Patterns of within-host genetic diversity in SARS-CoV-2
- Wellcome Sanger Institute, United Kingdom
- European Bioinformatics Institute, United Kingdom
- Department of Medicine, University of Cambridge, United Kingdom
- Department of Pathology, University of Cambridge, United Kingdom
- Cambridge Institute of Therapeutic Immunology and Infectious Disease, University of Cambridge, United Kingdom
- Public Health England, United Kingdom
- Nuffield Department of Medicine, University of Oxford, United Kingdom
Figures
Figure 1 with 4 supplements
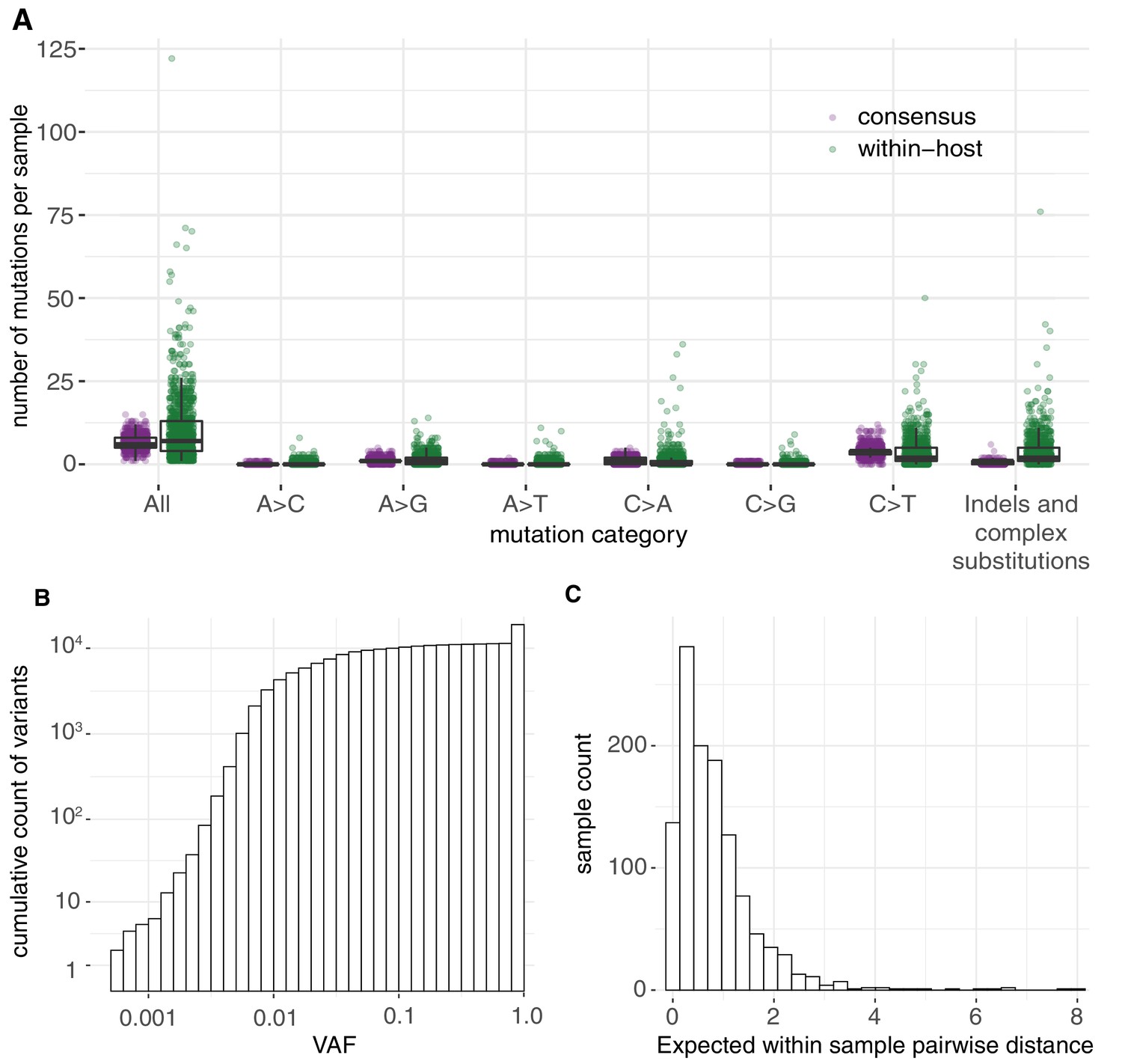
Allele frequencies and mutation burden.
(A) Number of variants per sample (y-axis) for each mutation type assuming the reference genome as the ancestral allele. (B) A cumulative histogram of the VAFs of all mutation calls. Note that variants shared across samples are counted multiple times and that the 7672 consensus variants correspond to 1079 different changes in 1063 different sites. (C) Histogram of the expected number of mutations separating two randomly sampled genomes for each sample (Materials and methods).
