Stable antibiotic resistance and rapid human adaptation in livestock-associated MRSA
- Department of Veterinary Medicine, University of Cambridge, United Kingdom
Figures
Figure 1 with 5 supplements
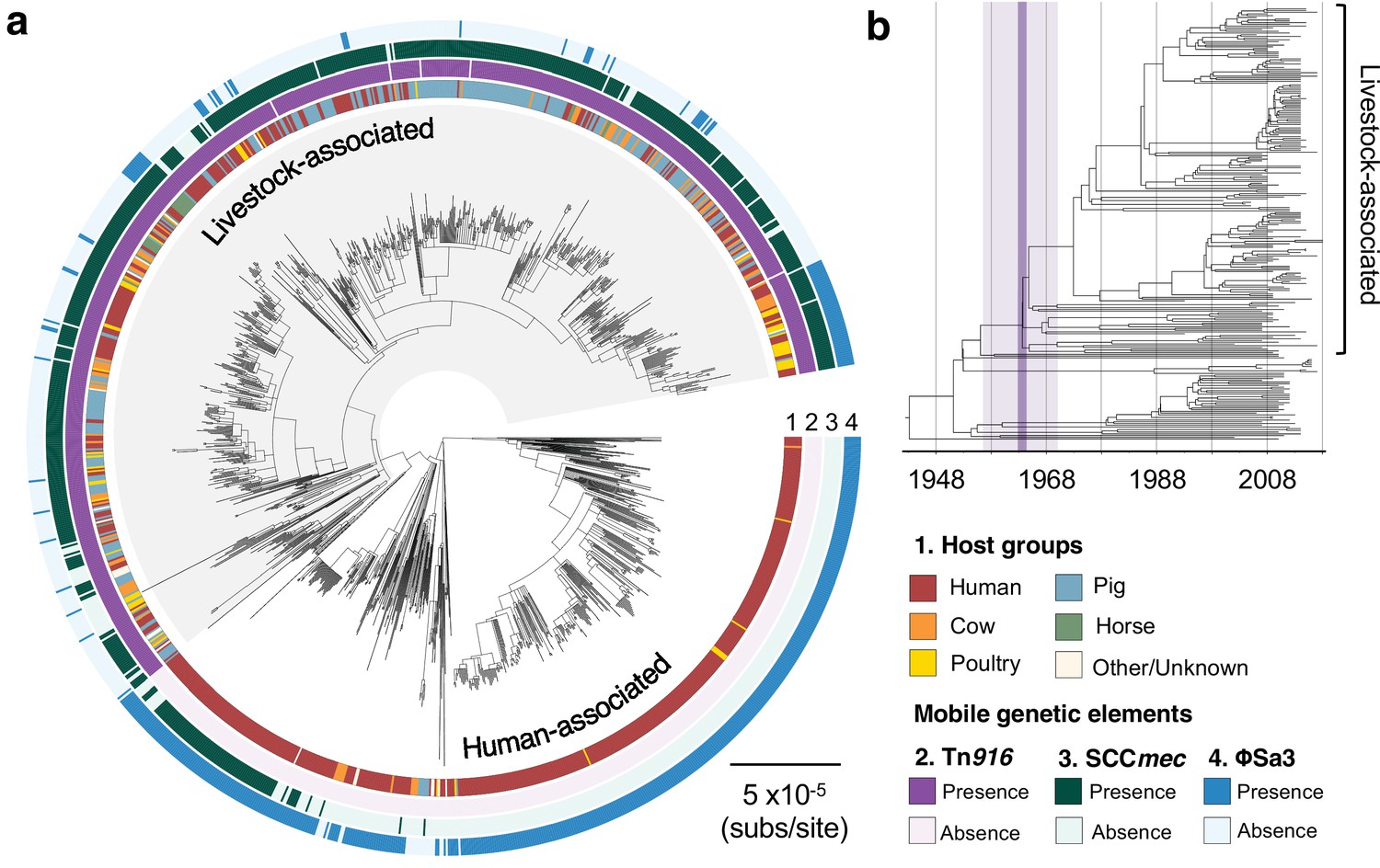
The transition to livestock association in the 1960s was accompanied by changes in the frequencies of three mobile genetic elements (MGEs).
(a) A maximum likelihood phylogeny of 1180 isolates of CC398, rooted using an outgroup from ST291. Grey shading indicates the livestock-associated clade. Outer rings describe (1) the host groups isolates were sampled from, and the presence of three MGEs: (2) a Tn916 transposon carrying tetM, (3) a SCCmec carrying mecA, and (4) a φSa3 prophage carrying a human immune evasion gene cluster. (b) A dated phylogeny of a sample of 250 CC398 isolates that shows livestock-associated CC398 originated around 1964 (95% HPD: 1957–1970).
Figure 1—figure supplement 1
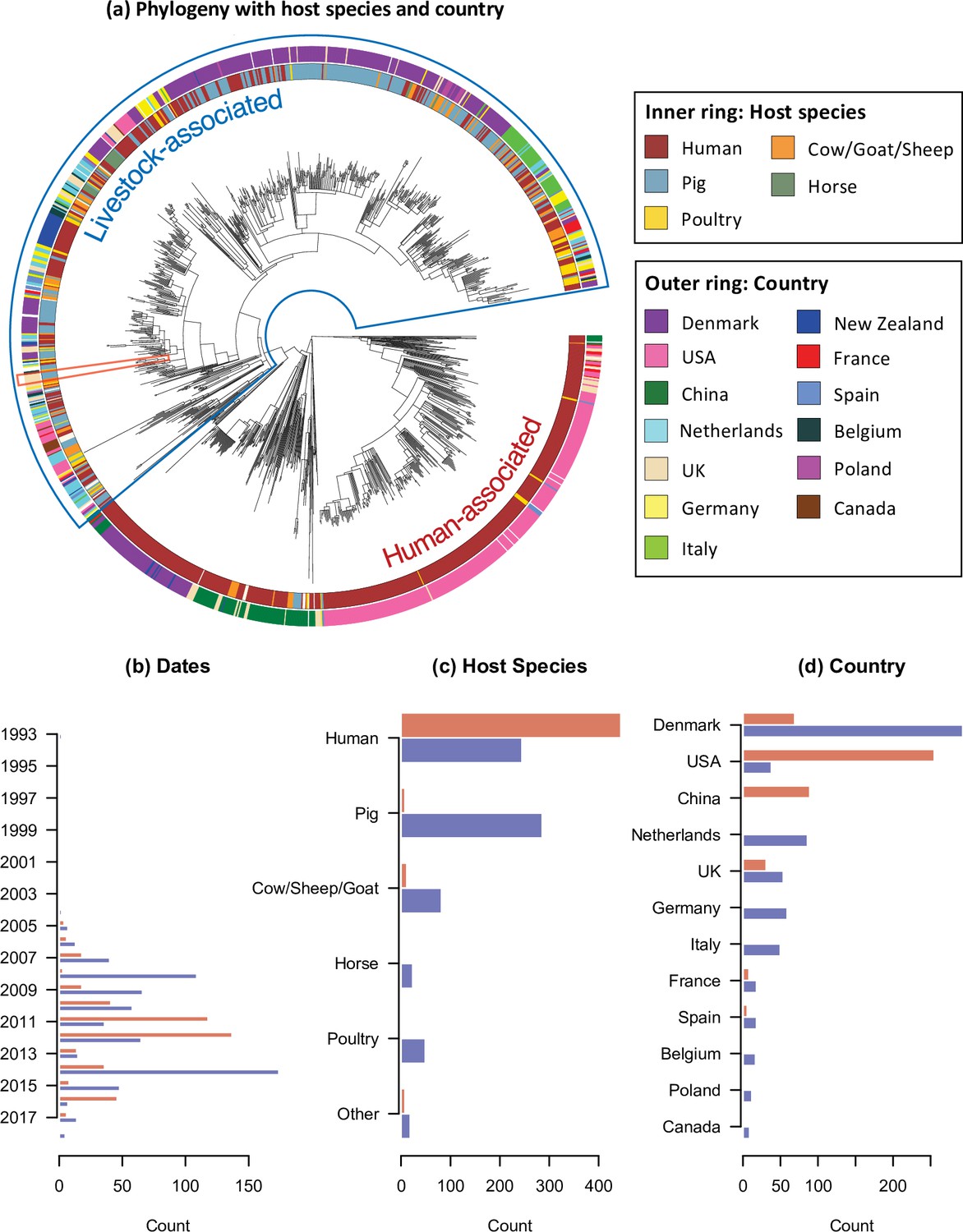
The temporal, host species, and geographic distribution of our collection of CC398 isolates.
(a) Phylogeny of CC398 with rings showing the host species and countries of origin for each isolate (groups with n < 10 not shown). The blue outline indicates the livestock-associated clade, and the red outline indicates the five most recent isolates in our collection (sampled in 2018), from pigs on UK farms. (b), (c), and (d) show the variation in sampling date, host species, and country across the livestock-associated (blue, lower) and human-associated (red, upper) clades.
