A computational screen for alternative genetic codes in over 250,000 genomes
- Department of Molecular and Cellular Biology, Harvard University, United States
- Howard Hughes Medical Institute, Harvard University, United States
- John A Paulson School of Engineering and Applied Sciences, Harvard University, United States
Figures
Figure 1 with 1 supplement

Schematic of the genetic code inference method implemented in Codetta.
(A) A fragment of the Mycoplasma capricolum genome is used to demonstrate alignment of a Pfam domain (PAD_porph) to a preliminary standard code translation of the input DNA sequence (one of six frames shown). All canonical stop codons, including UGA (TGA in genome sequence, reassigned to tryptophan in M. capricolum), are translated as ‘X’ in the preliminary standard code translation that hmmscan (program used to align Pfam domains) treats as an unknown amino acid. Each consensus column in the PAD_porph domain has a characteristic emission probability for each of the 20 canonical amino acids, represented by a heatmap. (B) Pfam consensus columns aligning to UGA codons across the entire genome comprise the set for UGA ( = 452 Pfam consensus columns). The Pfam emission probabilities for all 452 aligned consensus columns are used to compute the decoding probabilities . The most likely amino acid translation of UGA is inferred to be tryptophan, with decoding probability greater than the cutoff of 0.9999.
Figure 1—figure supplement 1
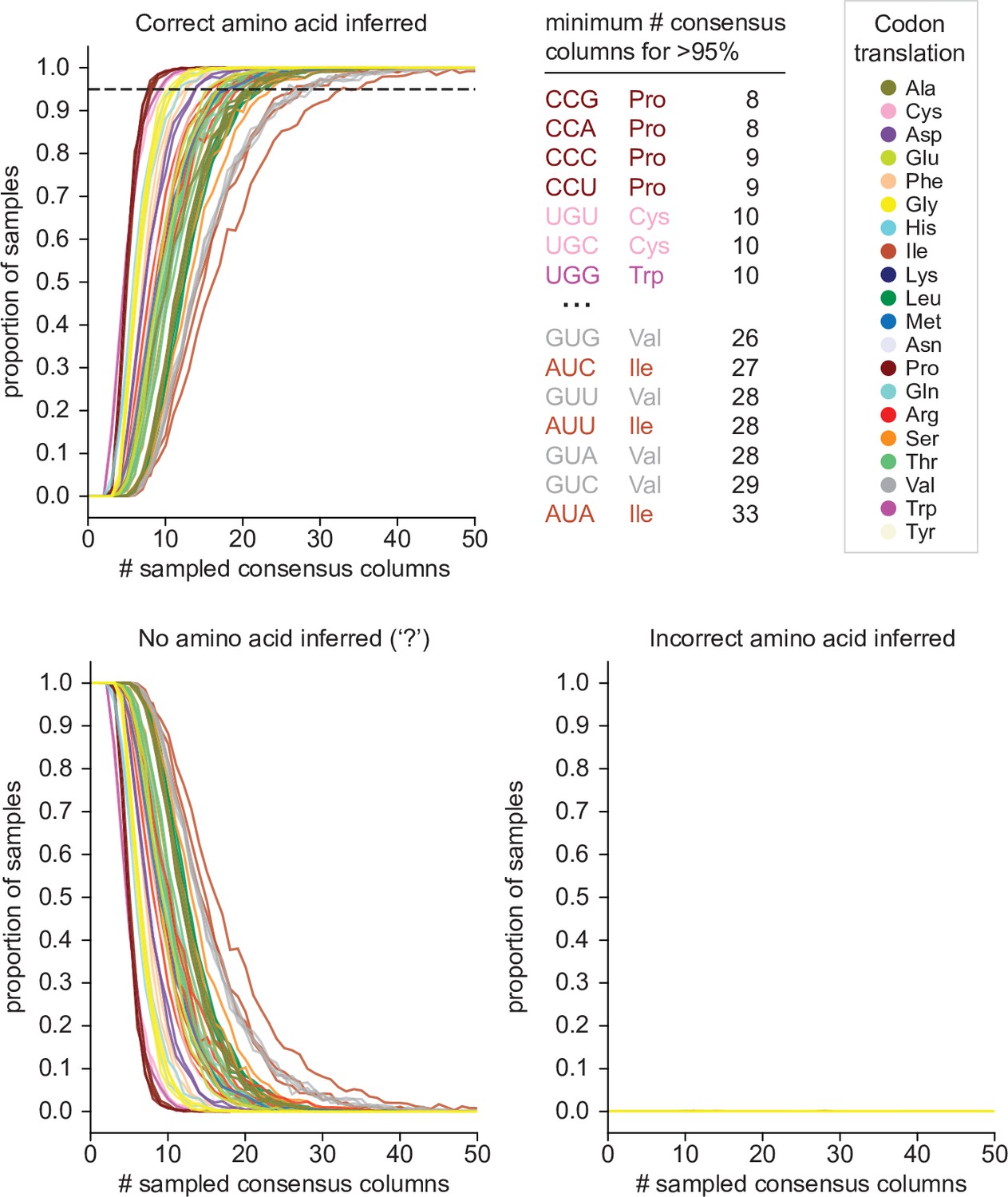
Estimates of error rate and power of Codetta inference.
Codetta error rate and power estimated by subsampling Pfam consensus columns aligned to an E. coli genome. Plots show the proportion of consensus column samples that resulted in the correct inference (power), incorrect inference (error rate), and no amino acid inference (‘?’ inference) under a decoding threshold of 0.9999 for the 61 sense codons, color-coded by the correct amino acid translation. The dashed line indicates 95% of samples with the correct amino acid inferred. The codons that required the fewest and greatest number of consensus columns for >95% correct inference are listed.
