The rise and fall of the ancient northern pike master sex-determining gene
- INRAE, LPGP, France
- Department of Ecology and Evolution, University of Lausanne, Switzerland
- Swiss Institute of Bioinformatics, Switzerland
- Department of Biology, Centre for Biomedical Research, University of Victoria, Canada
- Environmental and Fisheries Sciences Division, Northwest Fisheries Science Center, National Marine Fisheries Service, NOAA, United States
- Fisheries Aquatic Science and Technology Laboratory at Alaska Pacific University, United States
- Institut de Biologie Intégrative et des Systèmes (IBIS), Université Laval, Canada
- Fish and Wildlife Conservation Commission, Florida Marine Research Institute, United States
- School of Fisheries and Aquatic Sciences, Auburn University, United States
- Gulf Coast Research Laboratory, School of Ocean Science and Technology, The University of Southern Mississippi, United States
- Laboratoire de Biologie des organismes et écosystèmes aquatiques (BOREA), MNHN, CNRS, IRD, SU, UCN, Laboratoire de Biologie des organismes et écosystèmes aquatiques (BOREA), France
- Unité Mixte de Service Patrimoine Naturelle – Centre d’expertise et de données (UMS 2006 AFB, CNRS, MNHN), Muséum national d’Histoire naturelle, France
- Department of Biological Sciences, Northern Arizona University, United States
- Yellow Sea Fisheries Research Institute, CAFS, Laboratory for Marine Fisheries Science and Food Production Processes, Pilot National Laboratory for Marine Science and Technology (Qingdao), China
- College of Fisheries and Ocean Sciences Fisheries, United States
- Research Institute for Nature and Forest (INBO), Belgium
- Department of Marine Biology and Ecology, Institute of Oceanography, University of Gdansk, Poland
- INRAE, GABI, France
- GeT‐PlaGe, INRAE, Genotoul, France
- Beijing Fisheries Research Institute & Beijing Key Laboratory of Fishery Biotechnology, China
- U.S. Fish and Wildlife Service, United States
- Conservation Biology Division, Northwest Fisheries Science Center, National Marine Fisheries Service, National Oceanic and Atmospheric Administration, United States
- Institut de Génomique Fonctionnelle, IGF, CNRS, INSERM, Univ. Montpellier, France
- INRAE, Sigenae, Genotoul Bioinfo, France
- Tomsk State University, Russian Federation
- Institute of Molecular and Cellular Biology, Siberian Branch of the Russian Academy of Sciences, Novosibirsk State University, Russian Federation
- University of Wuerzburg, Developmental Biochemistry, Biocenter, 97074 Würzburg, Germany; and The Xiphophorus Genetic Stock Center, Texas State University, United States
- Institute of Neuroscience, University of Oregon, United States
Figures
Figure 1
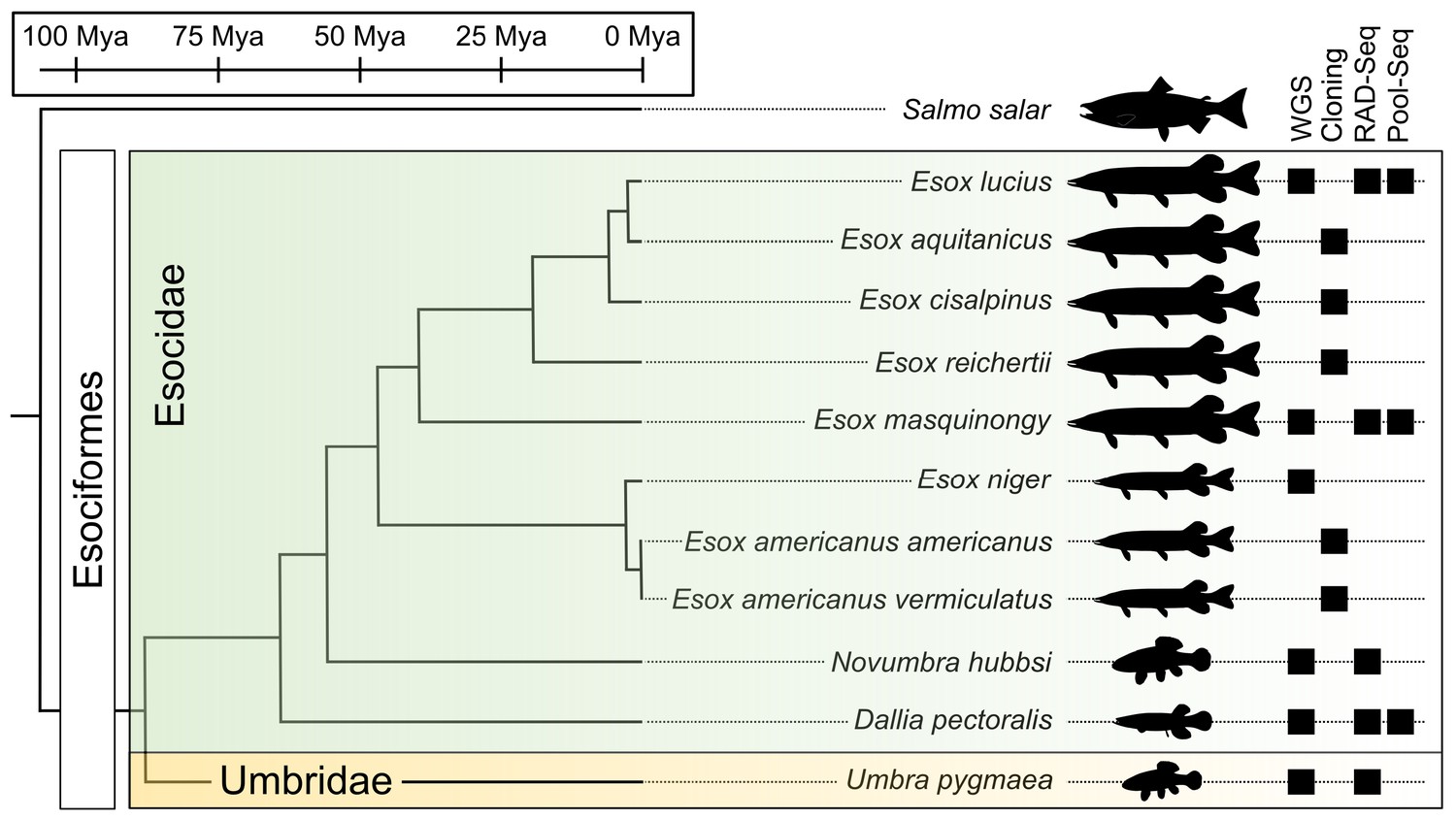
Species phylogeny with estimated divergence time (Campbell et al., 2013; Kumar et al., 2017) and technical approaches for investigating SD systems in Esociformes.
The two families within Esociformes, the Esocidae and the Umbridae, are highlighted with green and yellow background, respectively. Whole-genome sequencing (WGS), homology cloning (Cloning), restriction-site-associated DNA sequencing (RAD-Seq), and pooled-sequencing (Pool-Seq) approaches used in each species are shown by a black square on the right side of the figure. Fish silhouettes were obtained from phylopic.org.
Figure 2

Phylogenetic analysis of amh homologs from the Esociformes revealed an ancient origin of amhby.
(A) Phylogenetic tree of amh coding sequences built with the maximum-likelihood method. Bootstrap values are provided on each node of the tree. The amha ortholog cluster is highlighted with blue background and the amhby ortholog cluster is highlighted with red background. (B) Time-calibrated species phylogeny of the Esociformes (Campbell et al., 2013). The three putative SD transitions are shown by black stars. The presence of pre-duplication amh, as well as amha and amhby along with the amhby sex linkage, is represented by colored dots at the end of each branch. In E. cisalpinus and E. aquitanicus, sex linkage is not significant (NS) due to low sample size. The earliest duplication timing of amh is denoted by a black arrow at the root of the divergence of Esocidae.