Figure 1—figure supplement 2
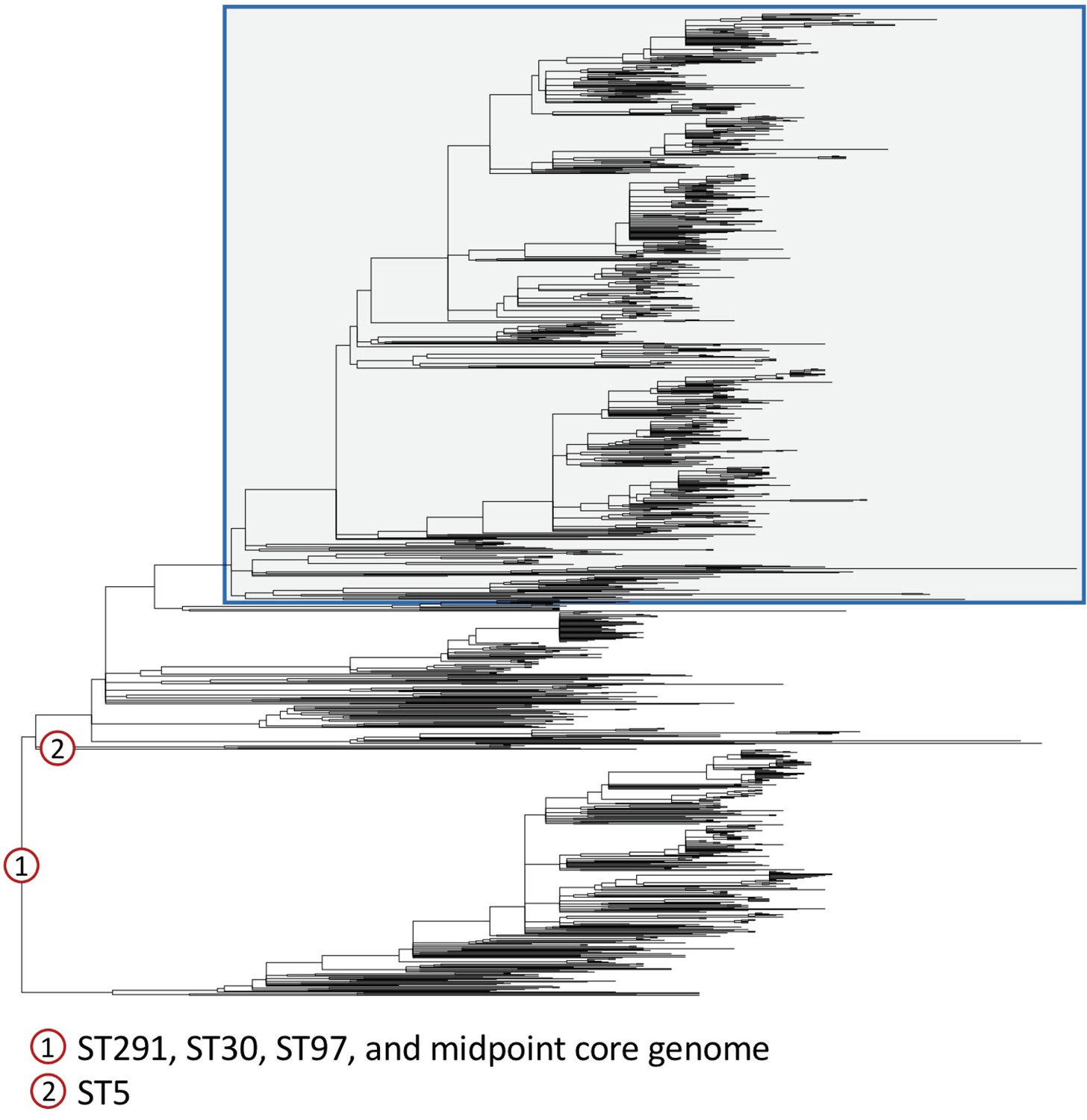
Different outgroups consistently identify the root of CC398 within human-associated CC398.
We constructed maximum likelihood phylogenies using a reference-mapped alignment of CC398 combined with four outgroups from sequence types (STs) 291, 30, 97, and 5; and using a core genome alignment of our de novo assemblies and a midpoint rooting. The outgroups we used covered a range of distances from the base of CC398: ST291 (ERR2729529) is ~0.005 subs/site, ST30 (ERS1420125) is ~0.012 subs/site, and ST97 and ST5 (ERR2729579 and SRS613151) are ~0.015 subs/site. The reference-mapped phylogenies that were rooted using ST291, ST30, and ST97, and the midpoint-rooted core genome phylogeny all showed a consistent root, which is shown in the figure (and indicated by 1). A different root was obtained when we used the outgroup of ST5 (location indicated by 2). This root is on a neighbouring branch to the root found in the four other reconstructions, and results in CC398 being rooted on the branch leading to a single isolate (ZTA09_03734_9HSA). Livestock-associated CC398 is indicated by a blue box with grey shading.
Figure 1—figure supplement 3
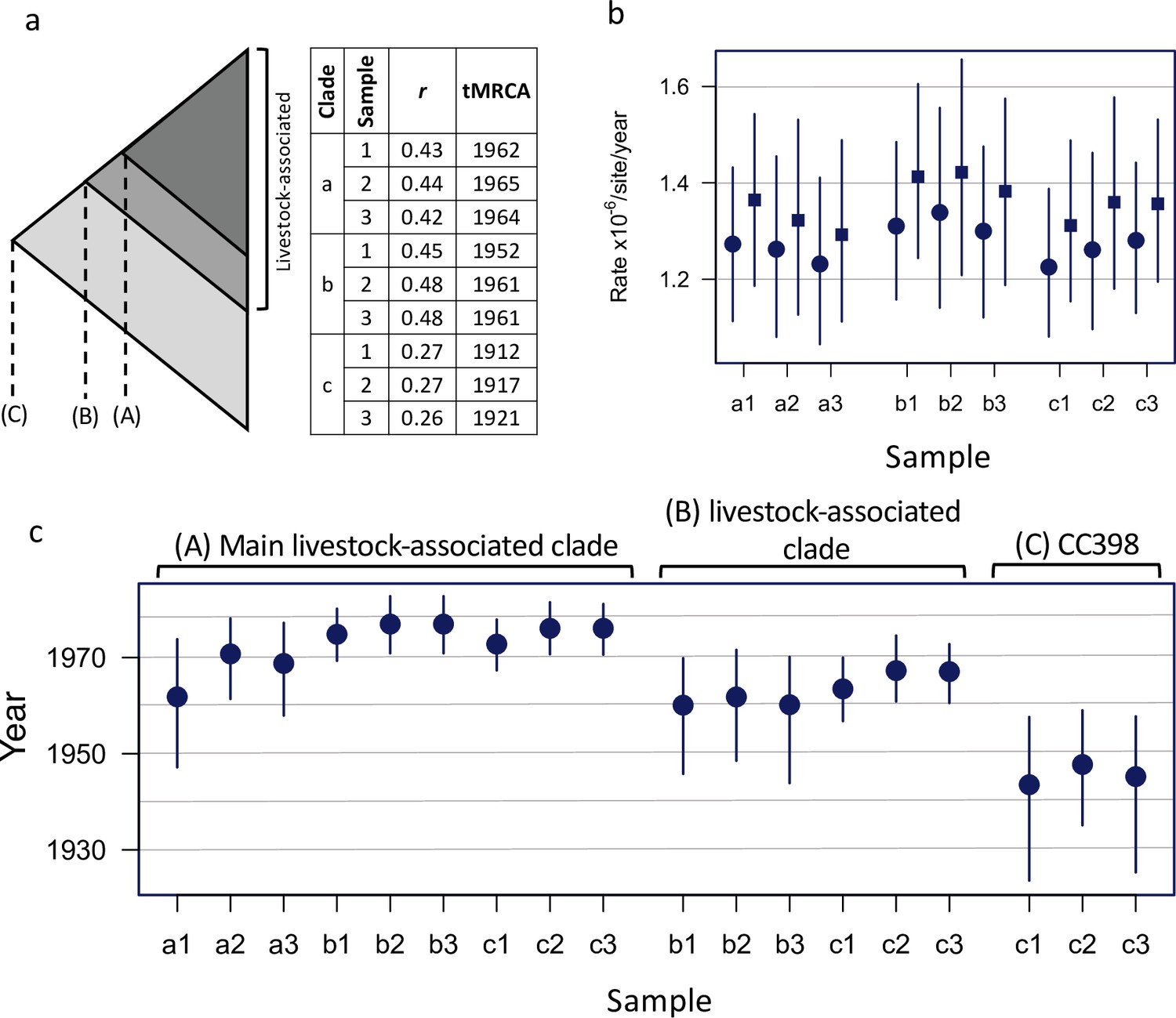
A consistent estimate of the age of the livestock-associated clade.
Results of BEAST dating analyses estimating (A) the origin of a shallower subclade within the livestock-associated clade, (B) the origin of the entire livestock-associated clade, and (C) the origin of CC398. (a) The figure shows a schematic representation of the CC398 phylogeny indicating the nodes of interest (A–C), and our sampling strategy. We randomly sampled our dataset three times to generate samples of 250 isolates (200 from the livestock-associated clade and 50 from the human-associated clade). Samples overlapped by only 30 isolates that represent the most divergent lineages of the livestock-associated clade, to ensure a consistent description of the most recent common ancestor. Each sample had the same range of sampling dates (1993–2018). (a) also describes the results of a regression of root-to-tip distance against sampling date (correlation coefficient and estimate of the date of the most recent common ancestor) for each sample (1–3), and subsamples that include isolates from (A) only the main livestock-associated clade, (B) only the livestock-associated clade, and (C) the entire sample. As we observed stronger temporal signal for A and B, than for C, we estimated dated trees using BEAST for each of these nine subsampled datasets. We observed consistent estimates of evolutionary rate across all these analyses (b). Rates at first/second codon positions are shown as circular points, and at third codon positions as square points. These analyses also returned broadly consistent estimates of dates (c). Although we found that estimates from subsamples that included outgroups of the node being dated returned more precise and marginally more recent estimates of age, likely due to more information about the location of the root.