Figure 1—figure supplement 1
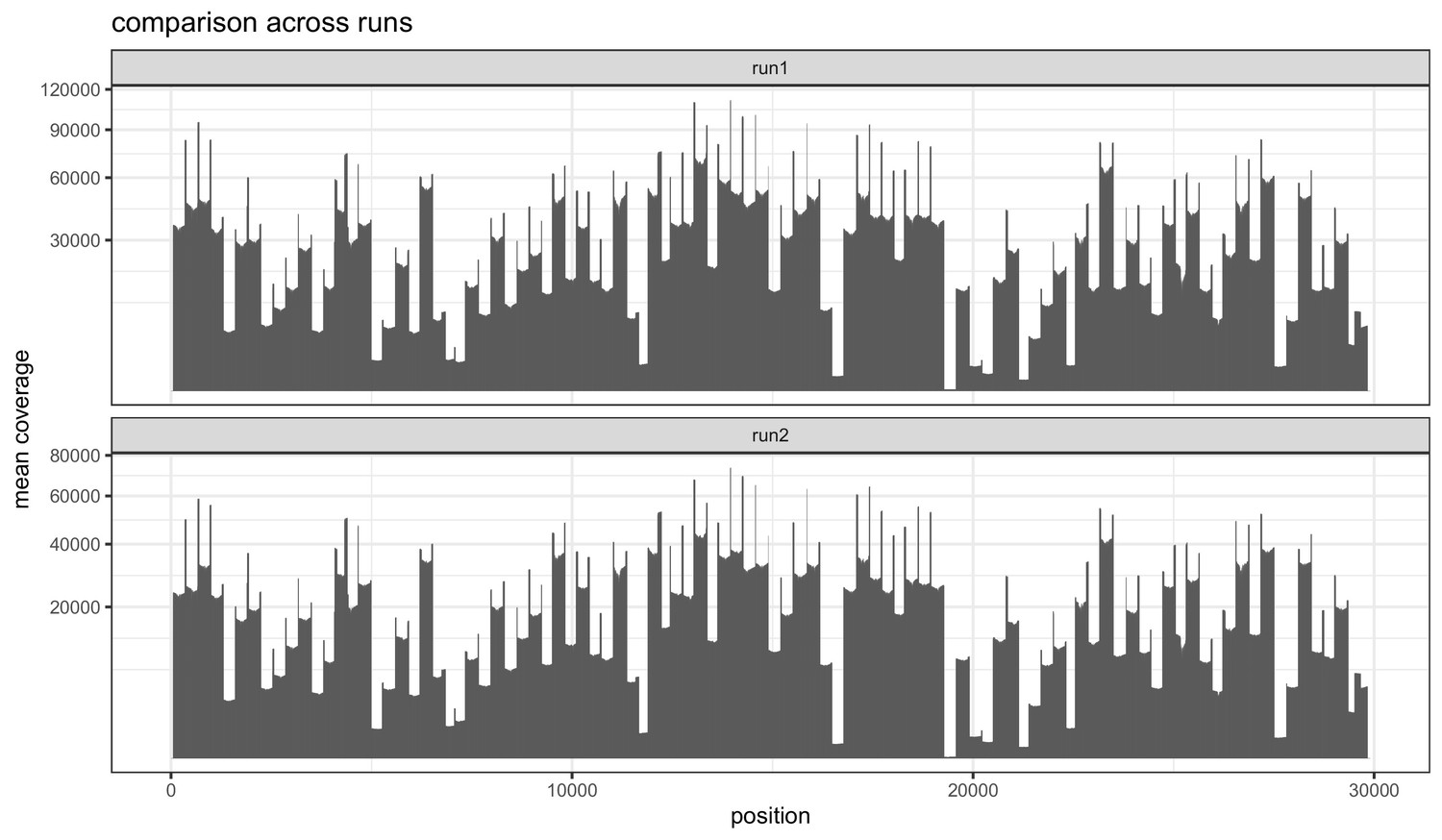
Barplots indicating the mean sequencing depth across the SARS-CoV-2 reference genome for the two replicate runs of the 1181 samples.
Figure 1—figure supplement 2

Dot plots indicating the concordance between variant allele frequency estimates across sequencing replicates in four samples.
Figure 1—figure supplement 3
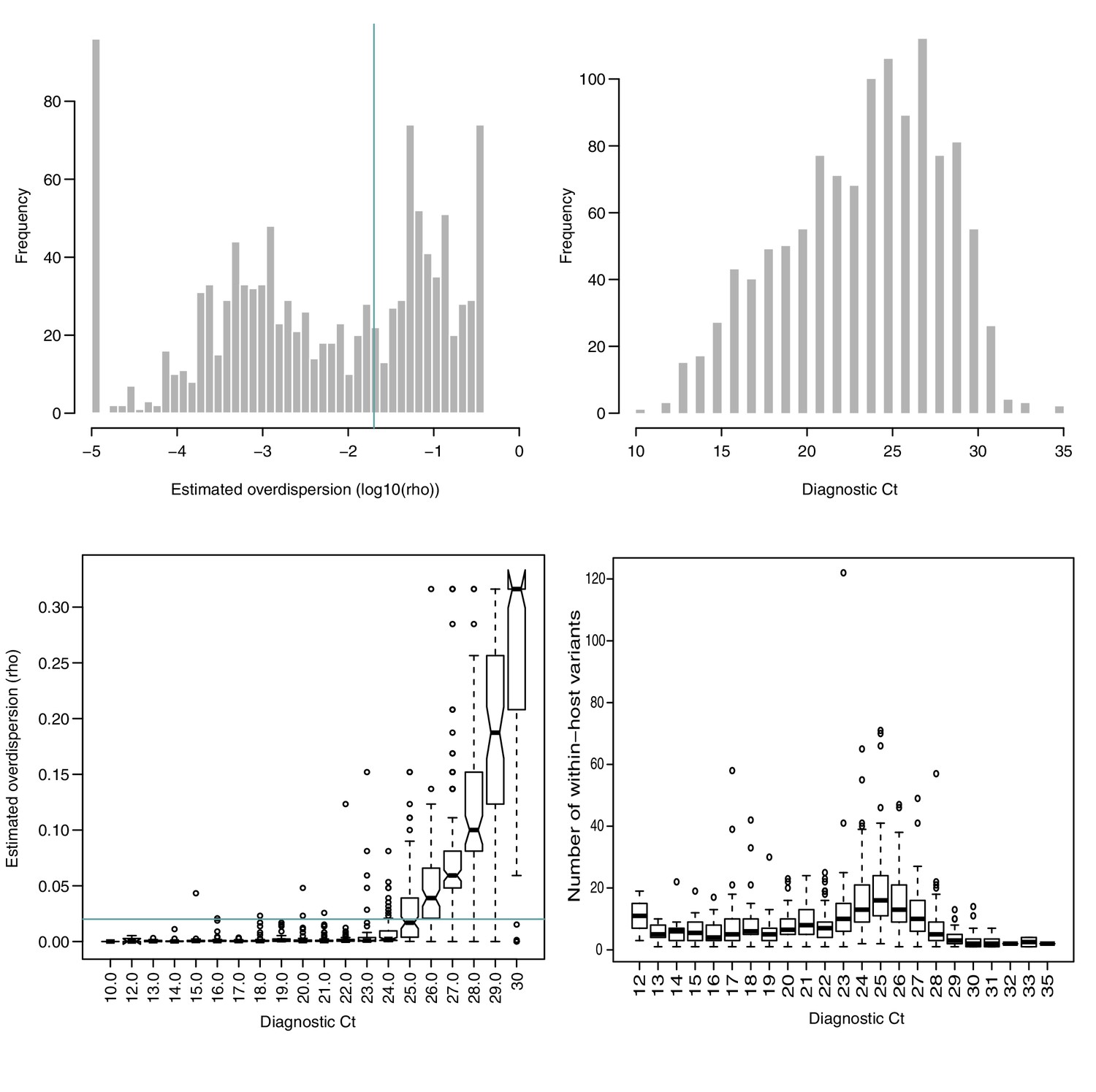
Estimated overdispersion of variant frequencies and the distribution of sample Ct values.
