Binding affinity landscapes constrain the evolution of broadly neutralizing anti-influenza antibodies
- Department of Organismic and Evolutionary Biology, Harvard University, United States
- NSF-Simons Center for Mathematical and Statistical Analysis of Biology, Harvard University, United States
- Quantitative Biology Initiative, Harvard University, United States
- Department of Physics, Massachusetts Institute of Technology, United States
- Department of Physics, Harvard University, United States
- Department of Applied Physics, Stanford University, United States
- Laboratoire de physique de ÍÉcole Normale Supérieure, CNRS, PSL University, Sorbonne Université, and Université de Paris, France
Figures
Figure 1 with 7 supplements
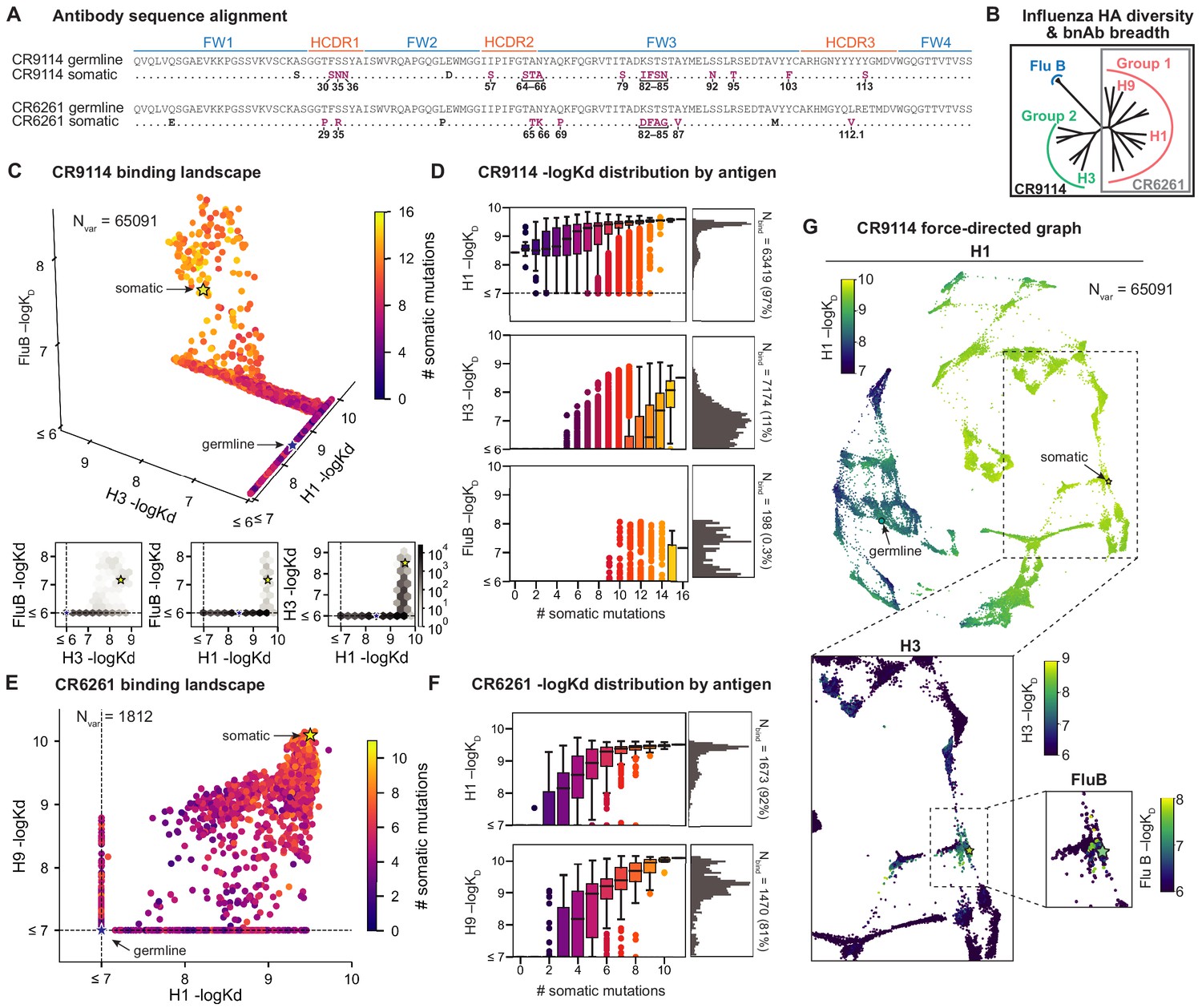
Binding landscapes.
(A) Sequence alignment comparing somatic heavy chains to reconstructed germline sequences. Mutations under study (purple, numbered) and excluded mutations (black) are indicated; residues are numbered by IMGT unique numbering. (B) Influenza hemagglutinin phylogenetic tree with selected antigens and breadth of CR9114 (black box) and CR6261 (gray box) indicated. (C, E), Scatterplots of the (C) CR9114 library binding affinities against three antigens, with 2D planes shown below, and (E) CR6261 library binding affinities against two antigens. (D, F) Distributions of library binding affinities for (D) CR9114 and (F) CR6261 for each antigen (gray histogram, right) separated by number of somatic mutations (boxplots, left). Numbers and percentages of variants with measurable binding are indicated at right. (G), Force-directed graph of CR9114 H1 –logKD. Each variant (node) is connected to its 16 single-mutation neighbors (edges not shown for clarity); edges are weighted such that variants with similar genotypes and –logKD tend to cluster. Nodes are colored by binding affinity to H1 (top; showing all 65,091 nodes), H3 (lower left inset; showing only the region containing nodes with –logKD > 6), and Flu B (lower right inset; showing only the region containing nodes with –logKD > 6).
-
Figure 1—source data 1
CR9114 library -logKD to H1, H3, and influenza B.
Biological triplicates, mean, and standard error reported.
- https://cdn.elifesciences.org/articles/71393/elife-71393-fig1-data1-v3.csv
-
Figure 1—source data 2
CR6261 library -logKD to H1 and H9.
Biological triplicates, mean, and standard error reported.
- https://cdn.elifesciences.org/articles/71393/elife-71393-fig1-data2-v3.csv
-
Figure 1—source data 3
Isogenic flow cytometry measurements of –logKD for select CR9114 and CR6261 variants.
Inferred –logKD and standard deviation for each replicate of isogenic FACS, alongside inferred –logKD mean and SEM from Tite-Seq using the mean bin and maximum likelihood (ML, shown only for CR9114) inference methods.
- https://cdn.elifesciences.org/articles/71393/elife-71393-fig1-data3-v3.xlsx
Figure 1—figure supplement 1
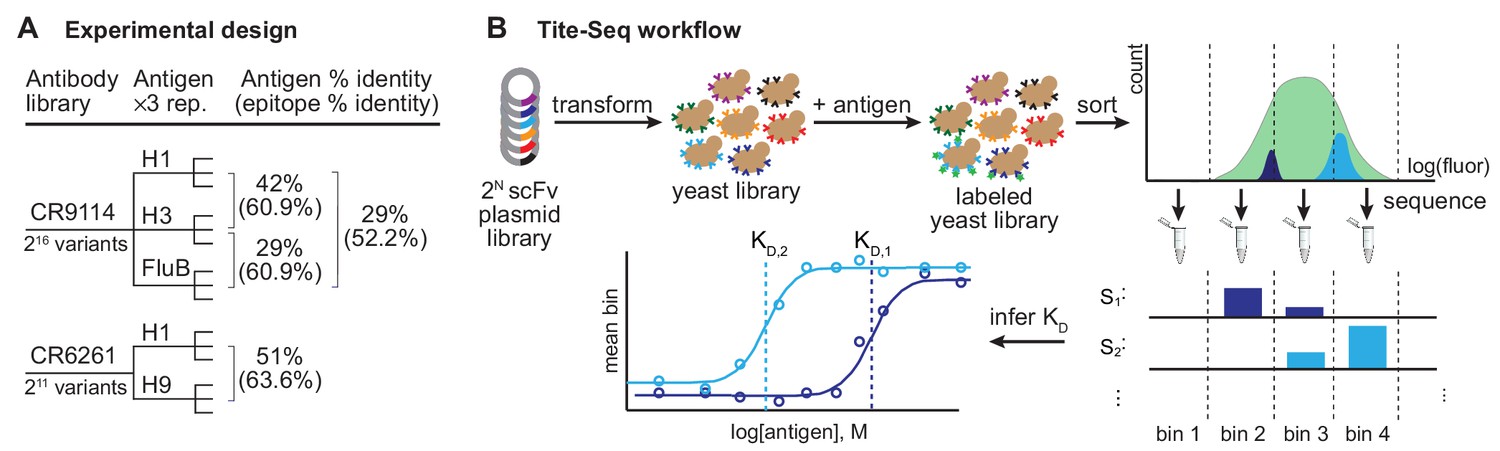
Experimental design and Tite-Seq workflow.
Experimental design and Tite-Seq workflow. (A) Experimental design. Amino acid sequence percent identity of the entire HA ectodomain and the stem epitope (Dreyfus et al., 2012) are shown between each pair of antigens tested for both antibodies. (B) Tite-Seq assay. Surface display single-chain variable fragment (scFv) libraries are transformed into yeast and labeled with fluorescent antigen, followed by FACS into bins and sequencing. Dissociation constants are inferred from changes in mean bin fluorescence across 12 antigen concentrations, see Materials and methods.
Figure 1—figure supplement 2
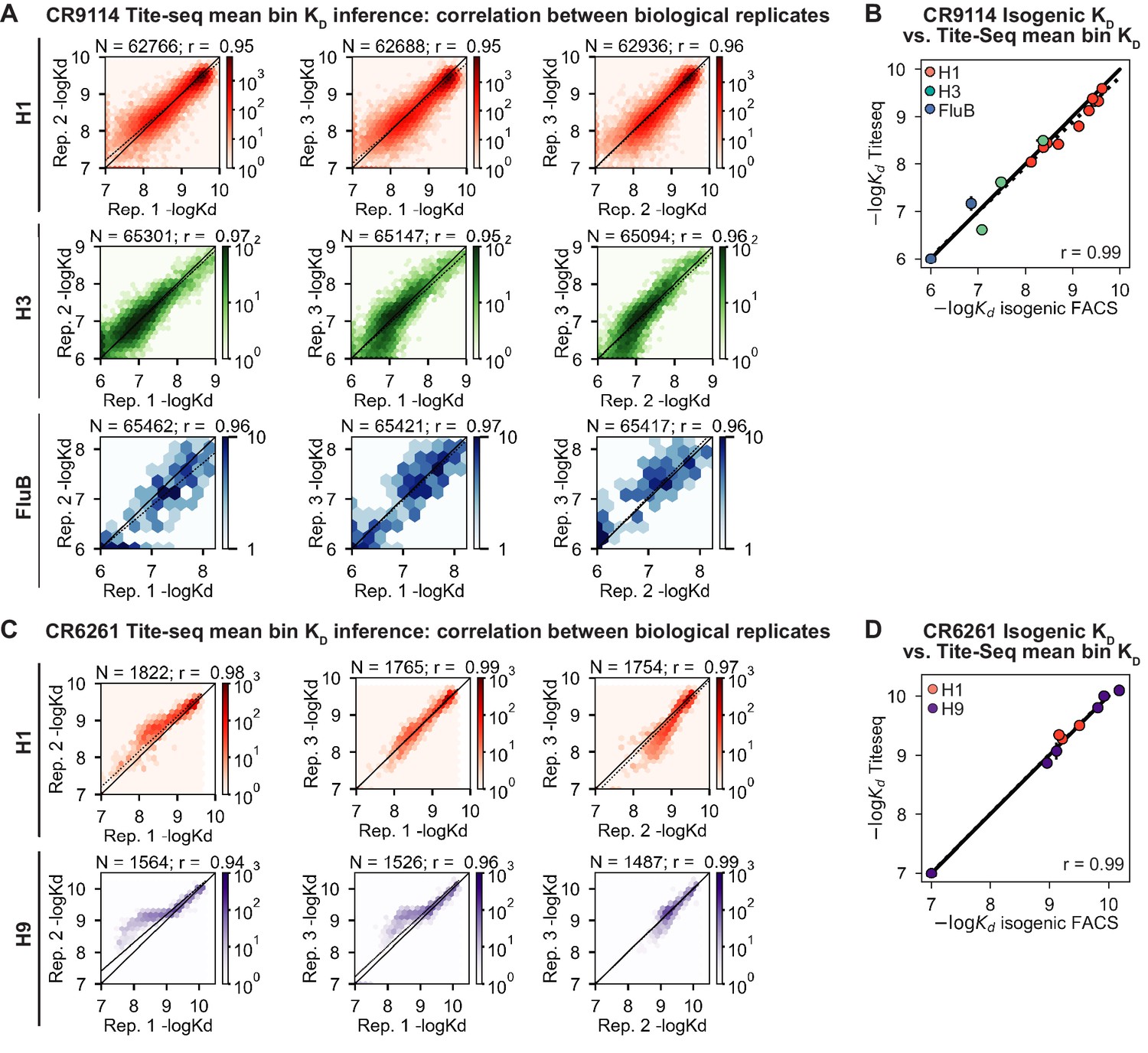
Tite-Seq data quality.