Figure 1—figure supplement 4
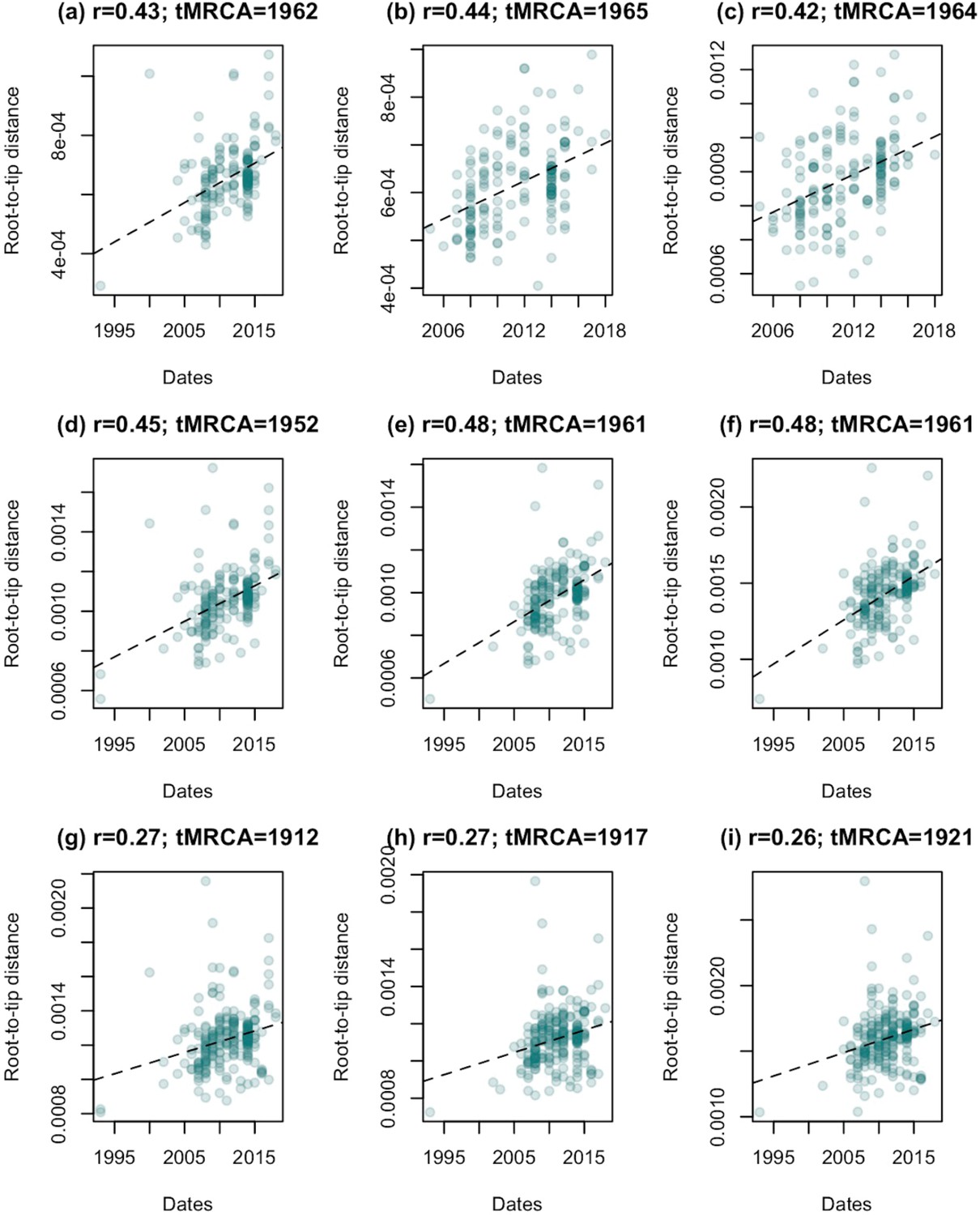
Evidence of temporal signal is present across in our subsampled datasets, but is stronger when isolates from the human-associated group are excluded.
Regressions of root-to-tip distance against sampling date for each of our datasets, rooted to minimise residual mean squares. (a)-(c) show the results for samples 1, 2, and 3 for clade a (described in Figure 1—figure supplement 3), (d)-(f) the results for samples 1, 2, and 3 for clade b, and (g)-(i) samples 1, 2, and 3 for clade c. For all datasets a randomisation test indicated that these correlations were unlikely to have arisen by change (p < 0.01). Correlation coefficients (r) and estimates of the time of the most recent common ancestor (tMRCA) based on the regression are shown for each dataset.
Figure 1—figure supplement 5
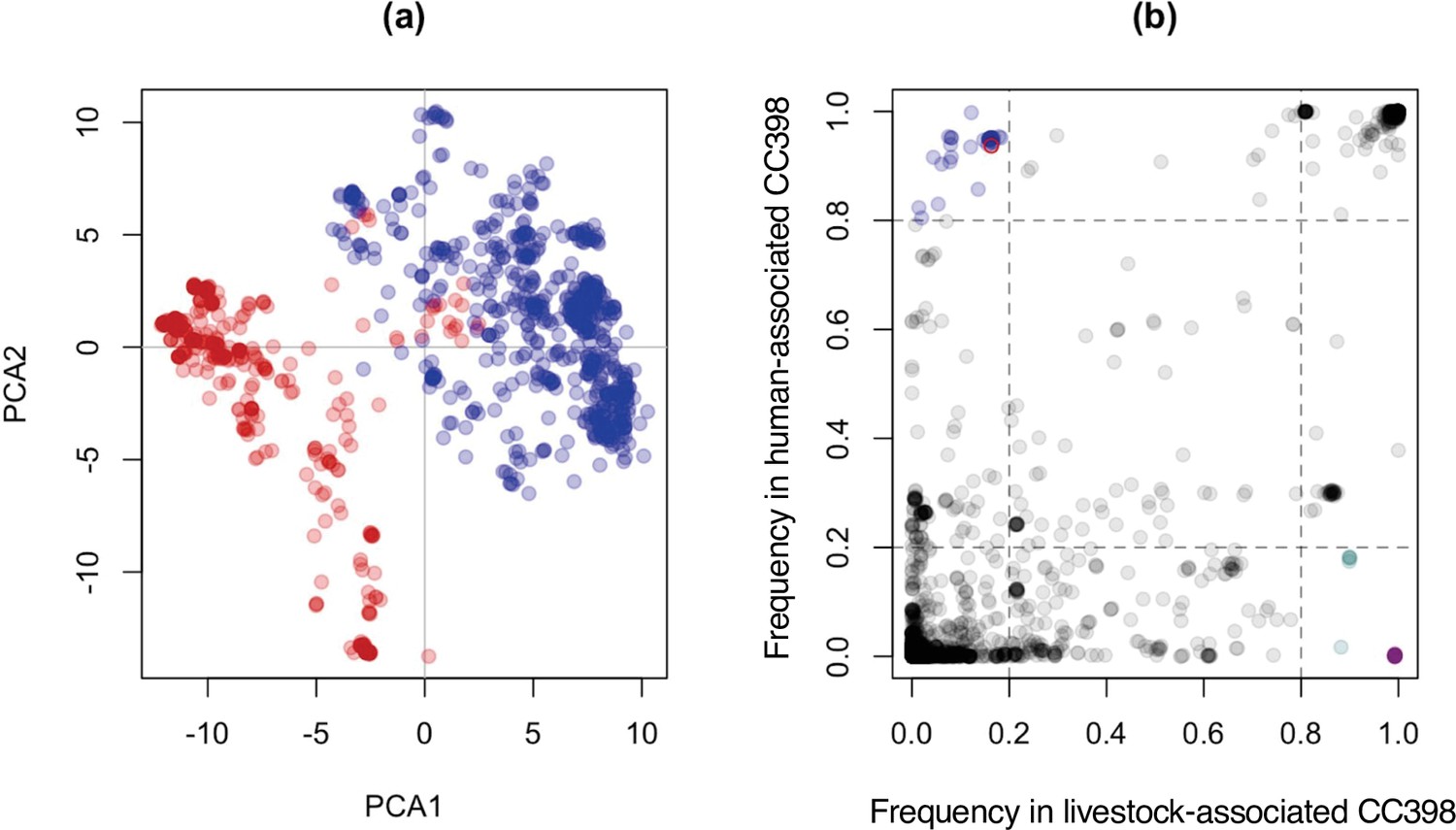
Livestock- and human-associated CC398 have divergent accessory genomes, and genes whose presence/absence most clearly distinguish these groups (except one) are associated with a Tn916 transposon, SCCmec, and φSa3 prophages.
(a) A plot of the first and second principal components of accessory genome content, with isolates from human-associated CC398 in red and isolates from livestock-associated CC398 in blue. This was constructed using the package adegenet in R (Jombart, 2008). (b) Comparison of gene frequencies across the human- and livestock-associated groups. Genes present in <20% of human-associated CC398 and >80% of livestock-associated CC398 and genes presence in >80% of human-associated CC398 and <20% livestock-associated CC398 are highlighted, with genes associated with the Tn916 transposon shown in purple (all are overlapping as they have identical frequencies), genes associated with SCCmec shown in turquoise, and genes associated with φSa3 prophages shown in blue. The one gene that distinguishes livestock-associated CC398 from human-associated CC398 that is not associated with one of these three mobile genetic elements (MGEs) is shown as a red circle (tatC).
