand, which is divergently expressed from the Hand2 protein-coding gene (Anderson et al., 2016). Loss of Upperhand transcription leads to a loss of histone acetylation upstream of Hand2, including at the cardiac enhancer. As a result, binding of GATA4 to its previously defined enhancer (McFadden et al., 2000) is reduced, and Hand2 expression in the heart is reduced as well. Hence, the Upperhand loss-of-function phenotype is similar to cardiac loss of Hand2 (Figure 3A). Additional mutants of Upperhand draw a more complicated picture of the role of Upperhand in activating Hand2. A complete deletion of the Upperhand transcription unit that encompasses all known regulatory regions of the Hand2 gene as well, causes loss of Hand2 5’UTR expression (Han et al., 2019). These findings assert the presence of important Hand2 activating genetic elements directly upstream of its TSS, independently of any RNA originating from this region. However, a promoter deletion of Upperhand causes a loss of its RNA while leaving all other elements in that region intact, but no effect on Hand2 expression was observed in this case. Furthermore, a deletion of the last two exons from Upperhand has a slight effect on Hand2 expression. There might be so far uncharacterized enhancer elements in the genomic region of these two exons and their deletion may influence Hand2 expression. In addition, although the Upperhand RNA is suggested to be not required for its in vivo function, the RNA generates peptides that might be functional (van Heesch et al., 2019). These somehow conflicting results underline the complexity of regulation of the Hand2 gene.
These examples highlight the importance of taking a careful look at the whole lncRNA locus that produces an RNA. The occurrence of an annotated regulatory element or the occupation of a genome regulating factor such as CTCF within the transcription unit can be an important indication to look for a genomic function of a lncRNA.
Gene regulation by lncRNA genes – the act of transcription is functional
The absence of a regulatory element within the transcription unit could be due to incomplete annotation or a yet unknown factor which binds there, or the act of transcription initiation or transcriptional elongation is important for the function of a lncRNA locus.
One example of such a regulation principle comes from work on the XIST lncRNA, which is one of the original lncRNAs that has been extensively studied (Brockdorff et al., 1992). While XIST acts via the produced RNA (Brannan et al., 1990; Brown et al., 1992), the regulation of XIST, at least in part, does not. The XIST lncRNA locus is flanked by many lncRNAs, and one of them is the Ftx locus found 140 kb upstream of Xist (Chureau et al., 2002). It was initially proposed that the Ftx RNA functions to regulate XIST (Chureau et al., 2011). However, detailed analysis uncovered that the transcription of Ftx, and not the produced RNA, is