(A) Histogram of estimated log10(ρ) values. ρ represents the dispersion parameter from the Beta-binomial model of variant frequencies. Green line represents in (A) and (C), as a suggested acceptable level of discordance between replicates. 58% of all samples in the cohort had . (B) Histogram of Ct values in the cohort. (C) Estimated ρ value as a function of Ct. (D) Number of within-host variants as a function of Ct.
Figure 1—figure supplement 4
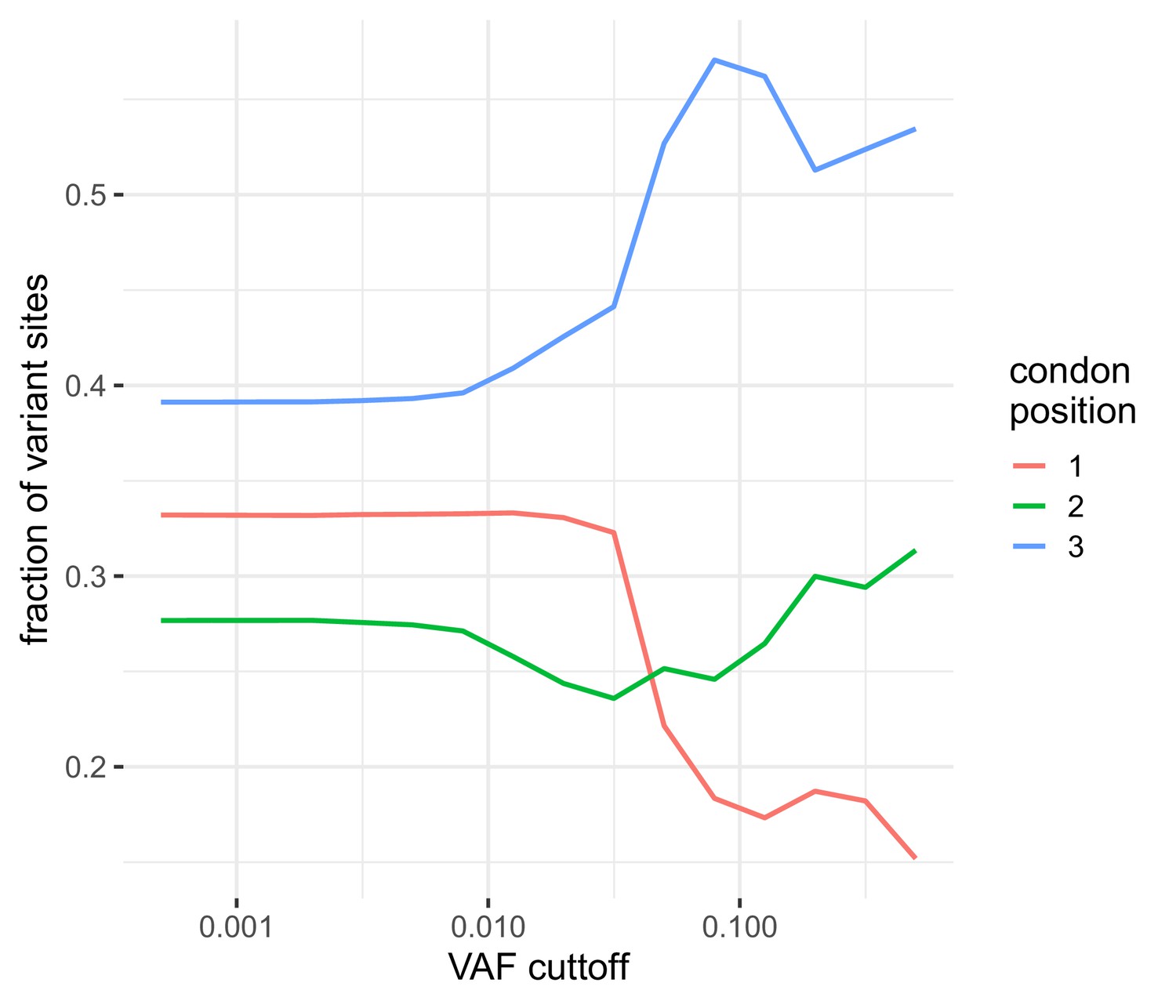
The distribution of the number of variable sites in coding regions among different coding positions.
Variable sites are dominated by those seen at the third codon position similar to that observed in Dyrdak et al., 2019. At higher frequencies, the reduction in the total number of variants leads to increased variabilitly.
Figure 2 with 1 supplement

Mutational spectra.
(A, C) Mutational spectra (without sequence context) of consensus (A) and within-host (C) variants, as mapped to the reference strand and normalised for the composition of nucleotides in the reference genome (MN908947.3). Rates reflect the fraction of the total number of mutations observed. Asymmetries suggest different mutation rates in the plus and minus strands. Error bars depict Poisson 95% confidence intervals. (B, D) Mutational spectra in a 96-trinucleotide context of consensus (B) and within-host (D) variants, as in Alexandrov et al., 2013. Mutations are represented as mapped to the pyrimidine base, depicted above the horizontal line if the pyrimidine base is in the reference (plus) strand and below it if the pyrimidine base is in the minus strand. Within-host variants observed across more than one sample can represent a single ancestral event or multiple independent events. To prevent highly recurrent events from distorting the spectrum, within-host variants observed across multiple samples were counted a maximum of five times in (C, D). (E) A diagram illustrating how asymmetrical mutation rates of C>U and G>A could be driven by viral sequences spending a longer time as plus strand molecules.
Figure 2—figure supplement 1

The mutational spectra in a 96-trinucleotide context of recurrent within-host variants.