(A, C) Correlation of (A) CR9114 and (C) CR6261 KD measurements between biological replicates. (B, D) Validation of (B) CR9114 and (D) CR6261 Tite-Seq KD measurements by isogenic flow cytometry measurements for a subset of variants and antigens.
Figure 1—figure supplement 3
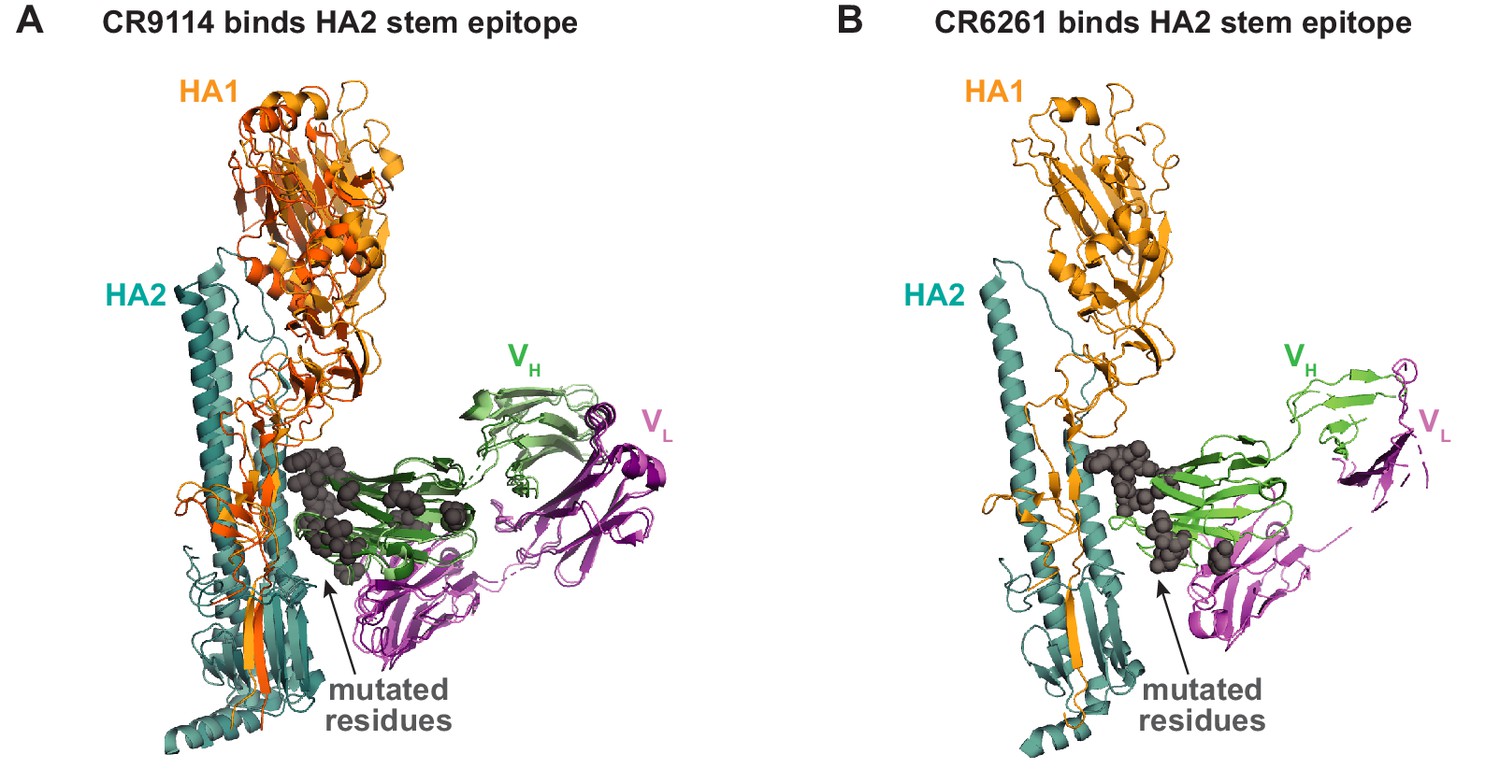
Antibody-antigen co-crystal structures.
(A) Alignment of co-crystal structure of CR9114 with H5 (light hues; PDB ID 4FQI) and CR9114 with H3 (dark hues; PDB ID 4FQY). Mutated residues shown as gray spheres. (B) Co-crystal structure of CR6261 with H1 (PDB ID 3GBN); mutated residues shown as gray spheres.
Figure 1—figure supplement 4
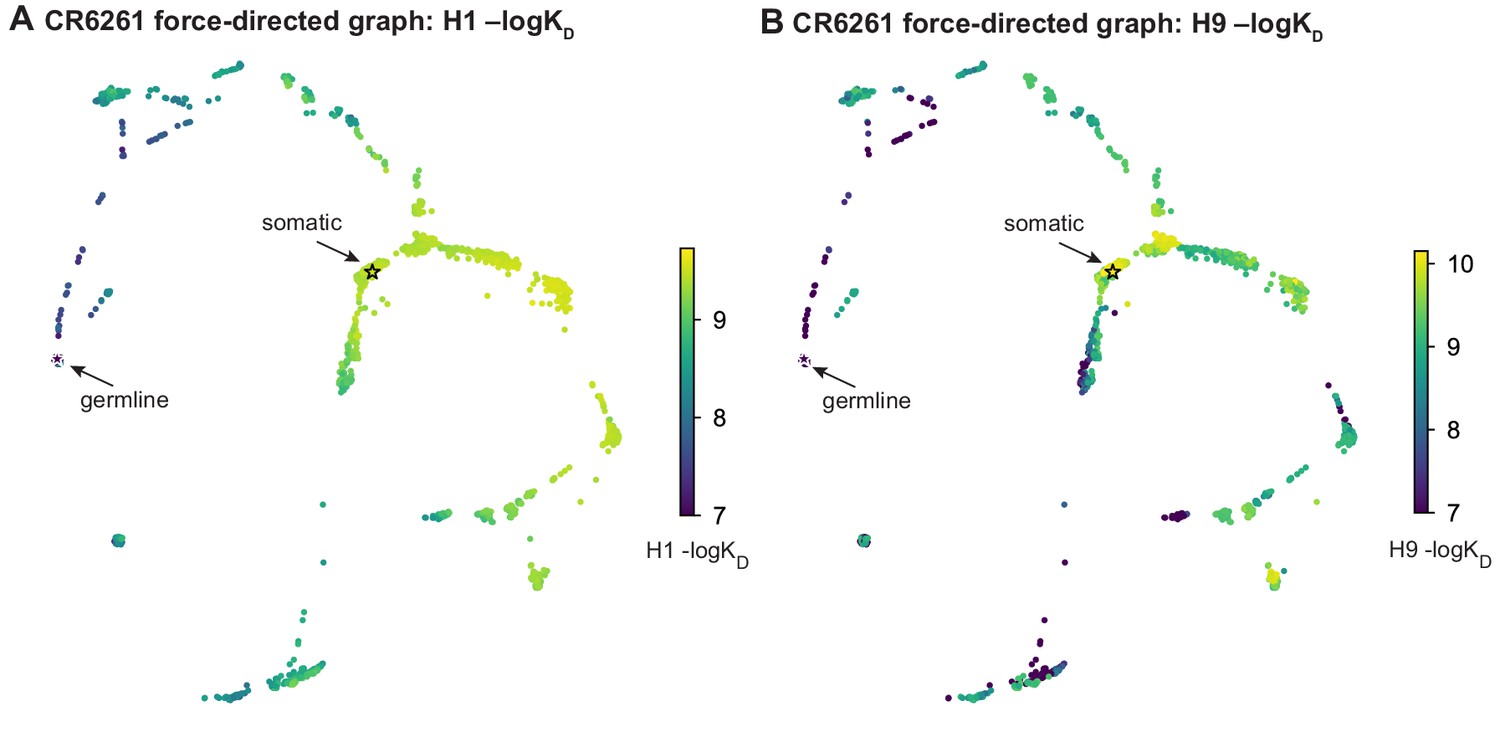
Force-directed graph for CR6261.
Force-directed graph for CR6261. (A, B) Force-directed graph for CR6261 H1 –logKD, as in Figure 1G. Nodes are colored by binding affinity to (A) H1 and (B) H9.
Figure 1—figure supplement 5
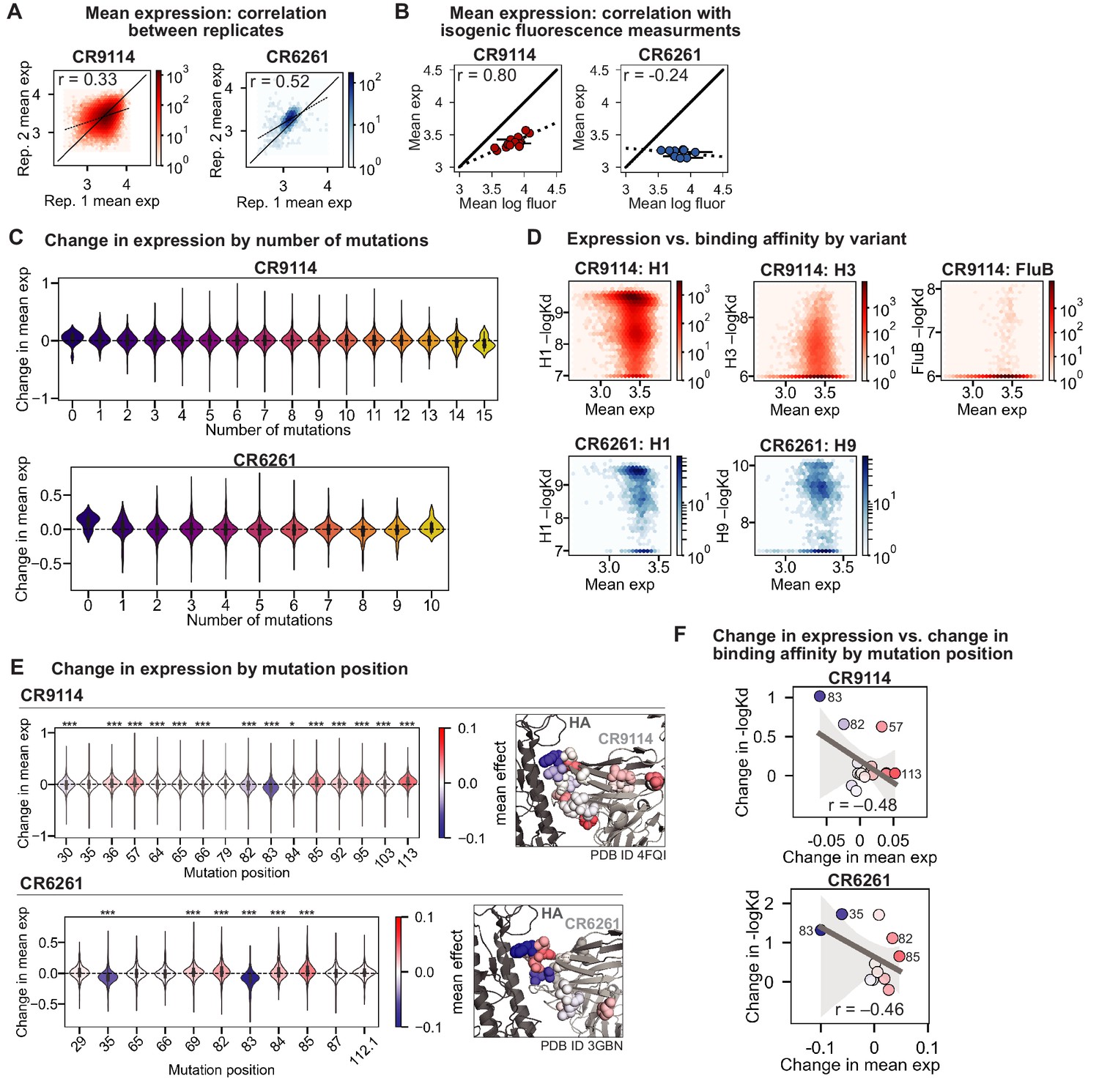
Expression of antibody libraries.
(A) Correlation of mean expression across Tite-Seq biological replicates for CR9114 library (left, red) and CR6261 library (right, blue). (B) Correlation between Tite-Seq mean expression and isogenic expression fluorescence for select CR9114 (left, red) and CR6261 (right, blue) variants. (C) Change in expression upon mutation for a given number of background somatic mutations. (D) Correlation between mean expression and –logKD. Average values across biological replicates (N logKD = 3) are plotted. (E) Change in expression upon mutation at a specific site. Violin plots (left) and residues in co-crystal structure (right) are colored by mean change in expression for each site. Asterisks above violins indicate p-values for two-sided t-test between the distribution means and zero ( (*), < 0.001 (**), < 0.0001 (***); , ). (F) Correlation between mean change in expression and mean change in –logKD (summed across all antigens) by mutation position. Select mutations with large impacts on expression and –logKD are labeled; all points are colored by mean change in expression, as in (F). Dark gray line indicates best-fit linear regression (95% confidence intervals in light gray).
