Applying causal discovery to single-cell analyses using CausalCell
- Bioinformatics Section, School of Basic Medical Sciences, Southern Medical University, China
- The Shraga Segal Department of Microbiology, Immunology and Genetics, Faculty of Health Sciences, Ben-Gurion University of the Negev, Israel
- Network Center, Southern Medical University, China
- Department of Pathology, School of Basic Medical Sciences, Southern Medical University, China
- Guangdong-Hong Kong-Macao Greater Bay Area Center for Brain Science and Brain-Inspired Intelligence, Southern Medical University, China
- Guangdong Provincial Key Lab of Single Cell Technology and Application, Southern Medical University, China
Figures
Figure 1
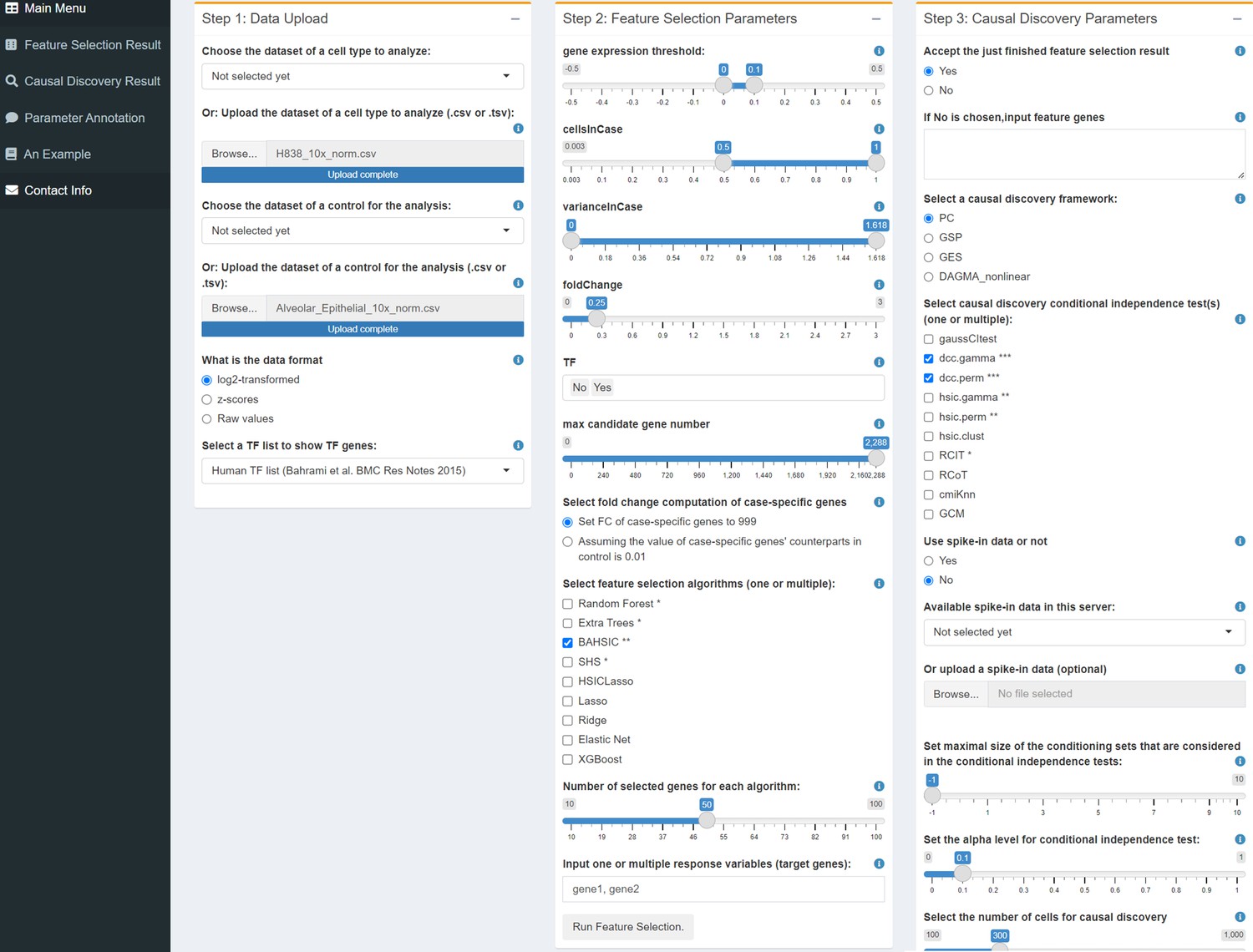
The user interface of CausalCell.
Multiple algorithms and functions are integrated and implemented to facilitate and compare feature selection and causal discovery.
Figure 2
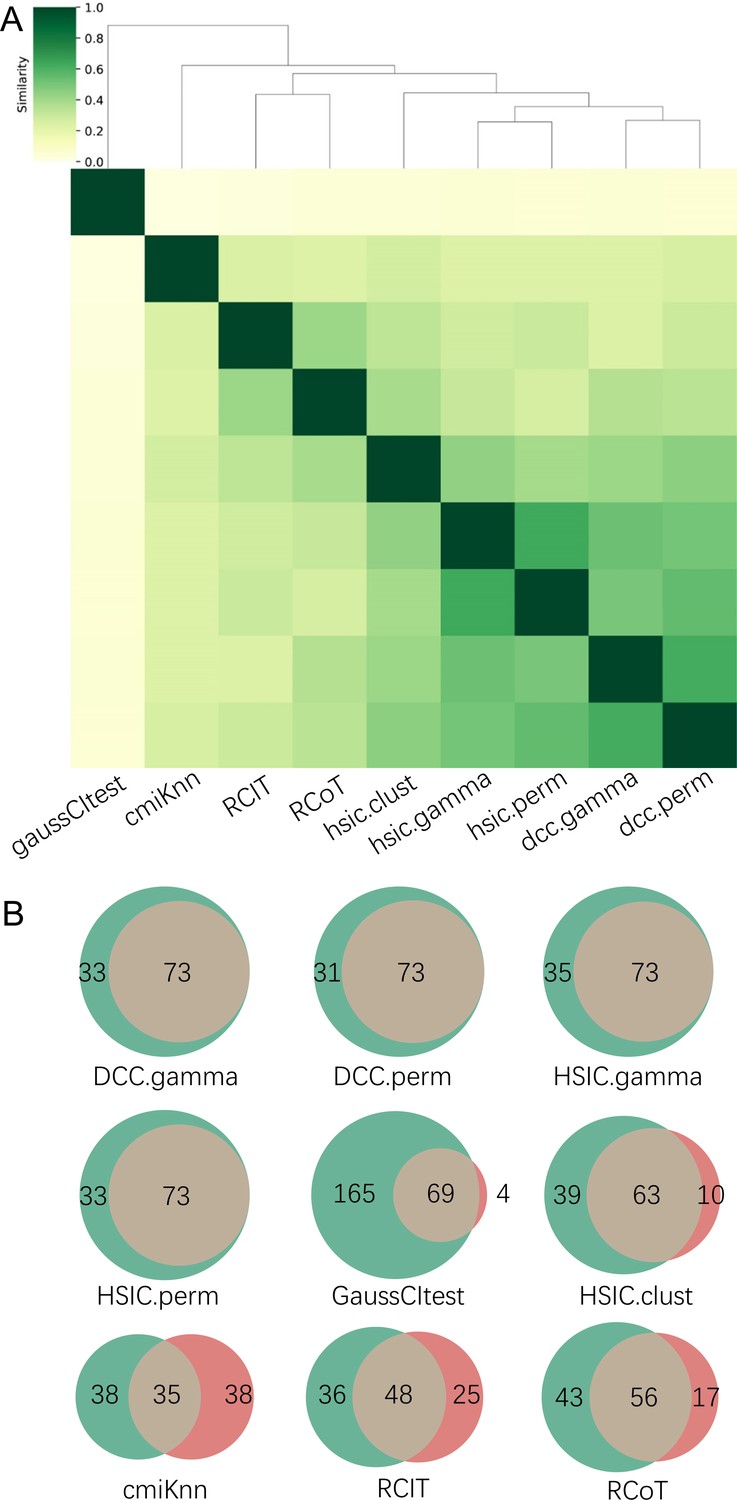
The accuracy of PC+nine CI tests was evaluated with four steps.
First, nine causal networks were inferred using the nine CI tests. Second, pairwise structural Hamming distances (SHD) between these networks were computed, and the matrix of SHD values was transformed into a matrix of similarity values (using the equation Similarity = exp(-Distance/2σ2), where σ=5). The networks of DCC.gamma, DCC.perm, HSIC.gamma, and HSIC.perm share the highest similarity. Third, a consensus network was built using the networks of the above four CI tests, which was assumed to be closer to the ground truth than the network inferred by any single algorithm. Fourth, each of the nine networks was compared with the consensus network. (A) The cluster map shows the similarity values (darker colors indicating higher similarity). (B) Shared and specific interactions in each algorithm’s network and the consensus network. In each panel, the gray-, green-, and pink-circled areas and numbers indicate the overlapping interactions, interactions identified specifically by the algorithm, and interactions specifically in the consensus network. There are 73 overlapping interactions between DCC.gamma’s network and the consensus network, and 33 interactions were identified specifically by DCC.gamma. Thus, the true positive rate (TPR) of DCC.gamma is 73/ (73+33)=68.9%. The TPRs of DCC.perm, HSIC.gamma, HSIC.perm, GaussCItest, HSIC.clust, cmiKnn, RCIT, and RCoT are 70.2%, 67.6%, 68.9%, 29.5%, 61.8%, 47.9%, 57.1%, and 56.6%, indicating that the two distance covariance criteria (DCC) CI tests perform better than others.
Figure 3
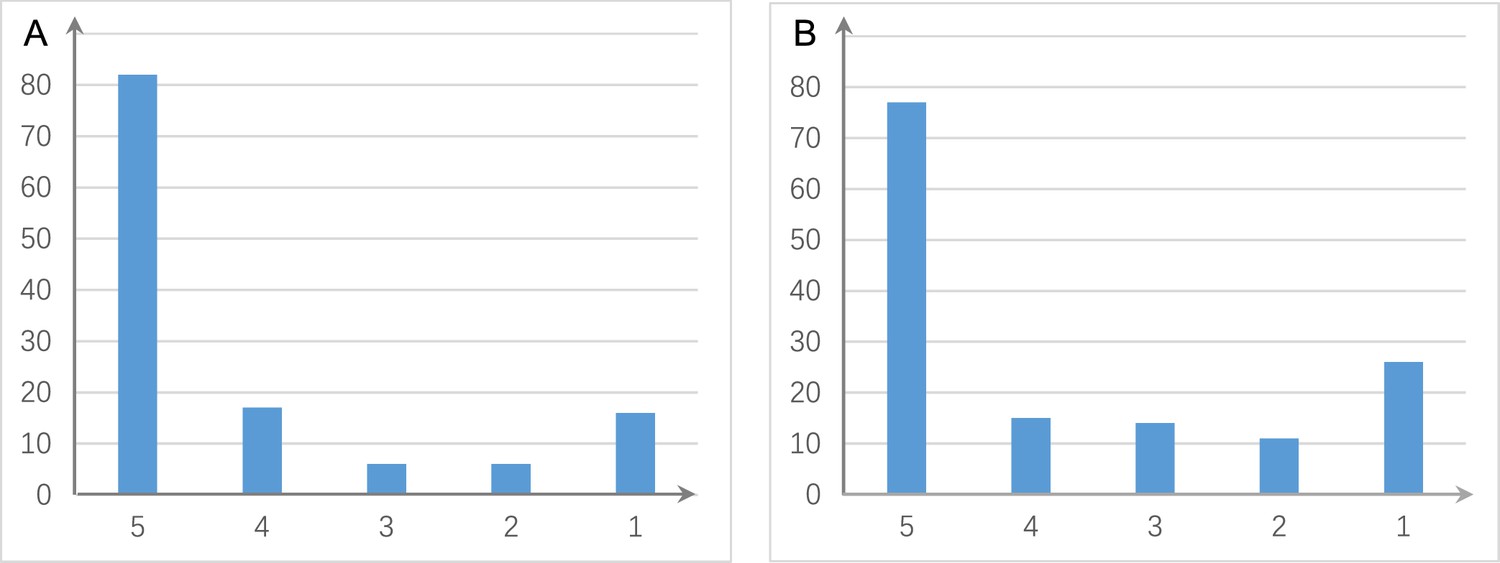
The shared and distinct interactions inferred by running causal discovery five times using the H2228 cell line dataset.