Figure 3 with 2 supplements
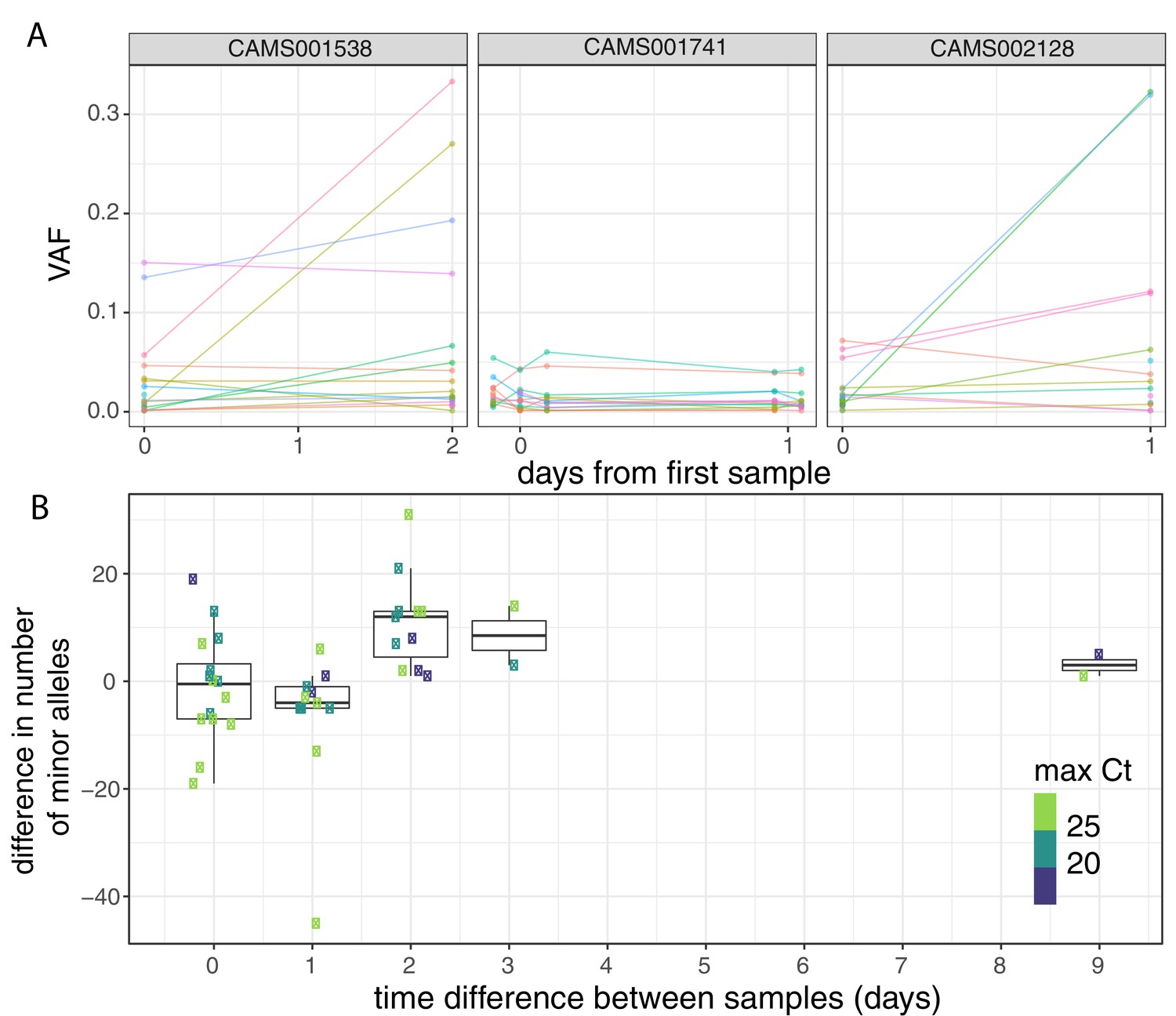
Longitudinal differences in within-host variant frequencies.
(A) Frequencies of within-host variants for three selected hosts where multiple samples were taken over consecutive days. Samples taken on the same day have been offset by a small distance. Plots for all hosts with multiple samples are given in Figure 3—figure supplement 1. (B) The difference in the number of within-host variants between pairwise combinations of samples taken from the same host. The order for samples taken on the same day was randomised, and the colour of the point indicates the maximum of the two Ct values for the corresponding samples.