Figure 2 with 1 supplement
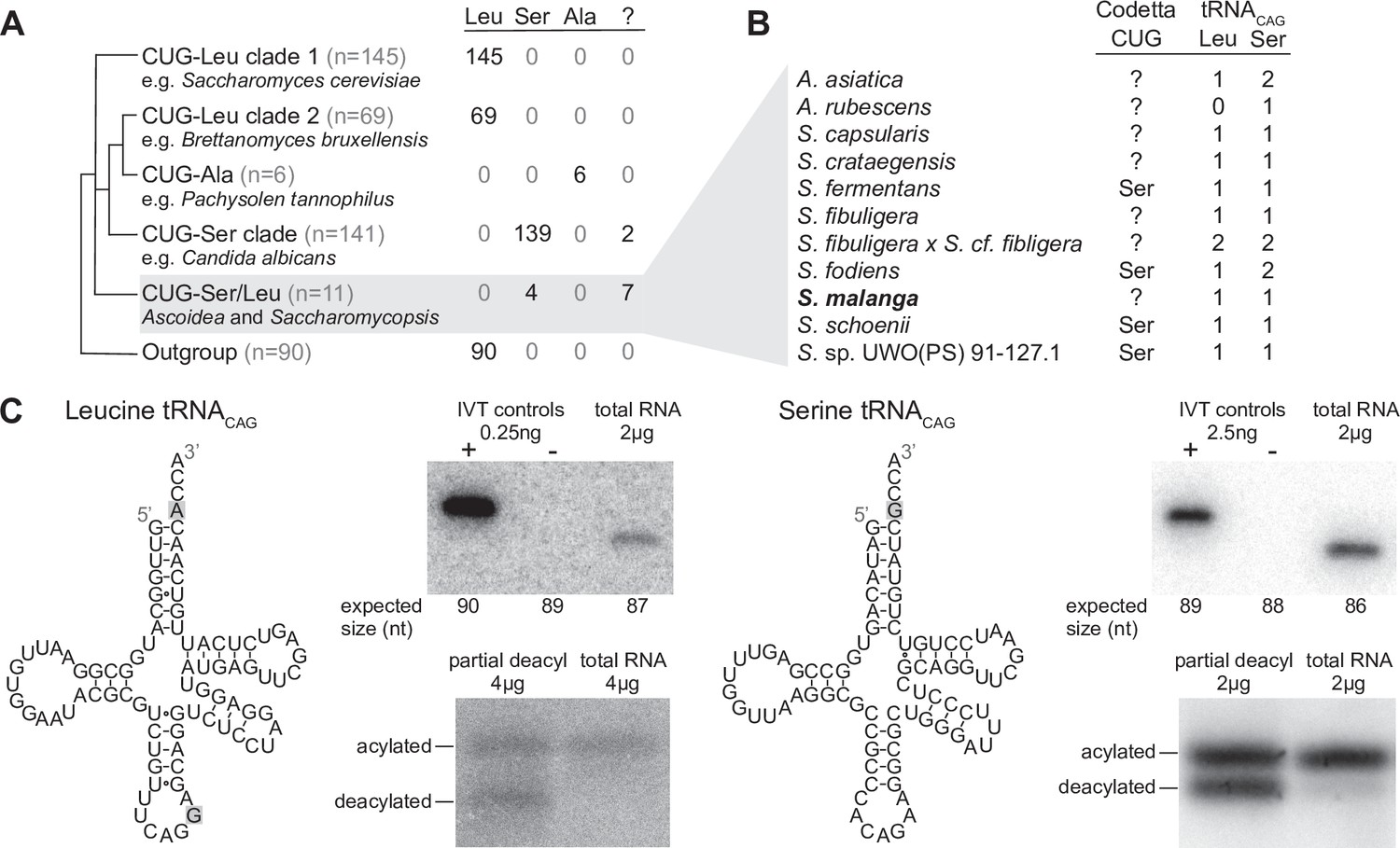
Analysis of CUG reassignments in yeast.
(A) CUG translation inferred by Codetta of 462 Saccharomycetes species, grouped by phylogenetic clade. Cladogram was adapted from Shen et al., 2018. Phylogenetic placement of CUG-Leu/Ser clade is unresolved, thus the three-way branch. (B) Codetta CUG inference and number of tRNACAG genes in Ascoidea and Saccharomycopsis genomes. tRNACAG genes were identified using tRNAscan-SE 2.0 and were classified as being serine-type or leucine-type based on the presence of tRNA identity elements. (C) Northern blotting to confirm expression and charging of leucine and serine tRNACAG genes in S. malanga. Probable secondary structures of the two S. malanga tRNACAG are shown with features used for leucine/serine classification highlighted in gray. In the tRNA expression blots, in vitro transcribed (IVT) versions of the target tRNA (+ control) and the most similar other tRNA (– control, as determined by sequence homology with the probe) were used as controls for probe specificity. In the tRNA charging blots, a partial deacylation control was used to help visualize the expected band sizes for acylated and deacylated versions of the probed tRNA.
-
Figure 2—source data 1
Table of all analyzed yeast genomes with phylogenetic grouping and Codetta CUG inference.
- https://cdn.elifesciences.org/articles/71402/elife-71402-fig2-data1-v3.xls
-
Figure 2—source data 2
Tarball of original blot images.
- https://cdn.elifesciences.org/articles/71402/elife-71402-fig2-data2-v3.gz
-
Figure 2—source data 3
Table of in vitro transcription DNA template sequences.