Numbers on the vertical and horizontal axes represent the percentages of interactions in 1, 2, 3, 4, and 5 networks, respectively. (A) The results of PC+DCC.gamma. (B) The results of PC+DCC.perm. These results indicate that 78% and 64.3% of interactions occurred stably in ≥4 networks, suggesting that the inferred networks are quite stable.
Figure 4
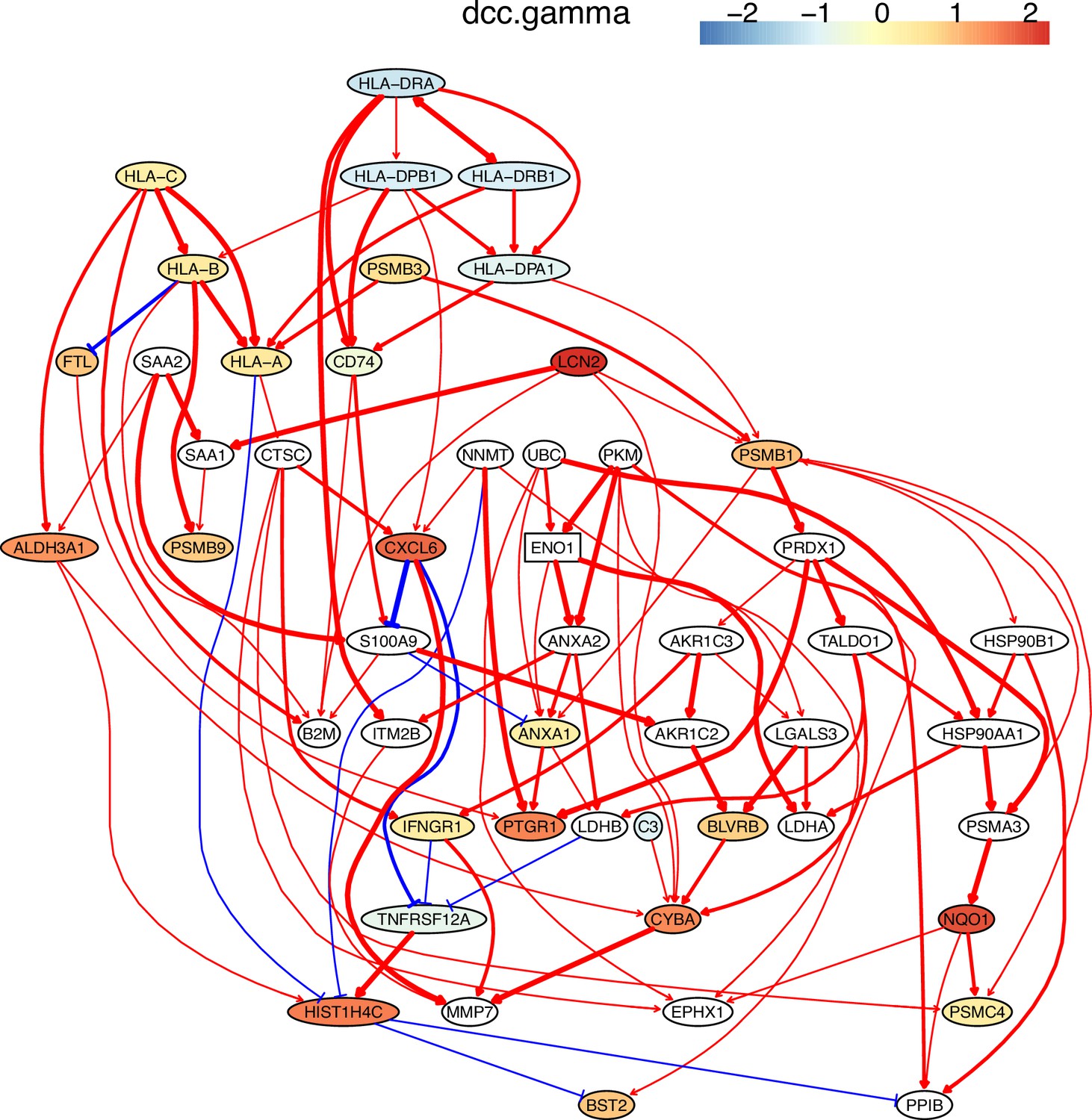
The network of the 50 genes inferred by DCC.gamma from the H2228 dataset (the alpha level for CI test was 0.1).
Red → and blue -| arrows indicate activation and repression, and colors indicate fold changes of gene expression compared with genes in the alveolar epithelial cells.
Figure 5
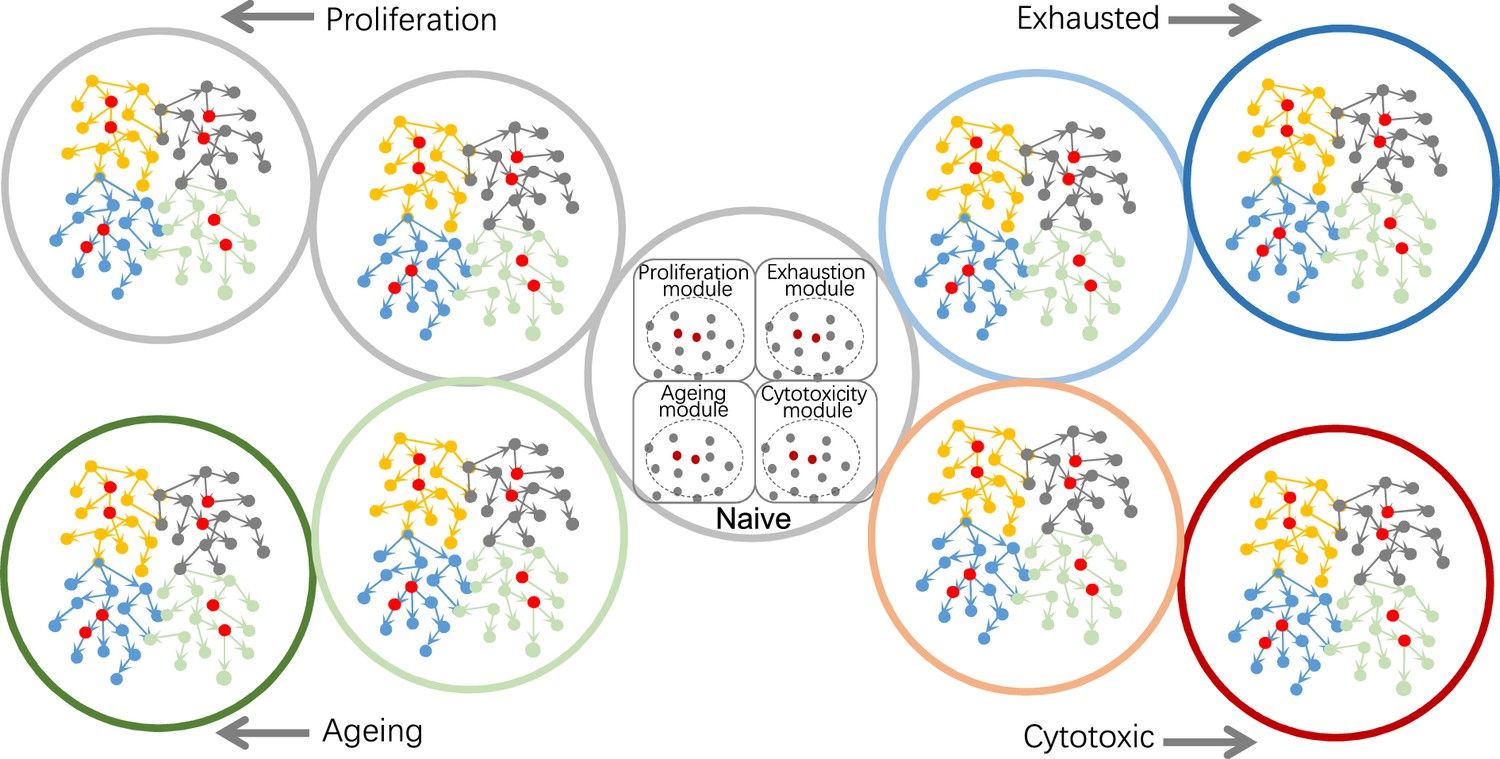
Using causal discovery to analyze different cells, cells at different stages, or different biological processes in cells.
The red and gray dots within the four circles in the central cell indicate the four modules' core genes and related genes. Genes in different modules should be chosen as target genes when exploring different biological processes.
Appendix 1—figure 1
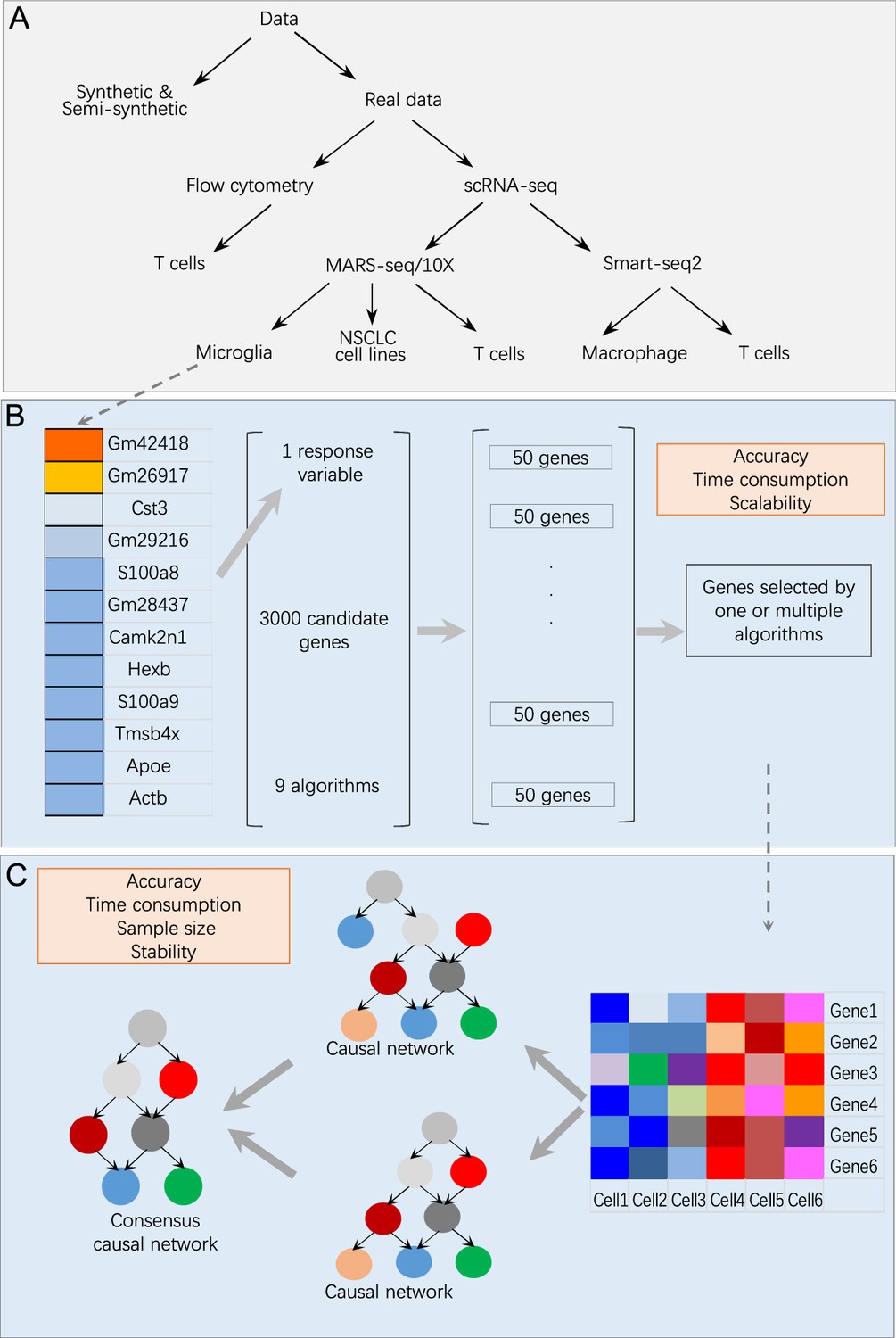
Overview of data and benchmarking.