Figure 3—figure supplement 1
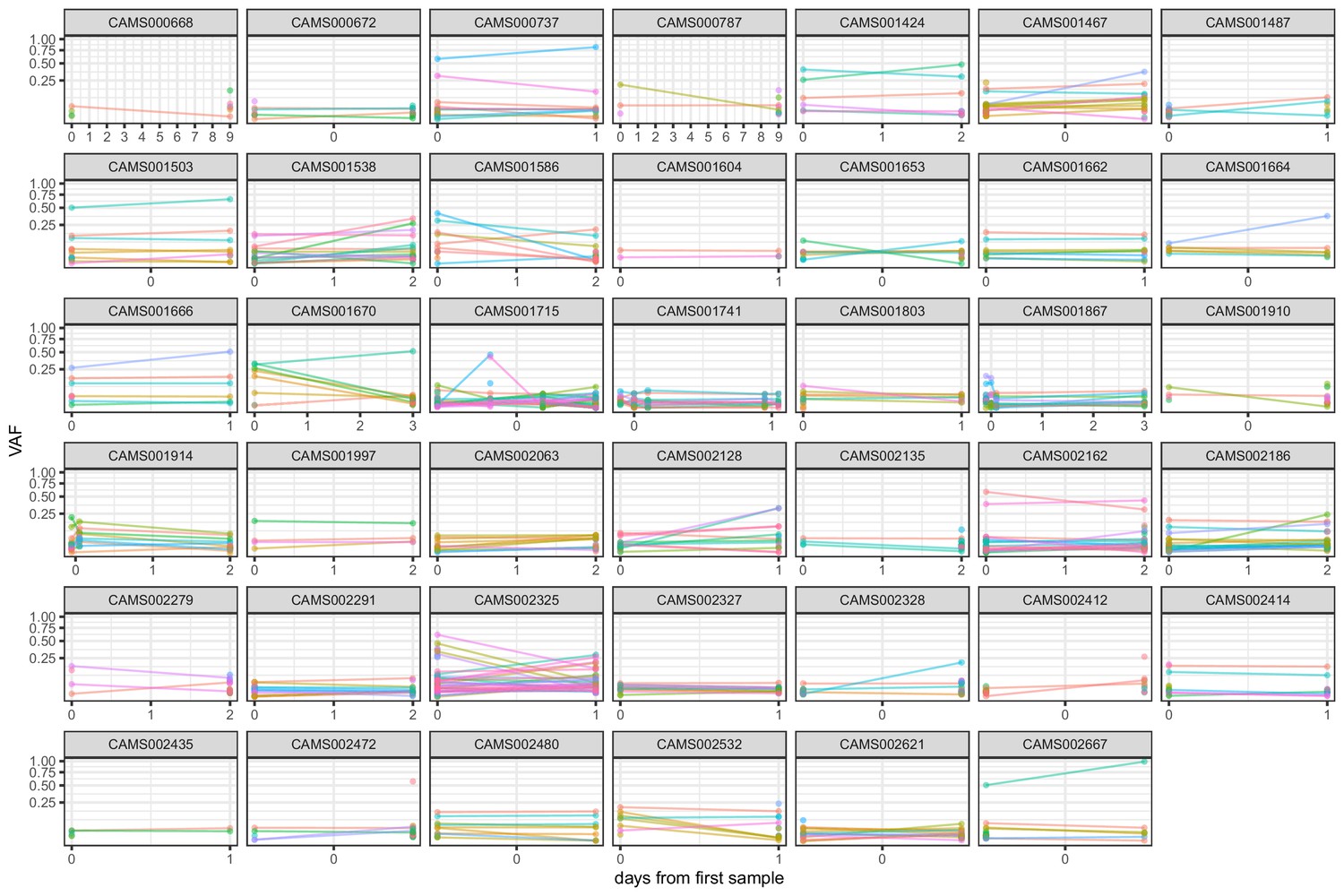
Frequencies of within-host variants for all hosts where multiple samples were taken over consecutive days.
Samples taken on the same day have been offset by a small distance to allow for comparison.
Figure 3—figure supplement 2
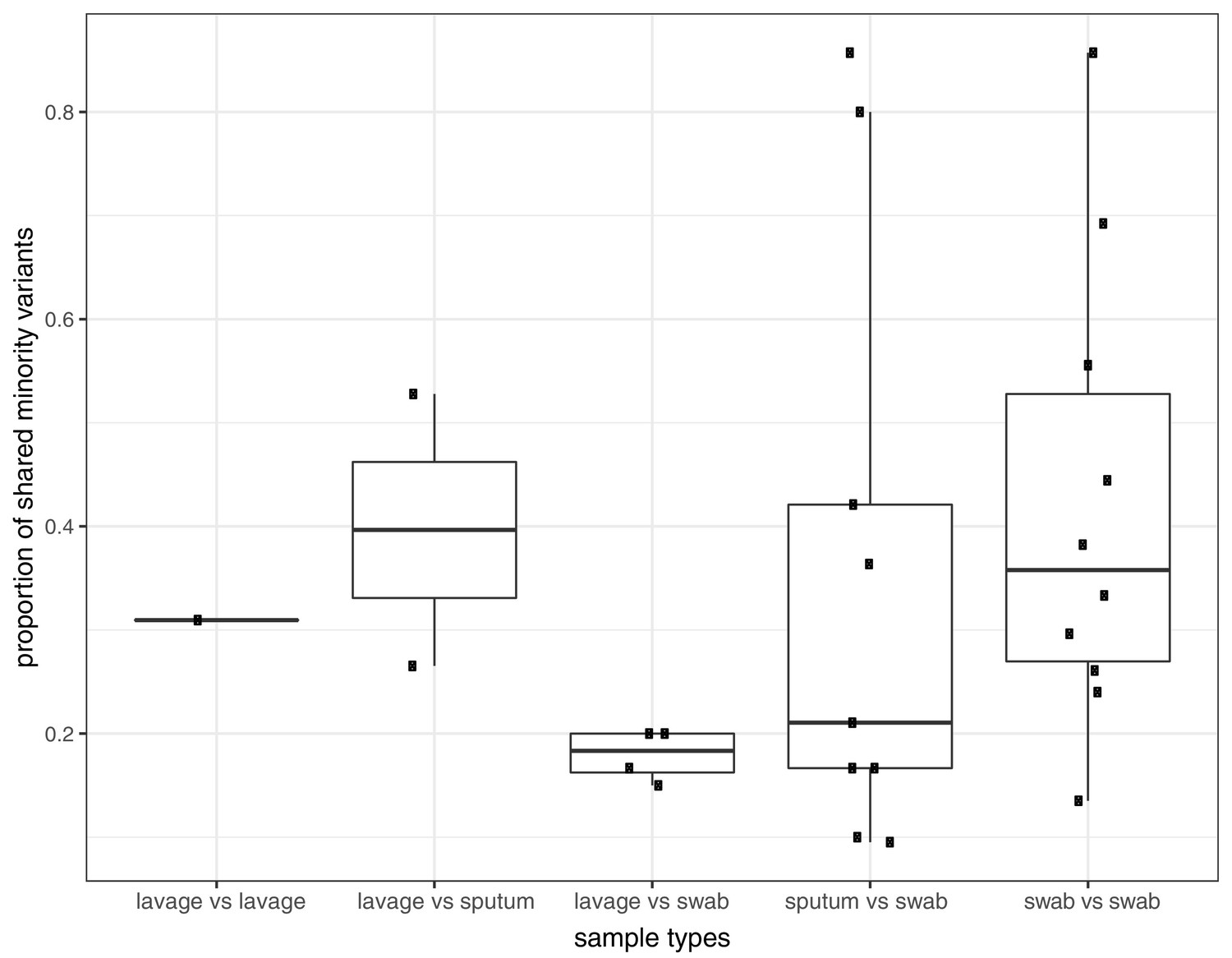
Proportion of shared variants between each pair of samples taken from the same host on the same day.
Pairs are split by sampling method, which included sputum, swabs, and bronchoalveolar lavage.