Figure 3
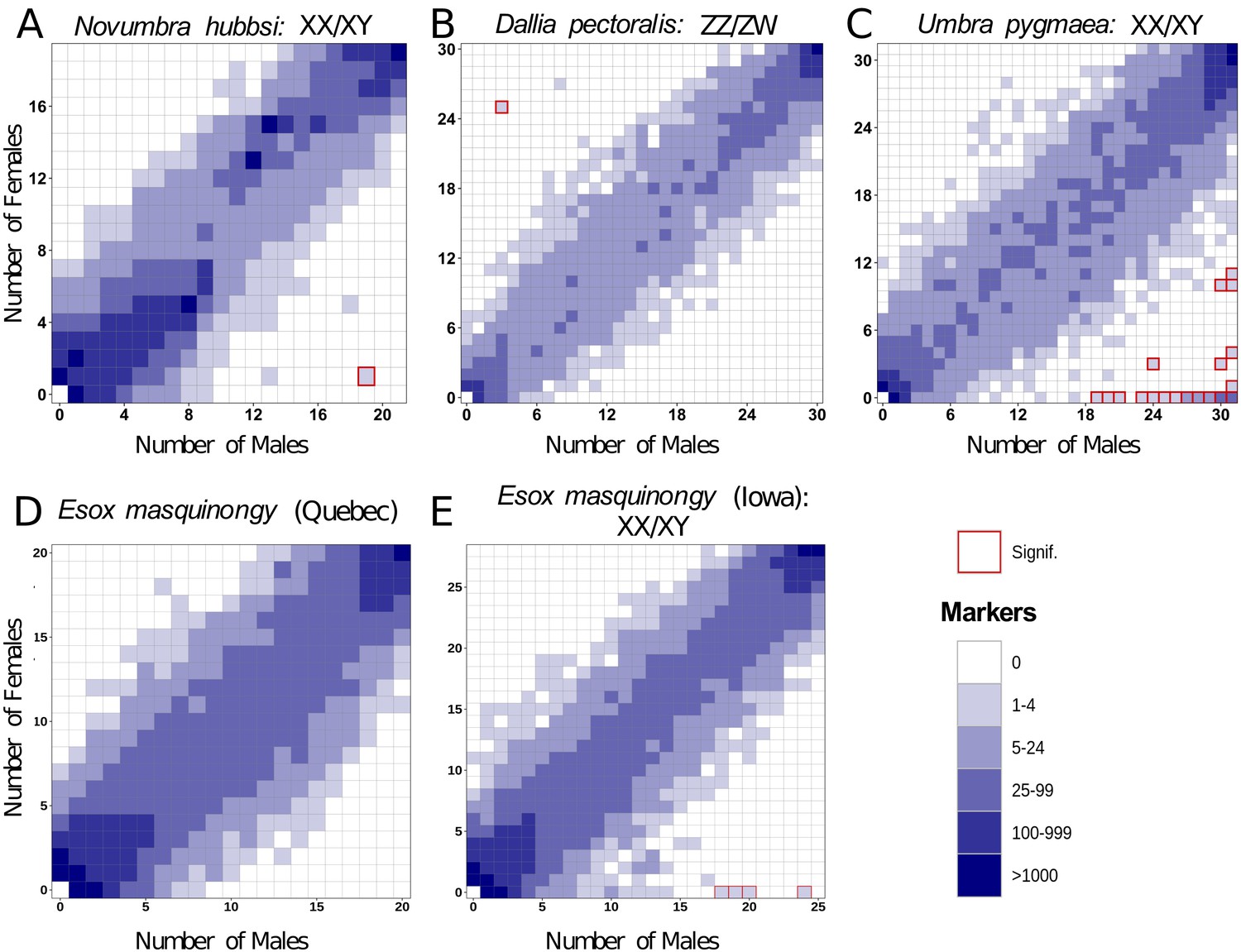
Characterization of sex determination systems in different Esociformes species through RAD-Seq analyses.
Each tile plot shows the distribution of non-polymorphic RAD-Seq markers shared between phenotypic males (horizontal axis) and phenotypic females (vertical axis). The intensity of color for a tile reflects the number of markers present in the respective number of males and females. Tiles that are significantly associated with phenotypic sex (chi-square test, p<0.05 after Bonferroni correction) are highlighted with a red border. (A) In N. hubbsi, two markers were present in 19 of 21 males and in 1 of 19 females, indicating a significant male association and thus an XX/XY SD system with a small sex locus. (B) In D. pectoralis, one marker showed a significant association with females, while no marker showed association with males, indicating a ZZ/ZW SD system. (C) In U. pygmaea, 140 markers showed a significant association with males, while no marker showed association with females, indicating a XX/XY SD system with a large non-recombining sex locus. (D) In the population of E. masquinongy from Quebec (Canada), no marker was associated with either sex. (E) In a population of E. masquinongy from Iowa (USA), five markers were significantly associated with male phenotype, indicating a XX/XY SD system.
-
Figure 3—source code 1
R Script to generate Figure 3.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig3-code1-v2.zip
-
Figure 3—source data 1
Distribution of RADsex markers of E. masquinongy from a Quebec population with a minimal marker depth of 10 reads.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig3-data1-v2.csv
-
Figure 3—source data 2
Distribution of RADsex markers of E. masquinongy from a Iowa population with a minimal marker depth of 10 reads.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig3-data2-v2.csv
-
Figure 3—source data 3
Distribution of RADsex markers of N. hubbsi a minimal marker depth of 10 reads.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig3-data3-v2.csv
-
Figure 3—source data 4
Distribution of RADsex markers of U. pygmaea a minimal marker depth of 10 reads.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig3-data4-v2.csv
-
Figure 3—source data 5
Distribution of RADsex markers of D. pectoralis a minimal marker depth of 10 reads.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig3-data5-v2.csv
Figure 4
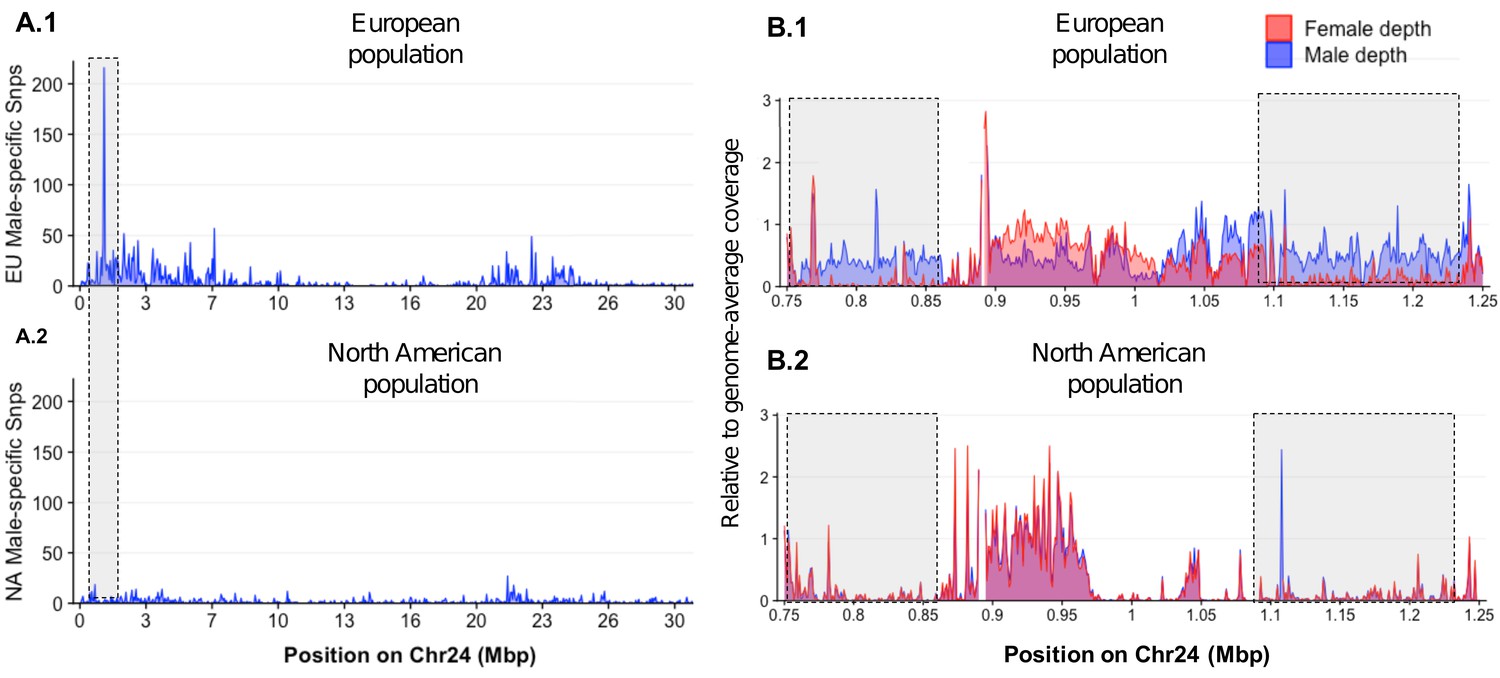
Loss of the ancestral sex locus and master sex determination gene in North American northern pike.
