Bacterial death and TRADD-N domains help define novel apoptosis and immunity mechanisms shared by prokaryotes and metazoans
- Computational Biology Branch, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, United States
Figures
Figure 1
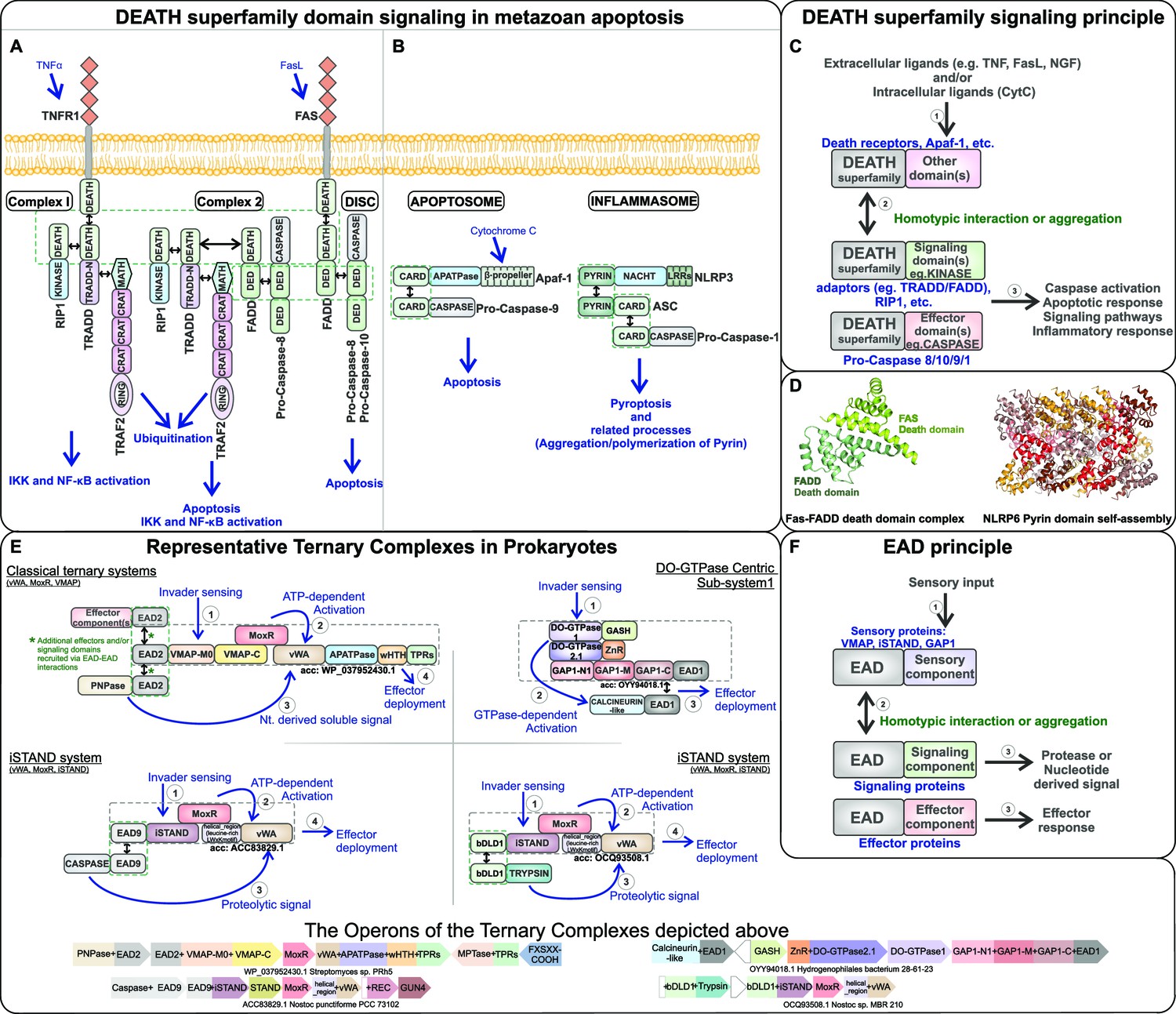
Signaling mechanism parallels between eukaryotic Death-superfamily domains and prokaryotic effector-associated domains (EADs) in their biological contexts.