Figure 1—figure supplement 6
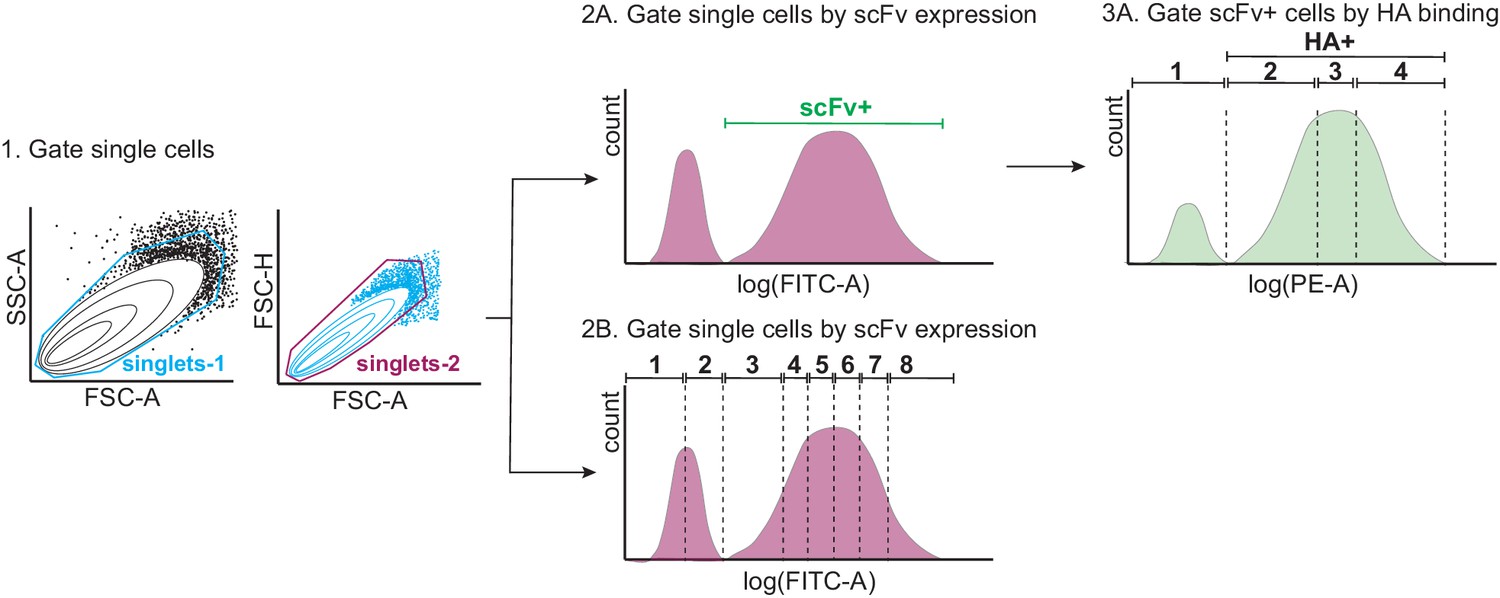
Tite-Seq gating strategy.
First, single yeast cells were gated by forward scatter (FSC) and side scatter (SSC) (step 1). Single cells were then either gated by scFv expression or HA binding. For the expression sort (step 2B), single cells were gated into eight bins along the log(FITC-A) axis, each containing 12.5% of the population. For the binding sort (steps 2A and 3A), scFv-expressing (scFv+) single cells were sorted into four bins along the log(PE-A) axis, with bin one comprising all HA- cells, and bins 2–4 each comprising 33% of the HA+ population.
Figure 1—figure supplement 7

Reversions of excluded mutations.
(A) Reversion of A24S and E46D in CR9114 (somatic-16) does not substantially impact binding affinity compared to the fully somatic version of CR9114 (somatic-18) to either H1 (orange) or H3 (turquoise); these mutations are thus excluded from the CR9114 library. (B) Reversion of Q6E, L50P, and V101M in CR6261 (somatic-11) does not substantially impact binding affinity compared to the fully somatic version of CR6261 (somatic-14) to either H1 (orange) or H9 (purple); these mutations are thus excluded from the CR6261 library. Measurements made in biological duplicate; mean +/- standard error shown.
Figure 2 with 1 supplement
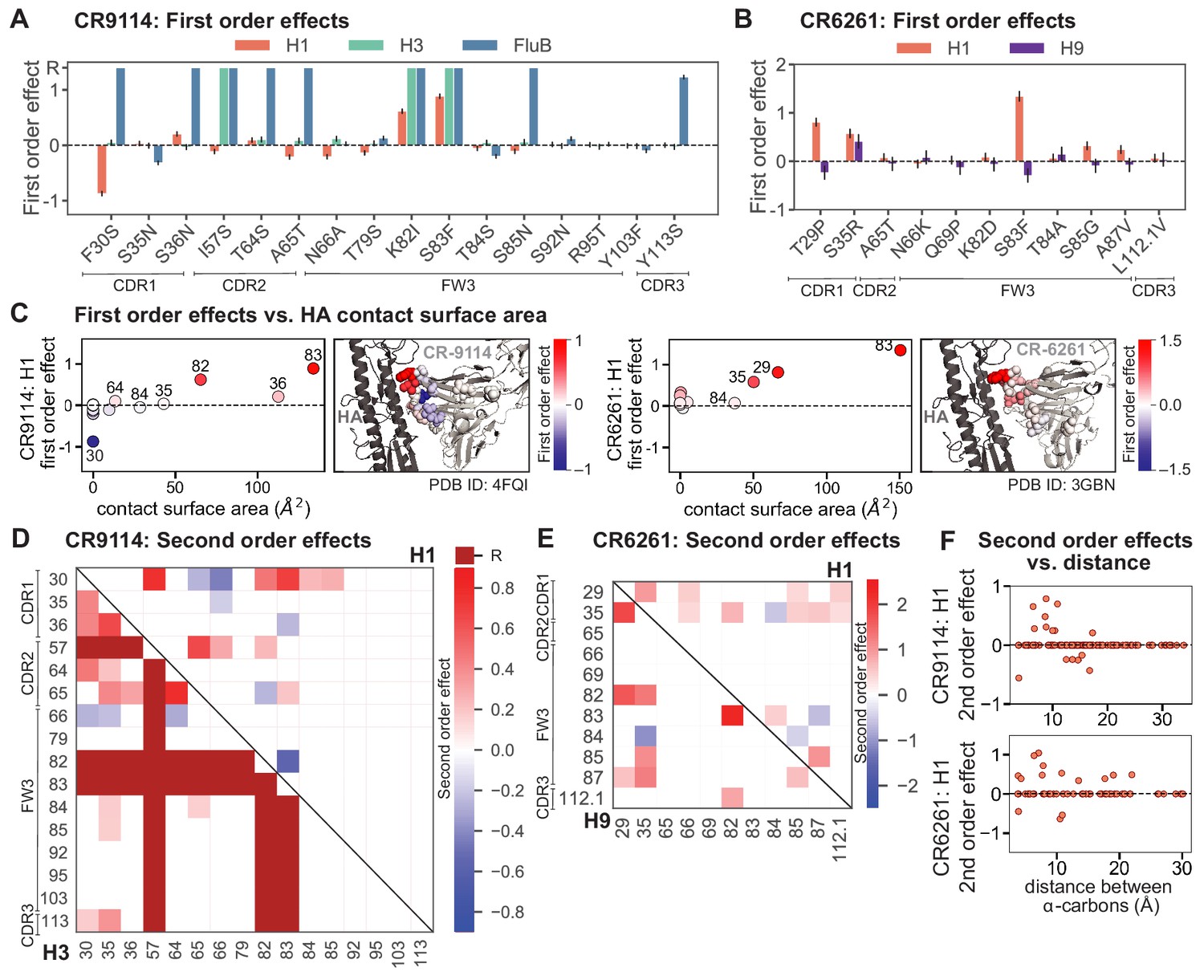
First- and second-order effects.
(A, B) First-order effects inferred in best-fitting epistatic interaction models for (A) CR9114 and (B) CR6261. Mutations required for binding (present at over 90% frequency in binding sequences) have effect sizes denoted as ‘R’ and are removed from inference. Error bars indicate standard error. (C) First order effects for each site plotted against the contact surface area between the corresponding somatic residue and HA (left, CR9114; right, CR6261). Sites with notable contact area or effect size are labeled. Cocrystal structures are also shown; mutations are colored by first-order effect size. (D) Significant second-order epistatic interaction coefficients for CR9114 mutations (bottom left, H3; top right, H1). Interactions involving required mutations are shown in dark red. (E) Significant second-order coefficients for CR6261 mutations (bottom left, H9; top right, H1). Significance in (D), (E) indicates Bonferroni-corrected p-value < 0.05, see Materials and methods. (F) Second-order coefficients for H1 –logKD plotted against the distance between the respective -carbons in the crystal structures.
-
Figure 2—source data 1
Interaction model coefficients for CR9114.
Coefficients are reported with standard errors, p-values, and confidence intervals (95% with Bonferroni correction by the number of parameters).
- https://cdn.elifesciences.org/articles/71393/elife-71393-fig2-data1-v3.csv
-
Figure 2—source data 2
Interaction model coefficients for CR6261.
Coefficients are reported with standard errors, p-values, and confidence intervals (95% with Bonferroni correction by the number of parameters).
- https://cdn.elifesciences.org/articles/71393/elife-71393-fig2-data2-v3.csv
-
Figure 2—source data 3
Tabulated contact surface area, number of HA contacts, and pairwise distances for mutations in CR9114 and CR6261.
For each mutated position, the contact surface area with HA (as plotted in Figure 2C and Figure 2—figure supplement 1) and the number of HA residues within six angstroms is tabulated for CR9114-H5 (4FQI), CR9114-H3 (4FQY), and CR6261-H1 (3GBN). Distances between alpha-carbons plotted in Figure 2F and Figure 2—figure supplement 1 are also tabulated here alongside the corresponding second order effects.
- https://cdn.elifesciences.org/articles/71393/elife-71393-fig2-data3-v3.xlsx
Figure 2—figure supplement 1

Structural context of first and second order effects.
(A) Left: first-order effects for each site, colored by effect size and plotted against the contact surface area between the corresponding somatic residue and HA (top, CR9114 with H3; bottom, CR6261 with H9); Right: co-crystal structures with mutation sites colored by first-order effects, as in Figure 2C. (B) Second-order coefficients for CR9114 with H3 (top) and CR6261 with H9 (bottom) plotted against the distance between the respective -carbons in the crystal structures, as in Figure 2F.
Figure 3 with 3 supplements
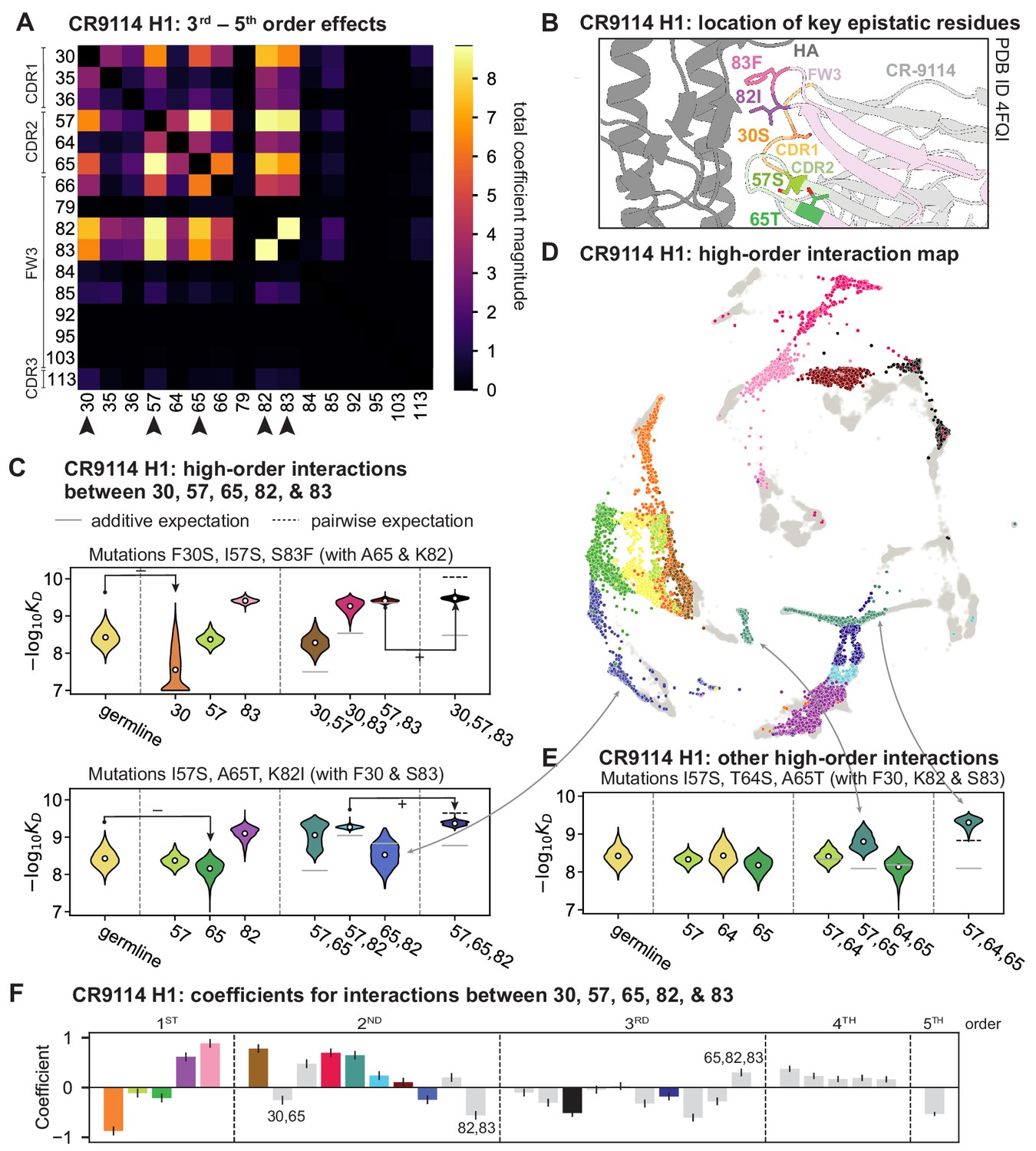
High-order epistasis for CR9114.