(A) The numbers of male-specific SNPs in 50 kb non-overlapping windows deduced from Pool-Seq data of a European population (A.1) and North American population (A.2) of E. lucius were mapped to the Y chromosome (LG24) of the sequence of E. lucius from a European male genome (SDAW00000000). The greatest number of male-specific SNPs is found in a single window located at the proximal end of LG24 (0.95 Mb–1.0 Mb) that contains 182 male-specific SNPs (highlighted by the gray box) in the European E. lucius population. The same region, however, shows no differentiation between males and females in the North American population. (B) Relative coverage of male and female Pool-Seq reads is indicated by blue and red lines, respectively, in contigs containing amhby in the European population (B.1) and in the North American population (B.2). Only zoomed view on the European population sex-specific region is presented to facilitate the visualization of the differences in coverage between two populations. We searched for Y-specific regions in 1 kb windows. A window is considered Y specific if it is covered by few mapped female reads (<3 reads per kb) and by male reads at a depth close to half of the genome average (relative depth between 0.4 and 0.6). Based on these depth filters, in the European population we identified 70 potential Y-specific 1 kb windows located between 0.76 Mb and 0.86 Mb and 70 Y-specific 1 kb windows located between 1.0 Mb and 1.24 Mb on LG24. This region is not covered by male or female reads in the North American population, indicating the loss of the entire Y-specific region, highlighted by the gray boxes. The region between 0.86 Mb and 1.0 Mb is not sex specific in both population and is likely an assembly artifact.
-
Figure 4—source code 1
R script to generate Figure 4.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig4-code1-v2.zip
-
Figure 4—source data 1
Pool-Seq comparison of sex-specific SNPs in windows of 50 kb between males and female from a European population of E. lucius.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig4-data1-v2.csv
-
Figure 4—source data 2
Pool-Seq comparison of sex-specific coverage in windows of 1 kb between males and female from a European population of E. lucius.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig4-data2-v2.csv
-
Figure 4—source data 3
Pool-Seq comparison of sex-specific SNPs in windows of 50 kb between males and female from a North American population of E. lucius.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig4-data3-v2.csv
-
Figure 4—source data 4
Pool-Seq comparison of sex-specific coverage in windows of 1 kb between males and female from a North American population of E. lucius.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig4-data4-v2.csv
-
Figure 4—source data 5
E. lucius chromosome length file for the R script.
- https://cdn.elifesciences.org/articles/62858/elife-62858-fig4-data5-v2.csv
Figure 5 with 1 supplement
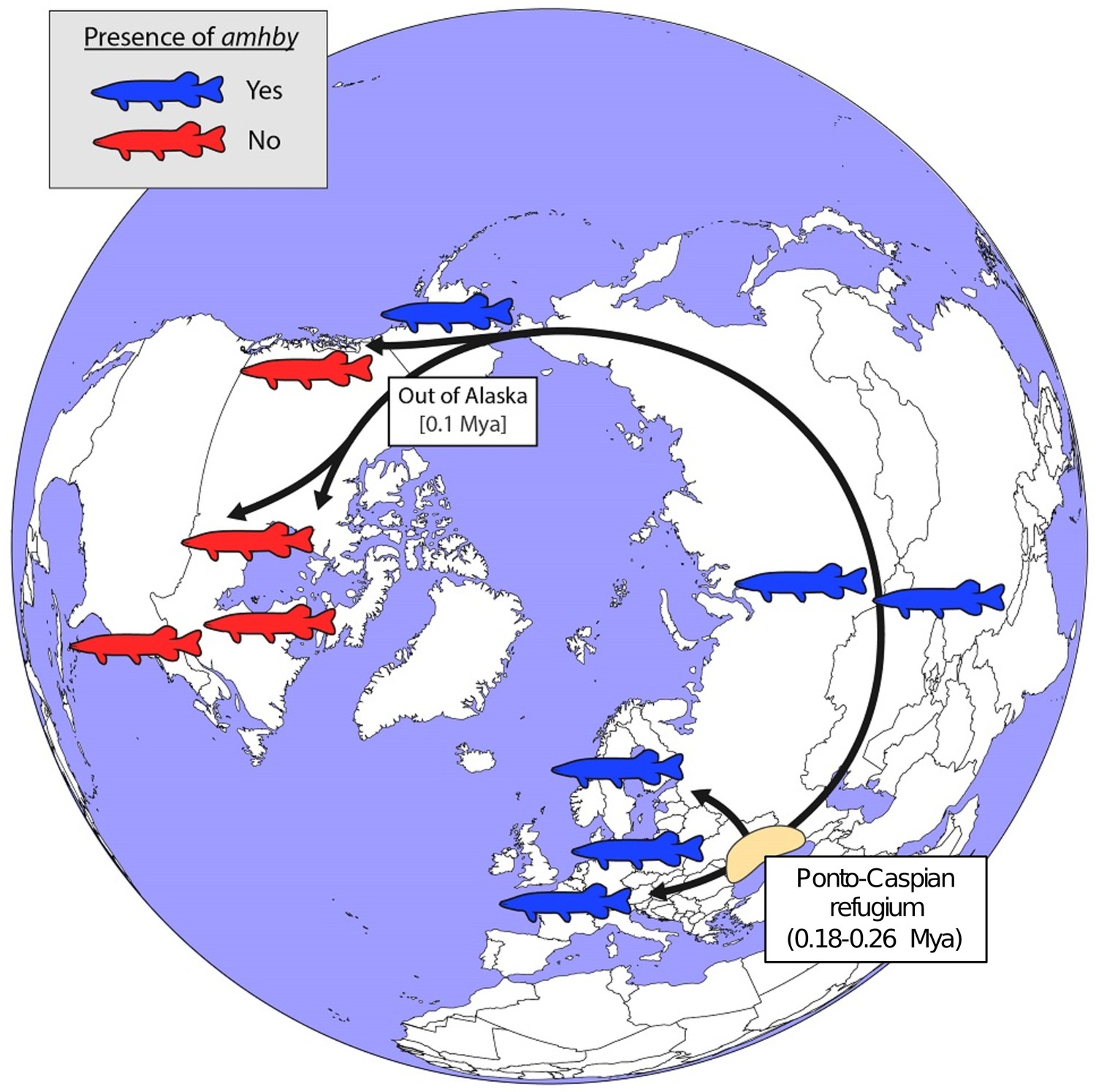
Schematic representation of a hypothesized route of post-glacial E. lucius expansion from a Eurasian refugium ~0.18 to 0.26 Mya.
The presence (blue silhouettes)/absence (red silhouettes) of amhby in different populations, showing that this MSD gene was lost in some North American populations during an out-of-Alaska dispersal around 0.1 Mya. The hypothesized refugium in the Ponto Caspian region is highlighted in beige. An alternative route of post-glacial dispersal is shown in Figure 5—figure supplement 1.