(A) Extrinsic and (B) intrinsic pathways in eukaryotic apoptosis signaling. (C) Schematic representation of the interactions mediated by Death domains in various metazoan signaling processes. (D) Cartoon structural representation of the Death-Death interaction in the FADD-FAS complex (PDB: 3EQZ) and the Pyrin domain-based protein-templated assembly of filamentous polymeric complexes in NLRP6 (PDB: 6NCV). (E) Representative ternary biological conflict systems where the EADs, predicted to perform roles comparable to eukaryotic Death domains, were discovered. (F) Schematic representation of the interactions mediated by the EADs in prokaryotic biological conflict systems that are predicted to lead to a highly regulated counter-invader response.
Figure 2
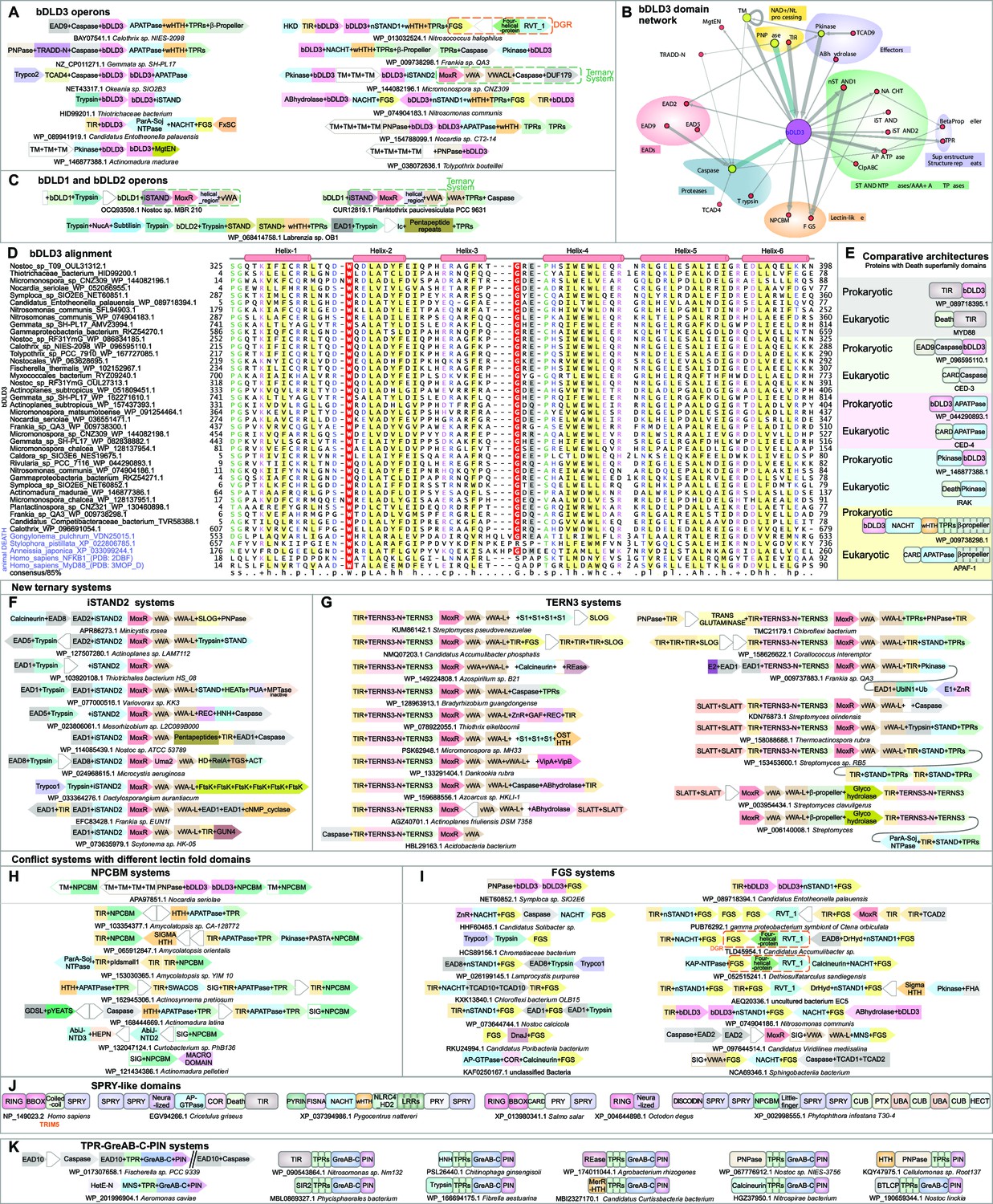
Domain and gene neighborhood contexts of the bacterial Death-like domains (bDLD).