(A) Total higher order epistatic contributions of CR9114 mutation pairs for binding H1. Color bar indicates the sum of absolute values of significant higher order interaction coefficients involving each pair of mutations; key epistatic residues indicated by arrows. Significance is given by Bonferroni-corrected p-value < 0.05, see Materials and methods. (B) Location of key epistatic residues in the CR9114 –HA co-crystal structure colored by region. (C) –logKD distributions for genotypes grouped by their identity at the five residues indicated in (A), (B), with means indicated as white dots ( genotypes per violin). Annotations indicate notable deleterious ('−') and beneficial ('+') mutational effects. (D) CR9114 force-directed graph from Figure 1G, colored as in (C) by the genotype at the five sites indicated in (A), (B). Genotypes not shown in (C) are shown in light gray. Data are also available in an interactive data browser at https://yodabrowser.netlify.app/yoda_browser/. (E), Third-order interaction involving site 64 accounts for distinct clusters (teal) corresponding to genotypes with mutations 57 and 65 in (D). Colors correspond to mutation groups in (C), (D) ( genotypes per violin). (F), Epistatic interaction coefficients among the five key sites from (A), (B). Colors for certain groups as in (C), (D); other groups denoted in gray, with notable terms labeled.
Figure 3—figure supplement 1
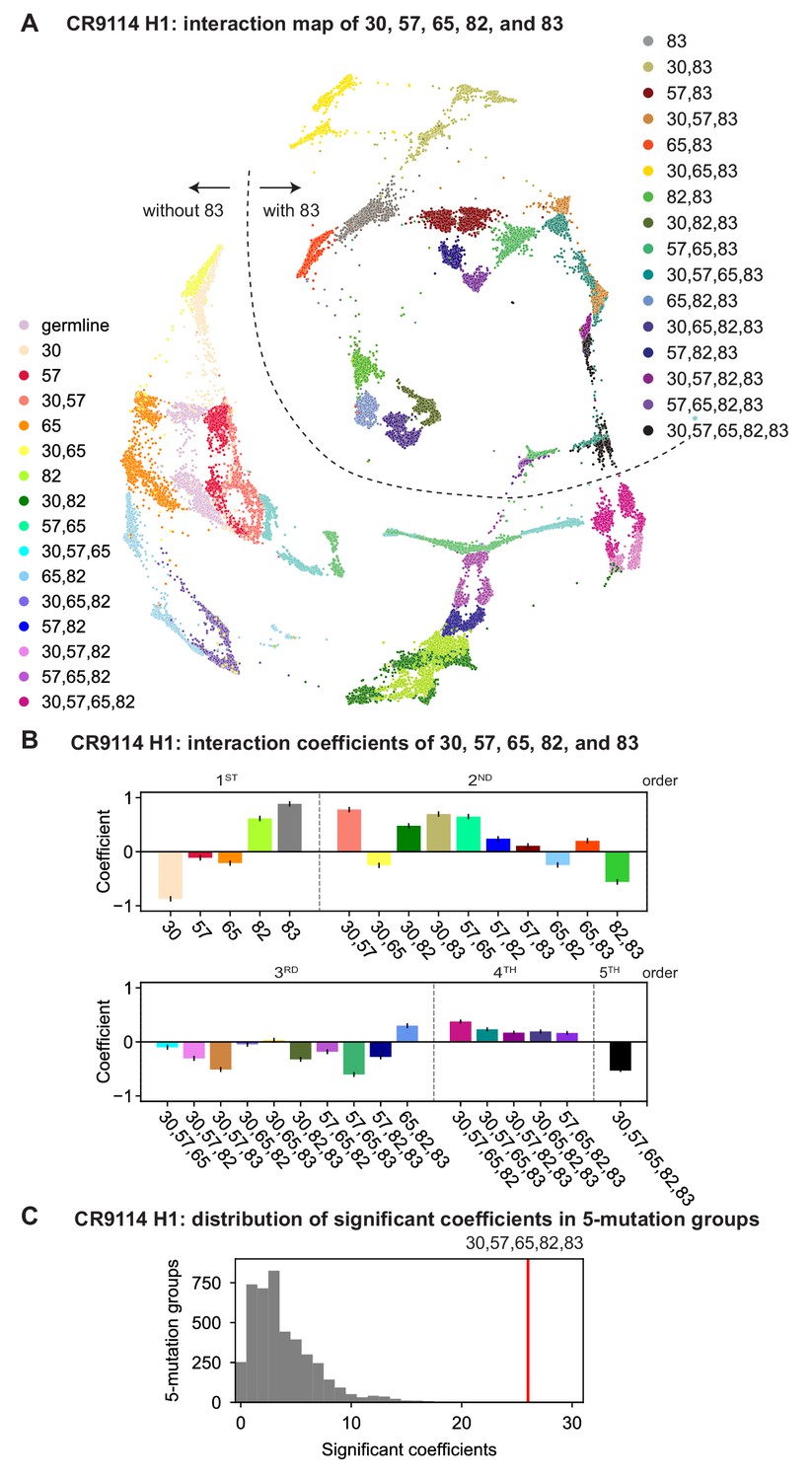
CR9114: interactions between five key sites.
(A) CR9114 force-directed graph, as in Figure 3D, colored by mutation groups of sites 30, 57, 65, 82, and 83 (32 total groups). The dashed line emphasizes the observed separation of genotypes with S83F (upper right) from those without S83F (lower left). (B) Coefficients for terms in the epistatic interaction model corresponding to mutation groups of sites 30, 57, 65, 82, and 83 (31 total groups, excluding the germline), colored according to (A) and grouped by order. Error bars indicate standard error. (C) Distribution of the number of significant coefficients for mutation groups in every possible set of five sites chosen from the 16 sites (up to 31 terms for each group, for 4368 groups). Significance is given by Bonferroni-corrected p-value lt0.05, see Materials and methods. The value for the group illustrated in (A), (B) is indicated in red (26 significant terms, empirical p-value ).
Figure 3—figure supplement 2
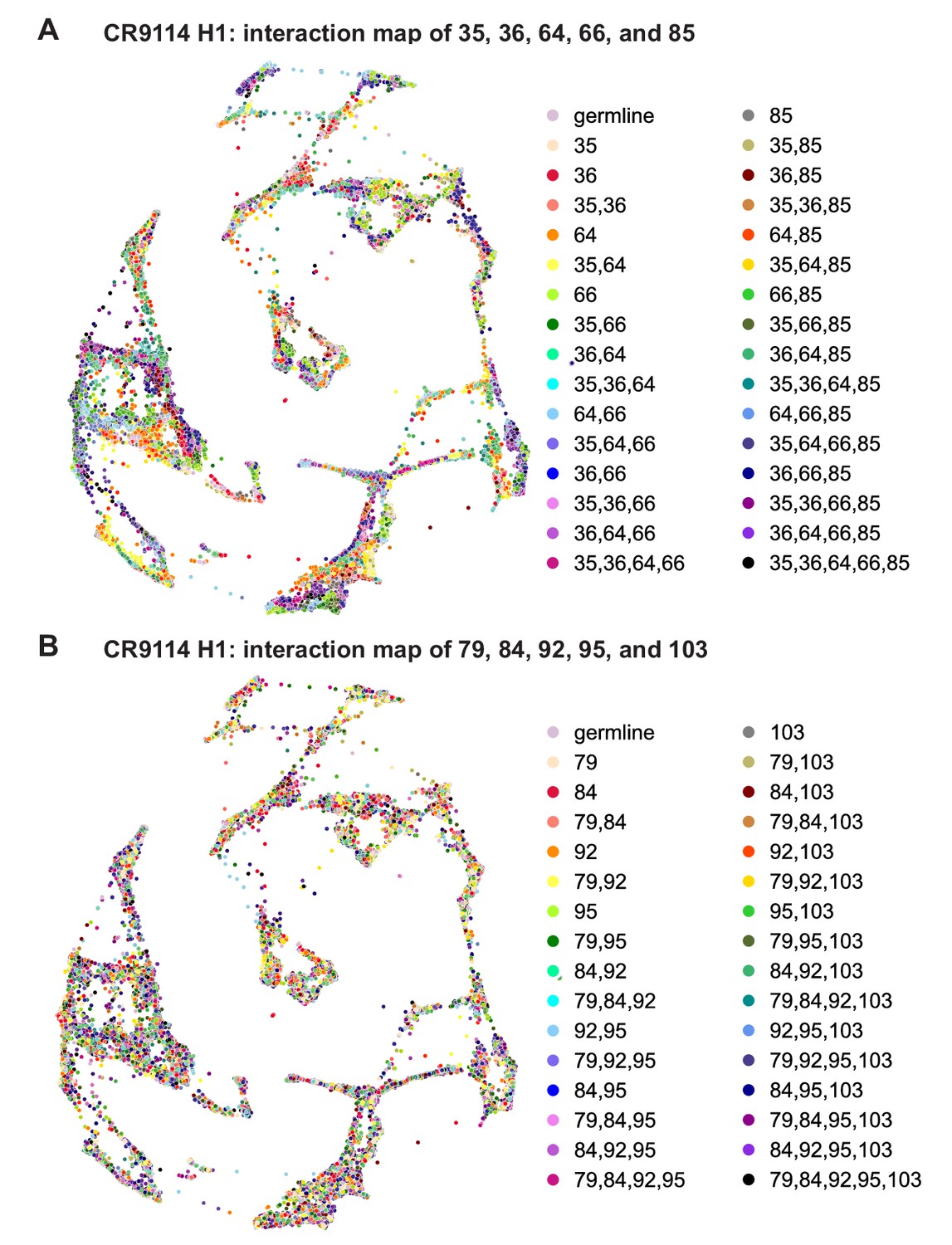
CR9114: interactions between other sets of five sites.
(A) CR9114 force-directed graph, as in Figure 3D, but colored by mutation groups of a different set of five sites with fewer strong epistatic interactions (35, 36, 64, 66, and 85). (B) CR9114 force-directed graph, colored by mutation groups of a different set of five sites with no strong linear contributions or epistatic interactions (79, 84, 92, 95, and 103).
Figure 3—figure supplement 3

High-order epistasis for CR9114 binding to H3.
(A) Higher order significant epistatic contributions of CR9114 mutation pairs, as in Figure 3A, for binding H3. Light yellow columns indicate required mutations (sites 57, 82, and 83). Significance is given by Bonferroni-corrected p-value < 0.05, see Methods.