Figure 5—figure supplement 1
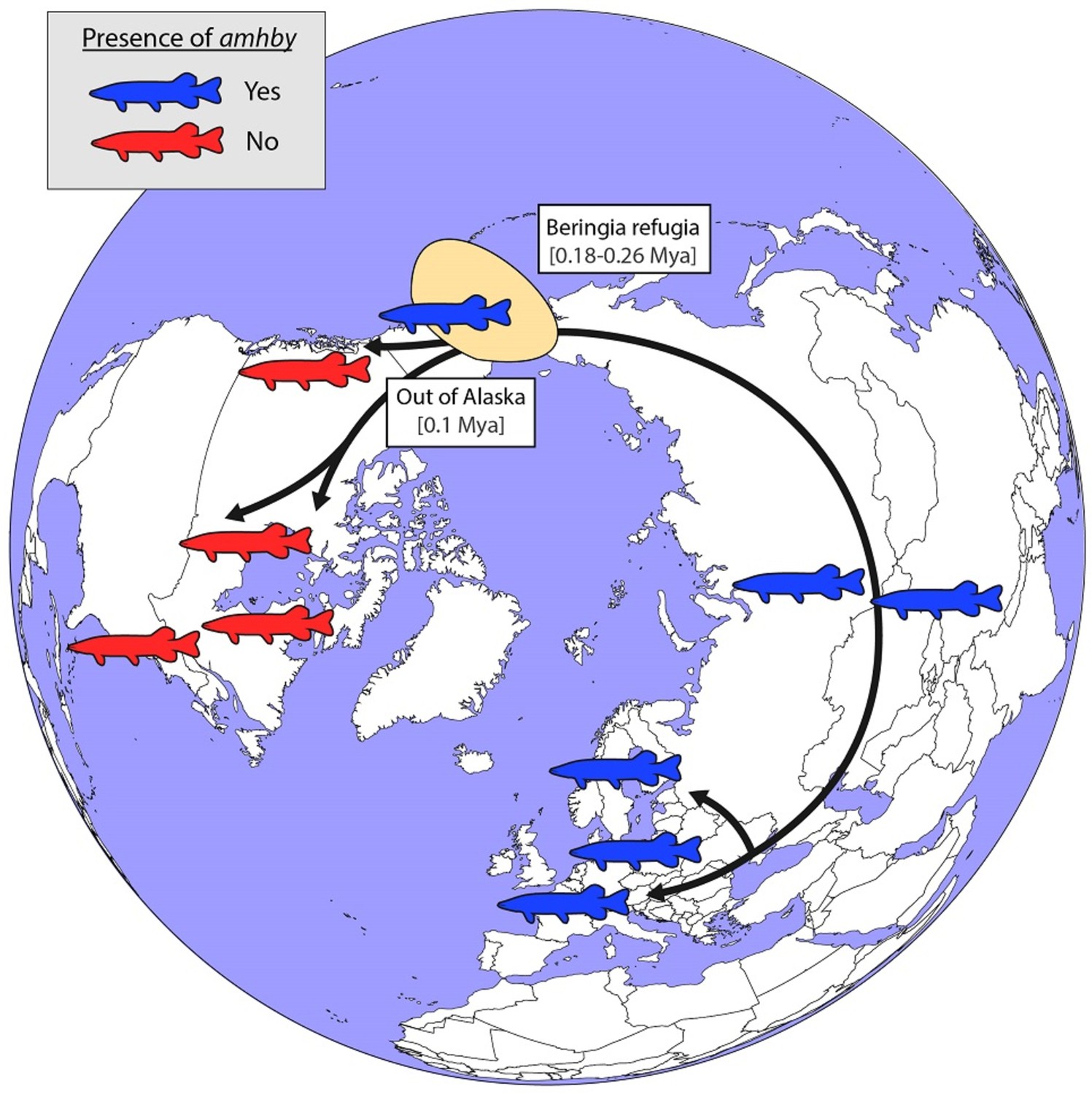
Schematic representation of an alternative hypothesized route of post-glacial E. lucius expansion from a Eurasian refugium ~0.18 to 0.26 Mya.
Appendix 1—figure 1
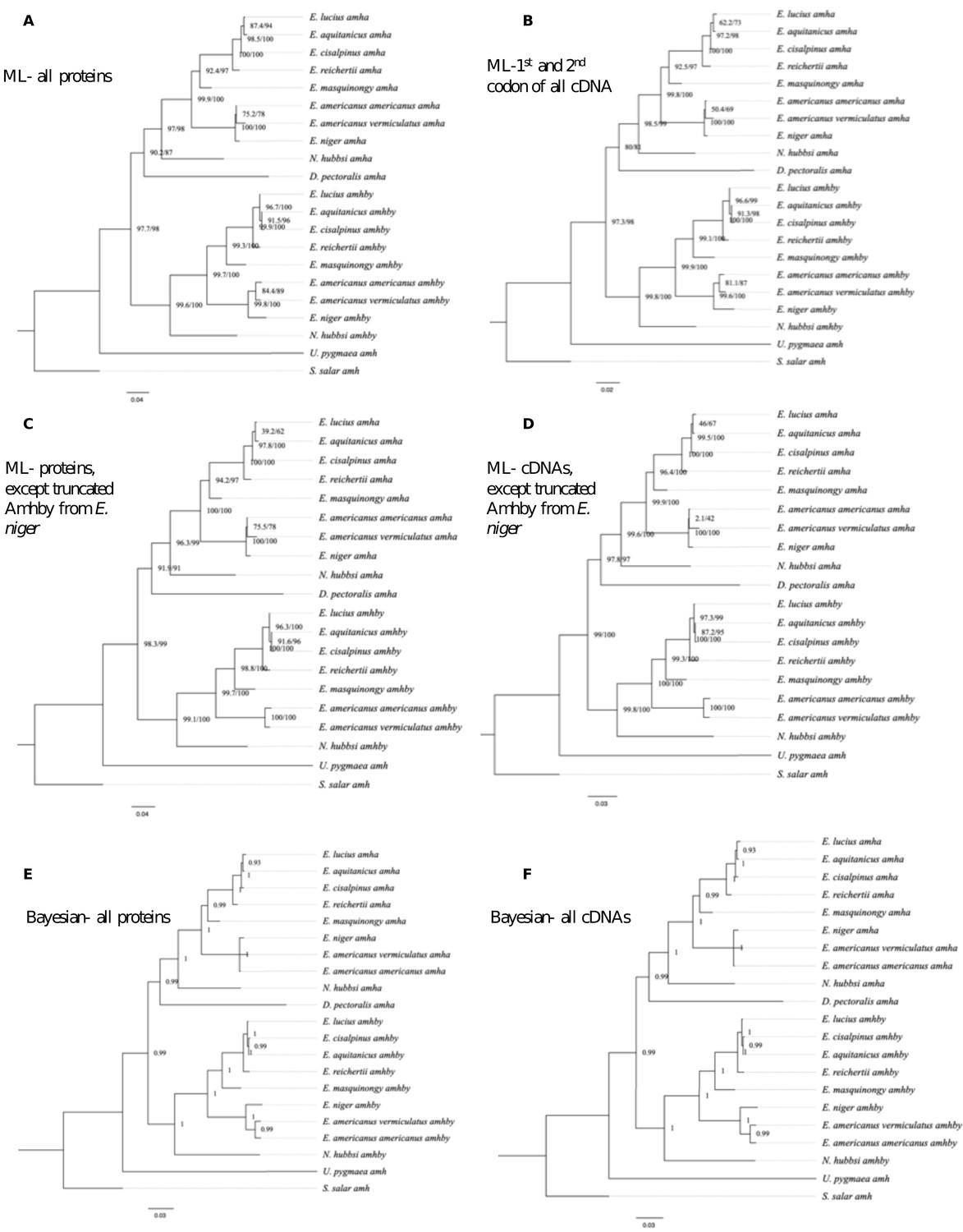
Additional phylogenetic reconstruction of amha and amhby orthologs from the Esociformes with amh sequence from Salmo salar as an outgroup.