- https://cdn.elifesciences.org/articles/71402/elife-71402-fig2-data3-v3.xlsx
Figure 2—figure supplement 1
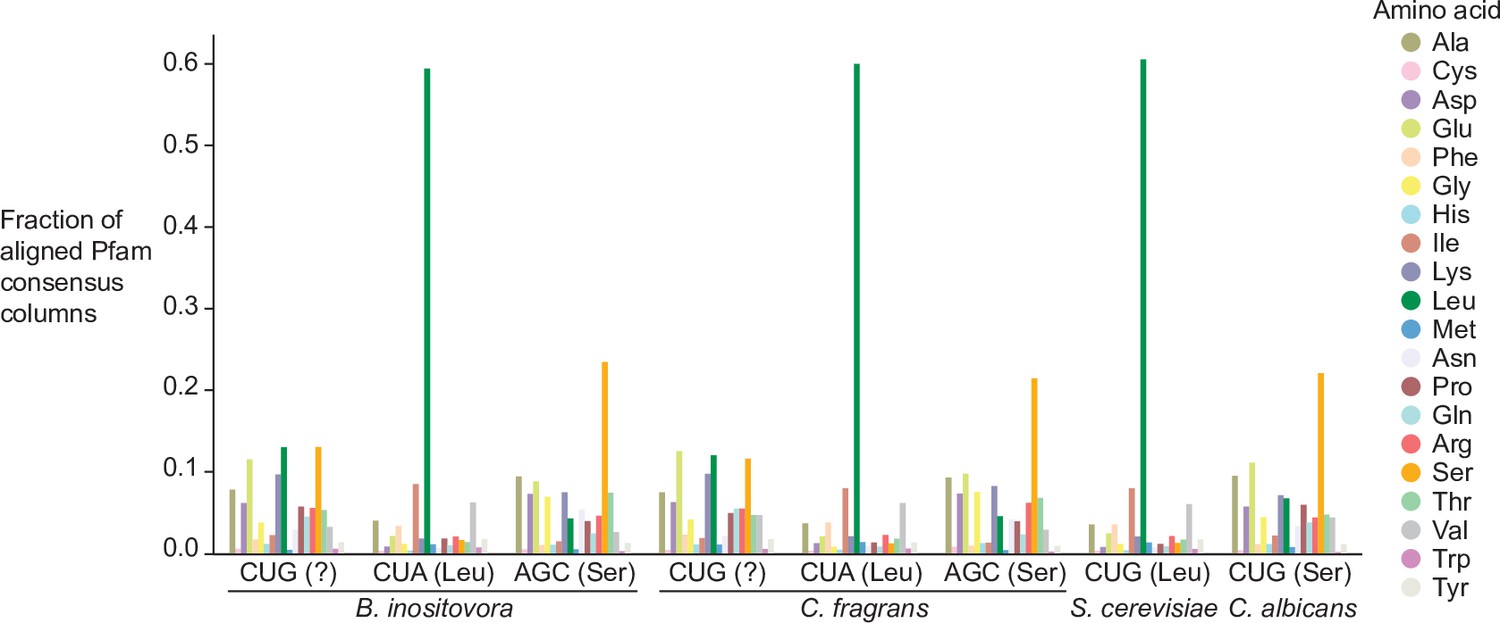
Distribution of Pfam consensus column amino acid support for CUG codons in B. inositovora and C. fragrans.
Distribution of the highest probability amino acid for all aligned Pfam consensus columns to CUG, CUA (rare leucine codon), and AGC (rare serine codon) in B. inositovora and C. fragrans and to CUG in S. cerevisiae and C. albicans. Codetta codon inference is labeled in parentheses.
Figure 3 with 1 supplement
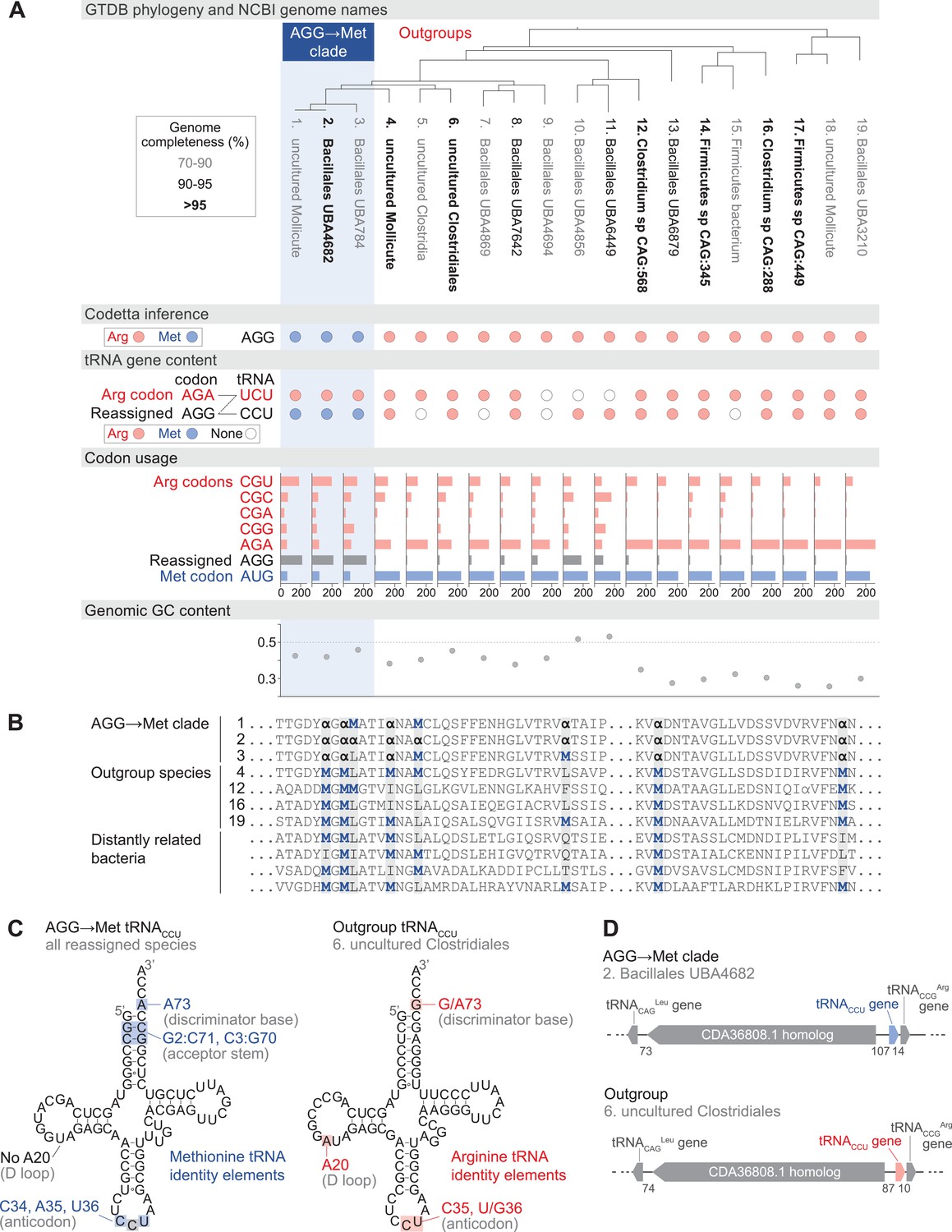
Reassignment of AGG from arginine to methionine in a clade of uncultivated Bacilli.