(A) Representative gene neighborhoods coding for bDLD3 proteins. (B) Domains architectural network of bDLD3. (C) Representative gene neighborhoods coding for bDLD1 and bDLD2 proteins. (D) Multiple sequence alignment (MSA) of bDLD3 and representative eukaryotic Death domains (in purple). Sequences are denoted by the organism name and NCBI protein accession number separated by an underscore. Domain limits are numbered. . The predicted secondary structure and the sequence consensus at 85% identity are depicted above and below the alignment, respectively. Coloring is as per the consensus abbreviation of residue type, where s: small; u: tiny; +: basic; h: hydrophobic; l: aliphatic; p: polar; b: big. In all figures, α-helices and β-strands in MSAs are depicted as cylinders and arrows, respectively. Insertions in the sequences are represented by the number corresponding to their length. (E) Comparable domain architectures of the bDLD3 and Death-superfamily domains. iSTAND2 (F) and TERNS3 (G) containing novel ternary conflict systems. Novel conflict systems utilizing the lectin fold domains NPCBM (H) and FGS (I). (J) Domain architectures of eukaryotic SPRY-like domains. (K) Novel conflict system with a constant core comprising TPR, GreA/B-C-terminal, and PIN domains.
-
Figure 2—source data 1
Comprehensive gene neighborhoods and domain architectures of systems described in the figure.
- https://cdn.elifesciences.org/articles/70394/elife-70394-fig2-data1-v2.pdf
-
Figure 2—source data 2
Multiple sequence alignments of novel domains described in this study.
- https://cdn.elifesciences.org/articles/70394/elife-70394-fig2-data2-v2.zip
Figure 3 with 1 supplement
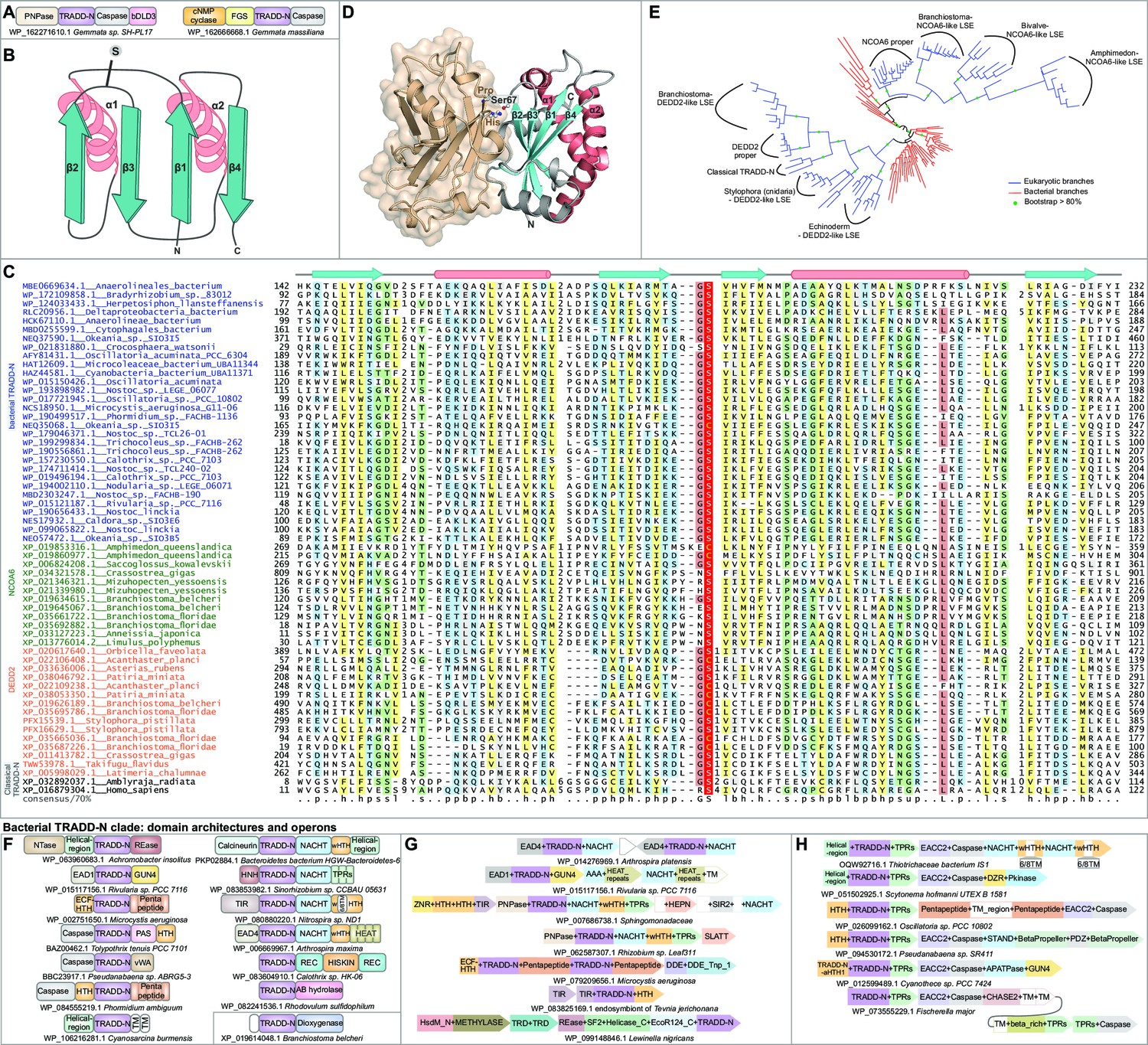
Structure, alignment, phylogeny, and contextual analysis of the TRADD-N domain.
(A) The bacterial TRADD-N domains that were initially recovered in searches. (B) Topology diagram of TRADD-N. Arrows and helices represent β-strand and α-helical regions, respectively. The conserved serine residue at the β2-β3 turn is indicated. (C) Multiple sequence alignment (MSA) of TRADD-N. Refer to Figure 2 legend for details of the MSA rendering. (D) The interaction of TRADD-N with the MATH domain (PDB: 1F3V). Residues mediating the non-covalent interactions are indicated. (E) Phylogenetic tree of representative TRADD-N domains showing the major clades . Domain architectures of (F) and gene neighborhoods coding for (G, H) bacterial TRADD-N domain proteins.
-
Figure 3—source data 1
Comprehensive gene neighborhoods and domain architectures of the bacterial TRADD-N domains.