Figure 4 with 2 supplements
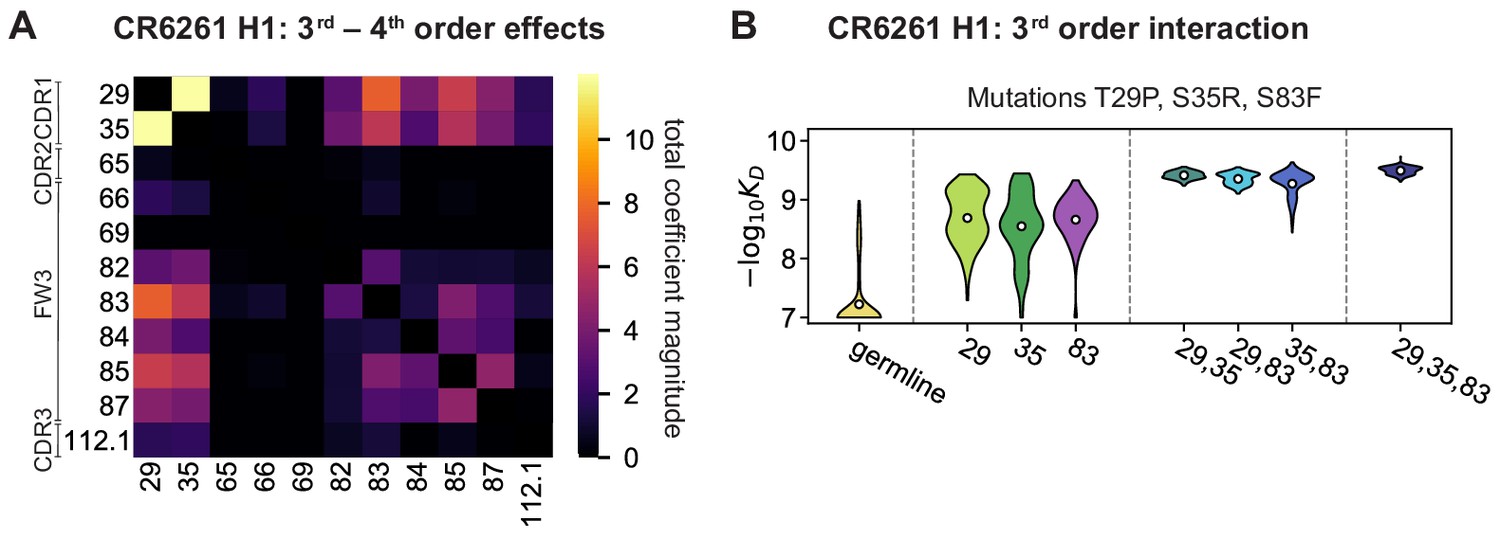
High-order epistasis for CR6261.
(A) Total significant epistatic contributions of CR6261 mutation pairs for binding H1, as in Figure 3A. Significance is given by Bonferroni-corrected p-value < 0.05, see Materials and methods. (B) Third-order interaction for CR6261 H1 binding between mutations T29P, S35R, and S83F ( genotypes per violin).
Figure 4—figure supplement 1
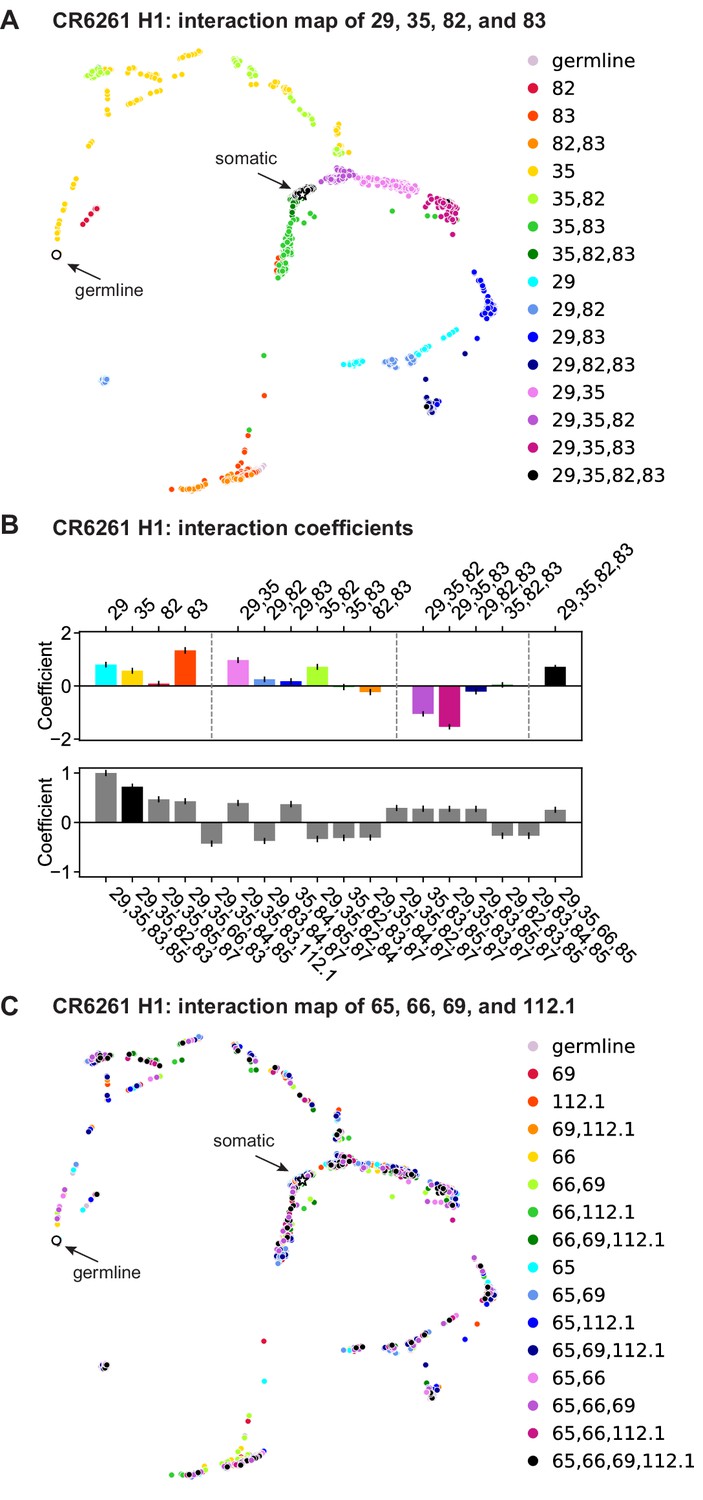
CR6261: interactions between four sites.
(a) CR6261 force-directed graph, as in Figure 1—figure supplement 4, colored by mutation groups of sites 29, 35, 82, and 83 (16 total groups). (B) Top, coefficients for terms in the epistatic interaction model corresponding to the mutation groups illustrated in (a) (15 total groups, excluding the germline), colored according to (a) and grouped by order. Bottom, the largest fourth-order coefficients observed in the epistatic interaction model, with sites indicated. In both, error bars indicate standard error. (C) CR6261 force-directed graph, colored by a different set of four sites with the fewest strong linear effects and epistatic interactions (65, 66, 69, and 112.1).
Figure 4—figure supplement 2

High-order epistasis for CR6261 binding to H9.
(A) Higher order significant epistatic contributions of CR6261 mutation pairs, as in Figure 4A, for binding H9. (B) Scatterplot of significant epistatic interaction model coefficients for binding to H1 and H9. Terms at different orders are colored and sized as indicated. Selected coefficients are annotated. Significance in (A), (B) is given by Bonferroni-corrected p-value < 0.05, see Methods.
Figure 5 with 3 supplements
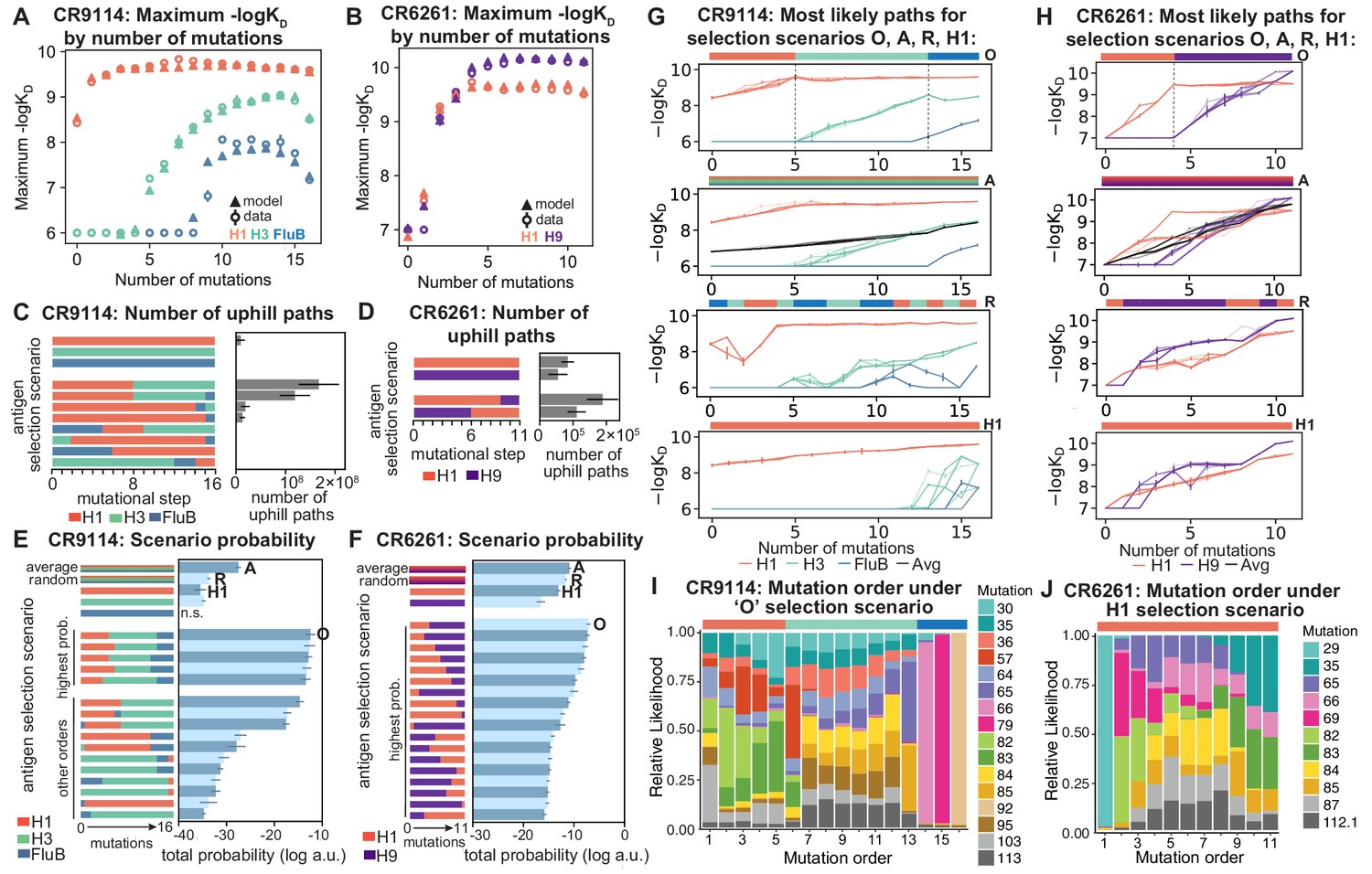
Antigen selection scenarios and likely mutational pathways.
(A, B) Maximum binding affinity achievable for sequences with a given number of mutations. For each antigen for (A) CR9114 and (B) CR6261, the maximum observed (circles) and model-predicted (triangles) affinity for each number of somatic mutations is shown. (C, D) Total number of 'uphill' paths for select antigen selection scenarios (colored bars) for (C) CR9114 and (D) CR6261. Error bars indicate standard error obtained through bootstrap, see Materials and methods. (E, F) Total log probability (in arbitrary units) of mutational trajectories from germline to somatic sequence for (E) CR9114 and (F) CR6261 under different antigen selection scenarios, in a moderate selection model. Error bars indicate standard error obtained through bootstrap, see Materials and methods. (G, H) 25 most likely paths for (G) CR9114 and (H) CR6261, from select scenarios in (E, F); –logKD plotted for each antigen. For the random mixed scenario ('R'), a representative case is shown. ’A’ indicates the average mixed scenario; ’O’ indicates the optimal scenario. (I, J) Probability of mutation order under optimal antigen selection scenario ‘O' for CR9114 (I) and H1 for CR6261 (J). Selection scenarios are as in (E, F) and shown in colored bar at top; the total probability (through all possible paths) for each mutation to occur at each mutational step is shown as stacked colored bars.
-
Figure 5—source data 1
Total probability of mutational trajectories for CR9114 under different antigen selection scenarios.
Mean and standard error across 10 bootstrap samples are reported for moderate, weak, and strong selection strengths.
- https://cdn.elifesciences.org/articles/71393/elife-71393-fig5-data1-v3.csv
-
Figure 5—source data 2
Total probability of mutational trajectories for CR6261 under different antigen selection scenarios.
Mean and standard error across 10 bootstrap samples are reported for moderate, weak, and strong selection strengths.