Figure 4
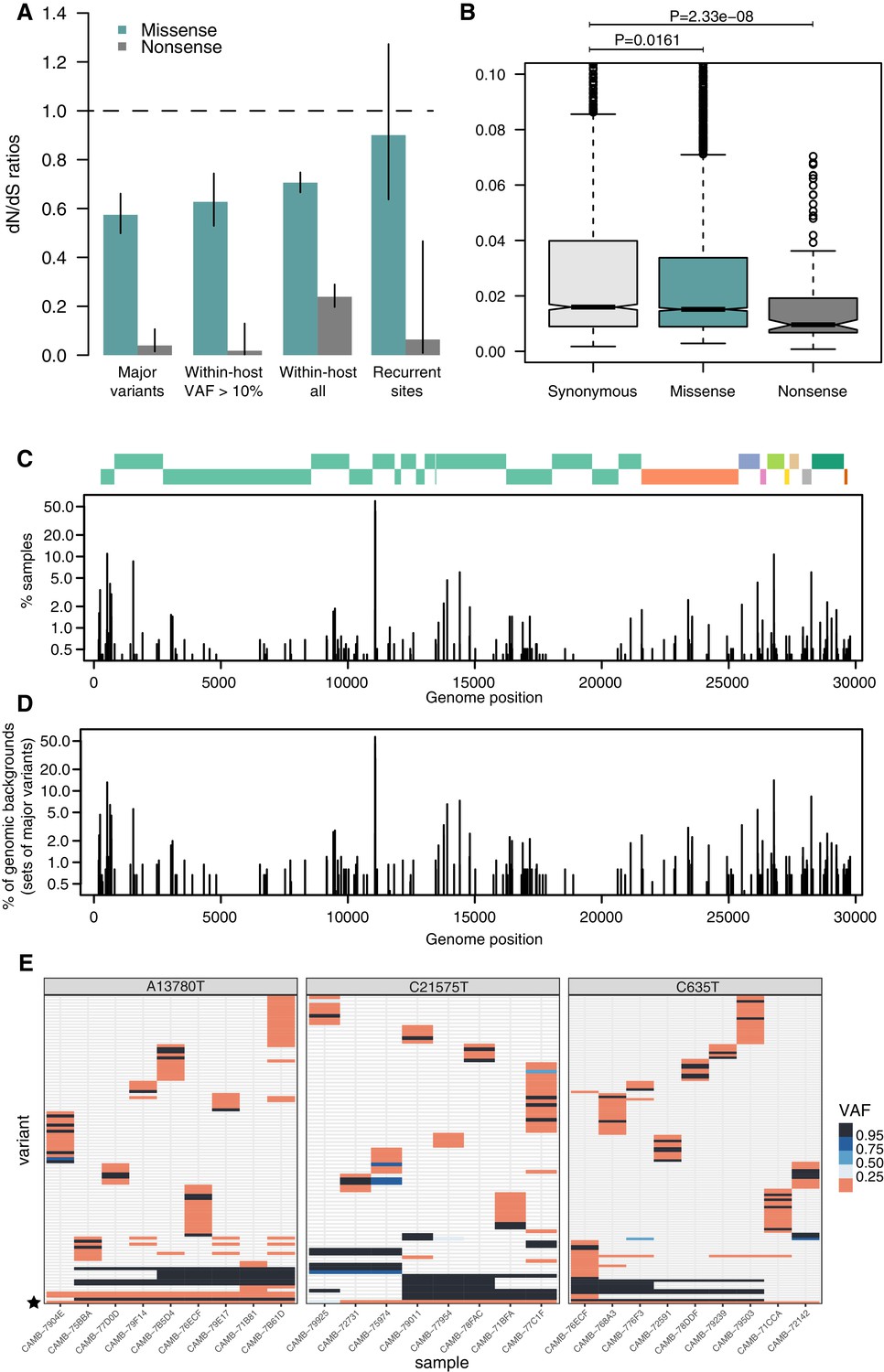
Patterns of selection and recurrent within-host variants.
(A) Genome-wide dN/dS ratios for missense and nonsense mutations (Materials and methods). Error bars depict 95% confidence intervals from the Poisson maximum-likelihood model. (B) ≥VAFs of within-host variants as a function of their predicted coding impact. p-values were calculated with Wilcoxon tests. (C) The top panel depicts the coordinates of the annotated peptides in the reference genome, coloured according to their ORF. The bottom panel depicts the frequency at which recurrent within-host variants (defined as those seeing in five or more samples) occur in the dataset. (D) Frequency of recurrent within-host variants (as in C) across different genomic backgrounds in the dataset (defined as the set of consensus variants in the sample). (E) Heatmaps of variant allele frequencies in samples containing three common within-host variants found at potential mutational hotspots are shown. The diversity of consensus variants with VAF ≥ 95% (black tiles) across samples is better explained by independent acquisitions of the minority variant rather than transmission.