(A) Phylogenetic tree built by Maximum likelihood method implemented in IQ-TREE putative protein sequences of all identified amh homologs. (B) Phylogenetic tree built by Maximum likelihood method with only the 1st and 2nd codon of putative coding sequence of all identified amh homologs. (C) Phylogenetic tree built by Maximum likelihood with putative protein sequences of amh homologs without one highly truncated sequence from E. niger. (D) Phylogenetic trees built by Maximum likelihood with putative coding sequences of amh homologs without one highly truncated sequence from E. niger. (E) Phylogenetic tree built by Bayesian method implemented in PhyloBayes with putative protein sequences of all identified amh homologs. (F) Phylogenetic tree built by Bayesian method with putative coding sequences of all identified amh homologs. Bootstrap values are given on each nod of the tree and all trees are rooted with amh sequence from S. salar.
Appendix 1—figure 2
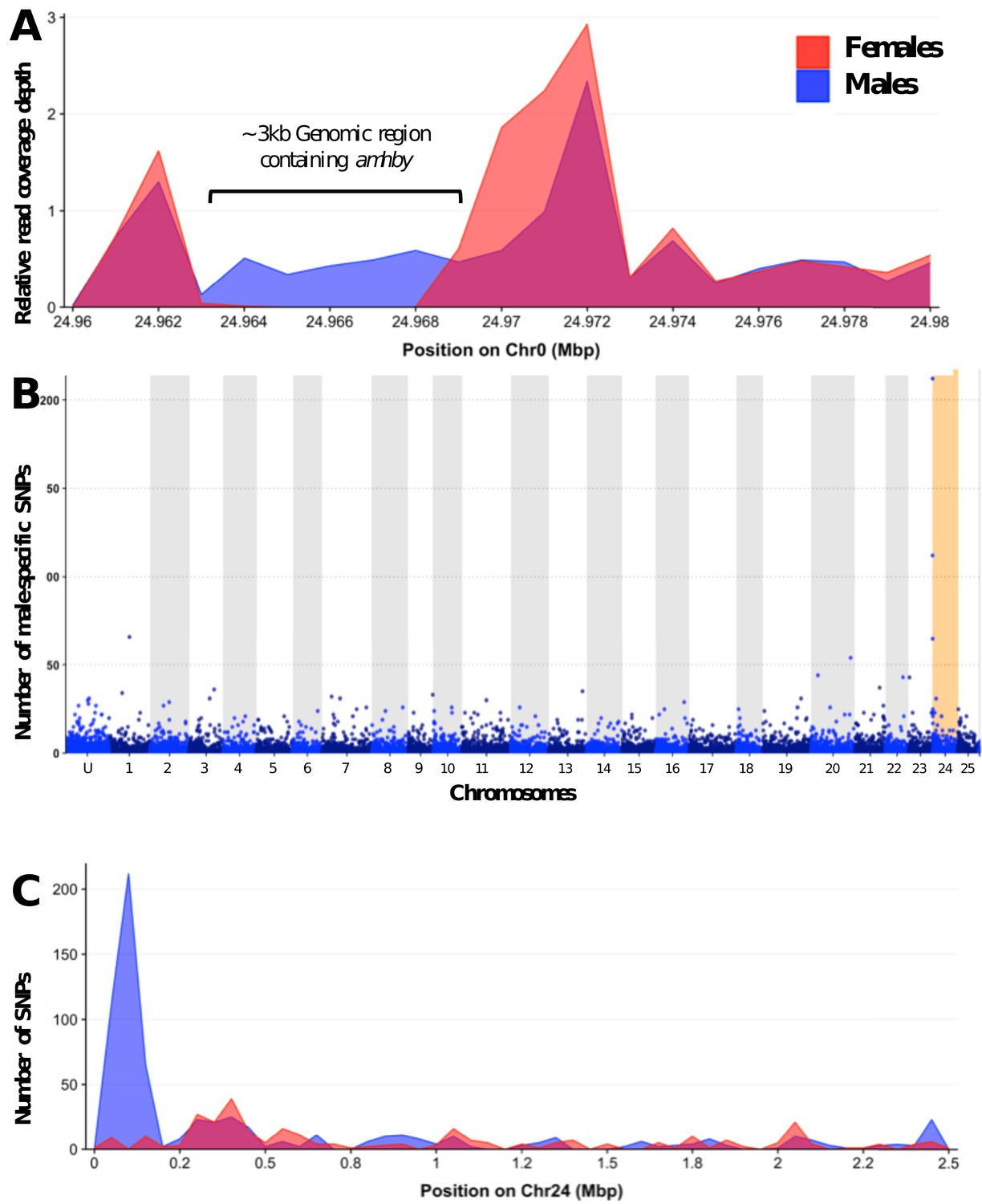
Analysis of sex differentiation in the genome of a male E. masquinongy with Pool-Seq data.
(A) The relative to genome average coverage depth of male and female Pool-Seq reads on the region containing amhby. The male data are represented in blue and female in red. The 3 kb region containing amhby (unplaced_scaffold_RaGOO_chr0: 24,967,172–24,968,938) is covered by virtually no female reads and by male reads at a depth about half of the genome average depth. (B) Number of male-specific SNPs in 50 kb non-overlapping windows in each linkage group in the male genome of E. masquinongy. The window containing the highest number of male-specific SNPs on LG24 is highlighted. (C) Number of male and female-specific SNPs from Pool-Seq in 50 kb non-overlapping windows is plotted along LG. we found the highest number of male-specific SNPs (MSS) in a single window located around 100–150 kb on LG24 that contained 212 MSS and zero female-specific SNPs, while genome average was 1.57 MSS per 50 kb window (3.17 MSS per window when excluding 50 kb windows without MSS).This result indicates that the sex locus in E. masquinongy is homologous to the proximal end of LG24 in E. lucius. Besides LG24, no other linkage group contained a window enriched with MSS. Overall, this Pool-Seq analysis confirms the XX/XY SD system with a low differentiation between the X and Y chromosomes identified with the sex linkage and RAD-Seq analyses, as observed in E. lucius (Pan et al., 2019) and N. hubbsi (see above), and supports the hypothesis that the sex locus of E. masquinongy is similar to that of E. lucius.
Appendix 1—figure 3
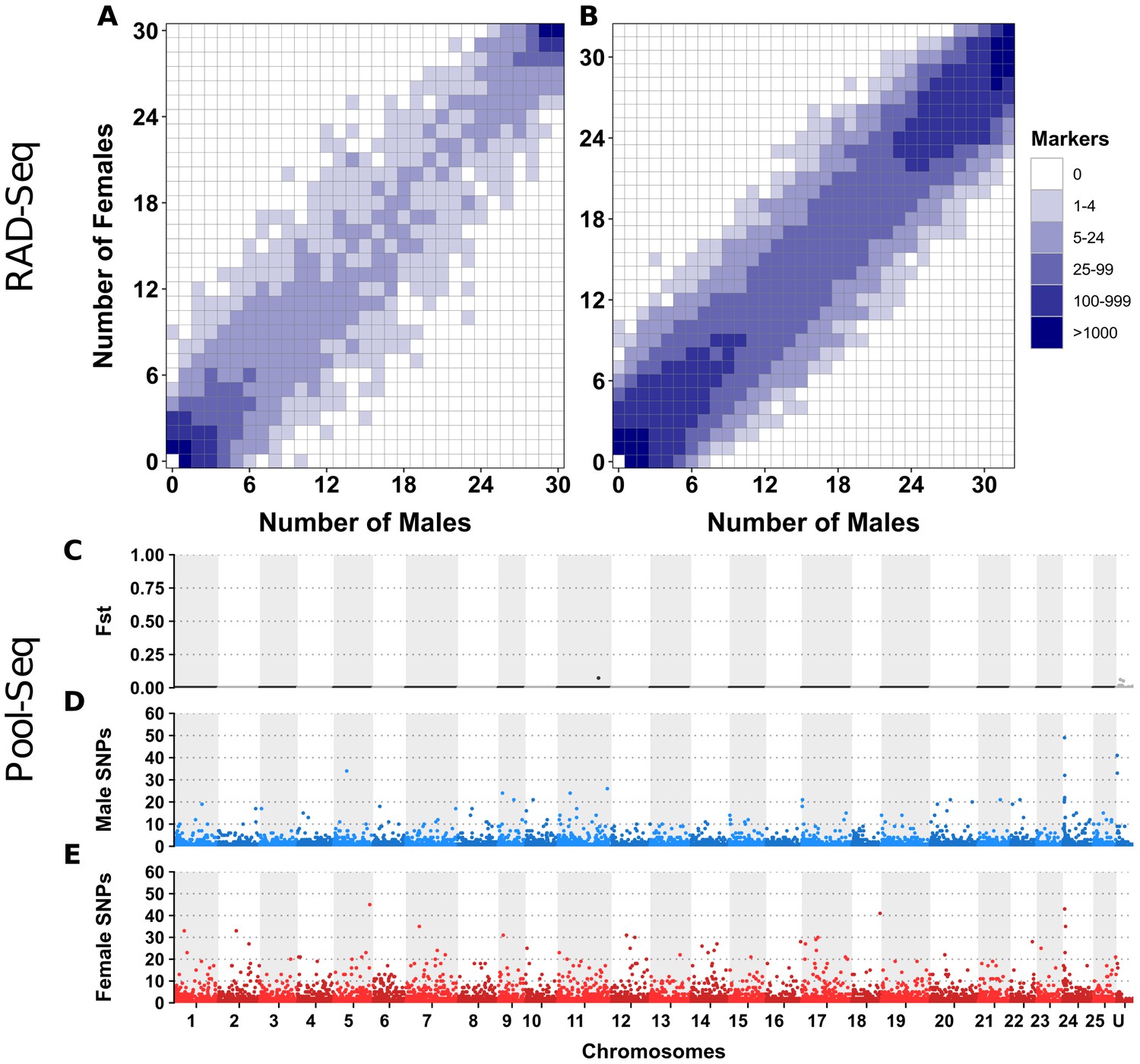
No differentiation between male and female genomes revealed by RAD-Seq and Pool-Seq in some North American populations of E. lucius.