(A) Genome Taxonomy Database (GTDB) phylogenetic tree of the Bacilli AGG→Met clade and closest outgroup genomes, with the annotated NCBI genome name shaded according to the GTDB CheckM estimated genome completeness. GTDB genus UBA7642 corresponds to species #1–9, and GTDB family CAG-288 corresponds to species #1–16. For each genome, the Codetta AGG inference is indicated by colored circles (red: arginine; blue: methionine). The presence of tRNA genes is also indicated by filled circles for tRNAUCU and tRNACCU, colored by the predicted amino acid charging based on known identity elements (see Materials and methods), or a white circle if no tRNA gene could be detected. The lines connecting codons and anticodons represent the likely decoding capabilities, with dashed lines representing likely weaker interactions. Codon usage is the frequency per 10,000 codons aligned to Pfam domains. (B) Multiple sequence alignment of uridylate kinase (BUSCO POG091H02JZ) from the reassigned species, selected outgroup species, and four more distantly related bacteria (Bacillus subtilis, Nostoc punctiforme, Chlamydia caviae, and Escherichia coli). All AGGs are represented by α. Alignment regions containing multiple nearby AGG positions in the reassigned species are shown. (C) A comparison of the AGG-decoding tRNACCU in the Bacilli AGG→Met clade (identical sequence in all genomes) and in an outgroup genome (#6, uncultured Clostridiales). tRNA sequence features involved in methionine identity in the reassigned clade tRNACCU and arginine identity in the outgroup tRNA are highlighted (Meinnel et al., 1993; Giegé et al., 1998), with nucleotide numbering following the convention of Sprinzl et al., 1998. The C35 anticodon nucleotide in the AGG→Met clade tRNA is highlighted in gray because it does not match the A35 methionine identity element. (D) The genomic context surrounding the tRNACCU gene in a member of the Bacilli AGG→Met clade (#2, Bacillales UBA4682) and in an outgroup species (#6, uncultured Clostridiales). Gene lengths and intergenic distances are drawn proportionally, with the number of base pairs between each gene indicated below.
-
Figure 3—source data 1
Table of genome accessions, Codetta AGG inference, tRNA gene presence, codon usage, and genome GC content for the reassigned AGG→Met Bacilli and outgroup species.
- https://cdn.elifesciences.org/articles/71402/elife-71402-fig3-data1-v3.xlsx
Figure 3—figure supplement 1
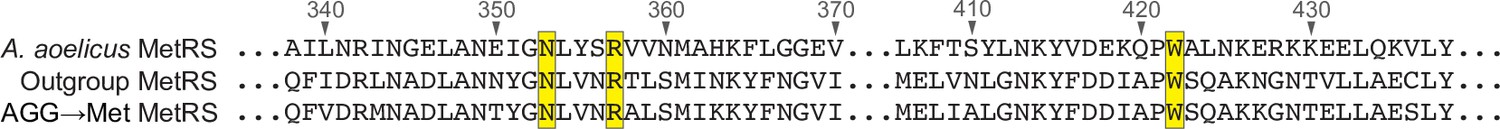
Alignment of MetRS sequences.
A multiple sequence alignment of the methionyl-tRNA synthetase (MetRS) sequence from Aquifex aeolicus and the predicted MetRS sequences from the outgroup species #6 (GCA_900316035.1) and Bacilli AGG→Met clade species #2 (GCA_002404995.1). Residues are numbered in reference to the A. aeolicus sequence. Residues in contact with the tRNA anticodon in the A. aeolicus crystal structure (PDB 2CSX/2CT8, Nakanishi et al., 2005) are highlighted in yellow.
Figure 4 with 4 supplements

Summary of GC content, codon usage, and tRNA genes of four CGA and/or CGG reassignments.