important to regulate Xist (Furlan et al., 2018). Knockdown of Ftx RNA does not cause a loss of Xist expression, but deletion of the promoter of Ftx, and the consequential loss of Ftx transcription, causes the loss of Xist expression. CRISPRi of Ftx similarly causes loss of Xist expression, suggesting that transcription of Ftx is the positive regulator of Xist expression. One possibility is that 3D genome architecture can be changed due to the transcriptional activity of a genomic locus (Figure 4). Strikingly, the promoter of Xist and Ftx are flanked by CTCF-occupied sites. However, deleting the CTCF-binding sites alone at the Ftx promoter has no effect on the expression level of Xist, arguing that genome folding induced by Ftx activity does not involve CTCF-binding.
Figure 4
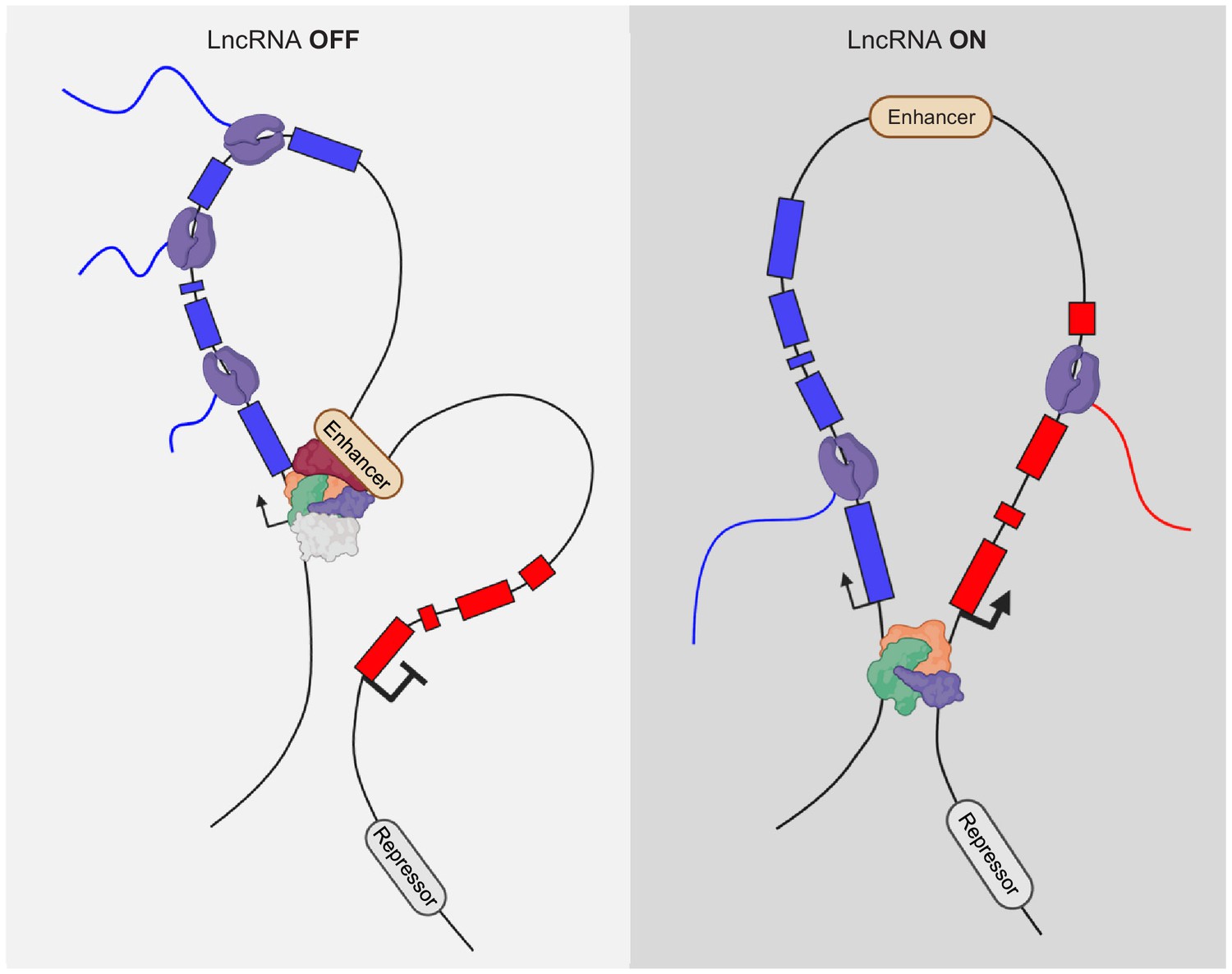
Alteration of genome interactions by lncRNA activity.
DNA:DNA contacts can change upon transcriptional activity of nearby, cis located lncRNA genes.
Another good example is the Chaserr lncRNA locus, which lies 16 kb upstream of the Chd2 protein- coding gene (Rom et al., 2019). Although, knock-down of Chaserr RNA does cause a slight increase in Chd2 expression, additional lines of evidence infer that the transcription of the lncRNA gene is likely the most important function of Chaserr in regulating Chd2 (Figure 4). In addition, the promoter of Chaserr interacts with the Chd2 promoter in chromosome conformation capture analysis. Upon deletion of the Chaserr promoter region, the Chd2 promoter increasingly interacts with other enhancer elements upstream. In contrast, if the gene body of Chaserr is deleted, leaving the promoter intact, these changes in enhancer/Chd2-promoter contacts are not observed. A plausible explanation is that the transcription initiation activity rather than the transcription elongation is important for regulation of Chd2 by Chaserr.
Similarly, transcription initiation is important for the PVT1 lncRNA locus. The Pvt-1 lncRNA was originally discovered as a genomic translocation that causes the activation of the Myc oncogene (Adams and Cory, 1985). Initially, it was suggested that miRNAs embedded in the lncRNA transcript of PVT1 are important for regulation of target genes (Wang et al., 2019). It turns out that PVT1 transcription has an RNA-independent function as well. The PVT1 locus encodes several transcripts with alternative start sites. The activity of its major TSS serves as a boundary element to shield the MYC promoter from over-activation by an enhancer located within the transcriptional unit of PVT1 (Cho et al., 2018; Figure 4). The transcriptional activity is important for this shielding capacity, but not the elongation of the transcription (Figure 4). This does not mean that the miRNAs produced by PVT1 do not serve a function, but it seems the major activity of the PVT1 lncRNA, and its effect on MYC is conveyed by the transcriptional activation of PVT1.