- https://cdn.elifesciences.org/articles/71393/elife-71393-fig5-data2-v3.csv
Figure 5—figure supplement 1
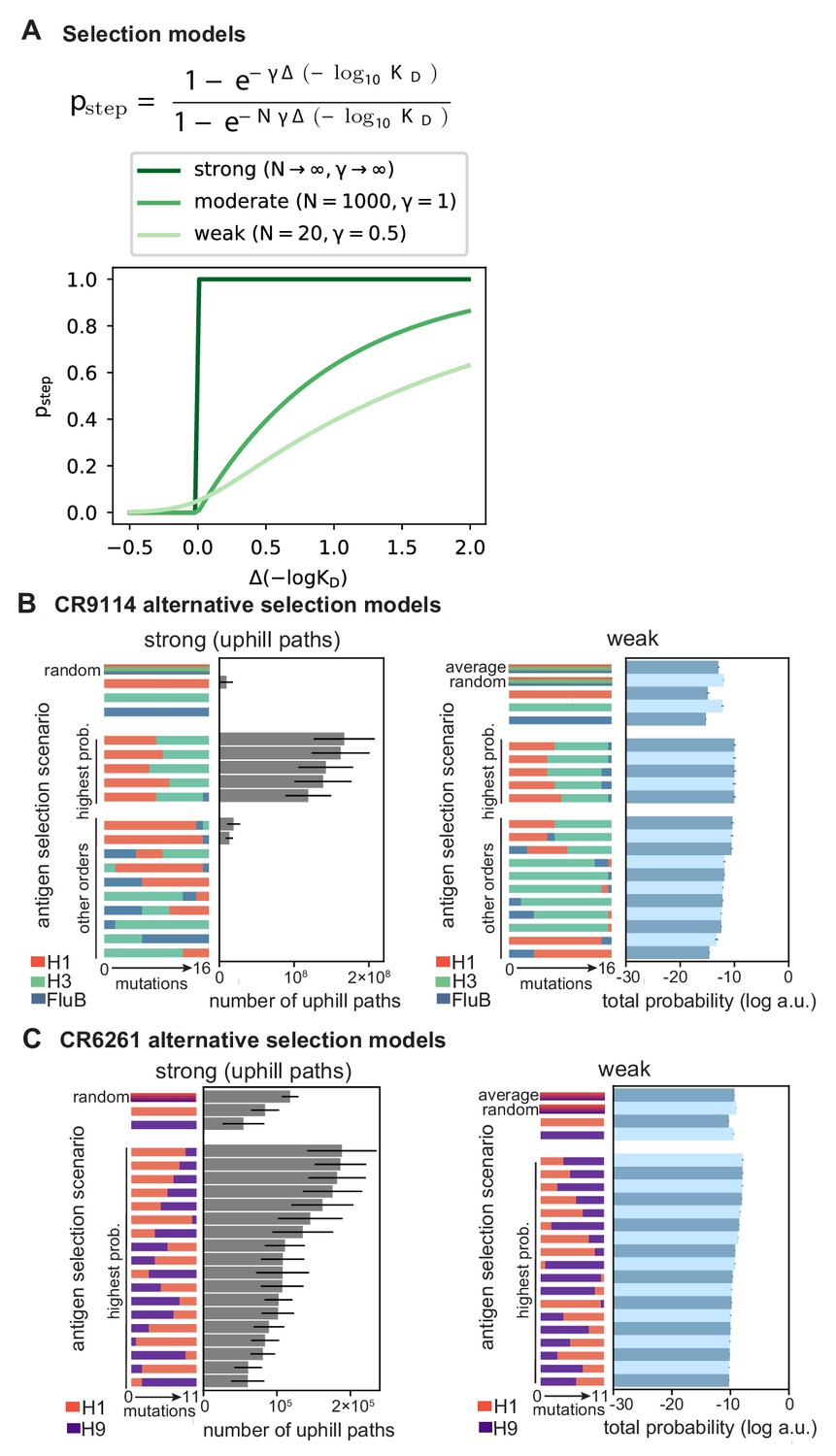
Selection models.
(A) Functional form of mutation step probability, illustrated for parameters chosen to represent strong, moderate, and weak selection models. (B, C) Total log probability of the mutational trajectories between germline and somatic sequences for (B) CR9114 and (C) CR6261 under different antigen selection scenarios, assuming strong (left) or weak (right) selection, as shown for moderate selection in Figure 5E,F. Strong selection scenarios are shown on a linear scale, as total probability is equal to the number of uphill paths. The ‘average’ mixed scenario is not evaluated for strong selection, as the quantitative effect of averaging is undone by the binarizing effect of the transition probability. Error bars indicate standard errors obtained through bootstrap, see Materials and methods.
Figure 5—figure supplement 2
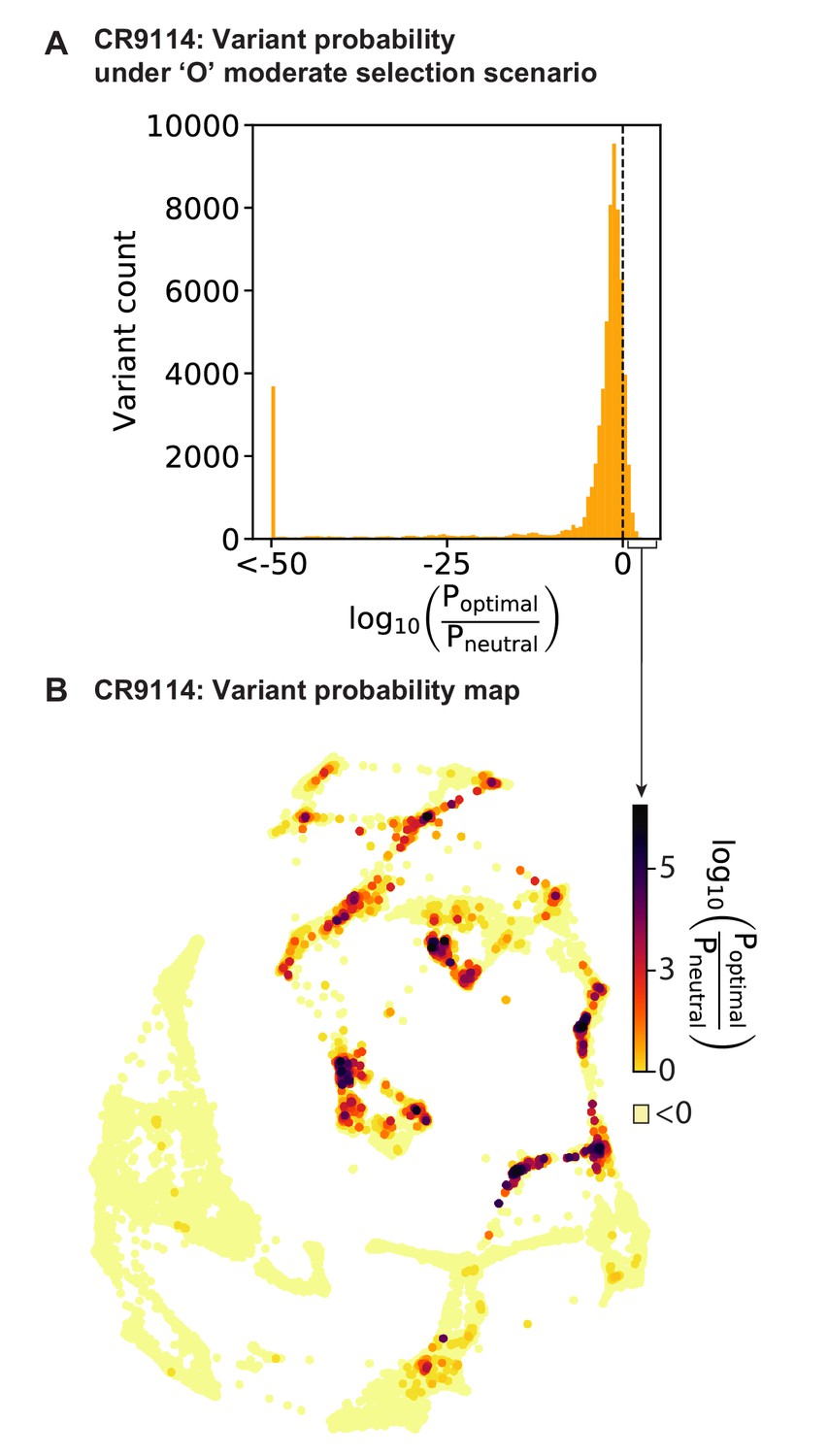
Variant probabilities for CR9114 under the optimal (’O’) selection model.
(A) Histogram of the total probability of all pathways passing through each variant in the optimal selection scenario, divided by the total probability in a model with no selection, transformed to log10 scale (see Materials and methods). Dotted line indicates the 11% of variants favored in the selective model (log probability ratio greater than zero). (B) Favored variants are shown on the force-directed graph for CR9114 H1 –logKD, as in Figure 1G, with darker color according to the log probability ratio. Other variants with log probability ratio less than zero are shown in light yellow.
Figure 5—figure supplement 3
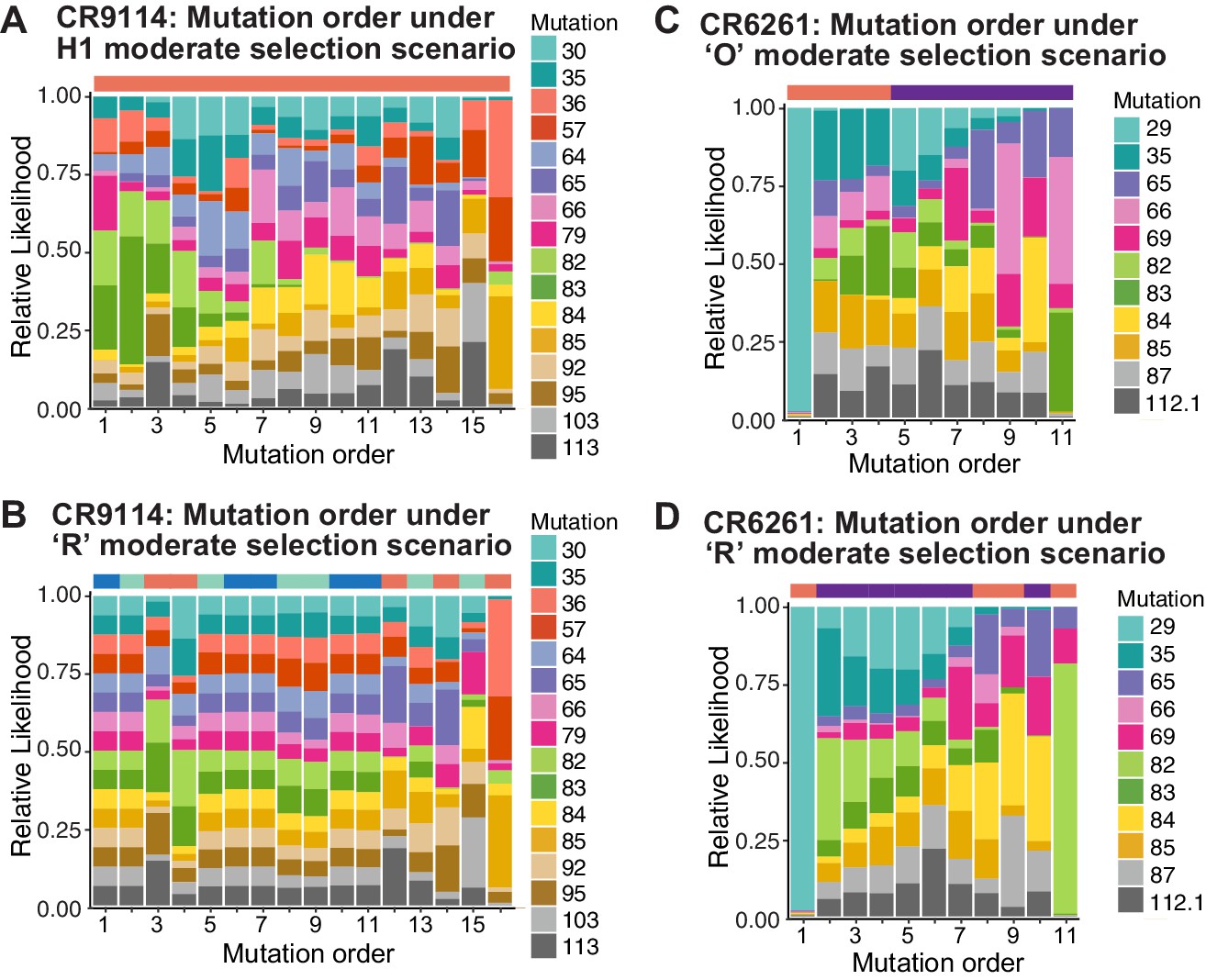
Probability of mutation order assuming moderate selection, under other antigen selection scenarios.
H1 (A) and 'R' (B) for CR9114 and 'O' (C) and 'R' (D) for CR6261, as in Figure 5I,J. For the random mixed scenario 'R', the representative cases from Figure 5G,H are shown.
Appendix 1—figure 1

Correlation between from ML inference on Tite-Seq data vs. from isogenic flow cytometry.
to H1 (salmon), H3 (green), and Flu B (blue) shown for select variants, identical to those shown in Figure 1—figure supplement 2B. Pearson’s r = 0.97.
Appendix 1—figure 2

Distributions of PE-A fluorescence (HA binding) for isogenic CR9114 strains incubated with H3.
PE-A fluorescence distributions from flow cytometry of isogenic CR9114 germline (left) and somatic (right) strains following incubation with 1 μM, 100 nM, and 10 nM H3, as described in Methods. Shape of distribution varies for different clones and is not strictly log-normal, hence deviating from assumptions made in the maximum-likelihood binding affinity inference.