(A) The single-cell RNA-sequencing (scRNA-seq) data were generated by different protocols and from different cell types (Appendix 1—table 1). (B) Illustration of feature selection benchmarking using data of microglia from humans and mice. Steps: (i) choose a target gene from a list of microglia biomarkers; (ii) let each algorithm select 50 genes from 3000 candidates expressed in most cells; (iii) merge the nine sets of feature genes into a superset; (iv) compare each selected feature gene set with the superset. (C) Illustration of causal discovery benchmarking using a set of feature genes. Steps: (i) use nine algorithms (PC+CI tests) to generate nine causal networks; (ii) generate a type 1 consensus network upon the networks of multiple algorithms (a type 2 consensus network is generated upon running an algorithm multiple times); (iii) compare each causal network with the consensus network.
Appendix 2—figure 1
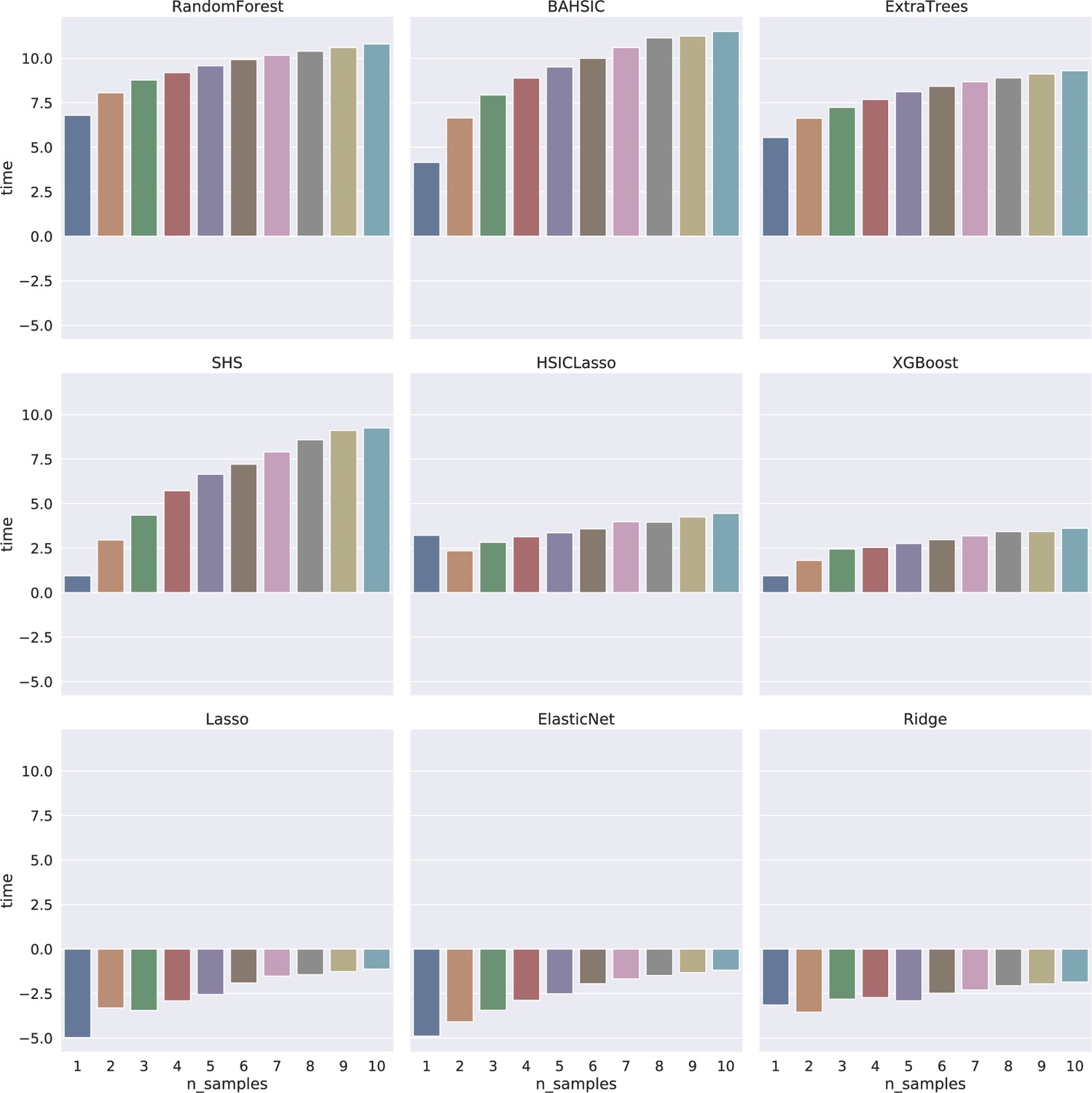
Time consumption of feature selection algorithms increased mildly when the sample size increased from 1000 to 10,000 (the X-axis indicates the sample size; 1–10 indicate 1000–10,000, respectively).
The Y-axis indicates time (s) in the log2 form. The log2 form makes the time consumption of Lasso, ElasticNet, and Ridge have negative values (<1 s).
Appendix 2—figure 2
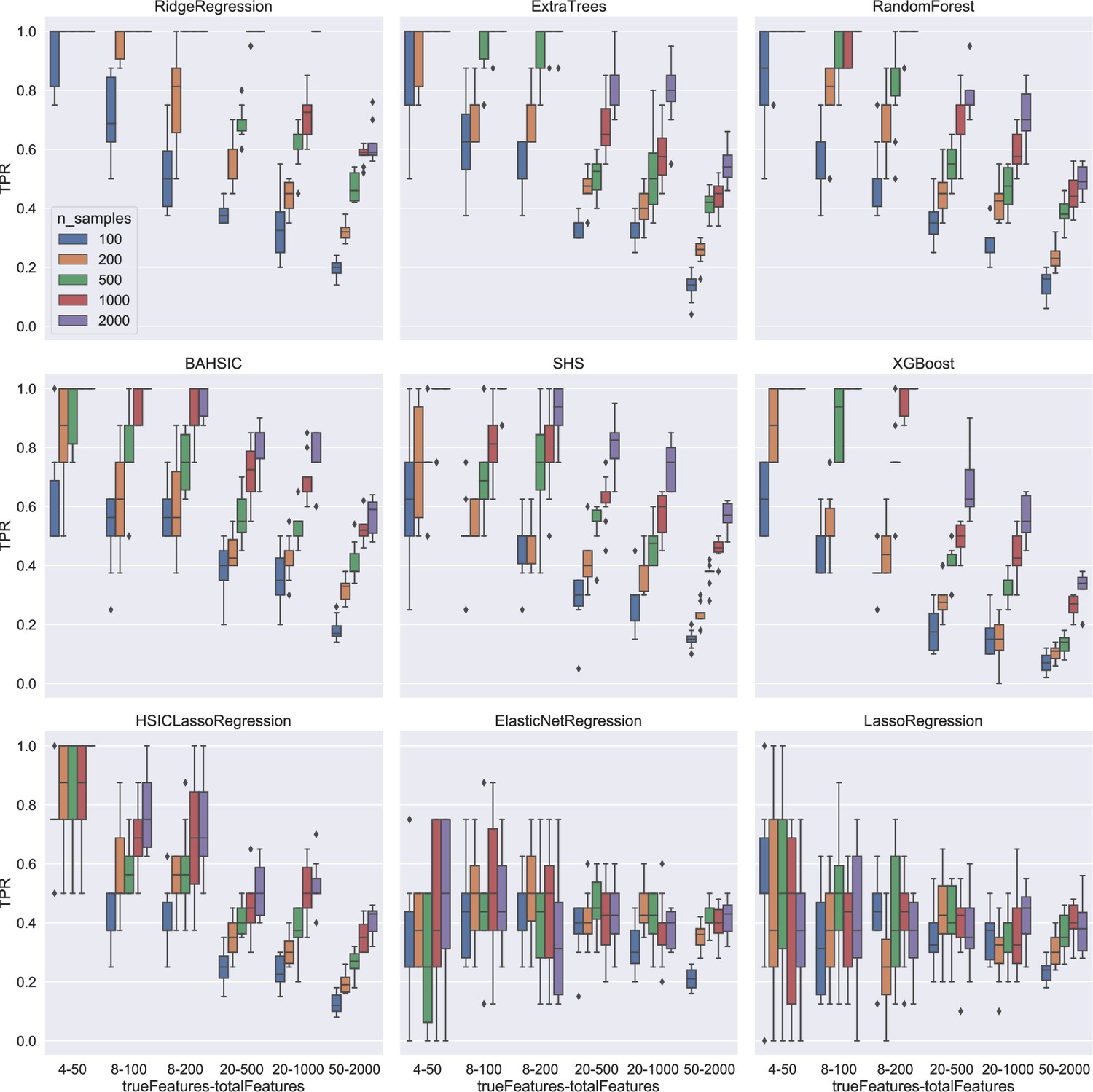
Feature selection results using synthetic data.
Colors indicate different sample sizes (see the inset in the top-left panel). The X-axis (depicted under the bottom panels) indicates different schemes. For example, 4–50 means that there are 50 candidates (i.e. total features), 4 of which are chosen randomly to generate the response variable, and feature selection should select the 4 features (i.e. true features) from the 50 candidates upon the response variable. The Y-axis indicates true positive rate (TPR). For simple schemes (e.g. 4–50), some algorithms reached a TPR of 1.0. For complex schemes (e.g. 50–2000, selecting 50 features from 2000 candidates upon the response variable), some algorithms (especially BAHSIC) reached a TPR of 0.6.