In addition to the Upperhand lncRNA upstream of Hand2 (see above), there are Hand2-regulating lncRNA loci downstream of Hand2. We initially characterized this locus and termed it Handsdown, due its location downstream of Hand2. The Handsdown locus is expressed in the same tissues as Hand2 but is most significantly expressed in the developing heart. We have shown that transcription of Handsdown is important to negatively regulate the expression of Hand2 (Figure 4). Moreover, the HAND2 TF binds two distinct sites around the TSS of Handsdown in the developing E9.5 heart (Laurent et al., 2017). This suggests that HAND2 activates its own suppressor region in a negative feedback loop to control its dosage. However, deletion of the TSS region of Handsdown, including only one of the HAND2 occupied sites, does not result in the expected upregulation of Hand2 (George et al., 2019). Multiple, potential TSS regions are present in at the 5’ region of Handsdown and the deletion of one or the major TSS can lead to the appearance of alternate transcripts (Lavalou et al., 2019). Therefore, it is plausible that the second HAND2 occupied site may be sufficient to instruct the transcription of an alternate Handsdown transcript. Hence, as long as transcriptional activity is present in the Handsdown region, Hand2 can be negatively regulated and its expression level adjusted. The dosage of Hand2 is particularly important as loss of one copy of the Hand2 gene, as well as the gain of an additional copy of the Hand2 gene, causes malformations during development (Tamura et al., 2014). In addition to these lncRNA loci flanking the Hand2 gene, additional putative enhancers are predicted up- and downstream of Hand2, underlining the complex regulome of this important gene in development.
While functions of lncRNAs on the transcript level are becoming increasingly understood, elucidating mechanisms of how such loci, whose function is based on the transcriptional level, exhibit their effect (Table 1) is still in its infancy. While this list is not saturated, the number of lncRNAs that at least partially act by such a mechanism will increase in the future. One very promising model of how they may act are functional microdomains. In such a scenario, these microdomains promote the co-operativity between interacting components such as TFs, co-factors, chromatin regulators, RNA polymerase II, and non-coding RNA, thereby governing basic processes of gene regulation. Such microdomains are favorably formed by super-enhancers that also often generate an RNA, but function on the transcriptional level. Hence, transcriptional activity itself can influence chromatin accessibility, DNA methylation, histone modification, and higher order chromatin structure.
Outlook
A core question for the near future is to define which of the lncRNA loci are functional on the transcript (RNA) level or on the transcriptional (genome) level and which loci may function on both levels.
The widespread use of the CRISPR toolbox does allow for the generation of targeted genomic modifications to dissect the mode-of-action of a lncRNA locus. With CRISPR/Cas9 deletions even in the mega-base range being feasible, the deletion of the entire transcription unit will allow one to determine if a lncRNA locus is functional at all (Barutcu et al., 2018; Kraft et al., 2015). This crude approach eliminates any transcript coming from the locus, also eliminating the possibility that degradation of any residual transcript does cause any effect by, for example, genetic compensation (El-Brolosy et al., 2019). Simply put: if the removal of a complete lncRNA locus does not result in even subtle effects on gene expression, this locus can be marked non-functional, at least in the analyzed biological system. Subsequently, the promoter encompassing the TSS can be removed to eliminate any transcriptional initiation of the transcript. A similar result can be achieved to use the CRISPRi (Ferreira et al., 2018) system to shut down the locus without removing any parts of the genome. It has to be kept in mind, that removing a TSS might trigger the emergence of new transcripts from secondary TSS-like sites in the vicinity (Lavalou et al., 2019). It is therefore important to evaluate this possibility and verify that no ‘novel’ transcripts arise. To interfere with transcriptional elongation and also study effects in regulatory elements within a gene body, the transcription can be terminated using a strong transcriptional stop signal. To allow for efficient targeting using the CRISPR toolbox, a short and powerful pA signal is preferred (Ballarino et al., 2018; Lavalou et al., 2019). In combination with an endogenous CRISPRa (Konermann et al., 2015) system, this meddling with the lncRNA does allow for a detailed assessment of its function. In particular, subsequent removal of parts of a lncRNA locus, for example, whole exons or potential regulatory elements within the transcription unit will allow one to define the functional elements on either the RNA or the locus.
The powerful tool of antisense oligonucleotide (ASO)-assisted knock-down of RNA can now help allow for a detailed assessment of RNA vs transcription-based function. Until now, if a lncRNA transcript was inhibited on the RNA level by antisense oligo methods, siRNA or locked nucleic acid (LNA)-based ASO, it was assumed that the RNA, rather than its transcription was important for the resulting phenotype. Initially, ASOs, which employ the endogenous RNAseH enzyme for target RNA degradation (Grünweller et al., 2003), were the method of choice, as they can target nuclear and cytosolic RNA similarly well. However, it turns out one must be a bit more cautious with this assumption. Several recent publications demonstrate that ASOs that target the 5’ end of an RNA can do this even on nascent RNA that is in the process of being transcribed (Eaton et al., 2020; Lai et al., 2020; Lee and Mendell, 2020). This premature cleavage of RNA leads to the recruitment of XRN2 and employs the torpedo mechanism to evict the POL II transcription machinery prematurely. Hence, a 5’ directed ASO mimics the loss of transcriptional elongation and may lead to confusion about potential lncRNA mechanistic function. To validate an RNA-based mechanism by ASOs, it is preferred to target the 3’ end of the RNA-of-interest. But, more importantly, this mechanism opens the possibility to target lncRNA (or any other locus) whose mechanism requires transcriptional elongation until the endogenous transcriptional termination site, independent of whether the RNA is functional. Carefully designed experiments can increase our understanding of which lncRNA functions are important for gene regulation, which can be beneficial in studying human disease involving dysregulation by such loci. Furthermore, this extends the repertoire of loci that can be targeted for studying the lncRNA genes and their therapeutic use. Now, not only genes that produce a functional RNA can be therapeutically targeted, but also any gene or regulatory locus that generates an RNA and has a gene or genome regulatory function via transcriptional elongation, per se, is amenable to ASO targeting.