- https://cdn.elifesciences.org/articles/70394/elife-70394-fig3-data1-v2.pdf
Figure 3—figure supplement 1
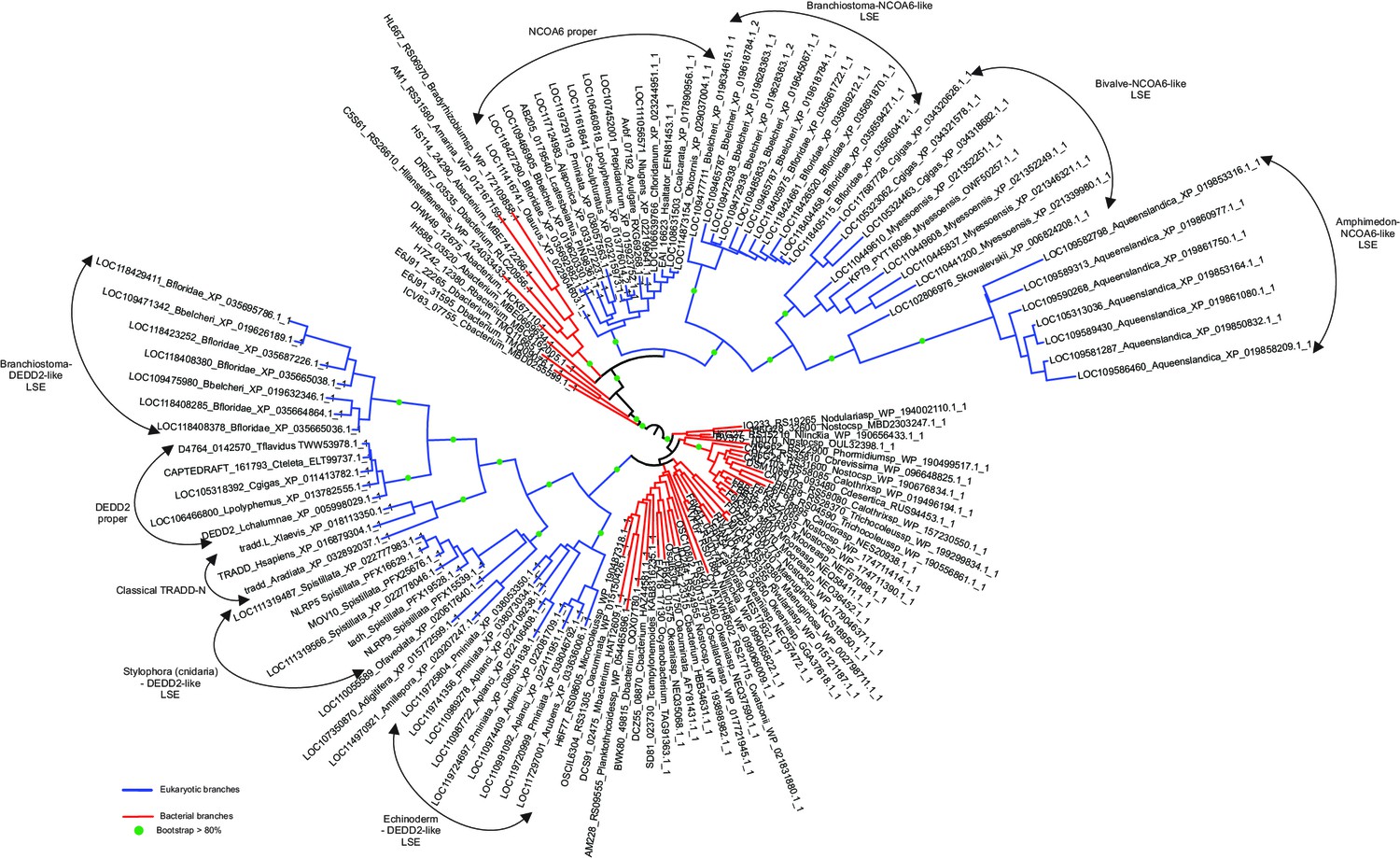
TRADD-N phylogenetic tree showing the names of the branches.