Appendix 2—figure 3
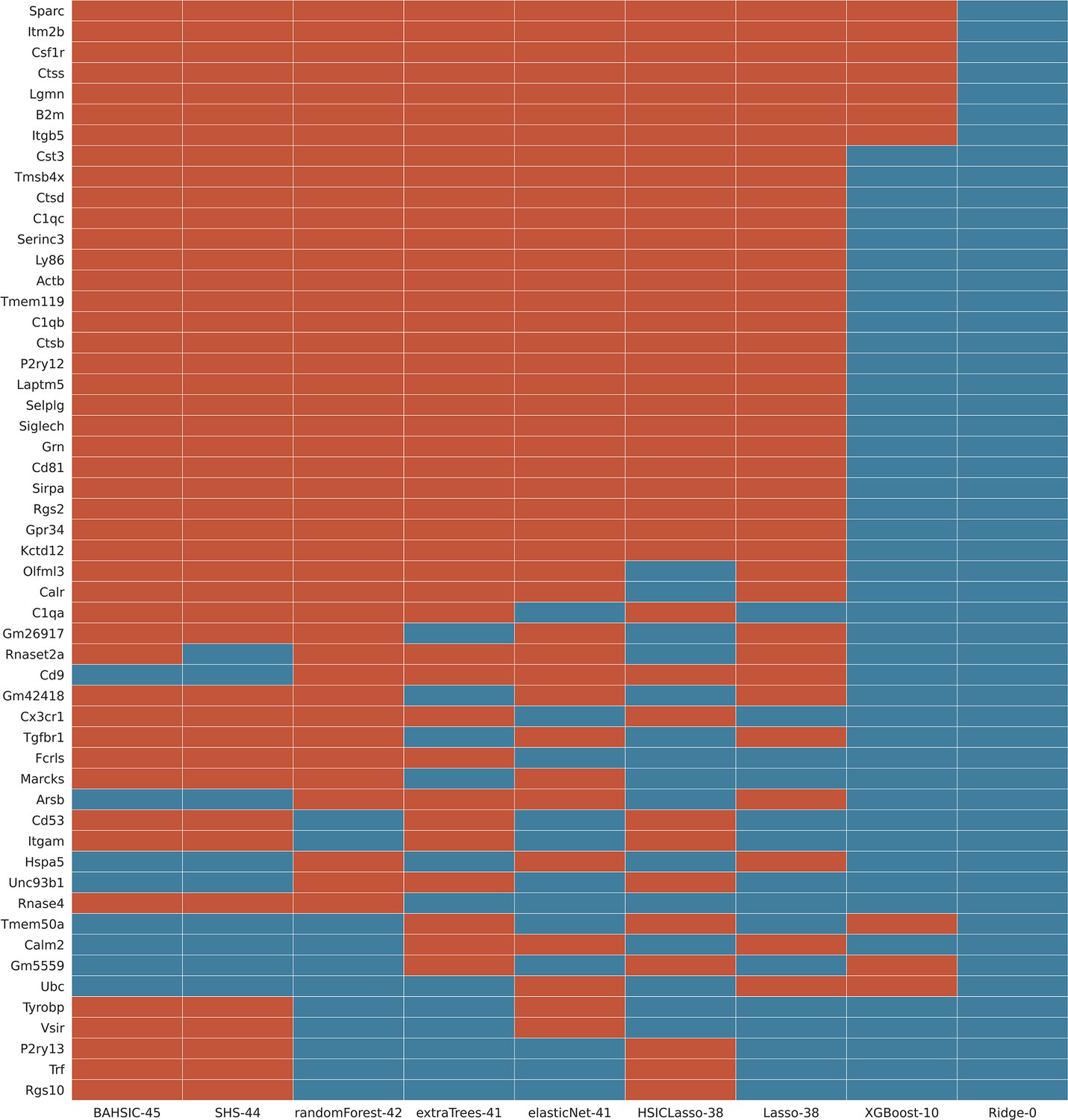
This screenshot showed a feature selection result (the superset of feature genes selected by ≥3 algorithms from the mouse microglia dataset) when all 9 algorithms were used.
The Hexb gene was the target gene, and each algorithm selected 50 feature genes from the 3000 candidate genes expressed in most cells. BAHSIC, SHS, RandomForest, ExtraTrees, ElasticNet selected highly overlapping feature genes, many of which are microglia biomarkers in mice (Appendix 2—figure 4). The numbers right side of algorithm names indicate genes overlapping with the superset.
Appendix 2—figure 4
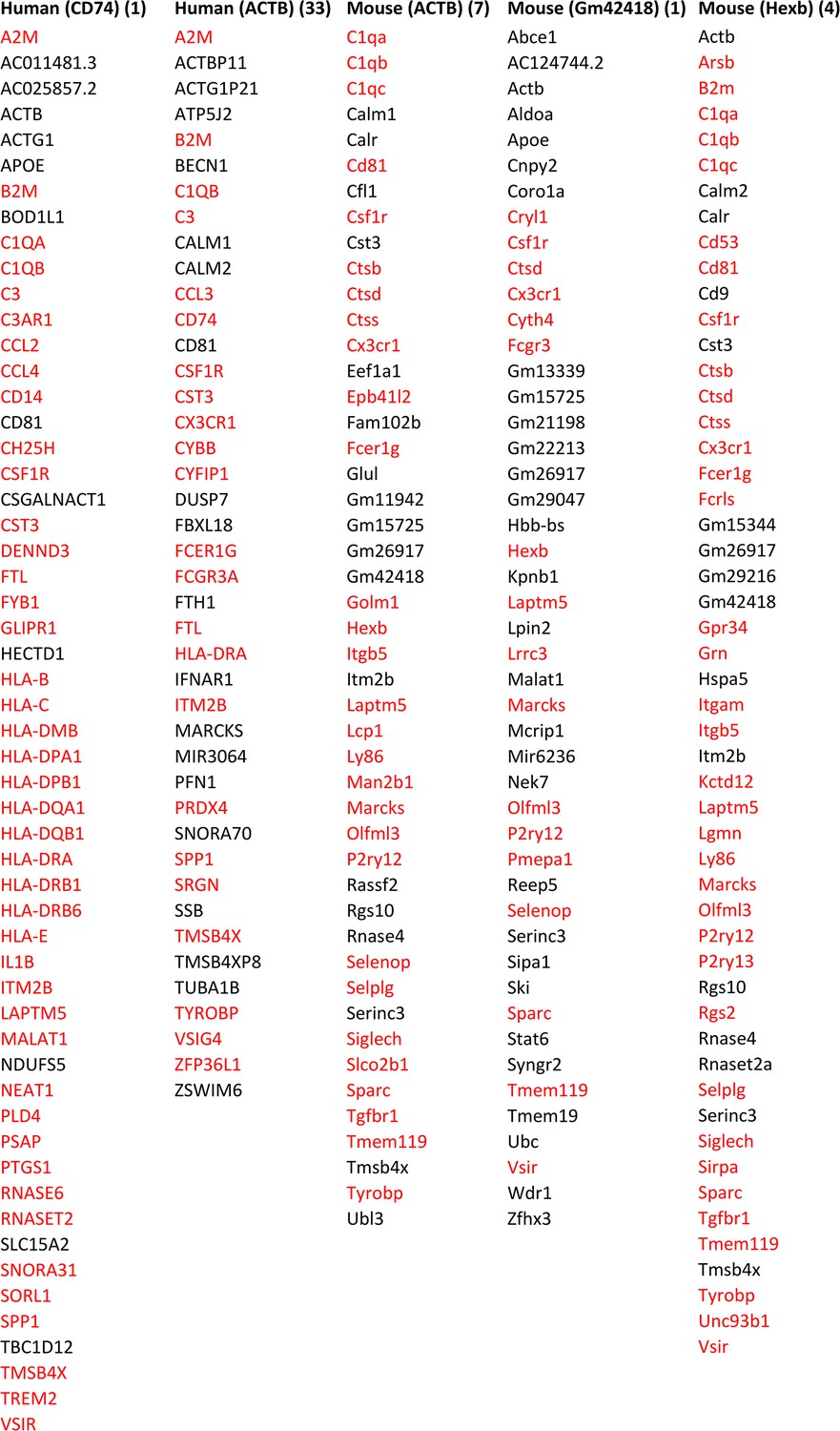
Feature genes selected by ≥3 algorithms in microglia from humans and mice using CD74, ACTB, ACTB, Gm42418, with Hexb as the target gene.
The number right side of each target gene indicates the rank of its transcript’s variance in the 3000 candidate genes. A large variance indicates that the gene may be important in the examined cells. Many feature genes are microglia biomarkers, indicating that feature genes selected by ≥3 algorithms are biologically rational. Annotated microglia biomarkers in humans and mice are marked in red (Patir et al., 2019).
Appendix 2—figure 5

Cell types and cells that express macrophage biomarker genes.
(A) The tSNE plot shows all cells isolated from the human glioblastoma (Neftel et al., 2019). Region 2 (the blue area in (B)) is macrophages. (B) The six macrophage biomarker genes were exclusively expressed in macrophages (the blue area).
Appendix 2—figure 6

These tSNE plots show that nearly all of the 50 feature genes were exclusively expressed in macrophages.
These feature genes were selected by BAHSIC using six macrophage biomarkers (CD14, AIF1, FCER1G, FCGR3A, TYROBP, and CSF1R) as the target genes. They include genes involved in macrophage activation (e.g. C1QA, CD74, TREM2) and multiple class II major histocompatibility complex (MHC) genes (e.g. HLA-DMA, HLA-DPA1). The interactions between CD74 and MHC-II genes (CD74 is an MHC class II chaperone) probably contribute to the co-selection of these genes.
Appendix 2—figure 7
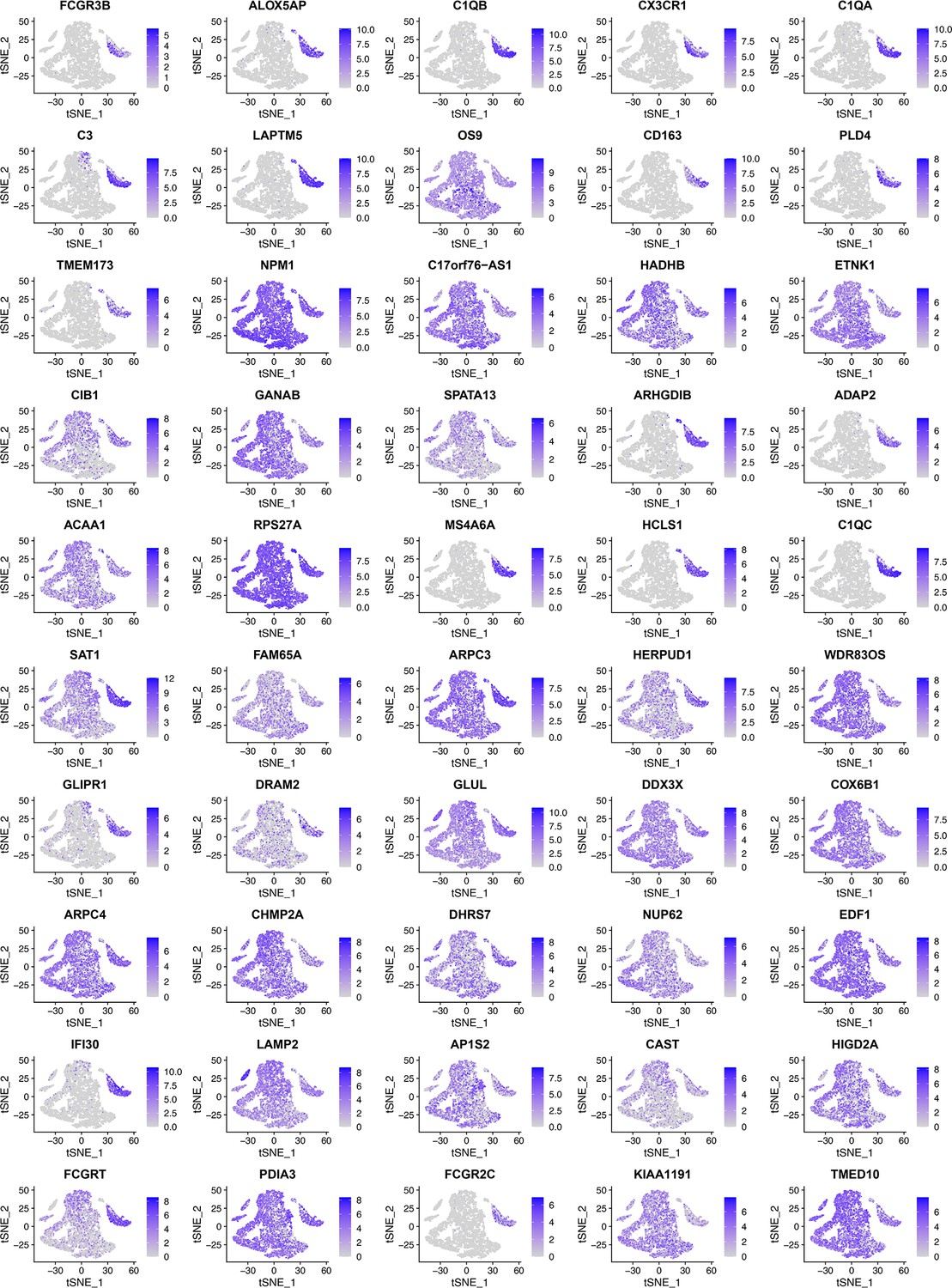
When RidgeRegression was used to select 50 feature genes using the same six macrophage markers (CD14, AIF1, FCER1G, FCGR3A, TYROBP, and CSF1R) as target genes, as these tSNE plots show, many feature genes were expressed in diverse cells instead of macrophages.