References
-
Myc oncogene activation in B and T lymphoid tumoursProc R Soc London Ser B, Biol Sci 226:59–72.https://doi.org/10.1098/rspb.1985.0079
-
Insulators and domains of gene expressionCurrent Opinion in Genetics & Development 37:17–26.https://doi.org/10.1016/j.gde.2015.11.009
-
A TAD boundary is preserved upon deletion of the CTCF-rich firre locusNature Communications 9:1444.https://doi.org/10.1038/s41467-018-03614-0
-
The product of the H19 gene may function as an RNAMolecular and Cellular Biology 10:28–36.https://doi.org/10.1128/MCB.10.1.28
-
Regulatory activities of transposable elements: from conflicts to benefitsNature Reviews Genetics 18:71–86.https://doi.org/10.1038/nrg.2016.139
-
CpG islands and the regulation of transcriptionGenes & Development 25:1010–1022.https://doi.org/10.1101/gad.2037511
-
Termination of transcription by RNA polymerase II: boom!Trends in Genetics 36:664–675.https://doi.org/10.1016/j.tig.2020.05.008
-
GENCODE reference annotation for the human and mouse genomesNucleic Acids Research 47:D766–D773.https://doi.org/10.1093/nar/gky955
-
PolyASite 2.0: a consolidated atlas of polyadenylation sites from 3’ end sequencingNucleic Acids Research 48:D174–D179.https://doi.org/10.1093/nar/gkz918
-
The role of 3D chromatin domains in gene regulation: a multi-facetted view on genome organizationCurrent Opinion in Genetics & Development 61:1–8.https://doi.org/10.1016/j.gde.2020.02.015
-
A GATA-dependent right ventricular enhancer controls dHAND transcription in the developing heartDevelopment 127:5331–5341.
-
Targeted RNA sequencing reveals the deep complexity of the human transcriptomeNature Biotechnology 30:99–104.https://doi.org/10.1038/nbt.2024
-
Unlinking an lncRNA from its associated Cis elementMolecular Cell 62:104–110.https://doi.org/10.1016/j.molcel.2016.02.029
-
The functions and unique features of long intergenic non-coding RNANature Reviews Molecular Cell Biology 19:143–157.https://doi.org/10.1038/nrm.2017.104
-
Principles of genome folding into topologically associating domainsScience Advances 5:eaaw1668.https://doi.org/10.1126/sciadv.aaw1668
-
Old cogs, new tricks: the evolution of gene expression in a chromatin contextNature Reviews Genetics 20:283–297.https://doi.org/10.1038/s41576-019-0105-7
-
The Hand2 gene dosage effect in developmental defects and human congenital disordersCurrent Topics in Developmental Biology 110:8.https://doi.org/10.1016/B978-0-12-405943-6.00003-8
-
The role of transcription in shaping the spatial organization of the genomeNature Reviews Molecular Cell Biology 20:327–337.https://doi.org/10.1038/s41580-019-0114-6
-
PVT1 promotes Cancer progression via MicroRNAsFrontiers in Oncology 9:609.https://doi.org/10.3389/fonc.2019.00609
Article and author information
Author details
Funding
Deutsche Forschungsgemeinschaft (SFB/Transregio TRR267)
- Tamer Ali
Deutsche Forschungsgemeinschaft (Excellence Cluster Cardio-Pulmonary System (Exc147-2))
- Phillip Grote
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We would like to thank the members of the lab for critical comments and discussions. Important input came from Arica Beisaw and Sandra Rogala, and we'd like to thank them for their important contribution to this review. Thanks also to Tracie Pennimpede for critical reading of the manuscript. Parts of the figures were created with BioRender.com.
Copyright
© 2020, Ali and Grote
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,628
- views
-
- 918
- downloads
-
- 225
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 225
- citations for umbrella DOI https://doi.org/10.7554/eLife.60583
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Beyond the RNA-dependent function of LncRNA genes
eLife 9:e60583.
https://doi.org/10.7554/eLife.60583




{kind=link}
{kind=link}
{kind=link}
{kind=link}