(A) Distribution of genomic GC contents across all species in the reassigned clades. (B) For each reassigned clade, we selected a representative species to show codon usage and tRNA decoding ability. Codon usage is plotted for the reassigned codon and for all other codons of the original and new amino acids in usage per 10,000 in Pfam alignments. Codons are colored by their Codetta inference and reassigned codons are bolded. The lines connecting codons and tRNA anticodons represent the likely decoding capabilities, with dashed lines representing likely weaker interactions. The anticodon ACG is presumed to be modified to ICG, and UCG is presumed to be modified in a way that restricts wobble to CGA and CGG, but could potentially recognize CGU and CGC as well depending on the true modification state. Anticodons in gray font are not expected to be found in the respective clade. Presence of tRNA genes is indicated by filled circles, colored by the predicted amino acid charging based on the identity elements in the key.
-
Figure 4—source data 1
Table for each CGA/CGG reassignment containing genome accessions, Codetta CGA/CGG inference, tRNA gene presence, codon usage, and genome GC content for the reassigned clade and outgroup species.
- https://cdn.elifesciences.org/articles/71402/elife-71402-fig4-data1-v3.xlsx
Figure 4—figure supplement 1
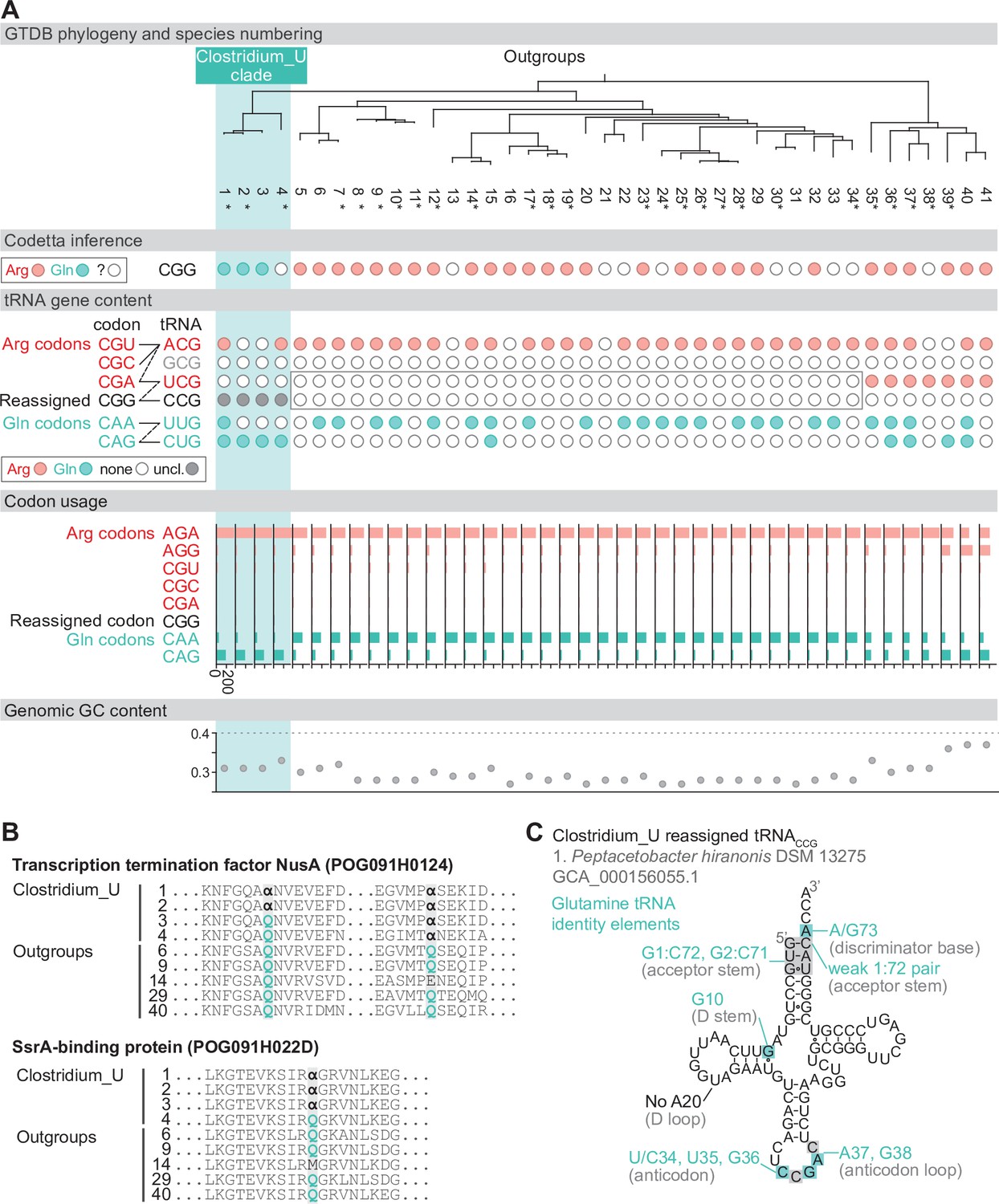
Reassignment of CGG→Gln in Peptacetobacter.
(A) Genome Taxonomy Database (GTDB) phylogenetic tree of Peptacetobacter (Clostridium_U in GTDB) and closest outgroup genomes. Species numbers can be cross-referenced with Figure 4—source data 1. We consider the entire Clostridium_U clade to have reassigned CGG to glutamine due to the presence of an almost identical CGG-decoding tRNACCG in all four species. Asterisks indicate genomes with GTDB CheckM estimated genome completeness >99%. For each species, the Codetta CGG inference is indicated by colored circles (red: arginine; light blue: glutamine; white: ‘?’). The presence of tRNA genes that recognize the CAR- and CGN-codons is indicated by filled circles, colored according to the predicted amino acid charging based on based on identity elements for tRNAs (see Materials and methods). A gray box outlines the inability to locate any CGG-decoding tRNAs in the Peptostreptococcaceae (species #5–34). The lines connecting codons and tRNA anticodons represent the likely decoding capabilities, with dashed lines representing weaker interactions. The anticodon ACG is presumed to be modified to ICG, and the U34 of UCG is presumed to be modified in a way that restricts wobble to CGA and CGG, but could potentially recognize CGU and/or CGC depending on the true modification state. Codon usage is the frequency per 10,000 codons aligned to Pfam domains. (B) Multiple sequence alignments of transcription termination factor NusA (BUSCO POG091H0124) and SsrA-binding protein (BUSCO POG091H022D) from the Clostridium_U clade and selected outgroup species. Alignment regions containing nearby CGG (α) positions are shown, with columns with CGG in Clostridium_U sequences highlighted. (C) The CGG-decoding tRNACCG from species #1 (Peptacetobacter hiranonis DSM 13275, GCA_000156055.1). tRNA sequence features involved in glutamine identity are highlighted (Jahn et al., 1991; Hayase et al., 1992), with nucleotide numbering following the convention of Sprinzl et al., 1998. Nucleotides highlighted in gray do not match the expected identity element.
Figure 4—figure supplement 2
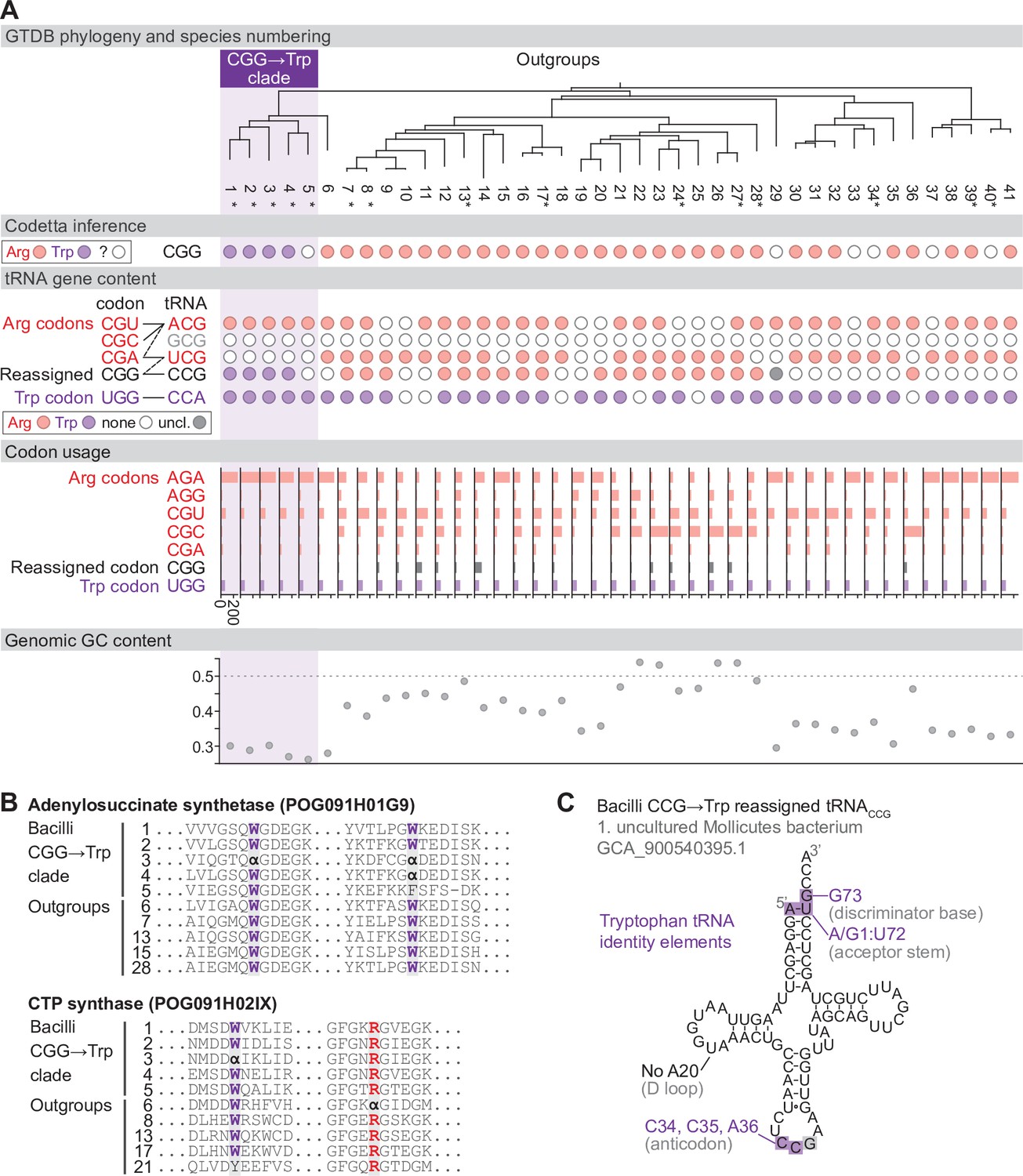
Reassignment of CGG→Trp in a clade of Bacilli.