(A-B) Distribution of RADSex markers in males and females of two populations of E. lucius from Canada. The distribution of markers in males and females was computed with RADSex with a minimum depth of 5 to consider a marker as present in an individual for both datasets. In each tile plot, the number of males and number of females are represented on the horizontal and vertical axes respectively, and intensity of color of a tile indicates the number of markers present in the corresponding number of males and females. In both populations, we did not find any sex-linked markers among a total of 8,440,899 and 4,526,552 markers identified in each population, indicating that if there is a new sex locus in these populations its detection escapes the resolution of RAD-Seq (31.8 RAD markers per Mb on average) due to a very low differentiation between the new sex chromosome pair. (C–E) Analysis of male/female differentiation across Canadian E. lucius genome with Pool-Seq reads from 30 males and 30 females mapped to the reference genome (Accession number: GCA_004634155.1). Between sex FST (C), female-specific SNPs (D) and male-specific SNPs (E) are computed for 50 kb non-overlapping windows across the 25 linkage groups (LGs) and unplaced scaffolds. We searched for 50 kb non-overlapping windows enriched with either male or female-specific SNP. Overall, the level of differentiation between males and females was low, and the highest Fst observed across the entire genome is 0.07 located between 0.397 Mb and 0.398 Mb on linkage group 11. Very low number of sex-specific SNPs were found for both male and female pools in this population, especially when comparing to the same analysis performed with data from an European population. In the European population, we found a genome average of 3.5 male-specific and 2.7 female-specific SNPs per 50 kb window, with peak windows containing 625 male-specific SNPs near the LG24 sex locus, and 217 female-specific SNPs at the proximal end of LG5. In comparison, the Canadian pools had roughly similar values of average genome sex-specific SNPs (3.3 male-specific and 2.9 female-specific SNPs per window), but with peak windows of sex-specific SNPs of 49 and 45 for males and females, respectively. Given this low amount of differentiation between the sexes, and that no particular chromosome was enriched with windows containing a high number of sex-specific SNPs, no region stood out to be the candidate region for the sex locus. Besides that, no 1 kb window with sex-specific read depth was found. Overall, no clear signal of a sex locus was observed across the entire genome supporting the hypothesis that the mainland USA and Canadian populations lack a well-differentiated sex determining region.
Appendix 1—figure 4

Sanger sequencing results for E. masquinongy one female and one male carrying two different alleles of amhby and one male carrying only one amhby allele.
The base with a bi-allelic SNP is highlighted with the red dashed-line box.
Appendix 1—figure 5
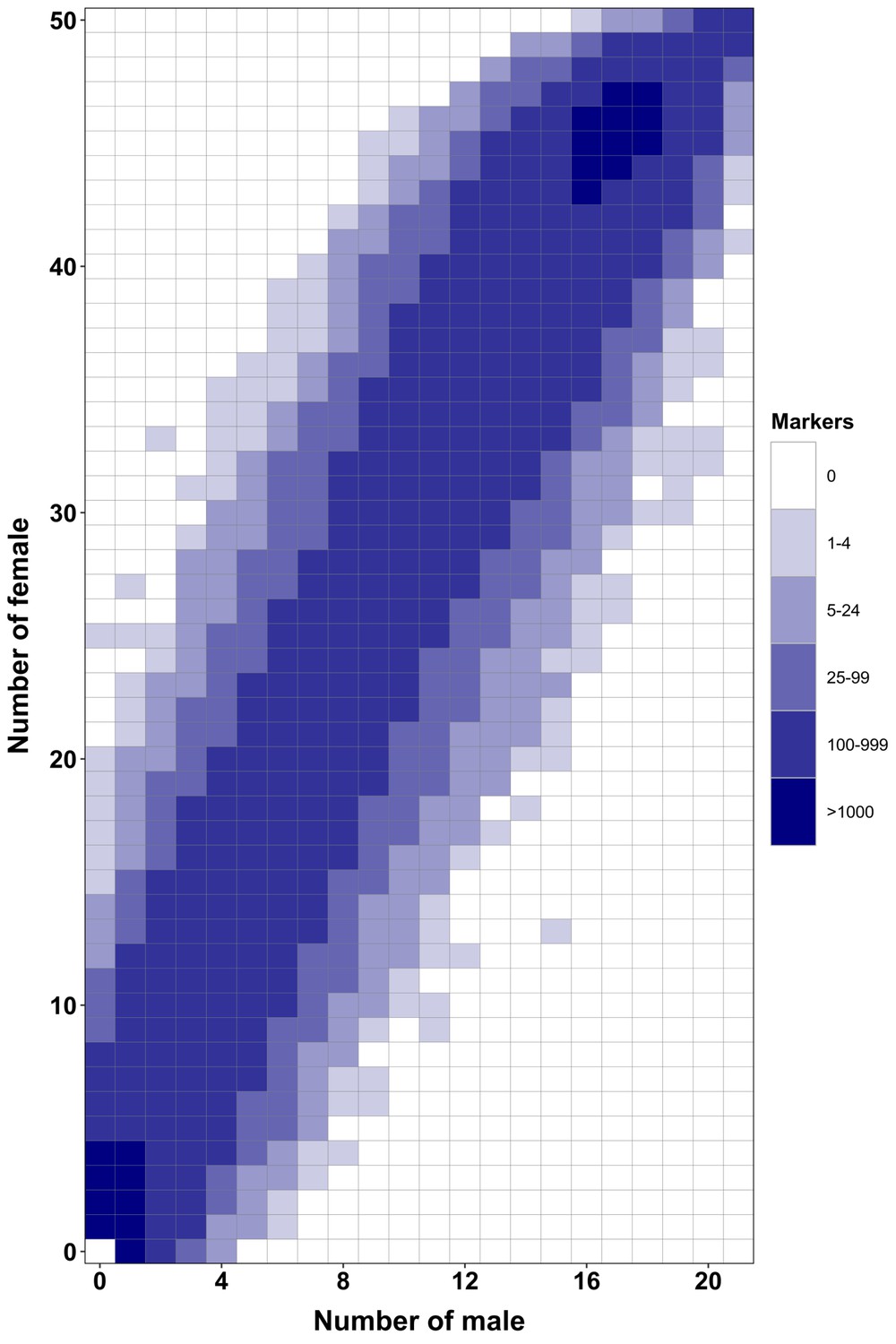
RAD-Seq analysis in a second Esox masquinongy population from Quebec (Canada) supports the lack of sex-specific marker in this population.