Figure 2 with 1 supplement
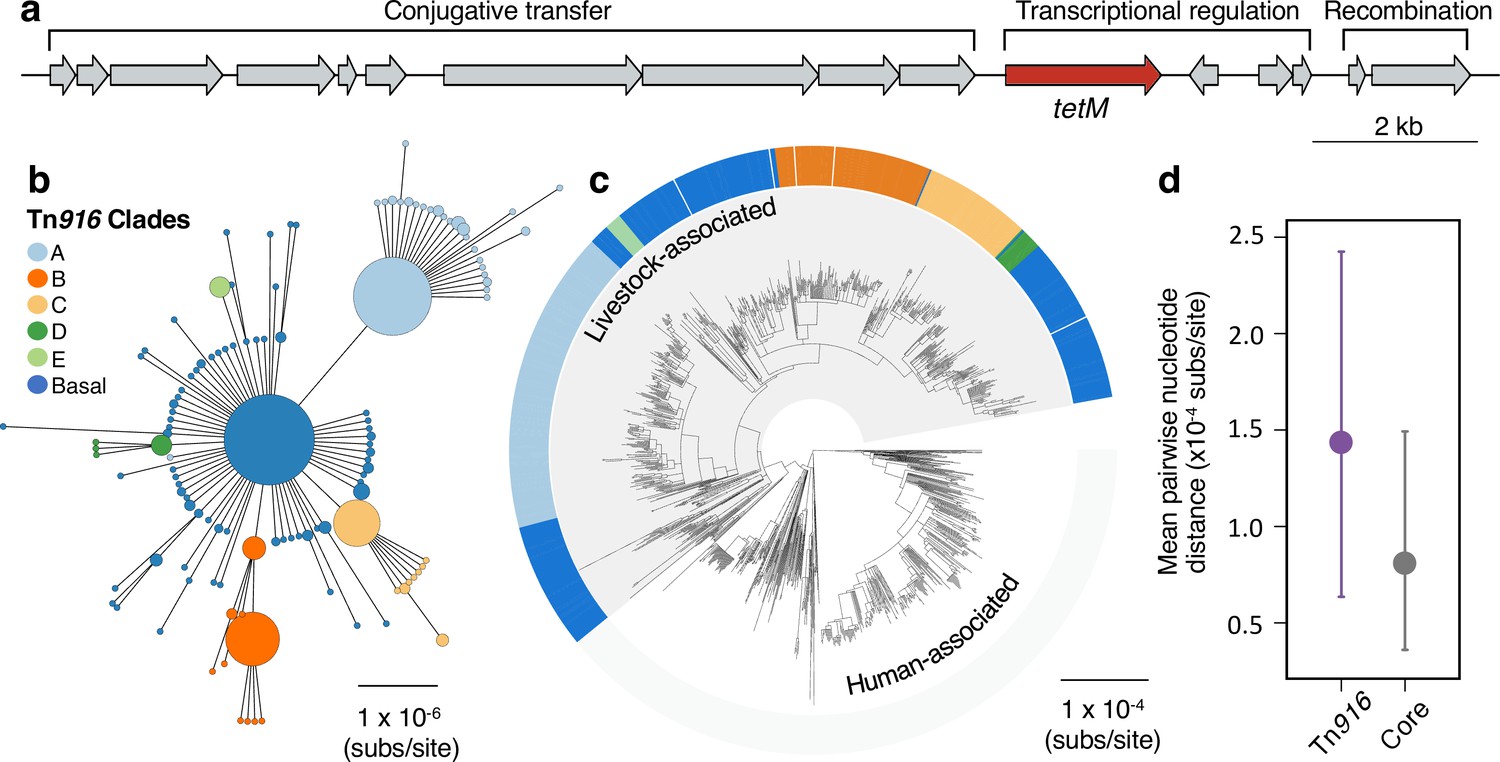
A Tn916 transposon carrying tetM has been stably maintained by livestock-associated CC398 since its origin.
(a) A gene map of the Tn916 transposon in CC398 (based on reference genome 1_1439), with annotations based on previous studies (de Vries et al., 2009; Roberts and Mullany, 2009). (b) A minimum-spanning tree of the element based on a concatenated alignment of all genes shown in (a). Points represent groups of identical elements, with point size correlated with number of elements on a log scale, and colours representing well-supported clades (>70 bootstrap support in a maximum likelihood phylogeny) that include >10 elements (smaller clades are incorporated into their basal clade). (c) These clades are annotated onto the CC398 phylogeny as an external ring. (d) Mean pairwise nucleotide distance between isolates carrying the Tn916 transposon based on genes in the Tn916 transposon and core genes, using bootstrapping to estimate error (see Materials and methods for details).
Figure 2—figure supplement 1
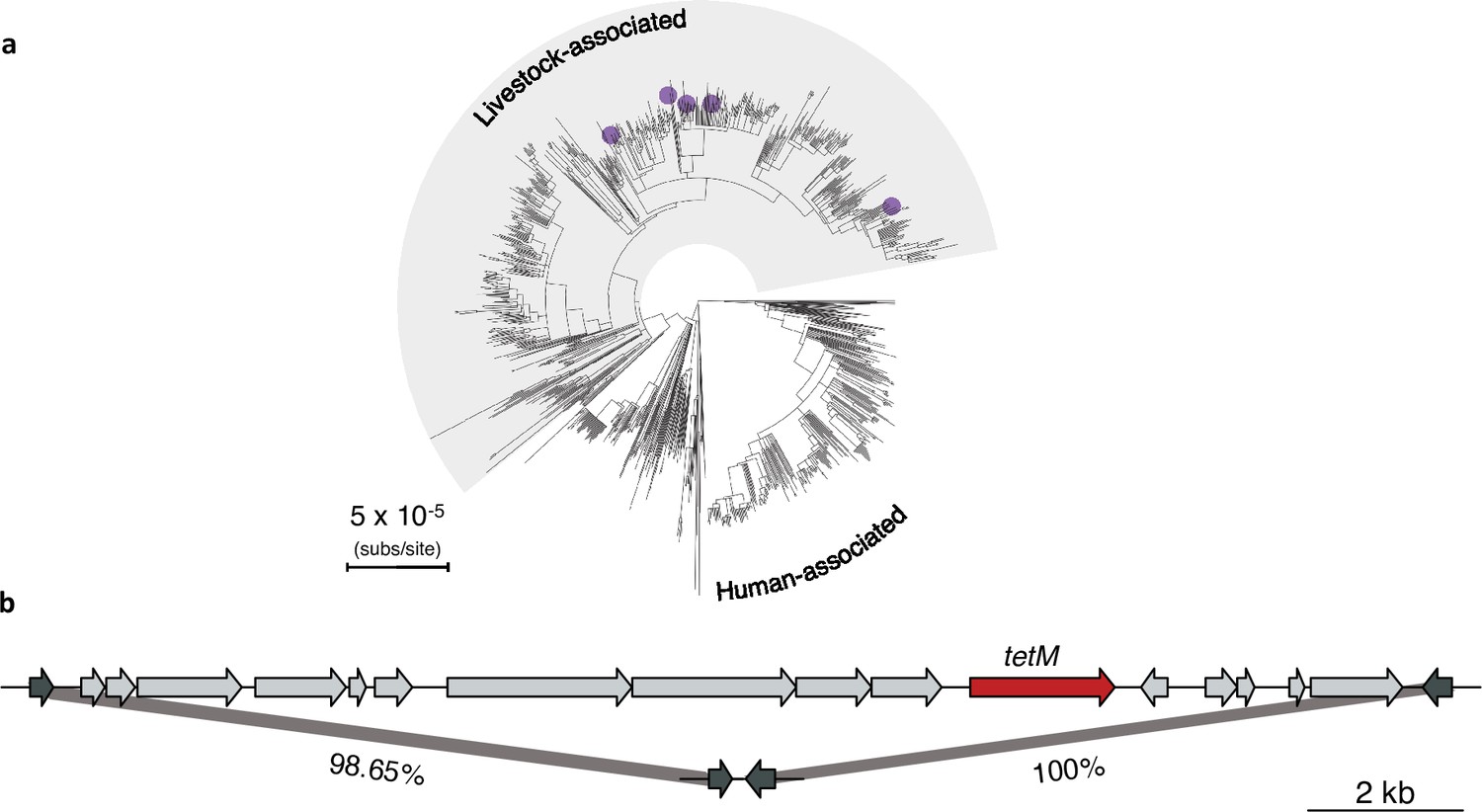
Evidence of repeated excision of Tn916 transposon in the livestock-associated clade.
(a) Purple points indicate which five isolates lack the Tn916 transposon in the livestock-associated clade. (b) Top: a gene map showing the Tn916 transposon from the 1_1429 reference strain and the two flanking genes (dark grey). Bottom: the integration site in the 62,951 strain within which the transposon is absent. The percentage nucleotide identity between the two flanking genes is indicated below the bars connecting the top and bottom gene maps.