Appendix 3—figure 1
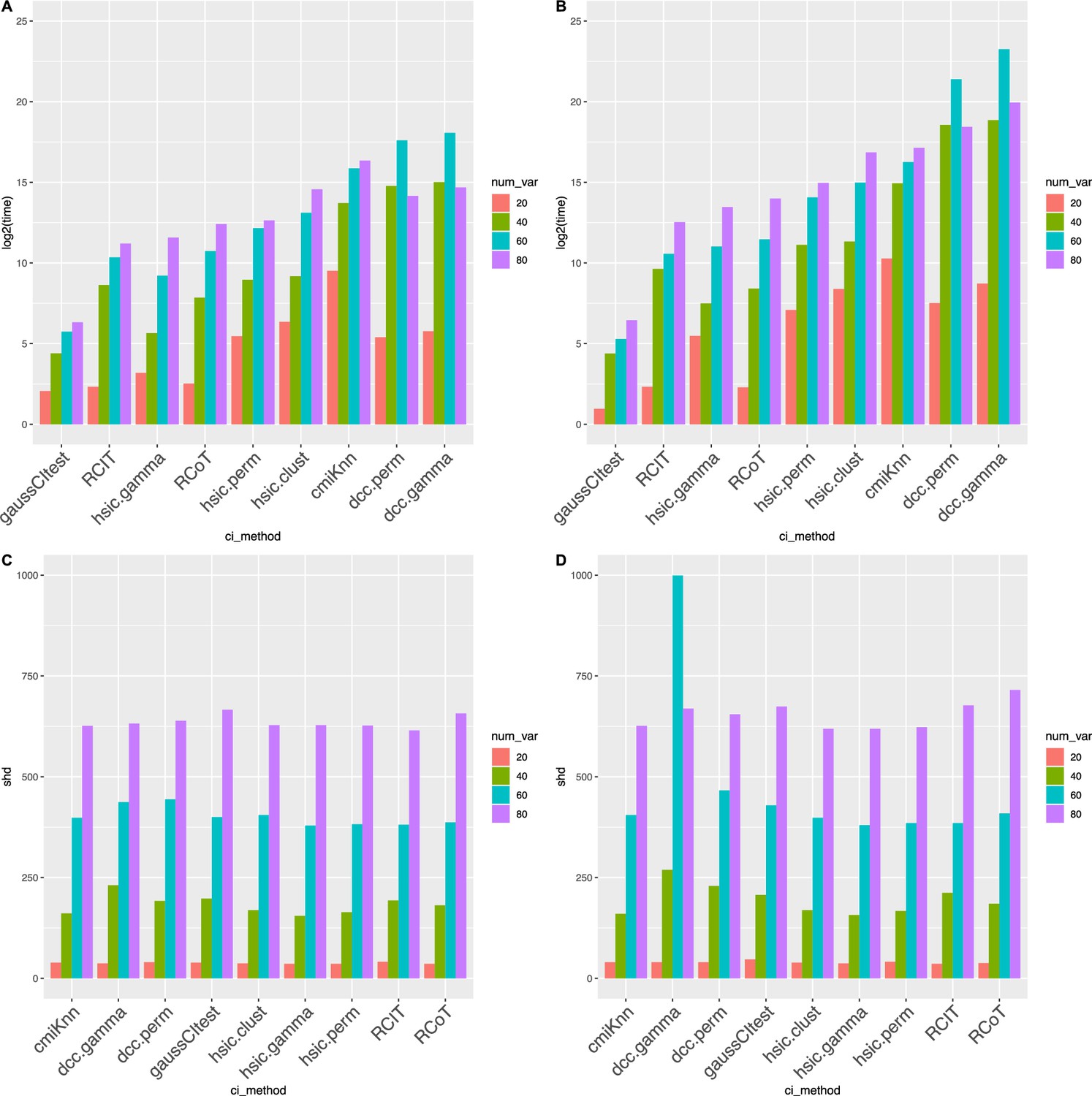
Causal discovery performance that was tested using synthetic data.
DCC.perm and HSIC.gamma are the most time-consuming algorithms, but all algorithms have similar accuracy. The sample size is 500 (AB) or 1000 (CD), and the variable number ranges from 20 to 80. (AC) show the time consumption (in second) of different algorithms, and (BD) show structural Hamming distance (SHD) values. ‘*‘ in (CD) indicates that the algorithm did not finish running in 6 weeks. These two cases, and the two cases in (A) where DCC.perm and DCC.gamma took more time when there were 60 variables than when there were 80 variables, were anomalies caused by synthetic data. When testing using real scRNA-seq data, no such anomalies occurred.
Appendix 3—figure 2

Consistency between each causal network and the consensus network.
(A) In the cluster map of different CI tests, darker colors indicate a higher similarity of networks. The networks of HSIC.gamma, HSIC.perm, HSIC.gamma, and HSIC.perm have the highest similarity values, thus sharing the most similar structures. We used the four networks to build a consensus network, which was assumed most close to the ground truth. (B) For interactions inferred by each algorithm (green circle), we checked how many interactions overlap the interactions in the consensus network (pink circle). The true positive rate (TPR) of DCC.gamma, DCC.perm, HSIC.gamma, HSIC.perm, GaussCItest, HSIC.clust, cmiKnn, RCIT, and RCoT were 62.7%, 63.4%, 63.4%, 70.3%, 14.6%, 40.5%, 26.7%, 50.8%, and 44.0%, respectively, confirming that Hilbert-Schmidt independence criterion (HSIC) and distance covariance criteria (DCC) are better than others and that it is reasonable to use the consensus network generated upon the four algorithms' networks to evaluate algorithms' performance.
Appendix 3—figure 3
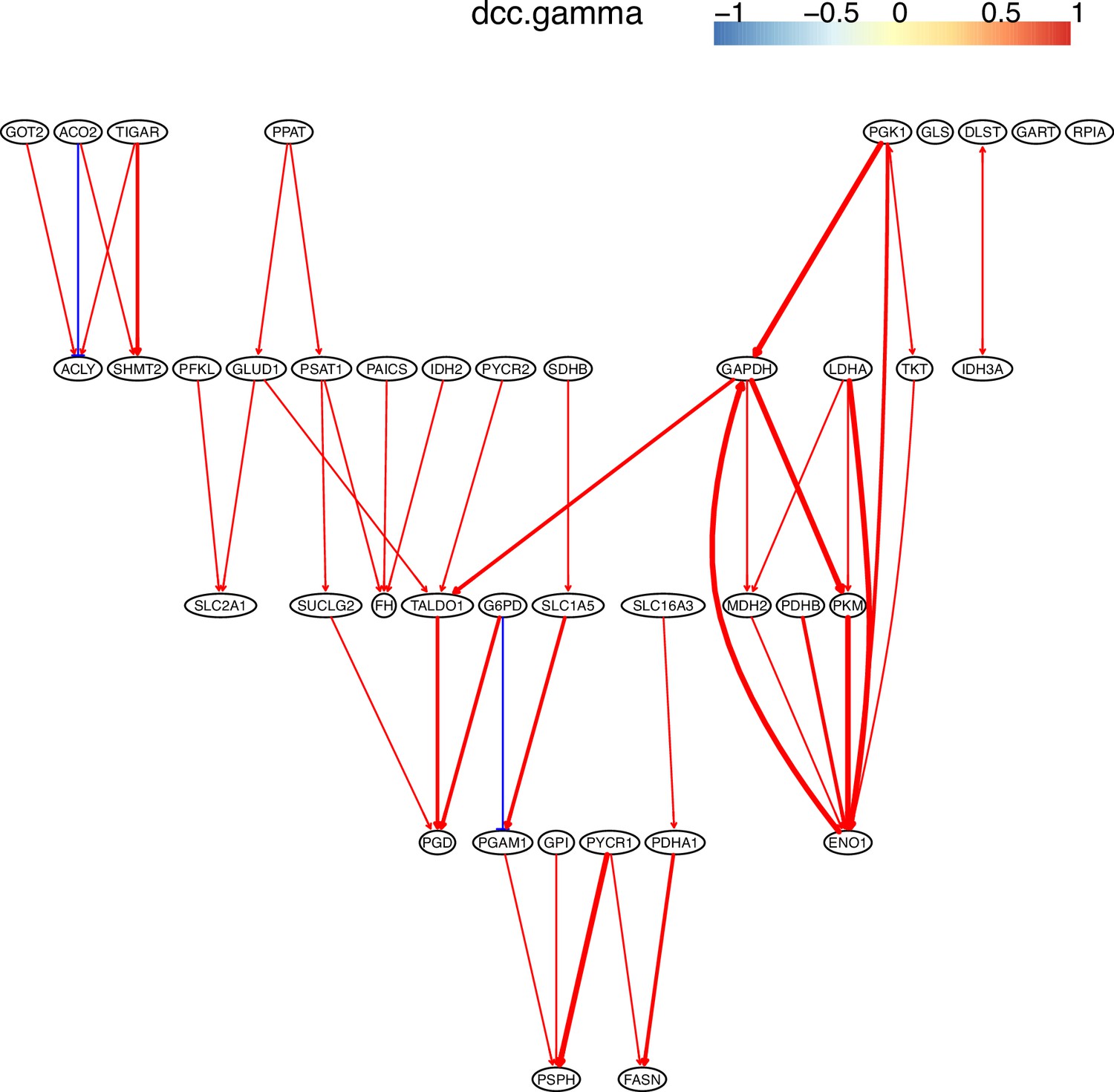
The causal relationships between genes in the WP4290 pathway in the cell line H838 inferred using 200 cells (alpha = 0.1).
The color bar indicates fold changes of genes in the case dataset compared with in the control dataset.