Appendix 2—figure 1
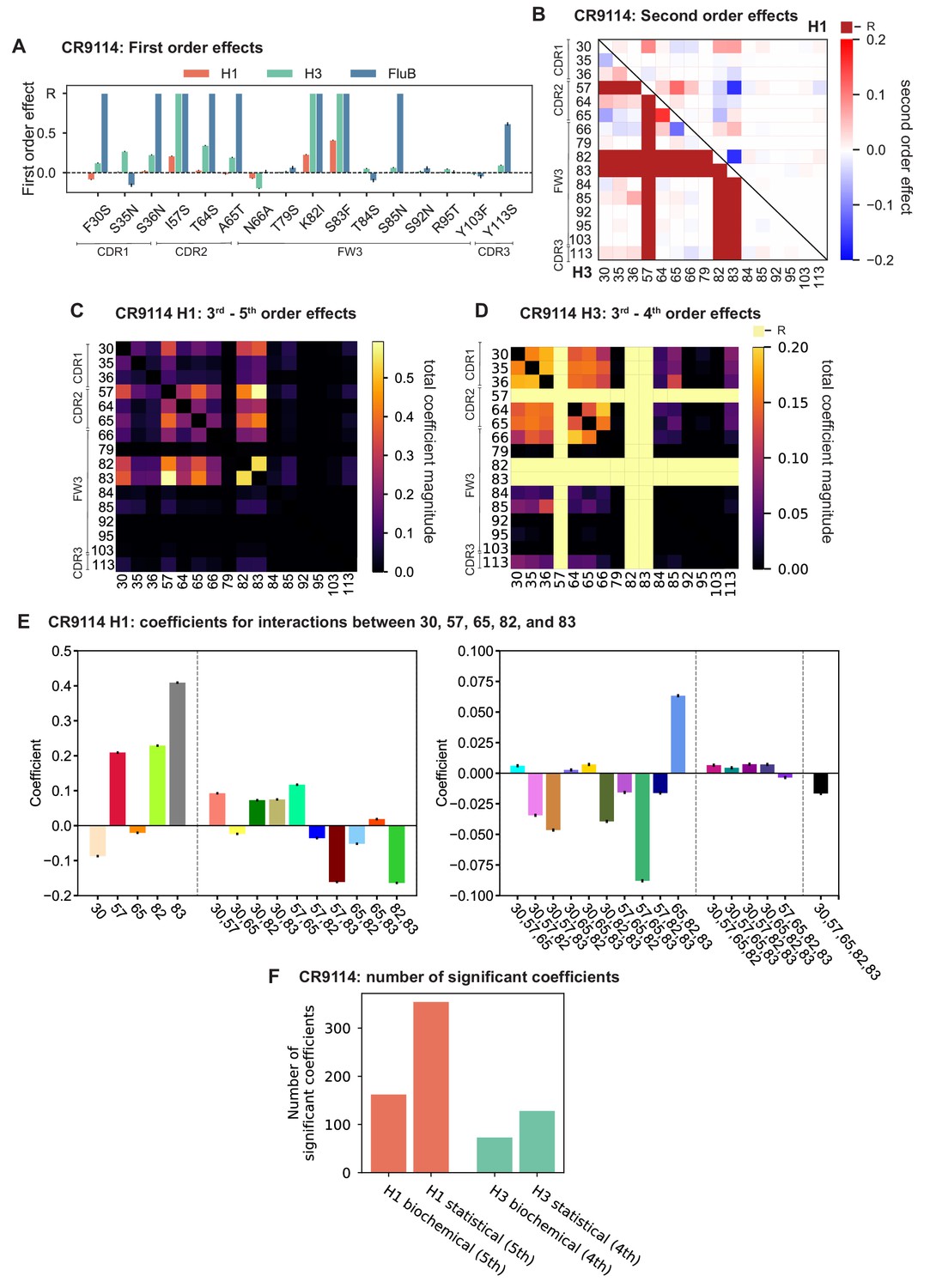
Results from statistical epistasis models for CR9114.
(A) First-order effects, as in Figure 2A. ‘R’ indicates required mutations. (B) Second-order effects for H1 (top right) and H3 (lower left), as in Figure 2D. Interactions with required mutations for H3 are noted in dark red. (C) Cumulative higher-order effects for CR9114 binding to H1, as in Figure 3A. (D) Cumulative higher-order effects for CR9114 binding to H3, as in Figure 3—figure supplement 3. (E) Inferred interaction coefficients for the set of five key epistatic loci, as in Figure 3—figure supplement 1B with corresponding colors. Note the different y-axis scales for the two subplots. Different interaction orders are separated by dotted lines. (F) Number of significant coefficients at all orders for the biochemical and statistical epistasis models. The maximal order of interaction for each model is indicated in parentheses.
Appendix 2—figure 2
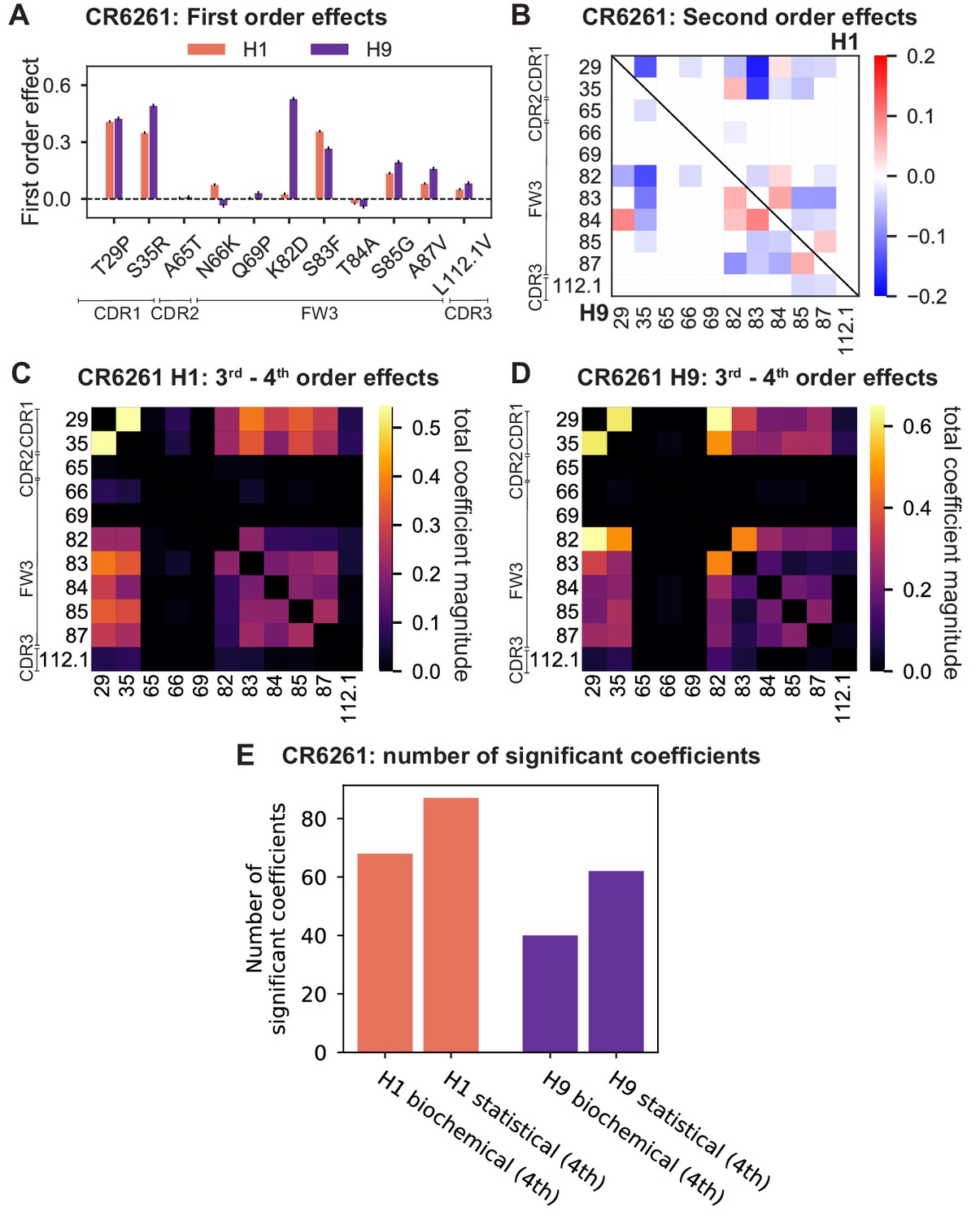
Results from statistical epistasis models for CR6261.
(A) First-order effects, as in Figure 2B. (B) Second-order effects for H1 (top right) and H9 (lower left), as in Figure 2E. (C) Cumulative higher-order effects for CR6261 binding to H1, as in Figure 4A. (D) Cumulative higher-order effects for CR9114 binding to H9, as in Figure 4—figure supplement 2A. (E) Number of significant coefficients at all orders for the biochemical and statistical epistasis models. The maximal order of interaction for each model is indicated in parentheses.
Appendix 2—figure 3
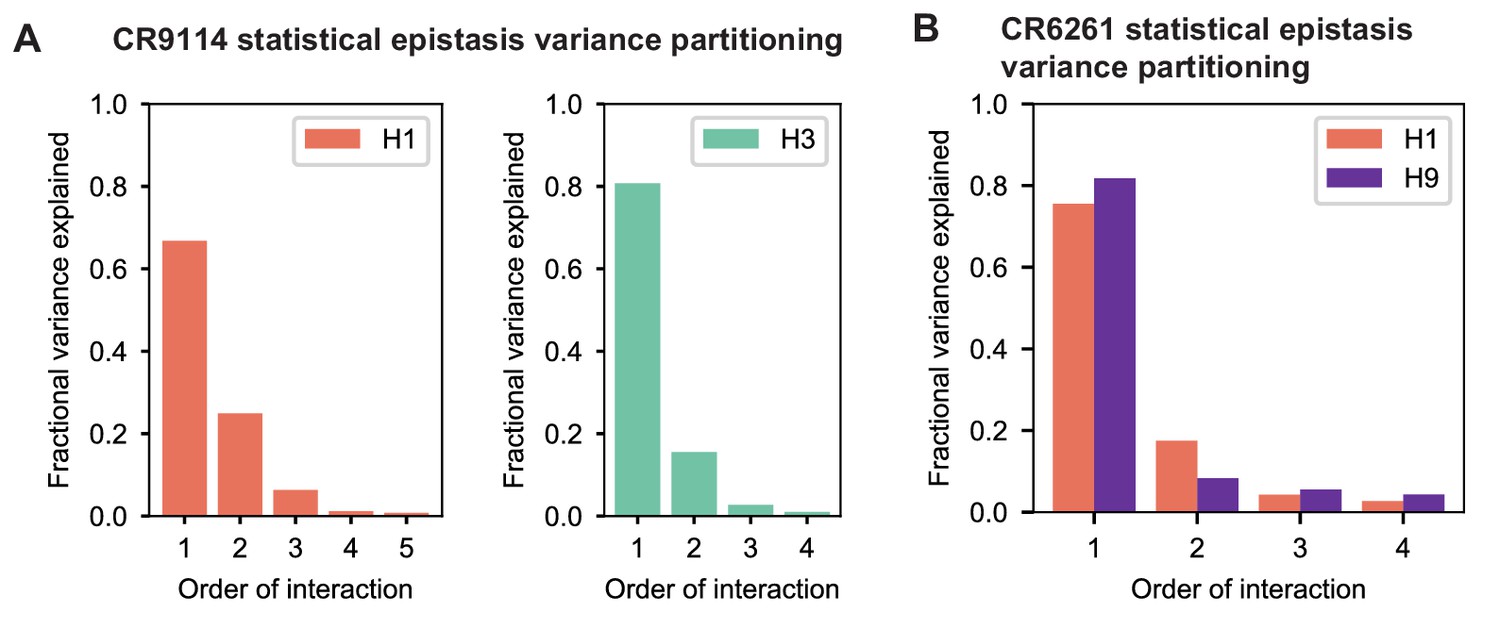
Variance partitioning of statistical epistasis models.
(A) Variance partitioning for CR9114 binding to H1 (left) and H3 (right). (B) Variance partitioning for CR9114 binding to H1 and H9, denoted by colors as indicated.