Figure 5
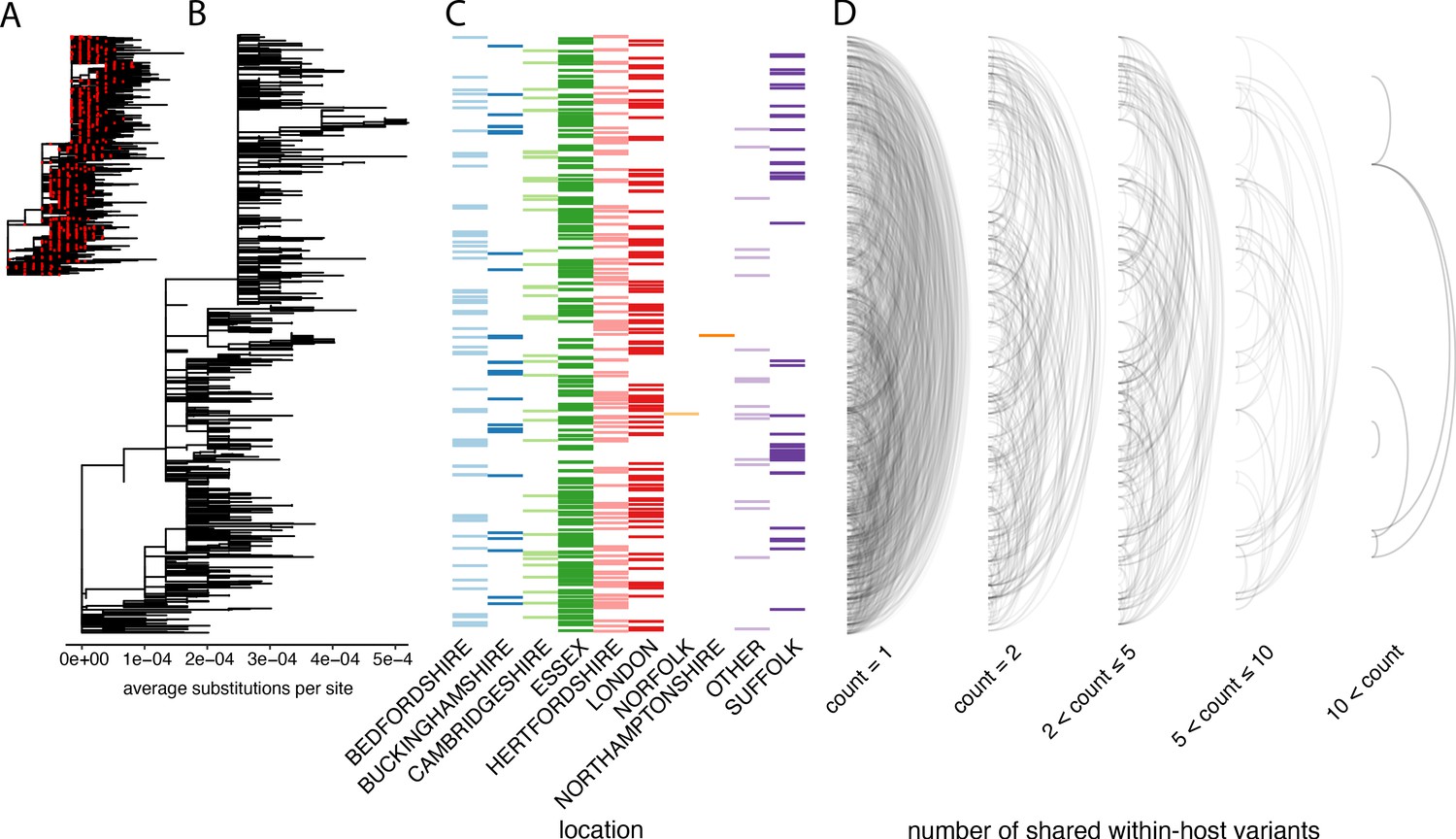
The distribution of shared within-host variants between samples with respect to the inferred consensus phylogeny.
(A) A maximum-likelihood phylogeny of all COG-UK consensus genomes available on 29 May 2020. Red dots indicate the location of those samples that were deep sequenced in replicate. (B) The same phylogeny restricted to those samples taken for deep sequencing. (C) The region each patient’s home address was located. (D) Links are drawn between tips of the phylogeny that share within-host minority. Links restricted to those variants seen in less than 2% of individuals and are separated based on the number of variants shared between samples.
Figure 6 with 1 supplement
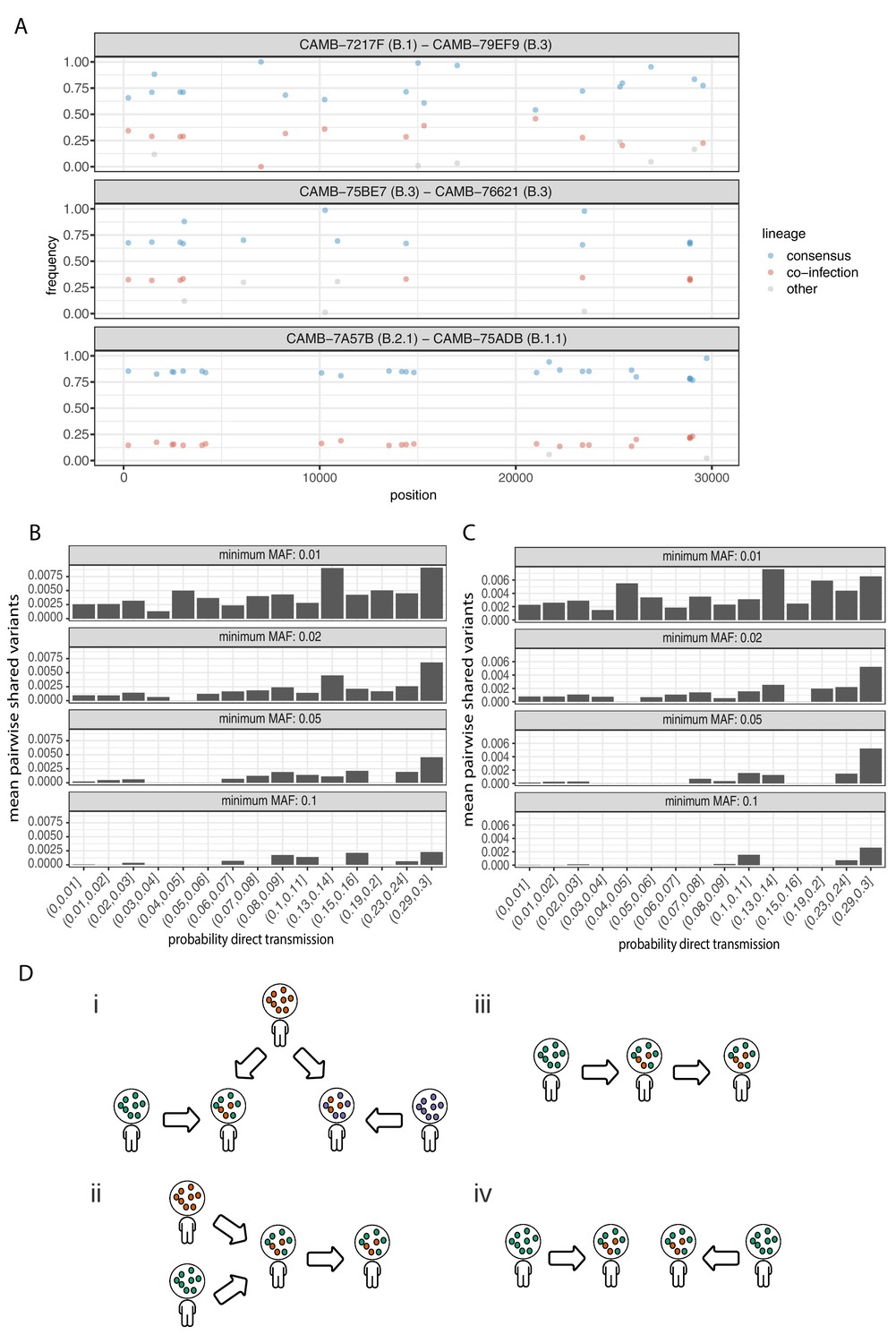
Potential mixed infections and the relationship between transmission and shared within-host variants.