Appendix 3—figure 4
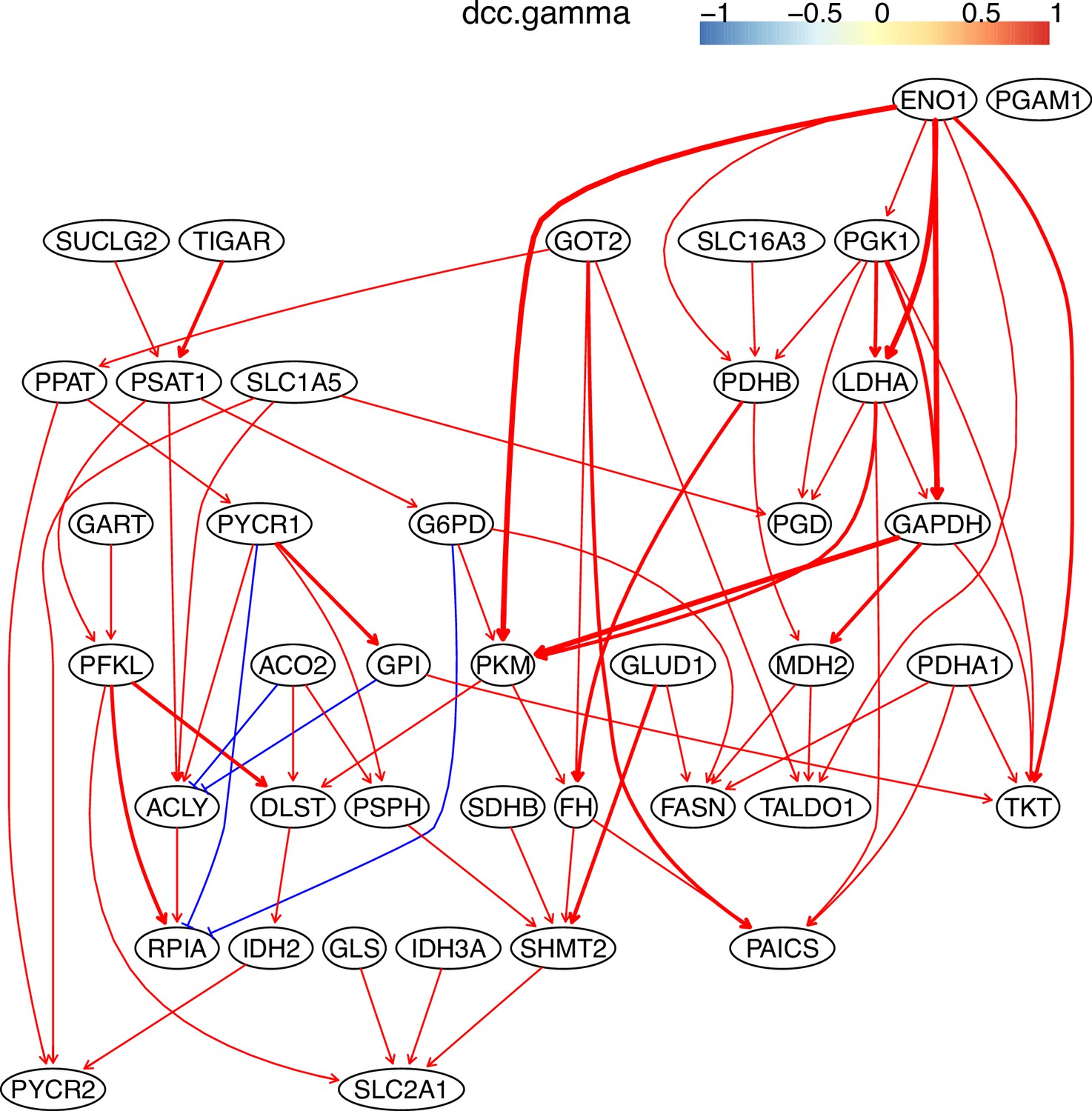
The causal relationships between genes in the WP4290 pathway in the cell line H838 inferred using 400 cells (alpha = 0.1).
Appendix 3—figure 5
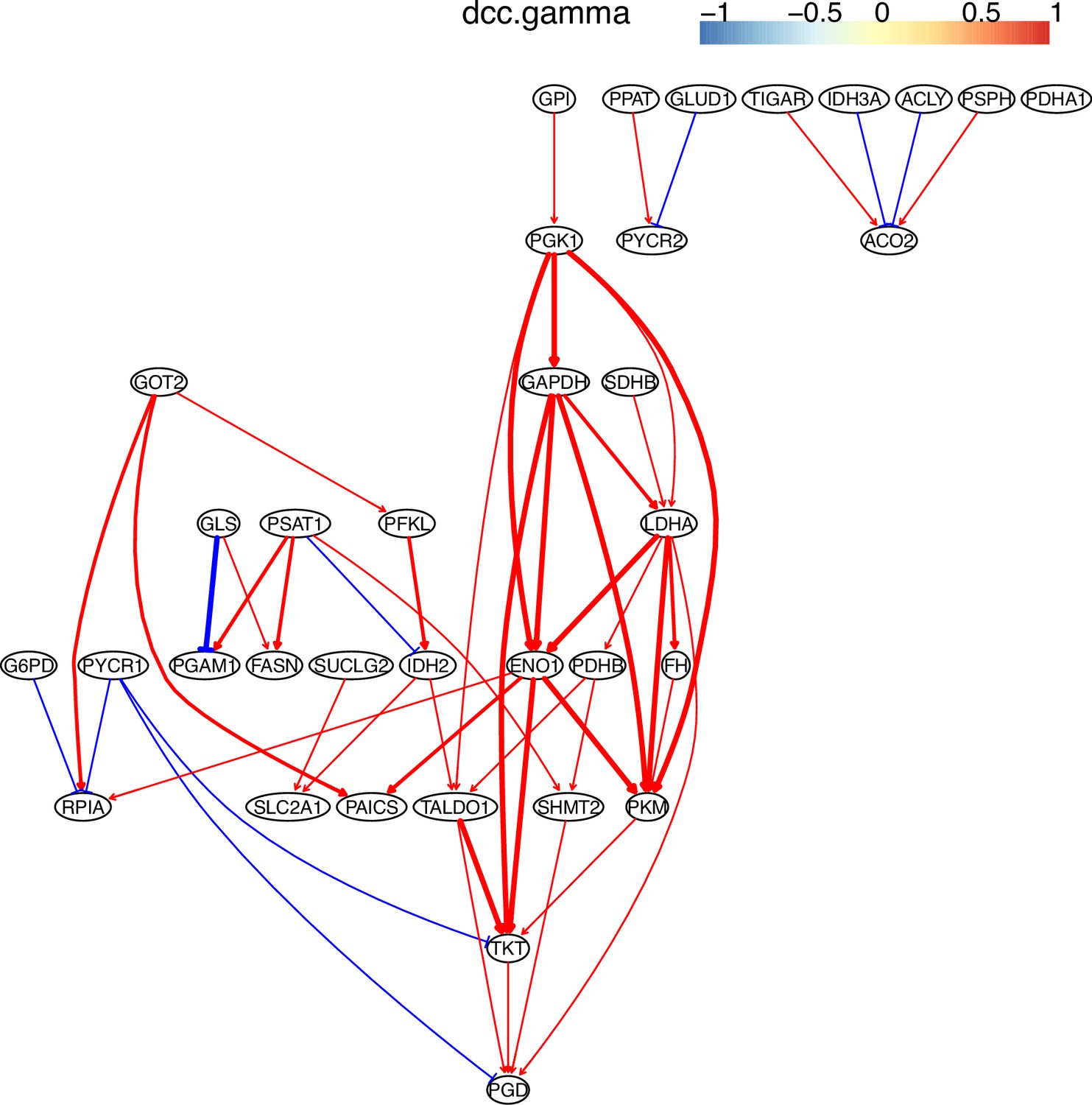
The causal relationships between genes in the WP4290 pathway in the cell line H838 inferred using 600 cells (alpha = 0.1).
Appendix 3—figure 6
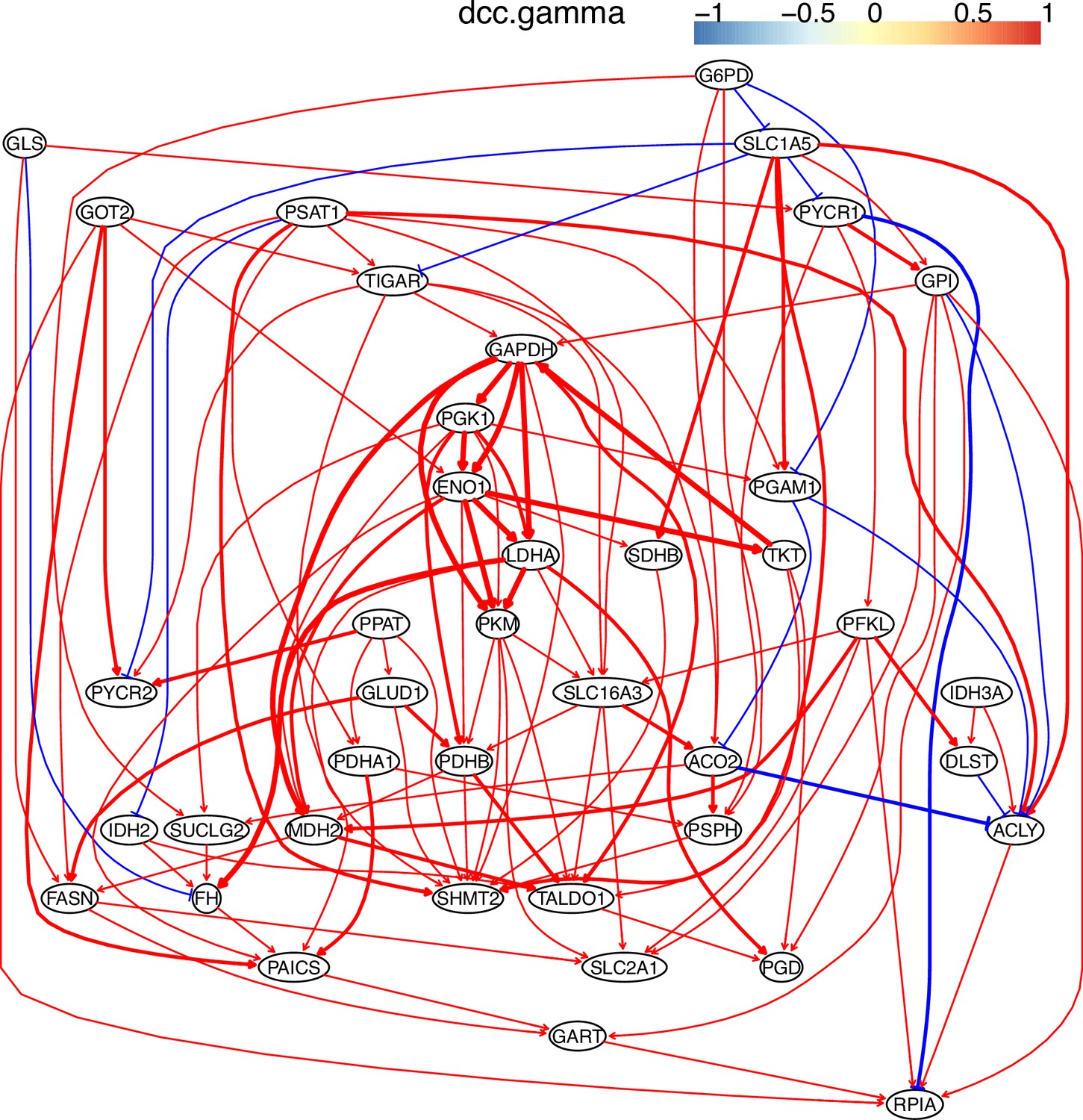
The causal relationships between genes in the WP4290 pathway in the cell line H838 inferred using 800 cells (alpha = 0.1).
More cells make more relationships be inferred, but relationships with high significance (with thick arrows) are stable.