(A) Genome Taxonomy Database (GTDB) phylogenetic tree of the Bacilli CGG→Trp clade and closest outgroup genomes. Species numbers can be cross-referenced with Figure 4—source data 1. We consider species #5 to be part of the reassigned clade due to the tree topology. Asterisks indicate genomes with GTDB CheckM estimated genome completeness >95%. For each species, the Codetta CGG inference is indicated by colored circles (red: arginine; purple: tryptophan; white: uninferred). The presence of tRNA genes that recognize the UGG and CGN-codons is indicated by filled circles, colored according to the predicted amino acid charging based on identity elements for tRNAs (see Materials and methods). The lines connecting codons and tRNA anticodons represent the likely decoding capabilities, with dashed lines representing weaker interactions. The anticodon ACG is presumed to be modified to ICG. The U34 of anticodon UCG is presumed to be modified in a way that restricts decoding to CGA and CGG, but could potentially recognize CGU and/or CGC depending on the true modification state. Codon usage is the frequency per 10,000 codons aligned to Pfam domains. (B) Multiple sequence alignments of adenylosuccinate synthetase (BUSCO POG091H01G9) and CTP synthase (BUSCO POG091H02IX) from the Bacilli CGG→Trp clade and selected outgroup species. Alignment regions containing CGG () at conserved positions are shown, with columns with CGG in Bacilli CGG→Trp clade and the closest outgroup (species #6) sequences highlighted. (C) The CGG-decoding tRNACCG from species #1 (uncultured Mollicutes bacterium, GCA_900540395.1). tRNA sequence features involved in tryptophan identity are highlighted (Giegé et al., 1998; Himeno et al., 1991), with nucleotide numbering following the convention of Sprinzl et al., 1998. Nucleotides highlighted in gray do not match the expected identity element.
Figure 4—figure supplement 3
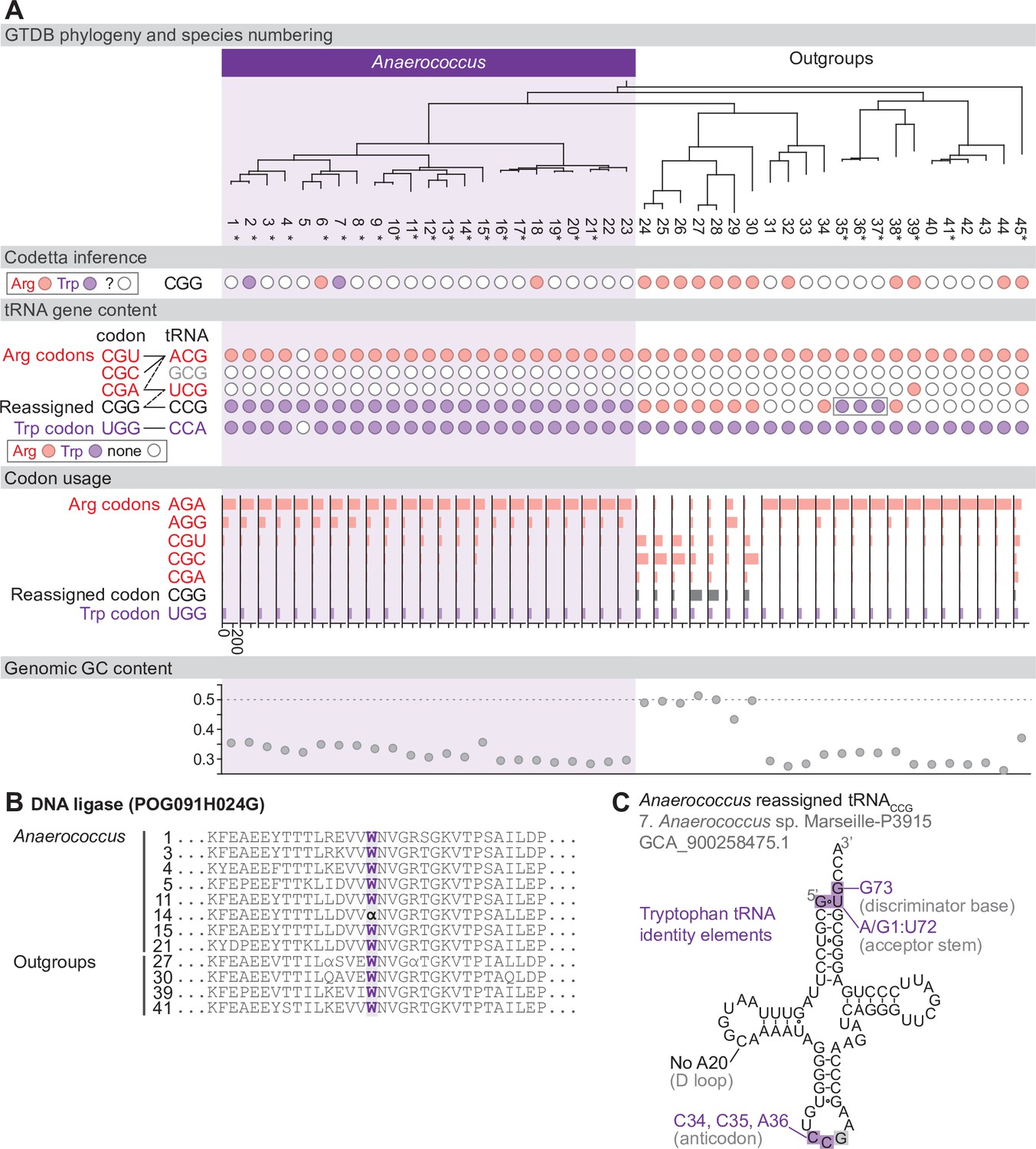
Reassignment of CGG→Trp in Anaerococcus.