Figure 3 with 4 supplements
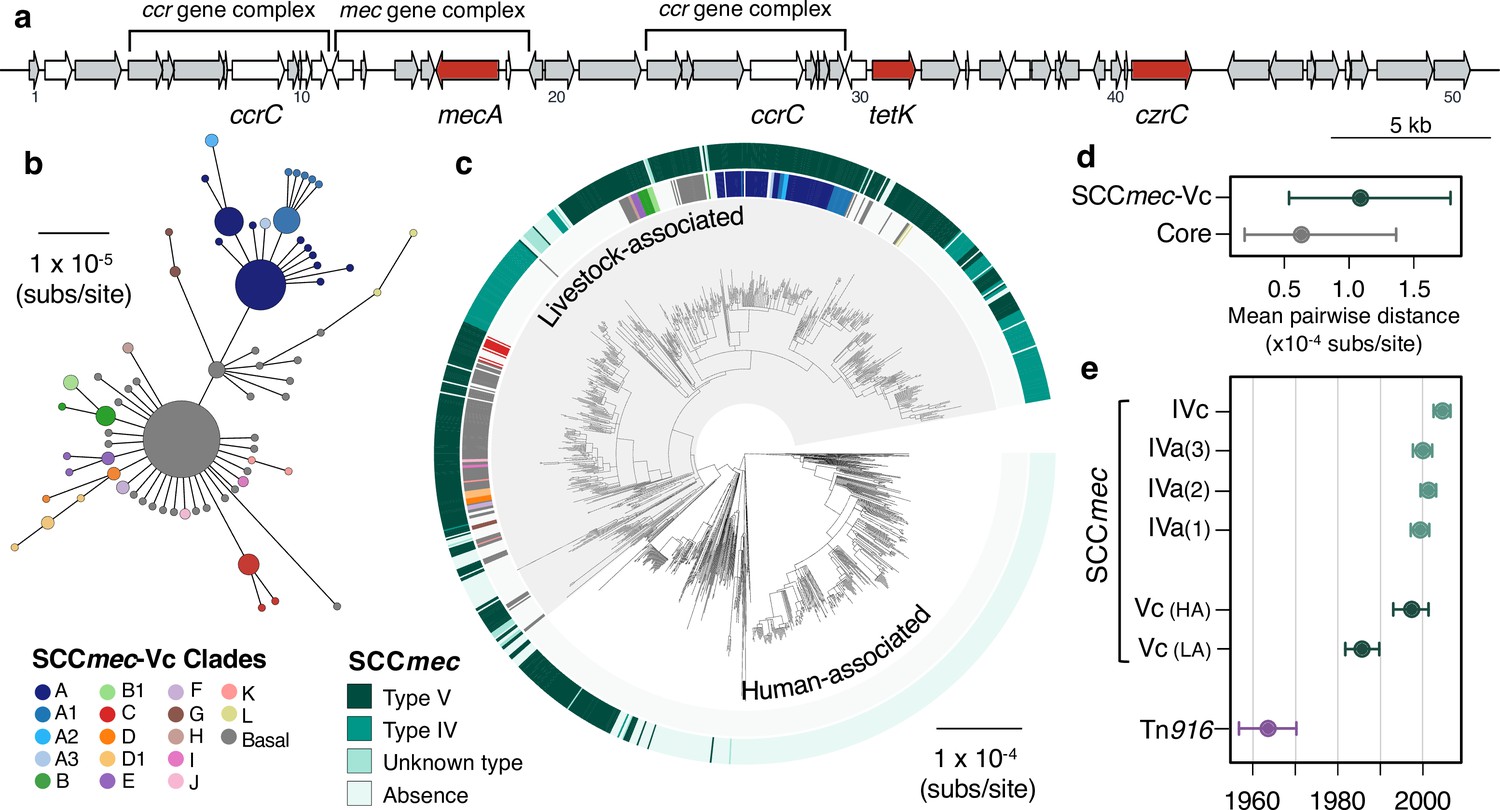
A type V SCCmec has been maintained since the 1980s, with occasional replacements.
(a) A gene map of the type Vc SCCmec element in CC398 (using the 1_1439 reference strain), with annotations from previous studies (Li et al., 2011; Vandendriessche et al., 2014). Genes in white were excluded from analyses of diversity within the element due to difficulties in distinguishing homologues. (b) A minimum-spanning tree of the type Vc SCCmec element based on a concatenated alignment of the genes (grey and red) in (a). Points represent groups of identical elements, point size correlates with group size on a log scale, and colours represent well-supported clades (>70 bootstrap support in a maximum likelihood phylogeny). (c) Well-supported clades and SCCmec type are annotated on the CC398 phylogeny in external rings. (d) Mean pairwise nucleotide distance between isolates carrying the SCCmec type Vc based on genes in the SCCmec type Vc and core genes, with error estimated by bootstrapping (see Materials and methods for details). (e) Acquisition dates for different SCCmec elements and Tn916 inferred from an ancestral state reconstruction over the dated phylogeny in 1 (b). Dates for type Vc are shown for both livestock- and human-associated CC398.
Figure 3—figure supplement 1
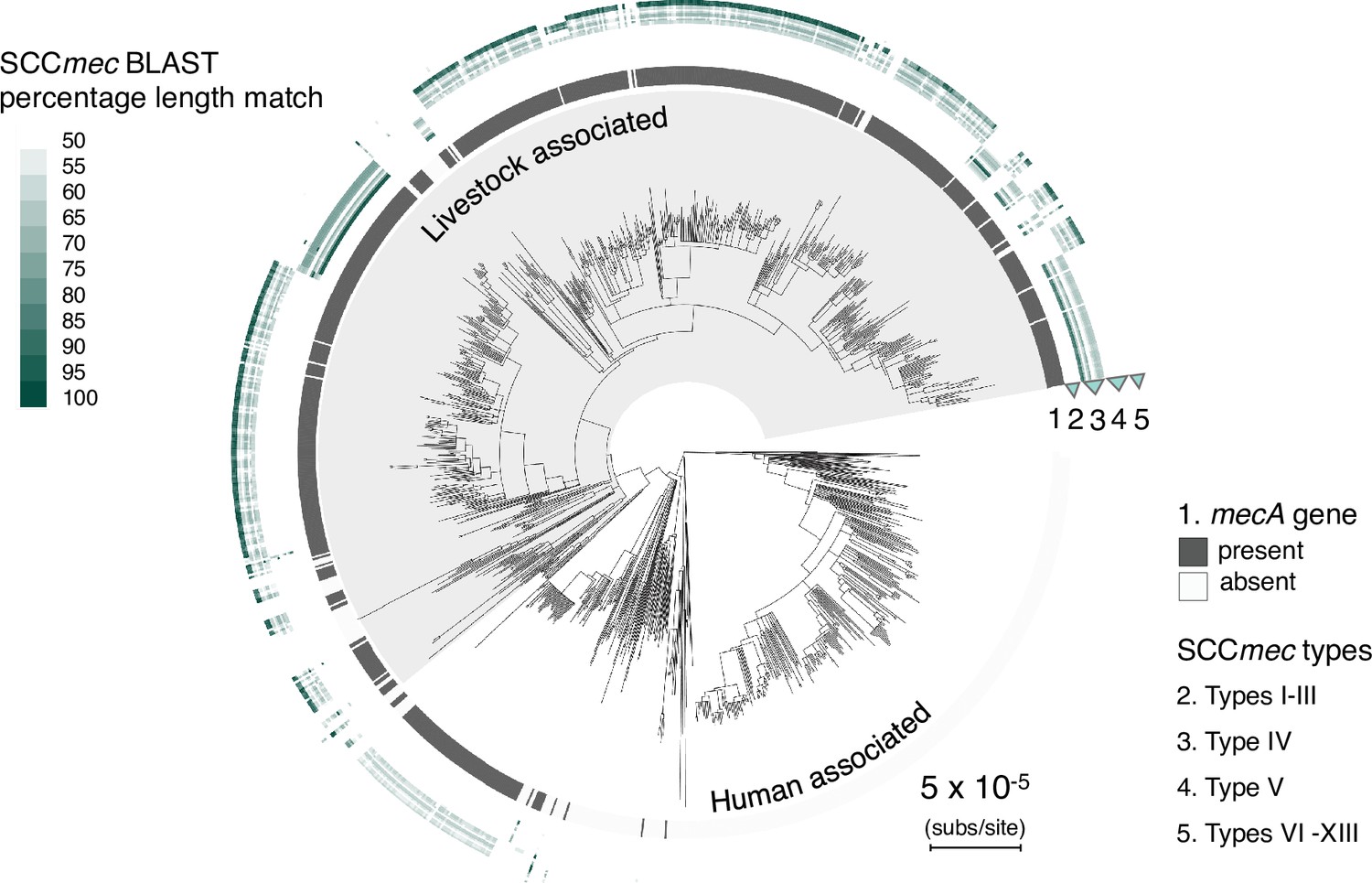
Type V and IV SCCmec elements identified in CC398 through a BLASTn search or representative types.
Initial identification of SCCmec types was carried out by BLASTn search of all SCCmec reference sequences on the SCCmecFinder extended database. As the SCCmec element in our dataset was commonly separated onto multiple contigs, we found that contiguous hits of the entire element were rare. Therefore, to determine the presence of a particular SCCmec type we considered the combined length of high-identity hits (hits that have >95% nucleotide identity and are ≥5% of the length of the element). We categorised elements into SCCmec types based on the overall length of the matched region, and described strains with best match lengths of <50% as unknown. The CC398 phylogeny is annotated with the results of this analysis. The innermost ring shows the presence/absence of mecA, and outer rings show the percentage length match for each type/sub-type (Table S7).