Appendix 3—figure 7
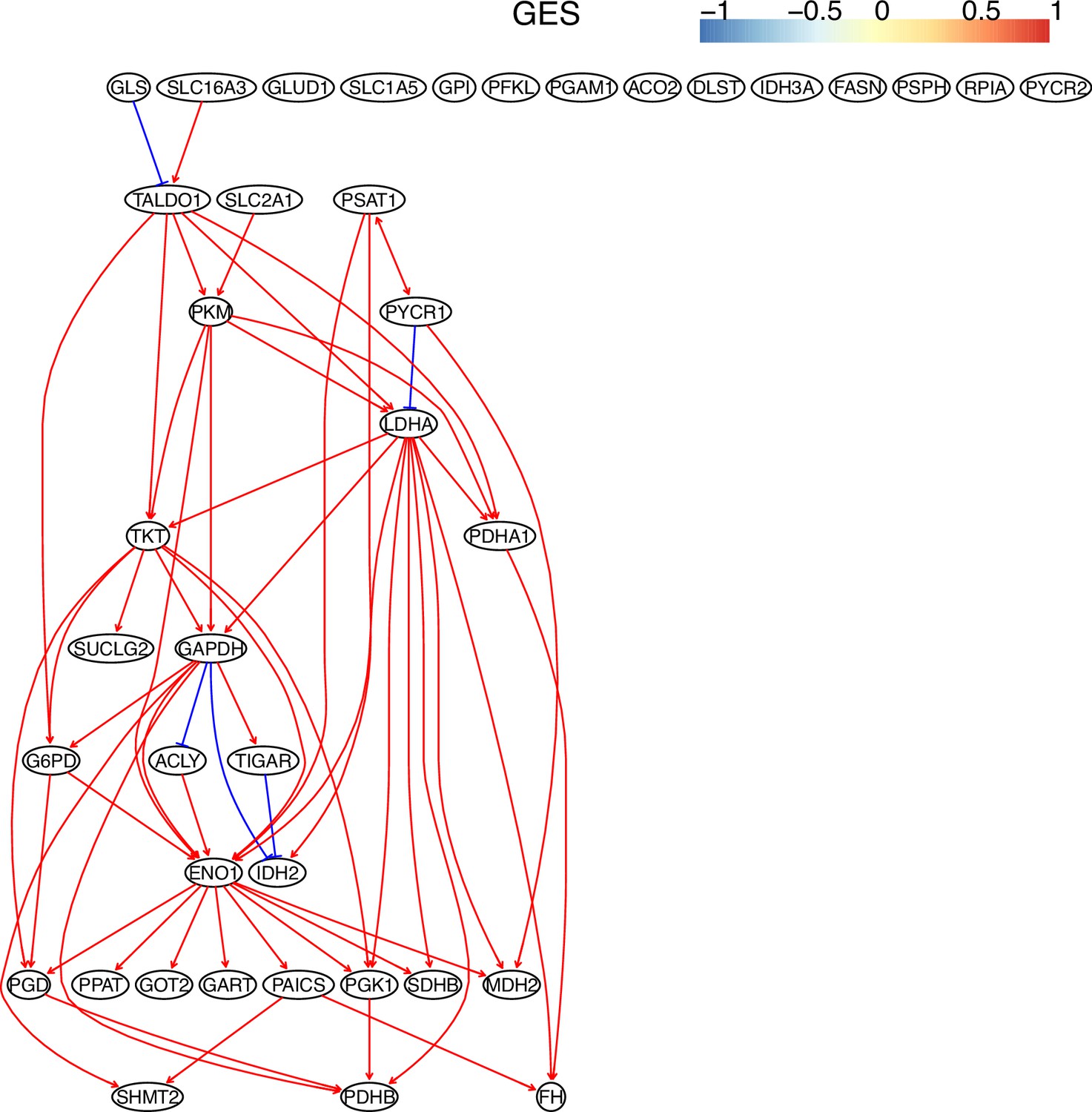
The causal relationships between genes in the WP4290 pathway in the cell line A549 inferred using the GES method and 600 cells (alpha = 0.1).
Compared with the networks in these cells inferred using PC+DCC.gamma, here there are more isolated nodes.
Appendix 3—figure 8
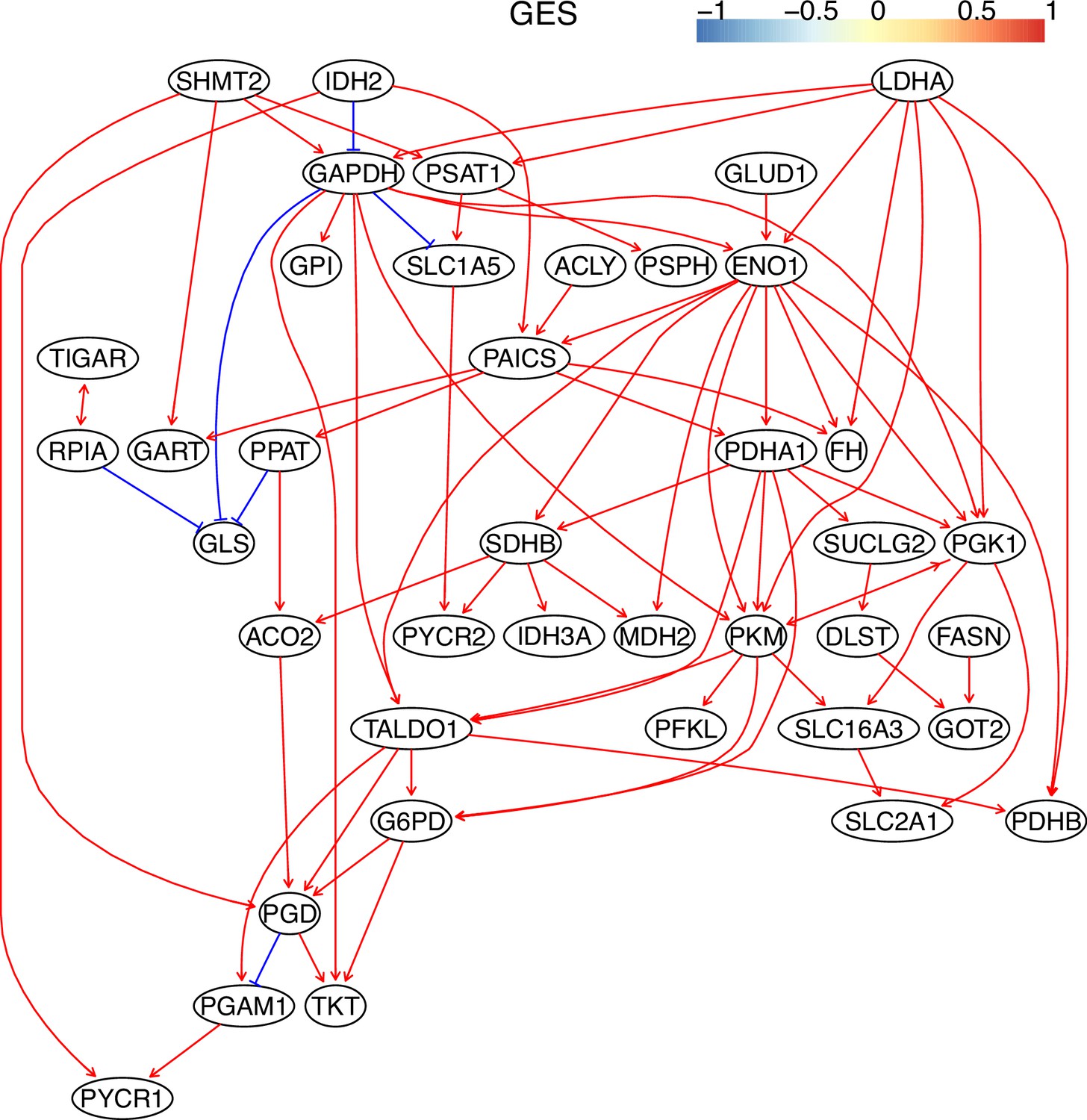
The causal relationships between genes in the WP4290 pathway in the cell line H2228 inferred using the GES method and 600 cells (alpha = 0.1).
Appendix 3—figure 9
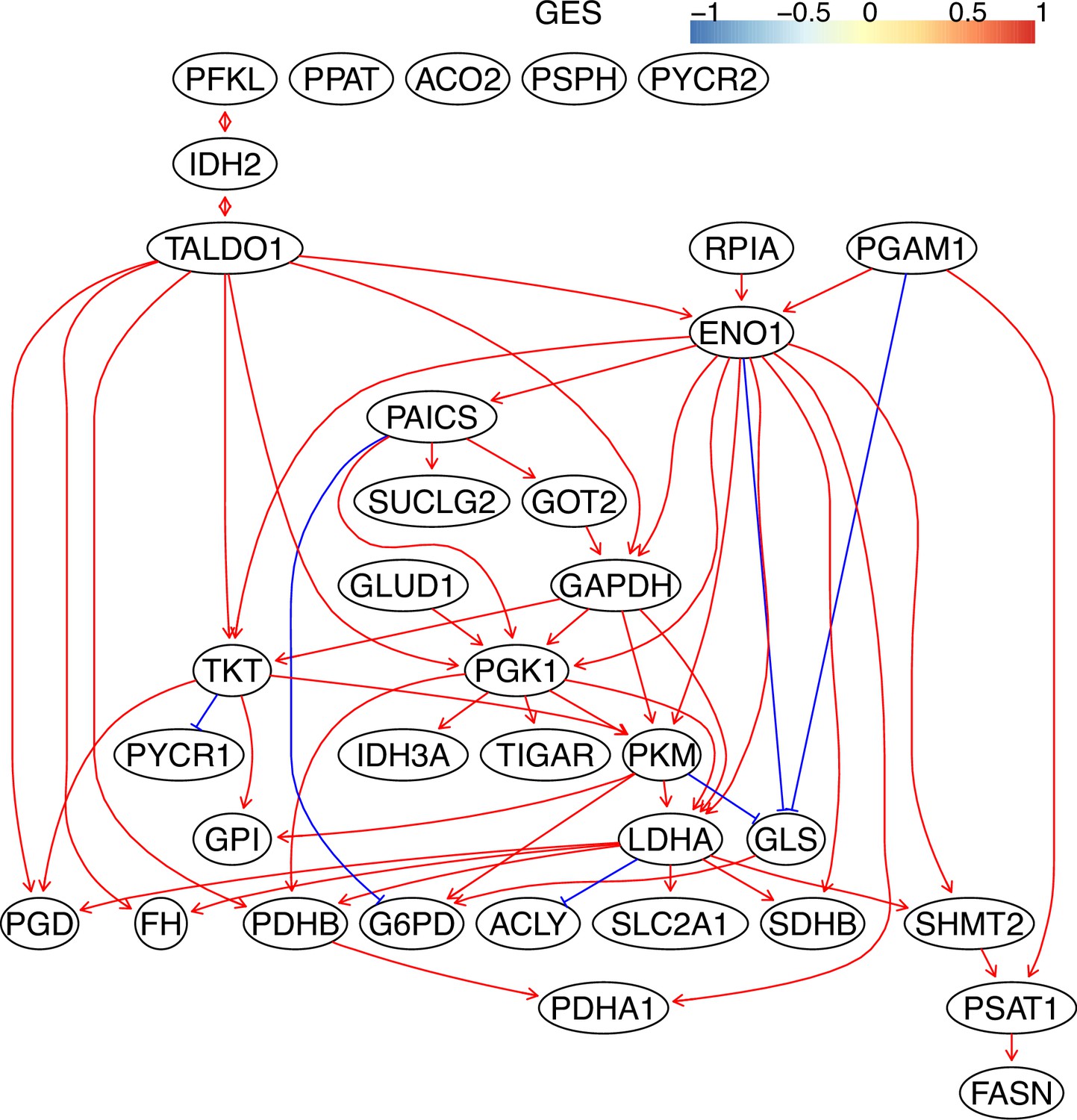
The causal relationships between genes in the WP4290 pathway in the cell line H838 inferred using the GES method and 600 cells (alpha = 0.1).