Figure 4
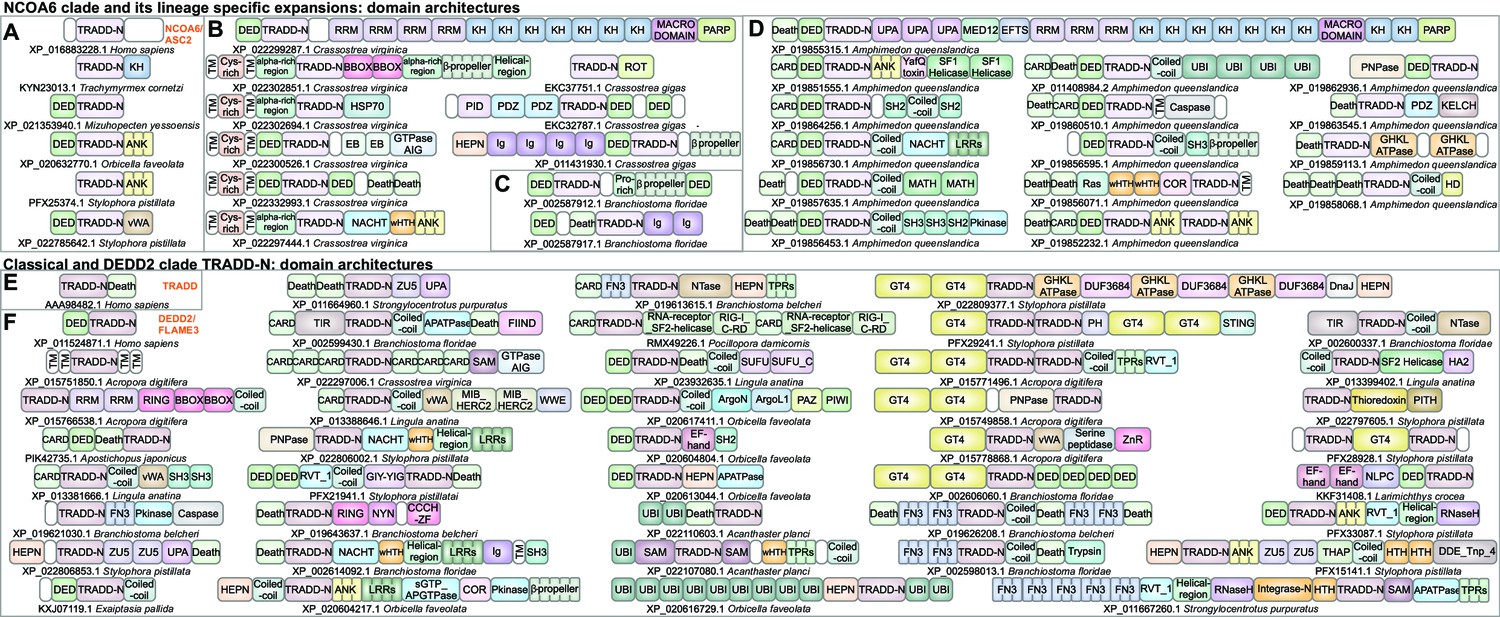
Domain architectures of NCOA6, classical TRADD-N, and DEDD2-like TRADD-N domains.
(A) Domainarchitectures of representative TRADD-N proteins from the NCOA6 clade. Lineage-specific domain architectures of NCOA6 clade TRADD-N proteins from (B) Crassostrea, (C) Branchiostoma, and (D) Amphimedon. (E) Domain architecture of the classical TRADD-N protein. (F) Domain architectures of representative DEDD2 TRADD-N proteins.
-
Figure 4—source data 1
Eukaryotic TRADD-N domain architectures and phyletic distribution.
- https://cdn.elifesciences.org/articles/70394/elife-70394-fig4-data1-v2.pdf
-
Figure 4—source data 2
Non-redundant counts of TRADD-N domains in eukaryotic lineages.