Figure 3—figure supplement 2
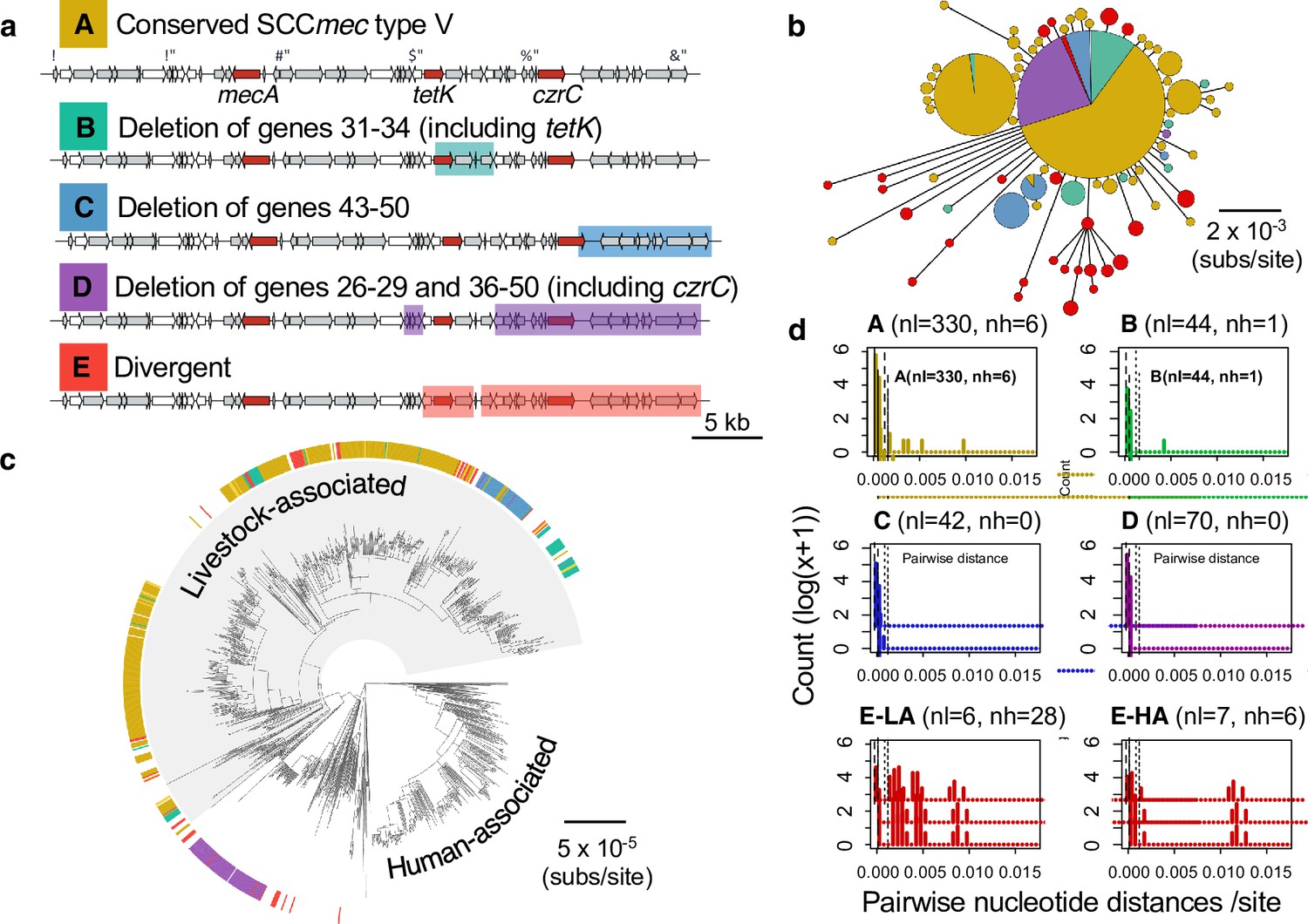
Most of the shorter versions of the type Vc SCCmec element in CC398 can be attributed to deletion events.
Of the 540 isolates that carry a type V SCCmec in CC398, 204 have >2 genes absent. There are 3 common forms of gene absence (B, C and D). (a) Gene maps showing common patterns of gene absence. (b) A minimum spanning tree of a concatenated alignment of the 40 genes associated with the full-length SCCmec type V. Points represent groups of elements that differ by a maximum of 1 SNP, with point size correlated with group size on a log-scale, and colours representing categories described in (a). (c) Annotation of the full-length and truncated categories onto the core genome phylogeny. (d) Histograms showing pairwise distances (per shared nucleotide site) between individual elements in each group and the least divergent member of Group A. Histograms are shown on a log(x+1) scale due to the high frequency of low divergence elements. The dashed vertical lines show the average pairwise distance between isolates within the livestock-associated clade based on a core gene alignment, and the dotted lines show 10-times this value. Groups A, B and C are only found in the livestock-associated clade. Due to the low level of diversity in group D, versions from the human-associated and livestock-associated clade are shown together, while for group E they are shown separately (HA/LA). There are 13 isolates from the human-associated clade in group E. Of these 13 isolates, only 3 show sufficiently low divergence to be consistent with a recent common ancestor with the majority of elements from Group A, within CC398 (for these three elements divergence is <1.7 x10-4 /site). The three isolates carrying these elements all fall within the recent Danish hospital outbreak clade, and show 100% identity with the majority of the elements within this clade (that fall within group D).
Figure 3—figure supplement 3

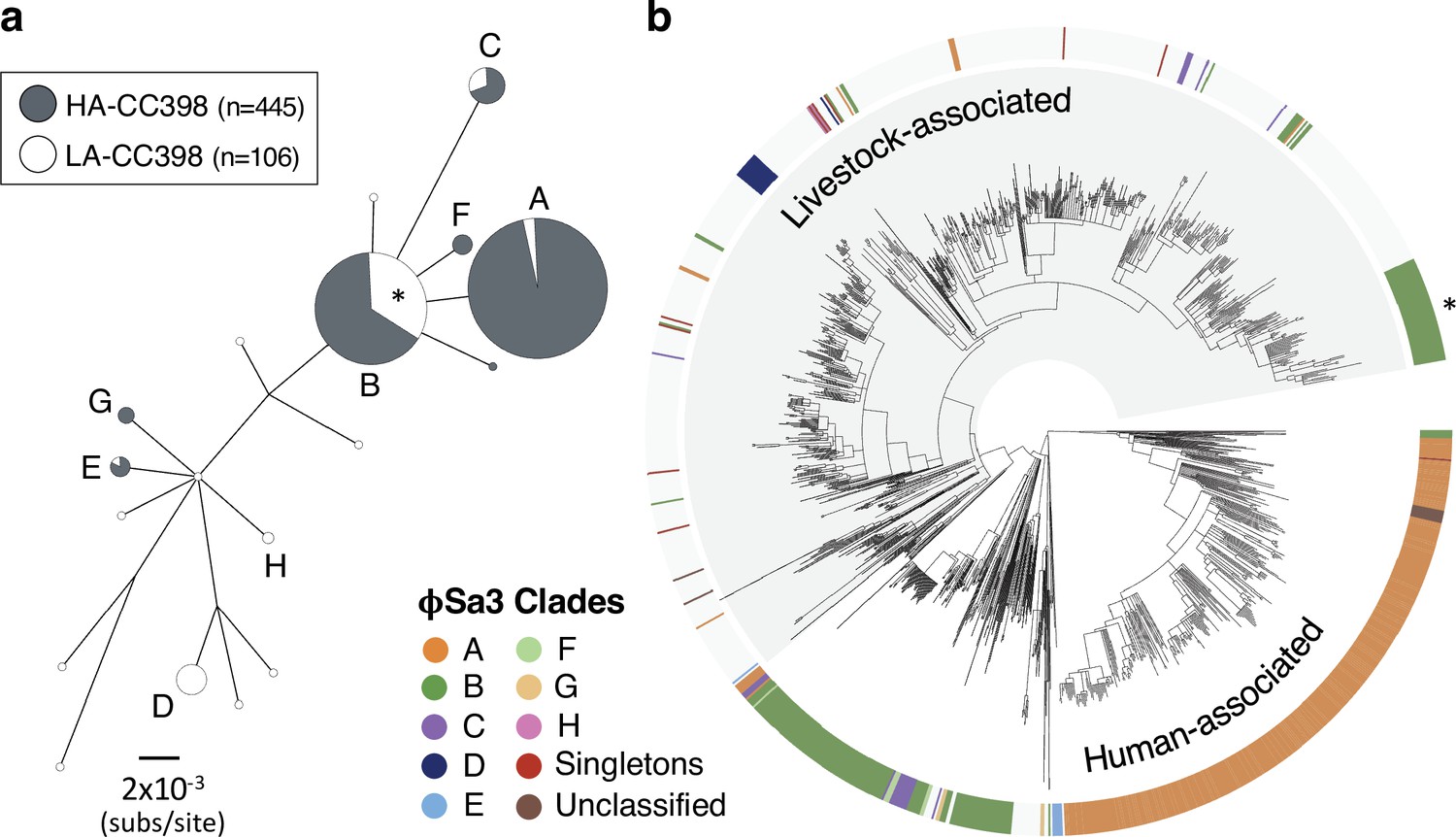
There have been at least four independent acquisitions of type IV SCCmec within livestock-associated CC398.
(a) An minimum-spanning tree based on a concatenated alignment of the 12 genes shared across all type IV SCCmec (n=148 isolates). Clades are distinguished based on nucleotide distances within these shared genes and by differences in the genes present in these elements. Clade A is IVa(1), Clade B is IVc, Clade C is IVa(2), and Clade D is IVa(3). IVa(2) and IVa(3) only differ by one SNP in the alignment of 12 shared genes, but they have different gene contents, suggesting divergent origins. (b) Clades of type IV elements mapped onto the core genome phylogeny.
Figure 3—figure supplement 4

Tn916 was acquired before the current complement of SCCmec elements in livestock-associated CC398, SCCmec type V was acquired before SCCmec type IV, and SCCmec type V was acquired by livestock-associated CC398 before human-associated CC398.
Date estimates based on ancestral state reconstructions with BEAST over dated phylogenies inferred from three subsampled data sets. (A) A dated phylogeny inferred from one subsample (no. 1) with branches coloured by inferred SCCmec element at their descendent node (>95% posterior support), and earliest nodes (>95% posterior support) for each element labelled. (B) Date estimates for the earliest nodes at which each SCCmec type is inferred to be present (>95% posterior support) across the three data sets. Points are median values and bars are 95% confidence intervals. Estimates are shown for three versions of SCCmec type IVa that were distinguished based on the divergence within this element (see Figure 3—figure supplement 3). Only one estimate is provided for SCCmec-IVa(3) because it is only observed in one of the subsampled data sets.
Figure 4 with 1 supplement
φSa3 prophages have been lost and acquired multiple times in both human- and livestock-associated CC398.