(A) Genome Taxonomy Database (GTDB) phylogenetic tree of Anaerococcus and closest outgroup genomes. Species numbers can be cross-referenced with Figure 4—source data 1. We considered the entire Anaerococcus clade to have reassigned CGG to tryptophan due to the presence of a tryptophan-like tRNACCG in all Anaerococcus species. Asterisks indicate genomes with GTDB CheckM estimated genome completeness >98%. For each species, the translation of the reassigned codon CGG inferred by Codetta is indicated by colored circles (red: arginine; purple: tryptophan; white: ‘?’). The presence of tRNA genes that recognize the UGG and CGN-codons is also indicated by filled circles, colored according to the predicted amino acid charging based on identity elements for tRNAs (see Materials and methods). A gray box outlines the tRNACCG in Finegoldia, which has features of tryptophan identity. The lines connecting codons and tRNA anticodons represent the likely decoding capabilities, with dashed lines representing weaker interactions. The anticodon ACG is presumed to be modified to ICG. The U34 of anticodon UCG is presumed to be modified in a way that restricts decoding to CGA and CGG, but could potentially recognize CGU and/or CGC depending on the true modification state. Codon usage is the frequency per 10,000 codons aligned to Pfam domains. (B) Region of a multiple sequence alignment of DNA ligase (BUSCO POG091H024G) from Anaerococcus species and selected outgroup species, containing a CGG (α) at a conserved position in a single Anaerococcus species. (C) The CGG-decoding tRNACCG from species #7 (Anaerococcus sp. Marseille-P3915, GCA_900258475.1). tRNA sequence features involved in tryptophan identity are highlighted (Giegé et al., 1998; Himeno et al., 1991), with nucleotide numbering following the convention of Sprinzl et al., 1998. Nucleotides highlighted in gray do not match the expected identity element.
Figure 4—figure supplement 4
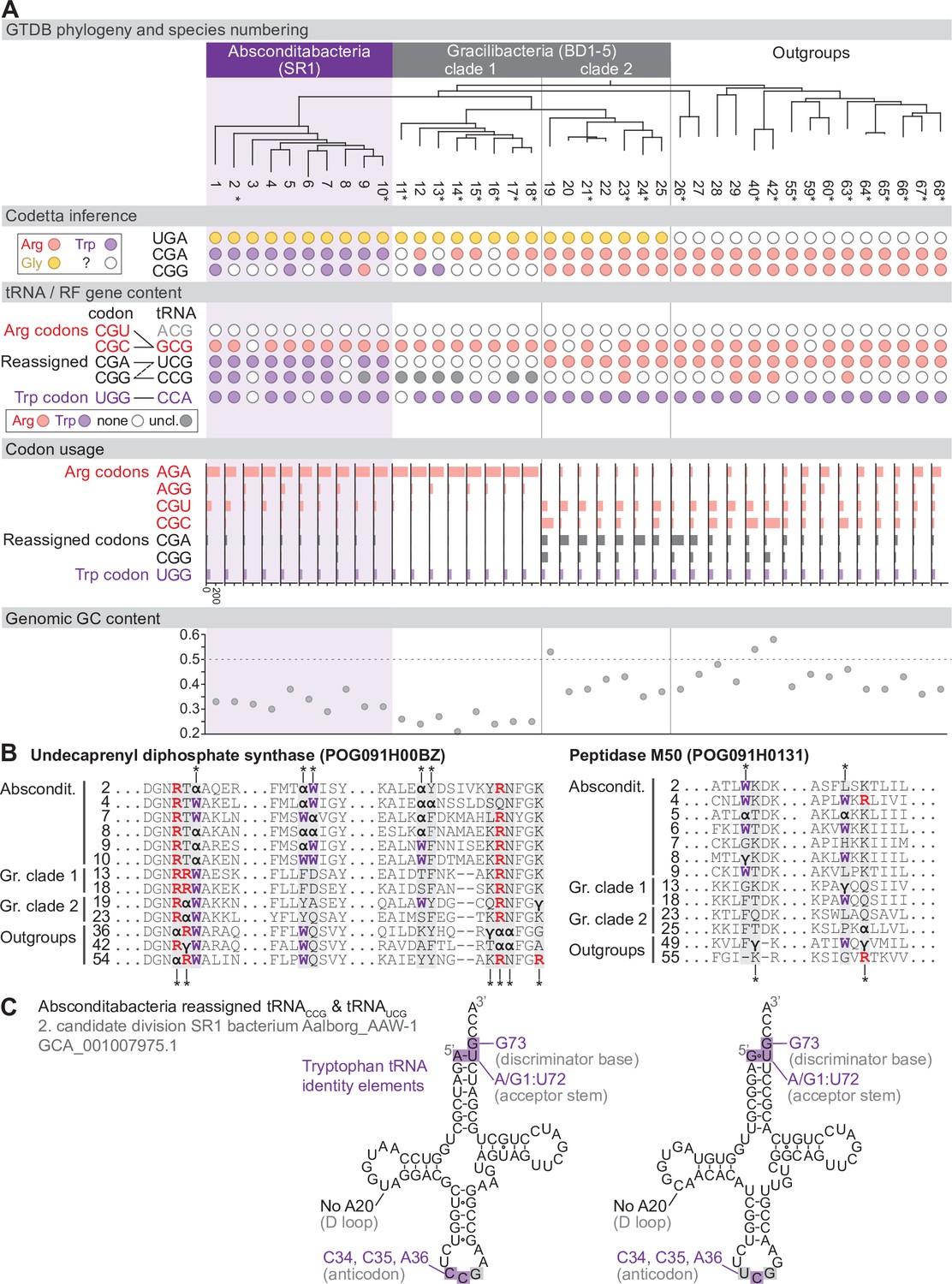
Reassignment of CGA and CGG→Trp in Absconditabacteria.