- https://cdn.elifesciences.org/articles/70394/elife-70394-fig4-data2-v2.pdf
Figure 5
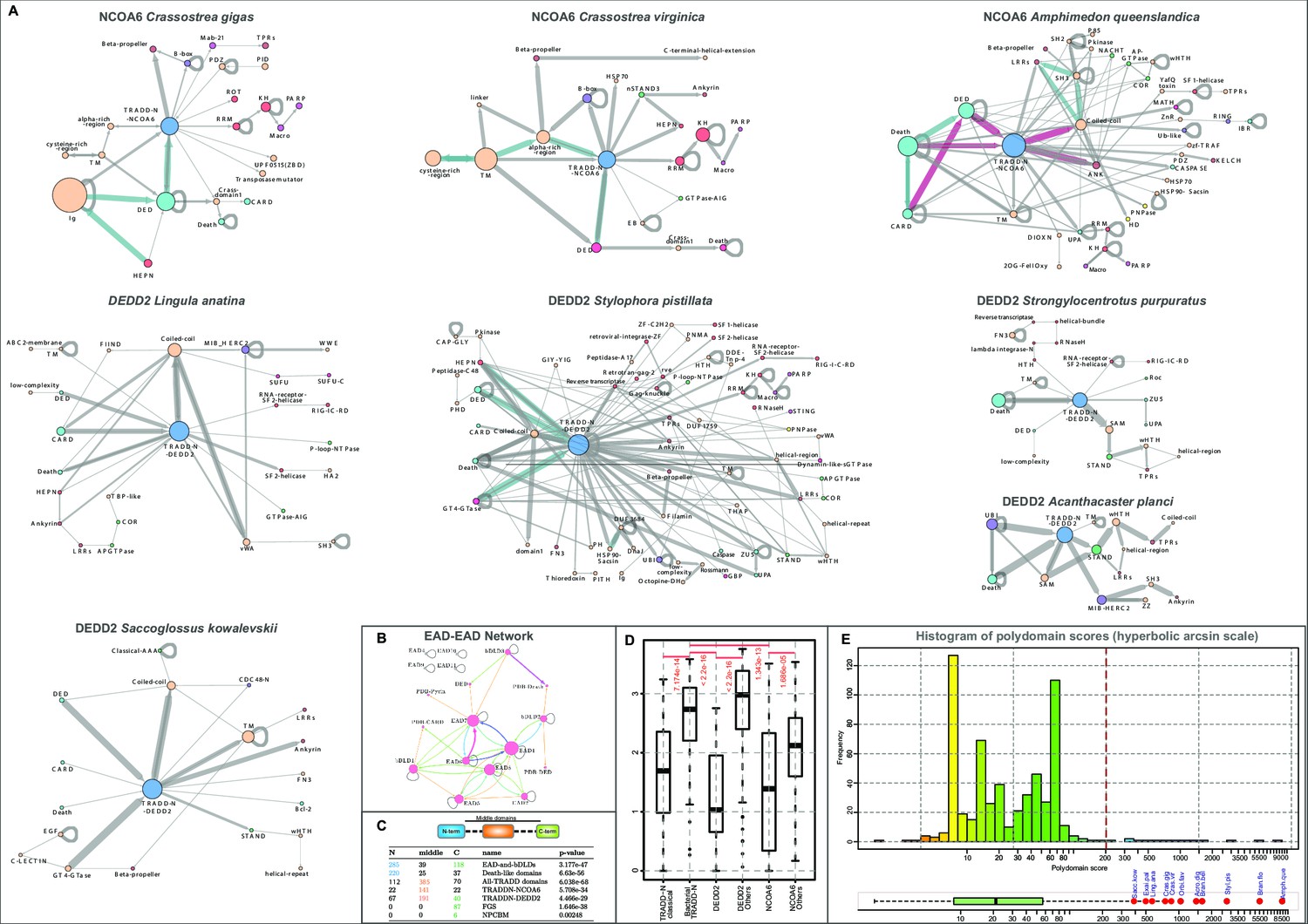
Domain architectures and analysis.