Appendix 2—figure 4
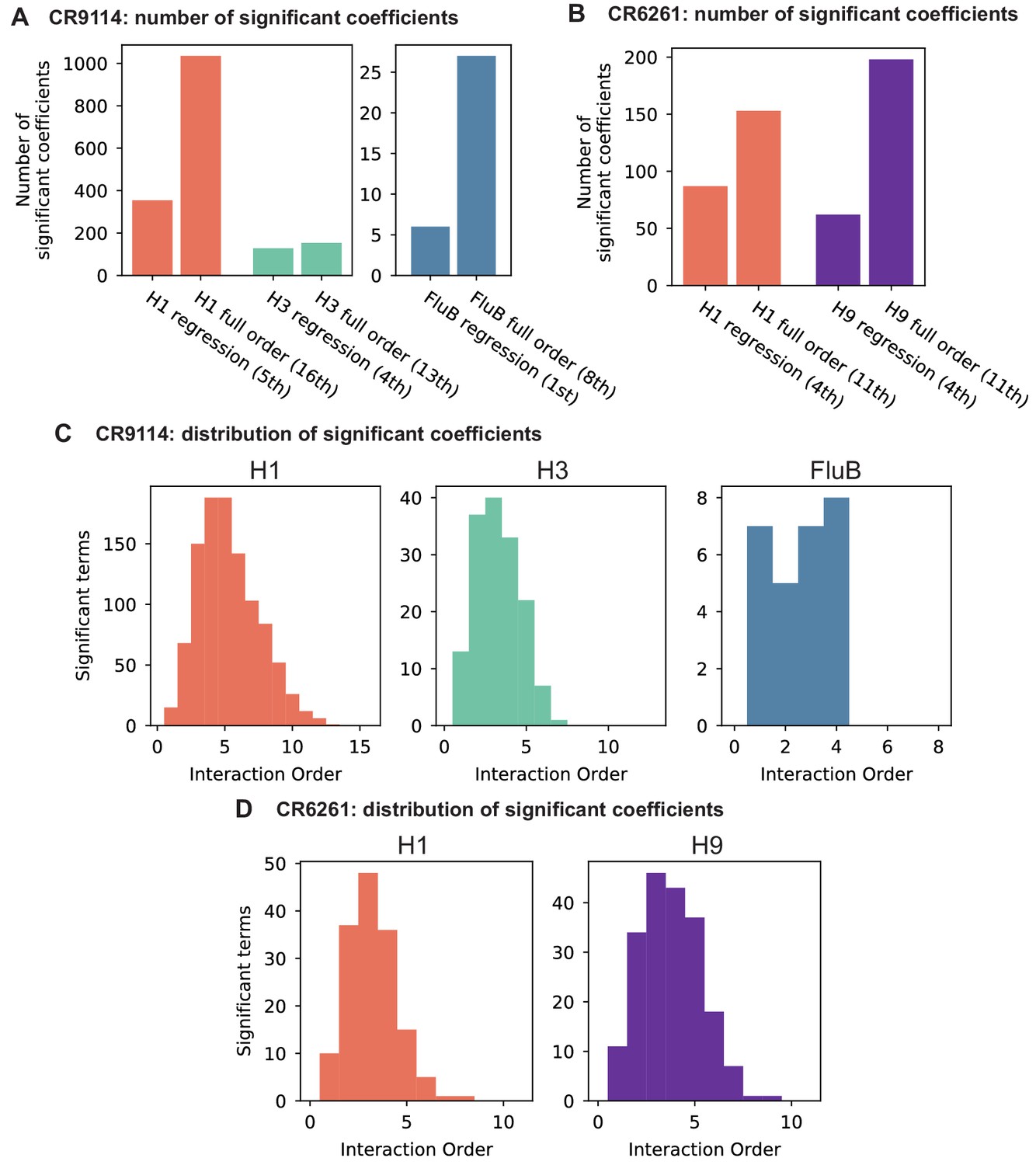
Epistasis inference at full order.
(A,B) Numbers of significant coefficients for the full-order inference compared to optimal truncated regression models for (A) CR9114 and (B) CR6261. Significance for both model types is determined by with Bonferroni correction by the number of model parameters. (C,D) Distribution of interaction orders of significant coefficients for (C) CR9114 and (D) CR6261.
Appendix 2—figure 5
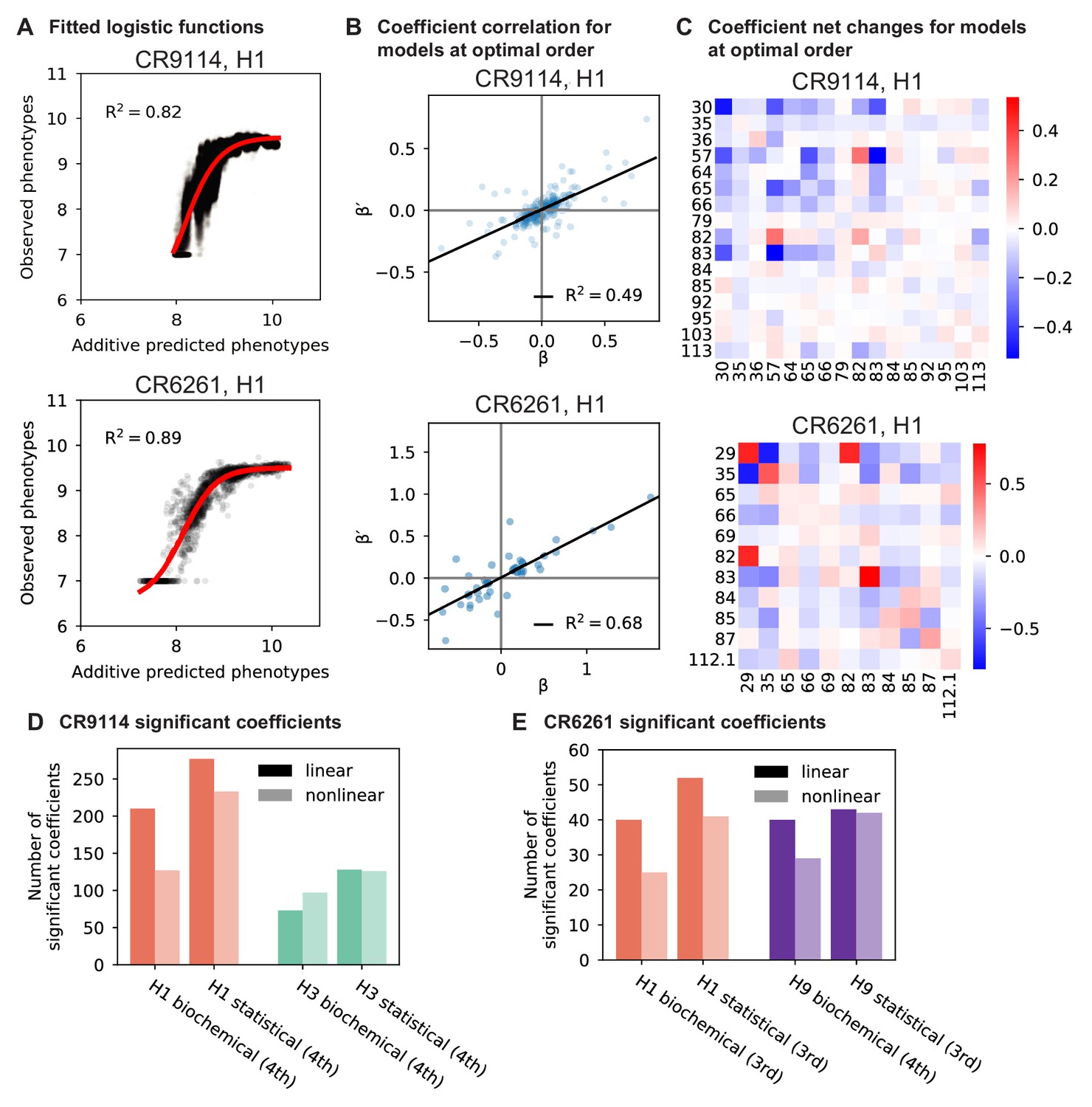
Results from epistasis models with nonlinear transformations.
(A), Fitting logistic functions to additive predicted phenotypes. Red lines indicate the optimized logistic function , with as indicated. (B), Scatterplot of coefficients from the optimal order model inferred on linearized data (after inverting the best-fit nonlinear transformation) against original coefficients for the model with the same maximum order. (C), Net changes of coefficients by site. Diagonal cells show changes in linear coefficients. Off-diagonal cells show the sum of changes over terms at all orders (2nd and above) in which the given pair of mutations is involved. For (A-C), we show two representative antibody-antigen combinations: CR9114 binding to H1, top, and CR6261 binding to H1, bottom. (D,E), Number of significant coefficients in optimal order models fit to phenotypes transformed by the inverse nonlinear function (light bars), compared to original coefficients from linear models with the same maximal order (dark bars), for (D) CR9114 and (E) CR6261. The epistasis type and model order are indicated on the x-axis.
Tables
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Saccharomyces cerevisiae) | EBY100 | ATCC | Cat#:MYA-4941 | |
| Cell line (Spodoptera frugiperda) | Sf9 | ThermoFisher | Cat#:B82501 | Cell line for production of baculovirus |
| Cell line (Trichoplusia ni) | High-Five | ThermoFisher | Cat#:B85502 | Cell line for HA expression |
| Antibody | Anti-cMyc-FITC (Mouse monoclonal) | Miltenyi Biotec | Cat#:130-116-485 | FACS (1:50) |
| Recombinant DNA reagent | pCT302 (plasmid) | Addgene | Cat#:41845 | |
| Recombinant DNA reagent | pCT302_CR9114 _germline (plasmid) | This paper | Plasmid map in Supplementary file 4 | |
| Recombinant DNA reagent | pCT302_CR9114 _somatic (plasmid) | This paper | Plasmid map inSupplementary file 5 | |
| Recombinant DNA reagent | pCT302_CR6261 _germline (plasmid) | This paper | Plasmid map inSupplementary file 6 | |
| Recombinant DNA reagent | pCT302_CR6261 _somatic (plasmid) | This paper | Plasmid map inSupplementary file 7 | |
| Recombinant DNA reagent | pET21a-BirA (plasmid) | Addgene | Cat#:20857 | |
| Sequence-based reagent | CR9114 golden gate dsDNA fragments | IDT | Sequences listed inSupplementary file 2 | |
| Sequence-based reagent | CR6261 Golden Gate primers | IDT | Sequences listed inSupplementary file 3 | |
| Sequence-based reagent | Illumina sequencing primers | IDT | Sequences listed inSupplementary file 1 | |
| Peptide, recombinant protein | Streptavidin-RPE | ThermoFisher | Cat#:S866 | FACS (1:100) |
| Peptide, recombinant protein | Biotinylated A/New Caledonia/99 (H1) ectodomain | This paper | Plasmid sequence inSupplementary file 8 | |
| Peptide, recombinant protein | Biotinylated A/Hong Kong/99 (H9) ectodomain | This paper | Plasmid sequence inSupplementary file 9 | |
| Peptide, recombinant protein | Biotinylated A/Wisconsin/05 (H3) ectodomain | This paper | Plasmid sequence inSupplementary file 10 | |
| Peptide, recombinant protein | Biotinylated B/Ohio/05 (Flu B) ectodomain | This paper | Plasmid sequence inSupplementary file 11 | |
| Commercial assay or kit | Bac-to-Bac Kit | ThermoFisher | Cat#:10359016 | |
| Commercial assay or kit | Zymo Yeast Plasmid Miniprep II | Zymo Research | Cat#:D2004 | |
| Software, algorithm | Custom code | This paper | https://github.com/klawrence26/bnab-landscapes (copy archived at swh:1:rev:61c1673a101ea739d5b7e9b282f6bcfad41d7e90, Phillips, 2021) | |
| Software, algorithm | Interactive CR9114 data browser | This paper | https://yodabrowser.netlify.app/yoda_browser/ |
Additional files
-
Supplementary file 1
Primer sequences for sequencing library preparation.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp1-v3.xlsx
-
Supplementary file 2
Fragment sequences for Golden Gate construction of the CR9114 library.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp2-v3.xlsx
-
Supplementary file 3
Primer sequences for Golden Gate construction of the CR6261 library.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp3-v3.xlsx
-
Supplementary file 4
Plasmid map of pCT302 with CR9114 germline sequence.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp4-v3.dna
-
Supplementary file 5
Plasmid map of pCT302 with CR9114 somatic sequence.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp5-v3.dna
-
Supplementary file 6
Plasmid map of pCT302 with CR6261 germline sequence.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp6-v3.dna
-
Supplementary file 7
Plasmid map of pCT302 with CR6261 somatic sequence.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp7-v3.dna
-
Supplementary file 8
Plasmid map of pFastBac with influenza A/New Caldeonia/1999 H1 ectodomain.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp8-v3.seq
-
Supplementary file 9
Plasmid map of pFastBac with influenza A/Hong Kong/1999 H9 ectodomain.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp9-v3.seq
-
Supplementary file 10
Plasmid map of pFastBac with influenza A/Wisconsin/2005 H3 ectodomain.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp10-v3.seq
-
Supplementary file 11
Plasmid map of pFastBac with influenza B/Ohio/2005 HA ectodomain.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp11-v3.seq
-
Supplementary file 12
Inferred CR9114 VH germline nucleotide sequence.
- https://cdn.elifesciences.org/articles/71393/elife-71393-supp12-v3.dna
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/71393/elife-71393-transrepform-v3.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Binding affinity landscapes constrain the evolution of broadly neutralizing anti-influenza antibodies
eLife 10:e71393.
https://doi.org/10.7554/eLife.71393




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}