(A) An example of three samples identified as potential mixtures. The consensus lineage is given first and coloured blue, while the potentially co-infecting lineage is given second and coloured red. Minority variants that do not match the co-infecting lineage are coloured grey. (B) The mean number of shared iSNVs shared by each pair of samples binned by the probability they were the result of a direct transmission according to the model of Stimson et al., 2019. Results, with a minimum minor allele frequency of 0.01, 0.02, 0.05, and 0.1 are shown in each of the facets. Within-host variants observed in more than 2% of samples were excluded. (C) The same plot as Figure 3B but having removed all samples that were inferred to be mixed infections. (D) A diagram demonstrating the four scenarios that can lead to shared within-host variants. (i) Superinfection of a common strain. (ii) Superinfection followed by co-transmission (iii) Transmission of the within-host variants through a large bottleneck. (iv) Independent de novo acquisitions of the same within-host variants. Shared within-host variants in scenarios (ii, iii) are concordant with the transmission tree, while (i, iv) are discordant, potentially confounding transmission inference efforts.
Figure 6—figure supplement 1
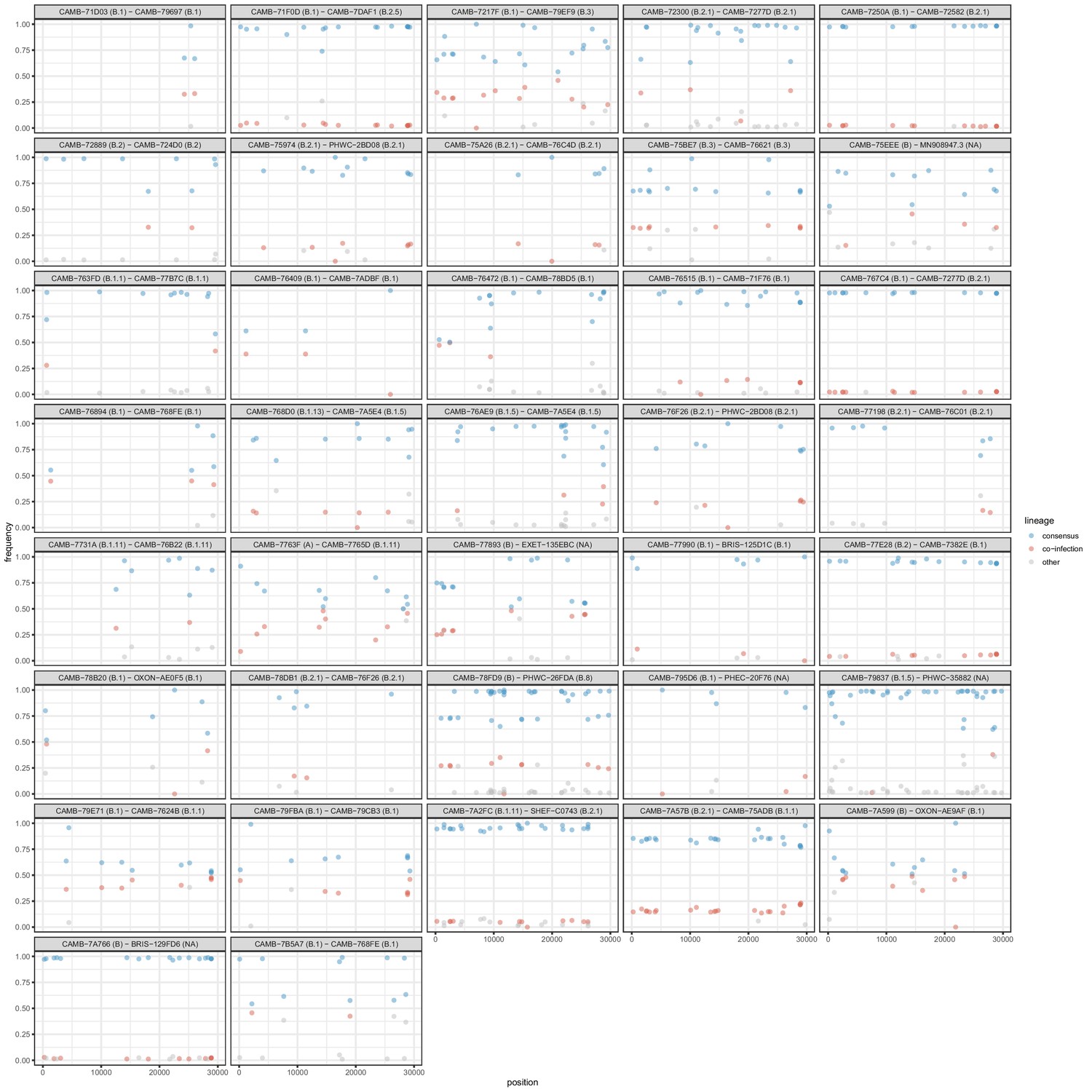
All samples identified as potential mixtures.
The consensus lineage is given first and coloured blue while the potentially co-infecting lineage is given second and coloured red. Minority variants that do not match the co-infecting lineage are coloured grey.
Additional files
-
Supplementary file 1
Sample IDs and metadata.
- https://cdn.elifesciences.org/articles/66857/elife-66857-supp1-v3.csv
-
Supplementary file 2
Shearwater within-host variant calls.
- https://cdn.elifesciences.org/articles/66857/elife-66857-supp2-v3.txt
-
Supplementary file 3
Sample specific sequence accession numbers.
- https://cdn.elifesciences.org/articles/66857/elife-66857-supp3-v3.csv
-
Supplementary file 4
Recurrent within-host mutations.
- https://cdn.elifesciences.org/articles/66857/elife-66857-supp4-v3.csv
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/66857/elife-66857-transrepform-v3.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Patterns of within-host genetic diversity in SARS-CoV-2
eLife 10:e66857.
https://doi.org/10.7554/eLife.66857




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}