(a) A maximum likelihood phylogeny based on 12 genes shared across the φSa3 prophages in our collection, with both low-support nodes (<70% bootstrap support) and branches <0.0018 subs/site collapsed. The latter cut-off is a conservative estimate of the maximum distance that could reflect divergence within CC398. It is the maximum pairwise distance between isolates carrying φSa3 prophages across 1000 estimates from random samples of a core gene alignment of the same number of sites as is in our φSa3 prophage alignment. Node size correlates with the number of elements on a log scale. Elements carried by isolates from human-associated CC398 (grey) and livestock-associated CC398 (white) isolates are indicated, and nodes that include multiple elements labelled (A–E). (b) These clades annotated on the CC398 phylogeny as an external ring. The element carried by the poultry-associated subclade of livestock-associated CC398 is indicated by *.
Figure 4—figure supplement 1
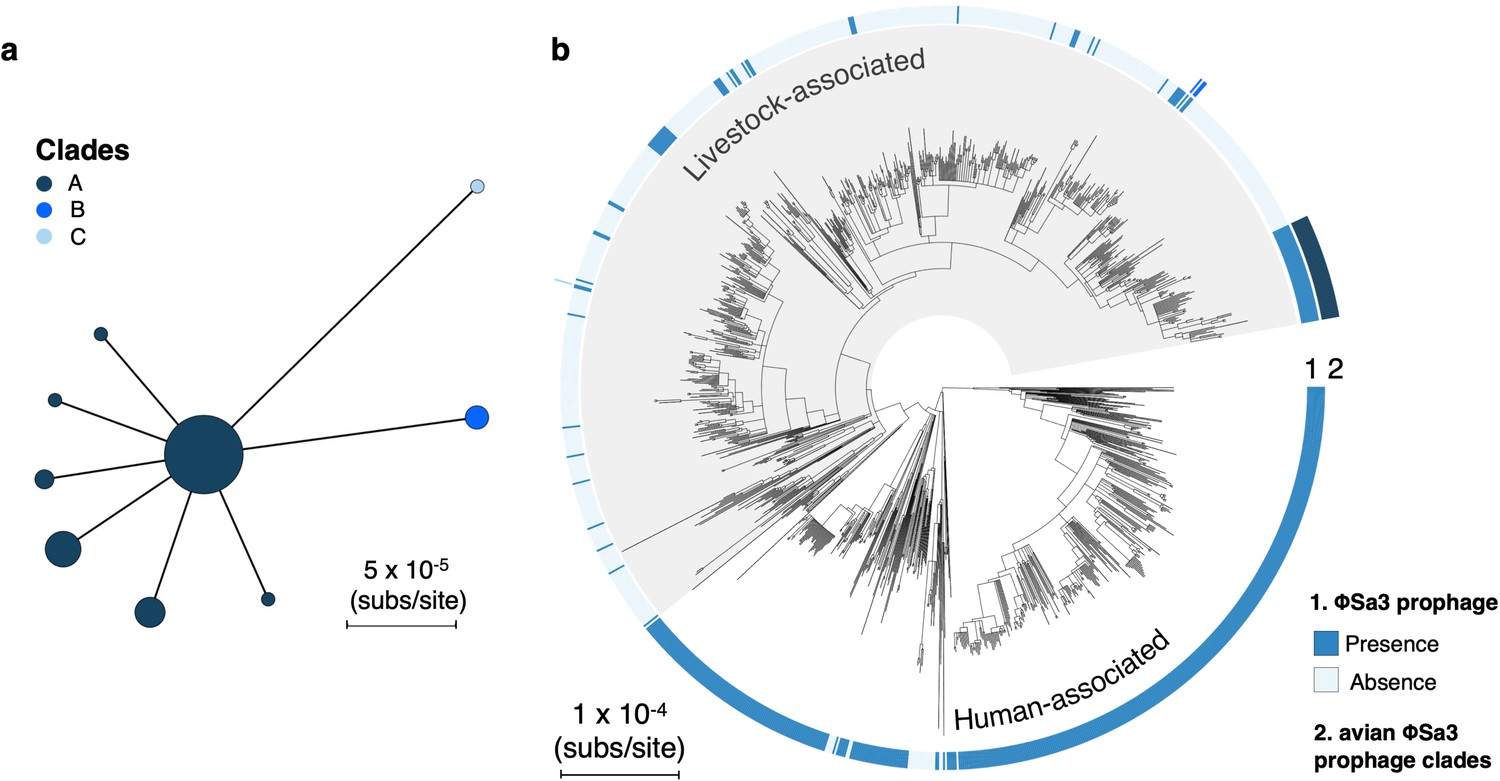
Maintenance of a φSa3 prophage in a poultry-associated clade of livestock-associated CC398 for around 21 years.
(a) A minimum-spanning tree of the type B φSa3 prophage present in the poultry-associated clade of livestock-associated CC398. Fifty-five isolates in our collection of CC398 carry a version of this element. We identified 34 genes shared across these 55 versions of the element. After pruning genes with homologues, and difficult to align regions 29/34 genes were used to construct the tree. Points represent groups of identical elements, with point size correlated with group size on a log scale, and colours representing clades that differ from the basal group by >1 SNP. (b) These clades annotated onto the CC398 phylogeny as an external ring. 51/55 isolates carrying this element fall within the poultry-associated clade and four other isolates are from other clades within livestock-associated CC398 from human hosts. Within the avian clade we see little diversity (maximum pairwise distance is 1 SNP). The elements from outside of this clade show greater divergence, but this could still represent divergence within livestock-associated CC398.
Figure 5 with 3 supplements
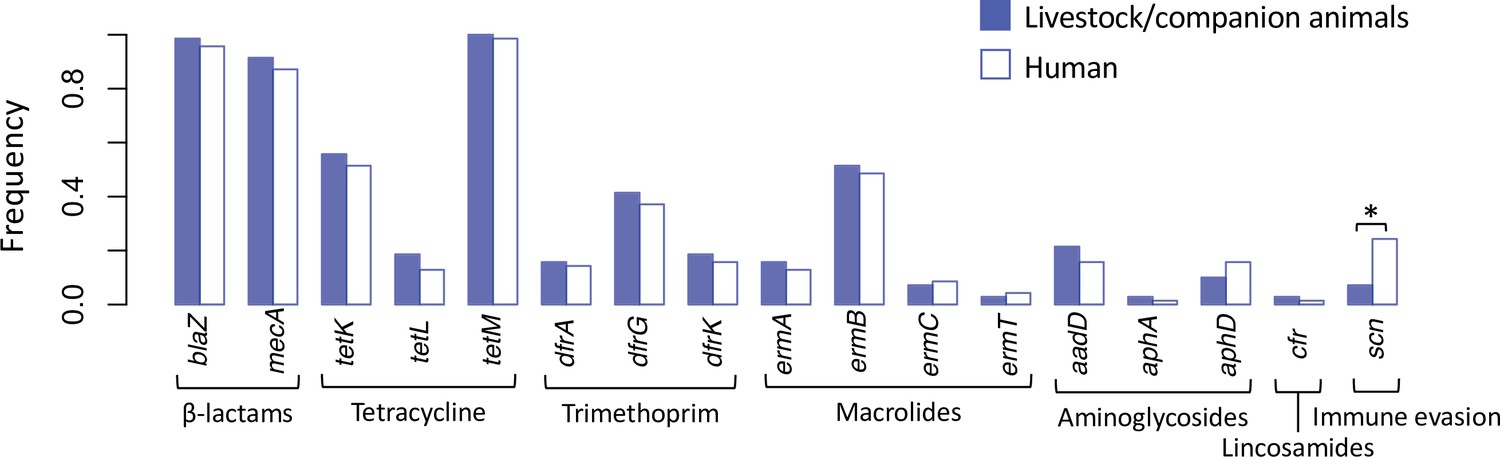
Spillover of livestock-associated CC398 into humans is associated with acquisition of human immune evasion genes.
Seventy phylogenetically independent clades that include isolates from both humans and other species were identified within the livestock-associated clade. The plot shows the frequency with which these genes were identified within isolates from humans (right, empty bars) and non-human species (left, filled bars) in these groups. An asterisk indicates a significant difference based on McNemar’s chi-squared test (p = 1.50 × 10−3). No resistance genes differed significantly in their frequency across the human and non-human hosts (p > 0.1). The scn gene is always present in the human immune evasion cluster carried by φSa3 prophages, and therefore represents the presence of this element.
Figure 5—figure supplement 1

Patterns in the presence/absence of antibiotic resistance genes vary across livestock- and human-associated CC398 in addition to the three genes associated with Tn916 and SCCmec.
Variation in presence of antibiotic resistance genes across CC398 mapped onto the core genome phylogeny. All genes that are present above a threshold of 5% are represented, with colours representing different antibiotic classes.