Each tile plot shows the distribution of non-polymorphic RAD-Seq markers between phenotypic males (horizontal axis) and phenotypic females (vertical axis). The intensity of color for a tile corresponds to the number of markers present in the respective number of males and females. No tiles for which the association with phenotypic sex is significant (chi-square test, p<0.05 after Bonferroni correction) were detected in this analysis. RAD-Seq reads data and samples information are found under NCBI bioproject PRJNA512459.
Appendix 1—figure 6
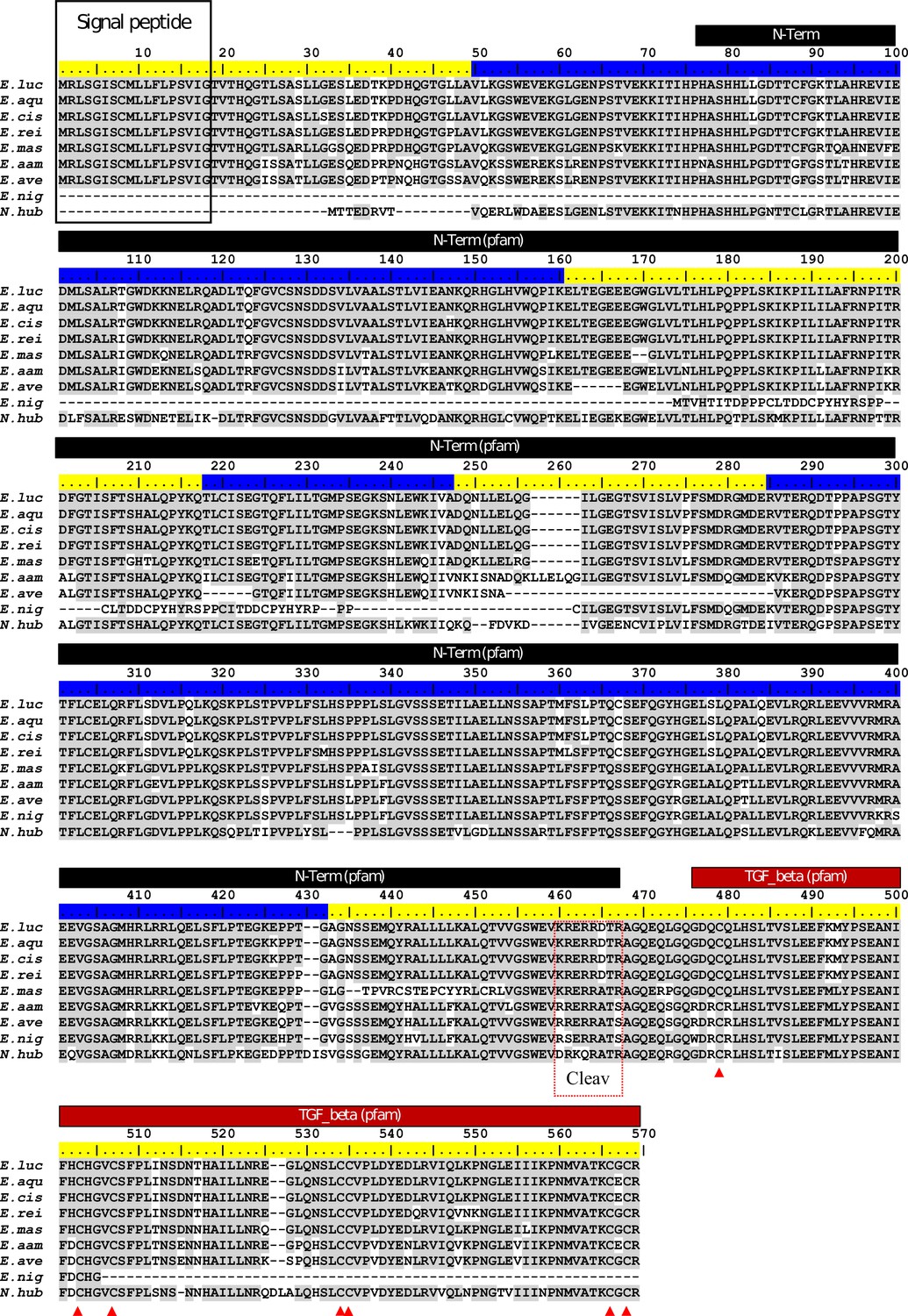
ClustalW alignment of Esocidae Amhby putative protein sequences.
The signal peptide was predicted with SignalP (Nielsen, 2017). No signal peptide was detected in the Amhby sequences of both E. niger and N. hubbsi. N-terminal region (indicated by a black bar) and the Transforming growth factor beta like domain (indicated by a red bar) were predicted with the Motif Scan tool at ExPASy (Gasteiger et al., 2003) with the Pfam motif database (Finn et al., 2014) optimized for local alignments (pfam-fs, Pfam 32.0, September 2018). The seven exons of amhby are shown by the alternating blue and yellow colors on the sequence ruler. The seven conserved cysteines of the TGF-beta domain are indicated by red arrowheads. The region containing the putative Amh cleavage site (Cleav) is boxed in red. E. luc (Esox lucius), E. aqu (E. aquitanicus), E. cis (E. cisalpinus), E. rei (E. reichertii), E. mas (E. masquinongy), E. aam (E. americanus americanus), E. ave (E. americanus vermiculatus), E. nig (E. niger) and N. hub (Novumbra hubbsi).
Appendix 1—figure 7

Hypothesized scenarios of sex determination system transition in North American populations of E. lucius.
In the ancestral population, Linkage group 24 (LG24) was the sex chromosome (Y). Males were the heterogametic sex carrying one Y chromosome with a small (~300 kb) sex-locus region (SLR) containing amhby and one X chromosome without the SLR and amhby, while females carried two X chromosomes. During post-glaciation re-colonization of North America, the Y chromosome with amhby gene was not carried by individuals that made up the small populations that dispersed out of Alaska. These populations could still produce phenotypic males in the absence of a master sex determining gene via environment-induced sex reversal, a well-documented phenomenon among teleosts. In this transitional state, both males and females carry two X sex chromosomes (LG24). The current sex determination mechanism in these North American populations is still unknown: it could rely entirely on environmental sex determination (ESD), random sex determination, or new male (XY) or female (ZW) heterogametic genetic sex determination system with highly undifferentiated sex chromosomes. SLR: sex-locus region. ESD: environmental sex determination. NA: North America. LGN: un-specified Linkage group, that is ancestral autosomes (A) that are not LG24.
Tables
Table 1
Summary of the identification of amhby and its association with sex phenotypes in 11 Esociformes species, including six populations of E. lucius and three populations of Esox masquinongy.