(A) Architectural networks of TRADD-N domains proteins from various metazoan lineages. Nodes represent domains and edges connecting them indicate their adjacency in the same polypeptide with the arrowhead pointing to the C-terminal domain . The thickness of the edge represents the frequency of co-occurrence of the connected nodes in different architectural contexts. (B) EAD-EAD search-retrieval network. The network was derived by using the results of various profile-profile (HHpred) searches using the individual nodes (EADs or Death-like domains) as queries. An edge was drawn between two nodes if they were recovered with p-values<0.0001 with the arrowhead pointing to the node recovered in the search. The edge thickness is scaled using the -log10(p-value). The network shows that several EADs recover each other and also Death-like domains in searches. (C) Chi-squared statistics of the positional bias of the specified domains in their domain architectures. The left three columns show the frequency of domain in the said positions in unique architectures, and the rightmost column gives the probability of this occurring by chance. . (D) Boxplot comparing position-specific entropy of sequences from MSAs of various TRADD-N clades. The t-test was used to determine the significance of the difference in mean entropy between the clades under comparison (indicated by the red line above the clades). (E) Histogram of the distribution of polydomain scores (PDS) of TRADD-N proteins. Outliers with high PDS, mostly marine organisms, are marked in red. Organism abbreviations are as follows: Sacc. kow.: Saccoglossus kowalevskii; Exai. pal.: Exaiptasia pallida; Ling. ana.: Lingula anatina; Crass. gig.: Crassostrea gigas; Crass. vir.: Crassostrea virginica; Orbi. fav.: Orbicella faveolata; Acro. dig.: Acropora digitifera; Bran. bel.: Branchiostoma belcheri; Styl. pis.: Stylophora pistillata; Bran. flo.: Branchiostoma floridae; Amph. que.: Amphimedon queenslandica.
Figure 6

Domain architectural associations of the GT4 glycosyltransferase domain in (A) representative metazoans and (B) prokaryotes.
(C) Predicted counter-invader two-gene systemwith each gene encoding a novel STAND NTPase (nSTAND2). (D) Prokaryotic counter-invader systems centered on the AP-GTPase proteins. (E) Topology diagram of the structural fold of an individual repeat unit of the vicinal oxygen chelate superfamily illustrating its core secondary structure units.
-
Figure 6—source data 1
Gene neighborhoods and domain architectures of the GT4 glycosyltransferases, the nSTAND2 and prokaryotic AP-GTPases.
- https://cdn.elifesciences.org/articles/70394/elife-70394-fig6-data1-v2.pdf
Additional files
-
Supplementary file 1
p-Values of profile-profile search (HHPRED) hits recovered with various effector-associated domains and bacterial Death-like domains as queries.
- https://cdn.elifesciences.org/articles/70394/elife-70394-supp1-v2.csv
-
Supplementary file 2
Hypergeometric distribution test for association of various conflict systems described in the text with multicellular prokaryotes.
- https://cdn.elifesciences.org/articles/70394/elife-70394-supp2-v2.csv
-
Supplementary file 3
Chi-squared statistics of the positional bias of major domains described in the study.
The left three columns show the frequency of domain occurrence in the various positions in unique architectures, and the probability of this occurring by chance is given in the rightmost column.
- https://cdn.elifesciences.org/articles/70394/elife-70394-supp3-v2.csv
-
Supplementary file 4
Position-specific entropy of sequences from multiple sequence alignments of various TRADD-N clades.
- https://cdn.elifesciences.org/articles/70394/elife-70394-supp4-v2.xlsx
-
Supplementary file 5
Polydomain scores of various TRADD-N-domain-containing species.
- https://cdn.elifesciences.org/articles/70394/elife-70394-supp5-v2.csv
-
Supplementary file 6
Multicellular status of all organisms included in analysis of the significance of systems overrepresentation in multicellular prokaryotic genomes.
The multicellular flag is defined as either True, False, or NA (not enough information available).
- https://cdn.elifesciences.org/articles/70394/elife-70394-supp6-v2.csv
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/70394/elife-70394-transrepform1-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Bacterial death and TRADD-N domains help define novel apoptosis and immunity mechanisms shared by prokaryotes and metazoans
eLife 10:e70394.
https://doi.org/10.7554/eLife.70394




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}