Figure 5—figure supplement 2
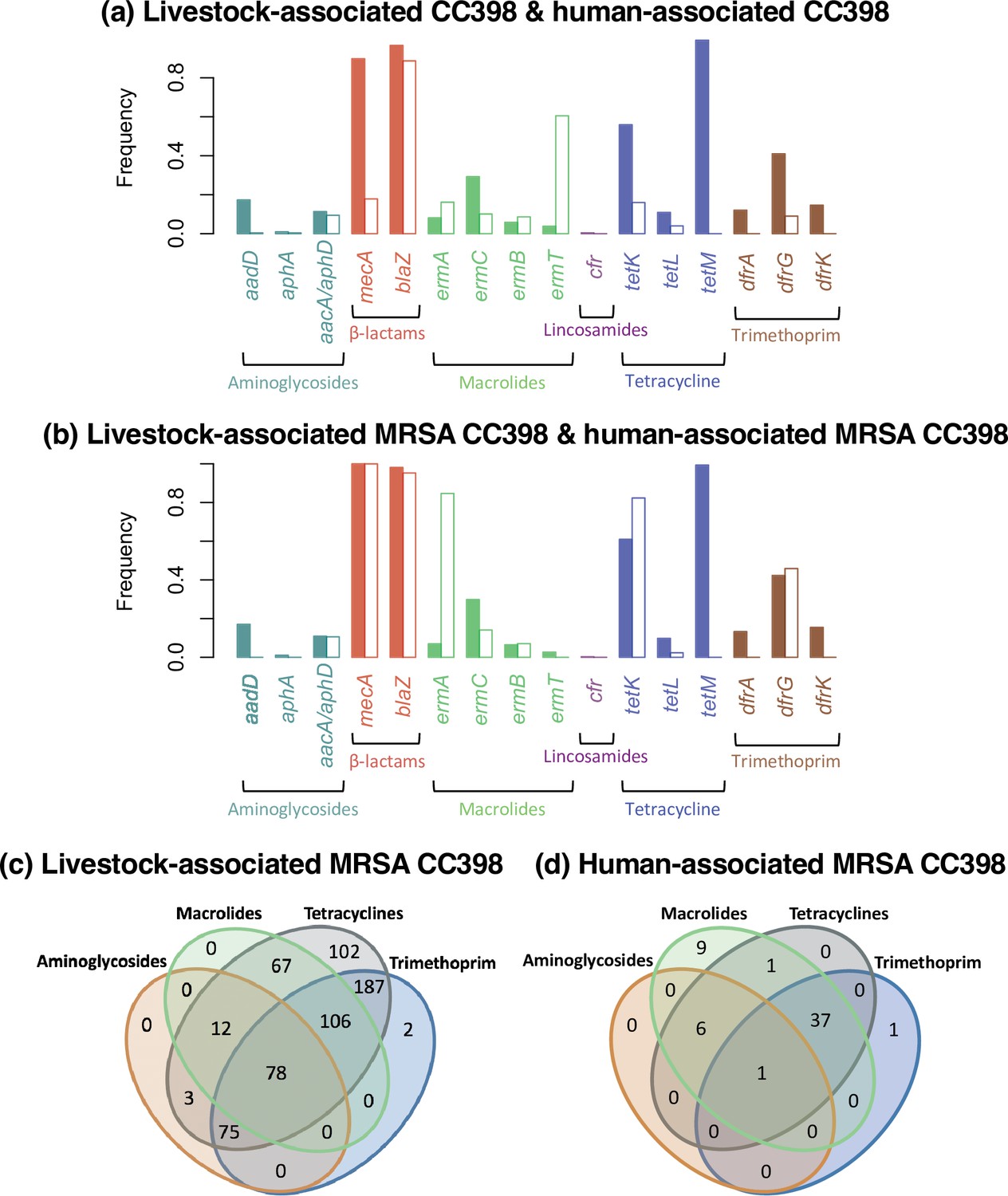
Differences in antibiotic resistance gene frequencies across human- and livestock-associated CC398.
(a) Comparison of the frequencies of resistance genes across clades, with the livestock-associated group shown in the filled bar on the left and the human-associated group the empty bar on the right. (b) Comparison of only isolates carrying mecA from the two groups. (c and d) Venn diagrams that show the relationship between the presence of genes associated with different antibiotic classes in methicillin-resistant Staphylococcus aureus (MRSA) isolates from both groups.
Figure 5—figure supplement 3
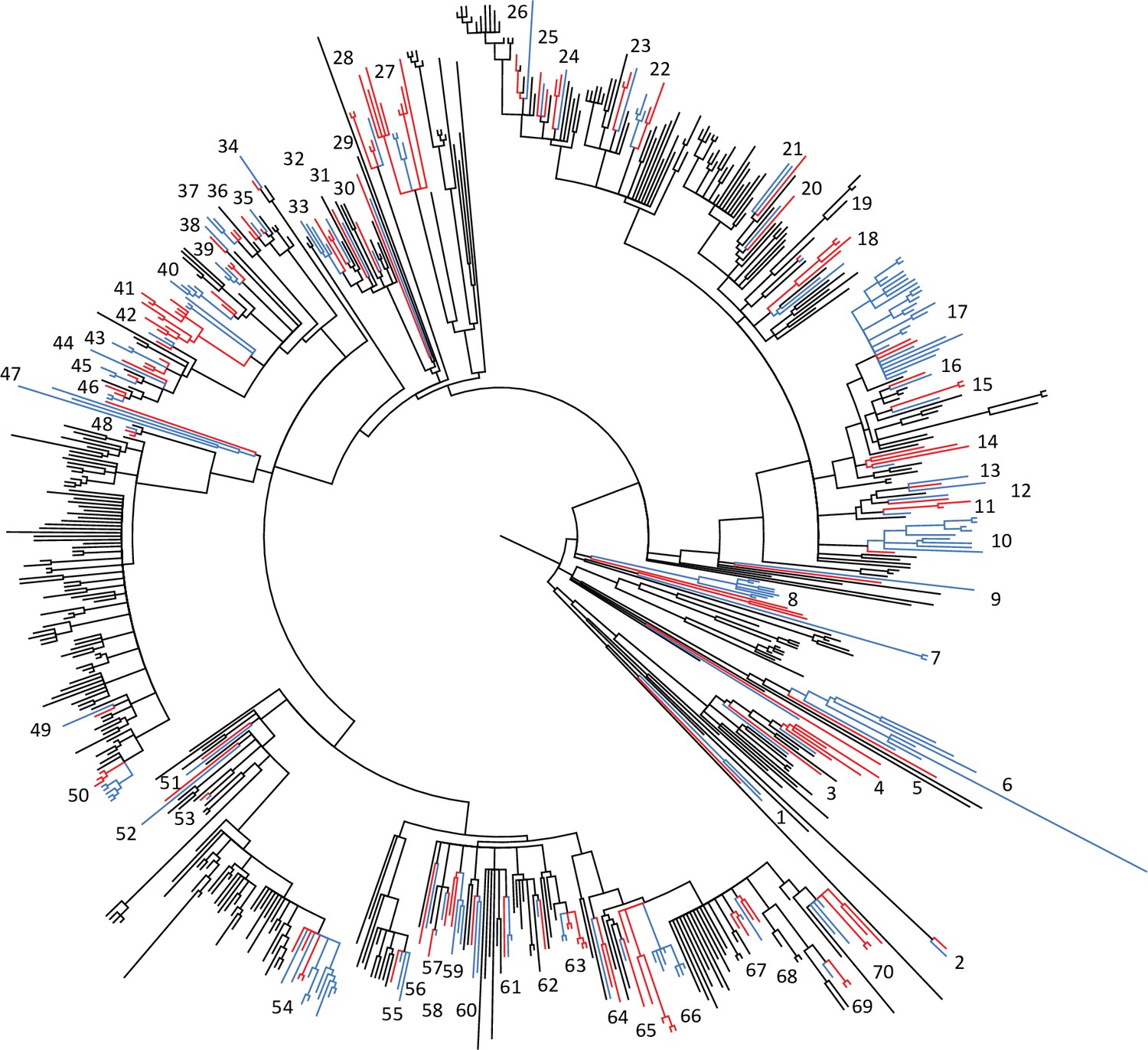
Phylogeny of the livestock-associated clade showing locations of 70 phylogenetically independent groups of isolates from human (red) and livestock/companion animal (blue) hosts.
The phylogeny is a maximum likelihood phylogeny of the livestock-associated clade, rooted using human-associated CC398, with nodes with <70% bootstrap support collapsed. Seventy well-supported independent clades that contain both isolates from human and livestock/companion animal hosts were identified across the livestock-associated clade, and were used to test for differences in the frequency of genes associated with both antibiotic resistance and adaptation to the human host across these two groups. The small number of isolates from wild or domestic animals (other than livestock and horses) was excluded from the analysis.
Additional files
-
Supplementary file 1
Strain names, country of origin, source (host species), year, accession numbers, and references for all isolates.
- https://cdn.elifesciences.org/articles/74819/elife-74819-supp1-v1.xlsx
-
Supplementary file 2
Genes that most strongly distinguish human- and livestock-associated CC398, and their association with mobile genetic elements.
Our gene identifiers and the gene locations and locus tags in published reference genomes are provided.
- https://cdn.elifesciences.org/articles/74819/elife-74819-supp2-v1.xls
-
Supplementary file 3
Description of the presence of mobile genetic elements (MGEs) and annotation of MGE types and clades.
The presence/absence of genes and MGEs is described by 1/0, and types and clades that are presented in the text are described.
- https://cdn.elifesciences.org/articles/74819/elife-74819-supp3-v1.xls
-
Supplementary file 4
Description of the genes in the Tn916 element.
The genes in the Tn916 element used in our analyses are described in the reference genome 1_1439.
- https://cdn.elifesciences.org/articles/74819/elife-74819-supp4-v1.xls
-
Supplementary file 5
Reference SCCmec elements used in BLAST typing.
- https://cdn.elifesciences.org/articles/74819/elife-74819-supp5-v1.xls
-
Supplementary file 6
Description of the genes in the type V SCCmec element.
The genes in the SCCmec type Vc element used in our analyses are described in the reference genome 12_LA_293.
- https://cdn.elifesciences.org/articles/74819/elife-74819-supp6-v1.xls
-
Supplementary file 7
AMR genes identified by PathogenWatch.
Gene presence/absence is described by 1/0.
- https://cdn.elifesciences.org/articles/74819/elife-74819-supp7-v1.xls
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/74819/elife-74819-transrepform1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Stable antibiotic resistance and rapid human adaptation in livestock-associated MRSA
eLife 11:e74819.
https://doi.org/10.7554/eLife.74819




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}