(A) Genome Taxonomy Database (GTDB) phylogenetic tree of Absconditabacteria, Gracilibacteria, and closest outgroup genomes. Species numbers can be cross-referenced with Figure 4—source data 1. We considered the entire Absconditabacteria clade to have reassigned CGA and CGG to tryptophan due to a combination of Codetta inference, phylogeny, and evidence from tRNA genes and multiple sequence alignments of BUSCO genes. We provisionally split the Gracilibacteria into two clades based on differences in Codetta CGG inference, tRNA gene content, codon usage, and GC content. Gracilibacteria clade 1 may have reassigned CGG to tryptophan, pending additional evidence. Asterisks indicate genomes with GTDB CheckM estimated genome completeness >75%. For each species, the Codetta inference of the three reassigned codons (UGA, CGA, and CGG) is indicated by colored circles (red: arginine; purple: tryptophan; yellow: glycine; white: ‘?’). The presence of tRNA genes that recognize UGG and CGN-codons is indicated by filled circles, colored according to the predicted amino acid charging based on identity elements for tRNAs (see Materials and methods). The lines connecting codons and tRNA anticodons represent the likely decoding capabilities, with dashed lines representing weaker interactions. The U34 of UCG is presumed to be modified in a way that restricts wobble to CGA and CGG, but could potentially recognize CGU and/or CGC depending on the true modification state. Codon usage is the frequency per 10,000 codons aligned to Pfam domains. (B) Multiple sequence alignments of undecaprenyl diphosphate synthase (BUSCO POG091H00BZ) and Peptidase M50 (BUSCO POG091H0131) from Absconditabacteria, Gracilibacteria clades 1 and 2, and selected outgroup species. Alignment regions containing nearby CGA (α) or CGG (γ) positions are shown, with columns containing CGA or CGG in Absconditabacteria or Gracilibacteria clade 1 sequences highlighted with an asterisk above, and columns containing CGA or CGG in Gracilibacteria clade 2 and outgroup sequences highlighted with an asterisk below. (C) The CGA- and CGG-decoding tRNAs (UCG and CCG anticodons) from species #2 (candidate division SR1 bacterium Aalborg_AAW-1, GCA_001007975.1). tRNA sequence features involved in tryptophan identity are highlighted (Giegé et al., 1998; Himeno et al., 1991), with nucleotide numbering following the convention of Sprinzl et al., 1998. Nucleotides highlighted in gray do not match the expected identity element.
Tables
Table 1
A summary of all bacterial clades previously known to use a codon reassignment.
For each clade, the NCBI taxonomic IDs (taxids) shown most closely correspond to the known phylogenetic distribution from the literature. For each codon reassignment, we show the number of sequenced species analyzed by Codetta and how many were inferred to use the expected amino acid or had no inferred amino acid. None of the analyzed species belonging to reassigned clades were predicted to use an unexpected amino acid at the reassigned codon. [1] Bové, 1993, [2] Volokhov et al., 2007, [3] McCutcheon et al., 2009, [4] Bennett and Moran, 2013, [5] McCutcheon and Moran, 2010, [6] Salem et al., 2017, [7] Rinke et al., 2013, and [8] Campbell et al., 2013.
| Reassigned codon | ||||||
|---|---|---|---|---|---|---|
| Phylogenetic distribution | NCBI taxids | Reference | N species | Codon reassignment | Expected amino acid | Uninferred (‘?’) |
| Entomoplasmatales and Mycoplasmatales | 186328, 264638, 2085 | [1, 2] | 199 | UGA Stop→W | 191 | 8 |
| Hodgkinia cicadicola | 573658 | [3] | 1 | UGA Stop→W | 1 | 0 |
| Nasuia deltocephalinicola | 1160784 | [4] | 1 | UGA Stop→W | 1 | 0 |
| Zinderia insecticola | 884215 | [5] | 1 | UGA Stop→W | 1 | 0 |
| Stammera capleta | 2608262 | [6] | 1 | UGA Stop→W | 1 | 0 |
| Gracilibacteria | 363464 | [7] | 15 | UGA Stop→G | 13 | 2 |
| Absconditabacteria | 221235 | [8] | 6 | UGA Stop→G | 6 | 0 |
Table 2
A summary of codon inferences from the bacterial and archaeal genomes analyzed by Codetta, dereplicated to one assembly per species.
The Codetta inference for each codon is compared against a genetic code annotation derived by layering the known bacterial genetic codes in Table 1 over the NCBI taxonomy. Reassigned stop codons are included with sense codons. Values can be calculated from Supplementary file 1.
| Bacteria | Archaea | ||||
|---|---|---|---|---|---|
| 46,384 species | 2309 species | ||||
| Sense | Total (N codons × N species) | 2,829,648 | 140,849 | ||
| Expected amino acid | 2,823,497 | 99.78% | 140,631 | 99.85% | |
| Other amino acid | 612 | 0.02% | 0 | 0.00% | |
| Uninferred (‘?’) | 5539 | 0.20% | 218 | 0.15% | |
| Stop | Total (N codons × N species) | 138,928 | 6927 | ||
| Amino acid | 290 | 0.21% | 9 | 0.13% | |
| Uninferred (‘?’) | 138,638 | 99.79% | 6918 | 99.87% | |
Additional files
-
Supplementary file 1
Table of predicted genetic codes by Codetta across all analyzed genome assemblies.
- https://cdn.elifesciences.org/articles/71402/elife-71402-supp1-v3.csv
-
Supplementary file 2
Table of all candidate novel genetic codes predicted by Codetta across all analyzed genome assemblies.
- https://cdn.elifesciences.org/articles/71402/elife-71402-supp2-v3.xlsx
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/71402/elife-71402-transrepform1-v3.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A computational screen for alternative genetic codes in over 250,000 genomes
eLife 10:e71402.
https://doi.org/10.7554/eLife.71402




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}