| Species | Sampling location | Geographic region | Males with amhby | Females with amhby | p-value of amhby association with sex |
|---|---|---|---|---|---|
| Esox lucius | France | Western Europe | 161/161 (100%) | 0/60 (0%) | <2.2E-16 |
| Esox lucius | Sweden | Northern Europe | 20/20 (100%) | 0/20 (0%) | <2.2E-16 |
| Esox lucius | Poland | Central Europe | 20/20 (100%) | 0/20 (0%) | <2.2E-16 |
| Esox lucius | Xinjiang, China | Eastern Asia | 5/5 (100%) | 0/5 (0%) | 0.0114 |
| Esox lucius | Continental USA and Canada | North America | 0/88 (0%) | 0/74 (0%) | amhby absent |
| Esox lucius | Alaska, USA | North America | 17/19 (89%) | 0/19 (0%) | 1.79E-07 |
| Esox lucius | Siberia, Russia | Northern Asia | 9/9 (100%) | 0/8 (0%) | 0.0003 |
| Esox aquitanicus | France | Western Europe | 1/1 (100%) | 0/2 (0%) | 0.665 |
| Esox cisalpinus | Italy | Western Europe | 2/2 (100%) | 0/2 (0%) | 0.317 |
| Esox reichertii | China | Eastern Asia | 11/11 (100%) | 0/10 (0%) | 3.40E-05 |
| Esox masquinongy | Iowa, USA | North America | 18/27 (66.7%) | 4/18 (22%) | 2.01E-08 |
| Esox masquinongy | Quebec, Canada | North America | 9/20 (45%) | 4/20 (20%) | 0.18 |
| Esox masquinongy | Wisconsin, USA | North America | 57/61 (93%) | 22/61 (36%) | 1.17E-10 |
| Esox americanus (americanus) | Mississippi, USA | North America | 6/6 (100%) | 0/4 (0%) | 0.0123 |
| Esox niger | Quebec, Canada | North America | 5/5 (100%) | 0/8 (0%) | 0.0025 |
| Novumbra hubbsi | Washington, USA | North America | 21/23 (91%) | 0/22 (0%) | 5.28E-09 |
| Dallia pectoralis | Alaska, USA | North America | 0/30 (0%) | 0/30 (0%) | amhby absent |
| Umbra pygmaea | Belgium | Western Europe | 0/31 (0%) | 0/31 (0%) | amhby absent |
Additional files
-
Source code 1
R script to generate Appendix 1—figure 3.
- https://cdn.elifesciences.org/articles/62858/elife-62858-code1-v2.zip
-
Source data 1
Poolseq comparison of sex-specific coverage in windows of 1 kb between males and female from a North American population of E. lucius.
- https://cdn.elifesciences.org/articles/62858/elife-62858-data1-v2.csv
-
Source data 2
Distribution of RADsex markers of a Canadian population of E. lucius with a minimal marker depth of 10 reads.
- https://cdn.elifesciences.org/articles/62858/elife-62858-data2-v2.csv
-
Source data 3
Distribution of RADsex markers of a second Canadian population of E. lucius with a minimal marker depth of 10 reads.
- https://cdn.elifesciences.org/articles/62858/elife-62858-data3-v2.csv
-
Source data 4
Poolseq comparison of sex-specific SNPs in windows of 50 kb between males and female from a Canadian population of E. lucius.
- https://cdn.elifesciences.org/articles/62858/elife-62858-data4-v2.csv
-
Source data 5
Poolseq comparison of sex-specific SNPs in windows of 50 kb between males and female from an Iowa population of E. masquinongy.
- https://cdn.elifesciences.org/articles/62858/elife-62858-data5-v2.csv
-
Source data 6
Poolseq comparison of sex-specific coverage in windows of 1 kb between males and female from an Iowa population of E. masquinongy.
- https://cdn.elifesciences.org/articles/62858/elife-62858-data6-v2.csv
-
Source data 7
E. masquinongy chromosome length file for the R script.
- https://cdn.elifesciences.org/articles/62858/elife-62858-data7-v2.csv
-
Supplementary file 1
Number of heterozygous sites on the amh region from Pool-seq reads (E. lucius and D. pectoralis) and whole-genome sequencing reads (U. pygmaea).
To verify that the two copies of amh were not collapsed into a single sequence during assembly, we computed the total number of apparent heterozygous sites on the amh region in population genomic data with the expectation that the presence of two divergent gene copies should result in high apparent heterozygosity when remapped on the single copy assembled in the genome. In total, 106 variants were observed in the pool of E. lucius males on the amha region located between 12,906,561 bp and 12,909,640 bp on LG08 of E. lucius (GCA_004634155.1: CM015581.1), resulting from the mapping of reads originating from both amha and amhby, while only 12 variants (true allelic variations) were observed in the same region when mapping reads from the female pool originating only from amha. With male Pool-Seq reads from D. pectoralis, we observed only four variant sites on the ~3 kb amh region located between 23,447 bp and 25,910 bp on the flattened_line_2941 contig and zero variant sites from the female Pool-Seq reads. Compared to the ‘control’ of E. lucius Pool-Seq reads where one amh gene from female pool result in 12 variant sites and two amh gene from the male pool result in 106 variant sites, the low number of variant sites in both male and female pool of D. pectoralis support that only one amh gene is present in the genome of D. pectoralis regardless of the phenotypic sex of the individuals. Although sex-specific Pool-Seq reads were not available for U. pygmaea, we performed the same analysis with reads from the single male individual used to assemble the genome. No variant was observed in the ~3 kb region containing amh located between 760,763 bp and 757,962 bp on contig 633485 of our draft genome of U. pygmaea, supporting that only one amh gene is also present in this species.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp1-v2.xlsx
-
Supplementary file 2
Estimated number of SbfI cutting sites and RAD-Seq marker frequency estimated for E. lucius, E. masquinongy, N. hubbsi, D. pectoralis, and U. pygmaea based on the size of draft genome assembly.
To help estimate the size of the sex locus from sex-specific RAD markers, we determined the number of potential SbfI cleavage sites based on our draft genome assemblies for each species. For E. lucius (Canadian population), E. masquinongy (Iowa population), N. hubbsi, and D. pectoralis, we predicted the number of RAD-Seq cleavage sites present in each genome by counting the number of unambiguous matches for sequence of SbfI (CCTGCAGG), the restriction enzyme used in RAD-Seq library preparation (Herrera et al., 2015). On average, we found 31.8 RAD markers per Mb in E. lucius, 38.4 in E. masquinongy, 41.6 in N. hubbsi, 30.8 in D. pectoralis, and 26.2 in U. pygmaea. Because we do not see large species differences (26–40 RAD markers/Mb) this suggests that, apart from potential local variations of the RAD markers density in sex loci, our RAD-Seq comparative analysis could to some extent be used to compare sex locus size within species on the basis of this number of sex-specific RAD markers. We are aware of the limitation that using the number of sex-specific marker usually lead to an overestimation of the size of the sex locus. For all of our species with the exception of U. pygmaea, we identified very few sex-specific markers, indicating very small sex locus. This simple calculation is only intended to helps provide a rough approximation of the size of the sex locus.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp2-v2.xlsx
-
Supplementary file 3
Assemblathon and BUSCOs metrics for the new genome assembly with additional Nanopore reads of a genetic European male of E. lucius.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp3-v2.xlsx
-
Supplementary file 4
dN/dS ratio between the amh paralogs in different Esociformes and amh of U. pygmaea.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp4-v2.xlsx
-
Supplementary file 5
Log-likelihood of different selection models tested on amha and amhby orthologs of the Esociformes.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp5-v2.xlsx
-
Supplementary file 6
Information on the different Esociformes species, sample collectors, sexing method, and experiments performed in this study.
*Samples from (Ouellet-Cauchon et al., 2014). **Sex was recorded in this E. masquinongy population based on the urogenital pores morphology. ***Sex was recorded in N. hubbsi based on the specific coloration of males during the breeding season. NR = phenotypes not recorded. NA = not applicable (sex phenotypes not recorded). Museum collection numbers are as follows: MNHN 2014–2719, MNHN 2014–2720, MNHN 2014–2721, MNHN 2014–2722, and MNHN 2014–2723 for E. cisalpinus and MNHN 2013–1246, MNHN 2013–1245, and MNHN 2013–838 for E. aquitanicus.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp6-v2.xlsx
-
Supplementary file 7
Sequencing information for the Pool-Seq and whole-genome sequencing (WGS) performed in this study.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp7-v2.xlsx
-
Supplementary file 8
Total number of reads and markers and range of markers among individuals for each RAD-Seq dataset.
The number of markers retained correspond to the number of markers present with depth higher than min. depth in at least one individual.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp8-v2.xlsx
-
Supplementary file 9
Primers used in this study to amplify amha and amhby sequences from the Esociformes.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp9-v2.xlsx
-
Supplementary file 10
Assemblathon and BUSCOs metrics for draft genome assembly for the Esociformes species.
- https://cdn.elifesciences.org/articles/62858/elife-62858-supp10-v2.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/62858/elife-62858-transrepform-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The rise and fall of the ancient northern pike master sex-determining gene
eLife 10:e62858.
https://doi.org/10.7554/eLife.62858




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}