Appendix 3—figure 10
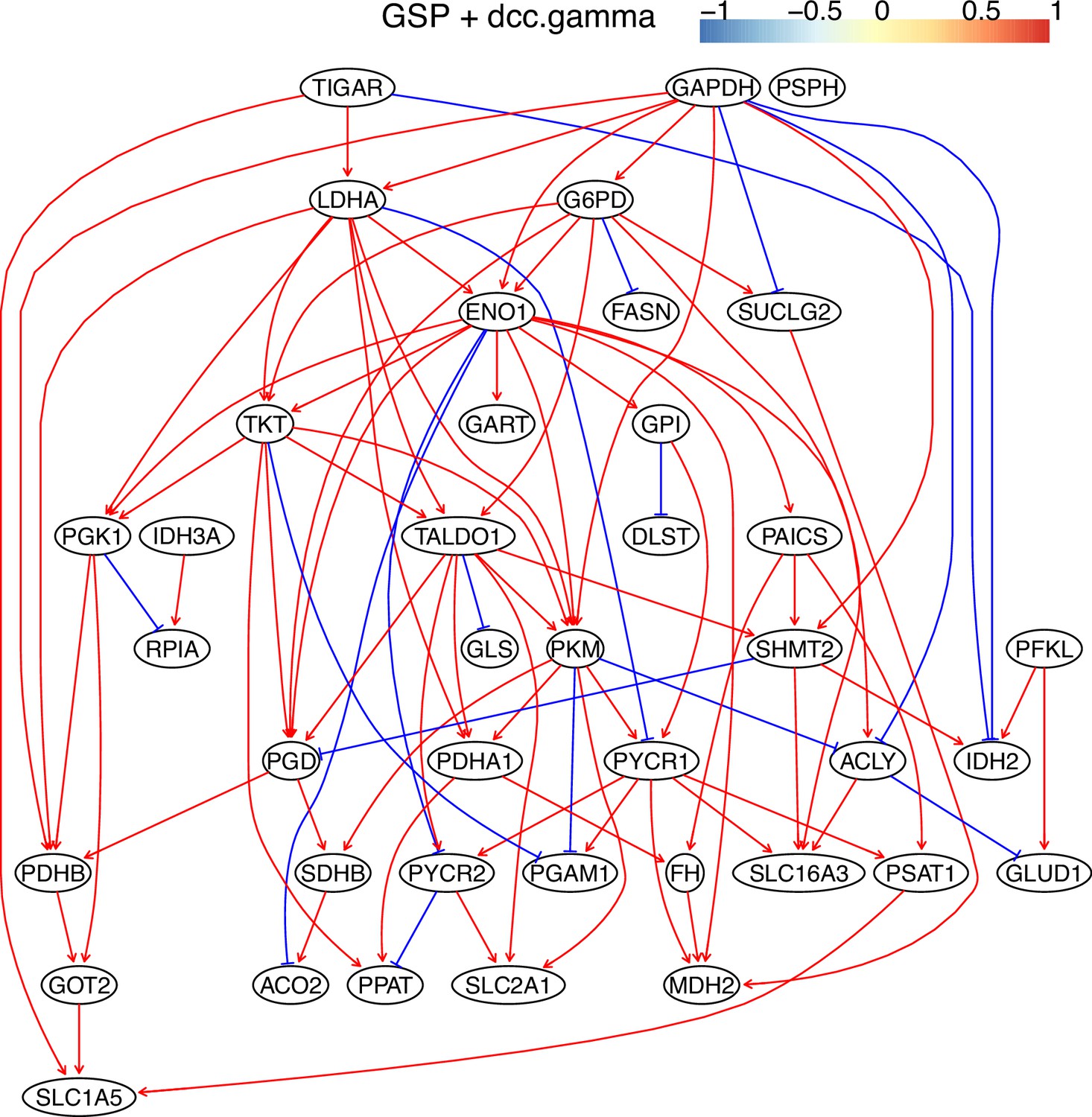
The causal relationships between genes in the WP4290 pathway in the cell line A549 inferred using the GSP+DCC.gamma and 600 cells (alpha = 0.05).
Compared with the networks in these cells inferred using PC+DCC.gamma (Appendix 4—figures 6–8), more relationships are inferred even if the significance level is 0.05. The key features of the reprogrammed glucose metabolism (as indicated in the inferred networks of PC+DCC.gamma, see Appendix 4) also occur in the network.
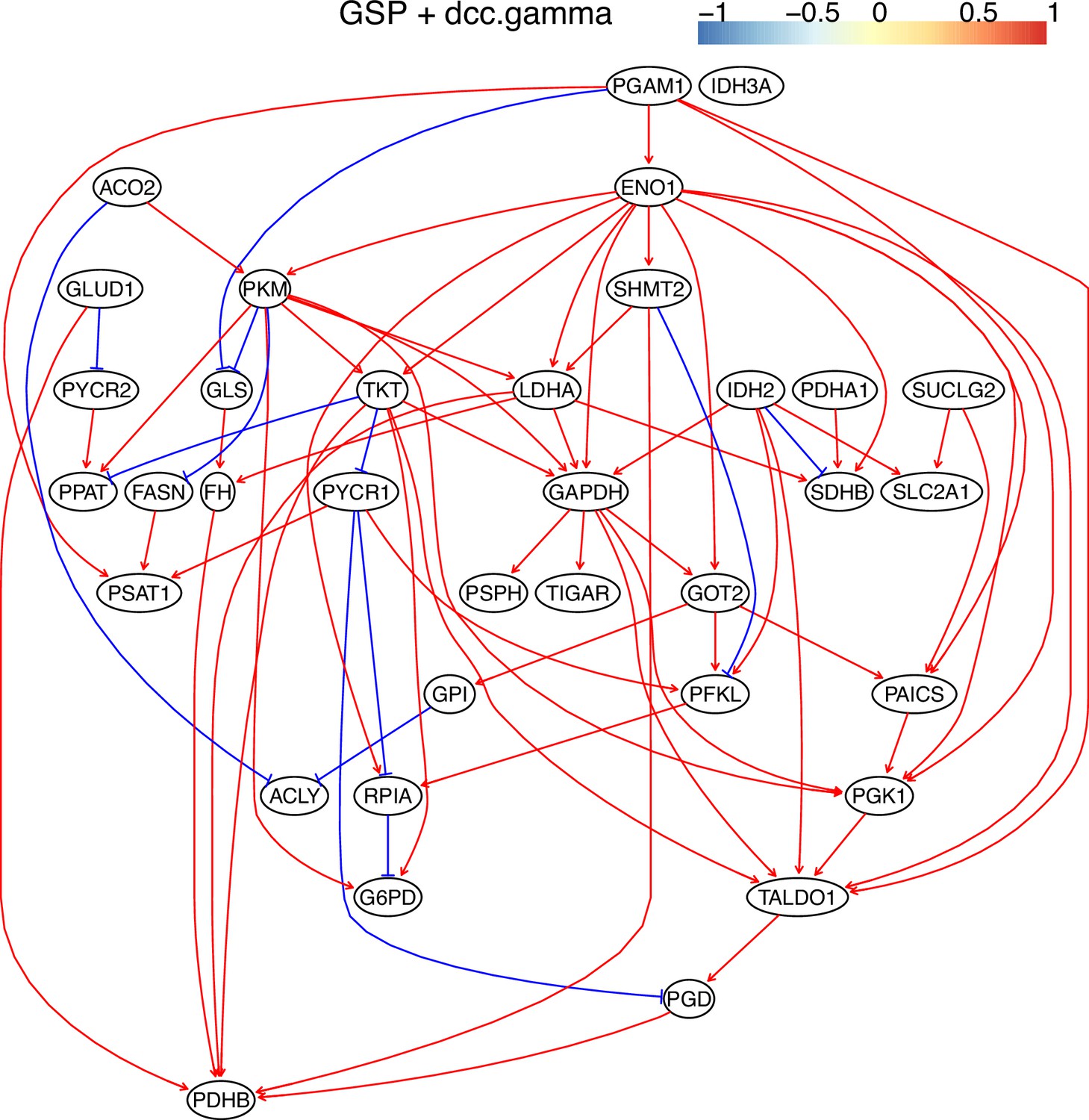
Appendix 3—figure 11
The causal relationships between genes in the WP4290 pathway in the cell line H2228 inferred using the GSP+DCC.gamma and 600 cells (alpha = 0.05).
Compared with the networks in these cells inferred using PC+DCC.gamma (Appendix 4—figures 6–8), more relationships are inferred even if the significance level is 0.05. The key features of the reprogrammed glucose metabolism (as indicated in the inferred networks of PC+DCC.gamma, see Appendix 4) also occur in the network.
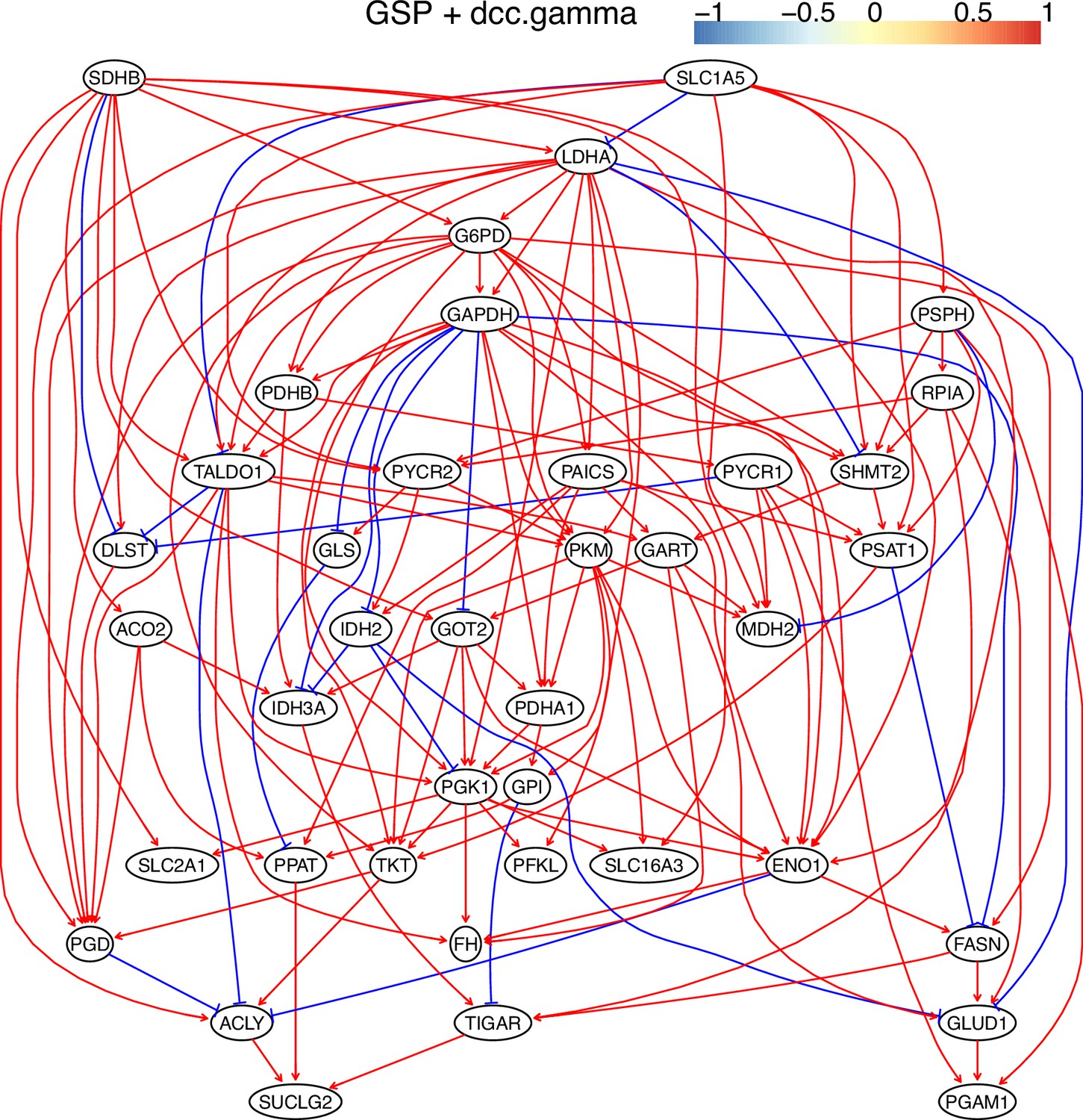
Appendix 3—figure 12
The causal relationships between genes in the WP4290 pathway in the cell line H838 inferred using the GSP+DCC.gamma and 600 cells (alpha = 0.05).
Compared with the networks in these cells inferred using PC+DCC.gamma (Appendix 4—figures 6–8), more relationships are inferred even if the significance level is 0.05. The key features of the reprogrammed glucose metabolism (as indicated in the inferred networks of PC+DCC.gamma, see Appendix 4) also occur in the network.
Appendix 4—figure 1
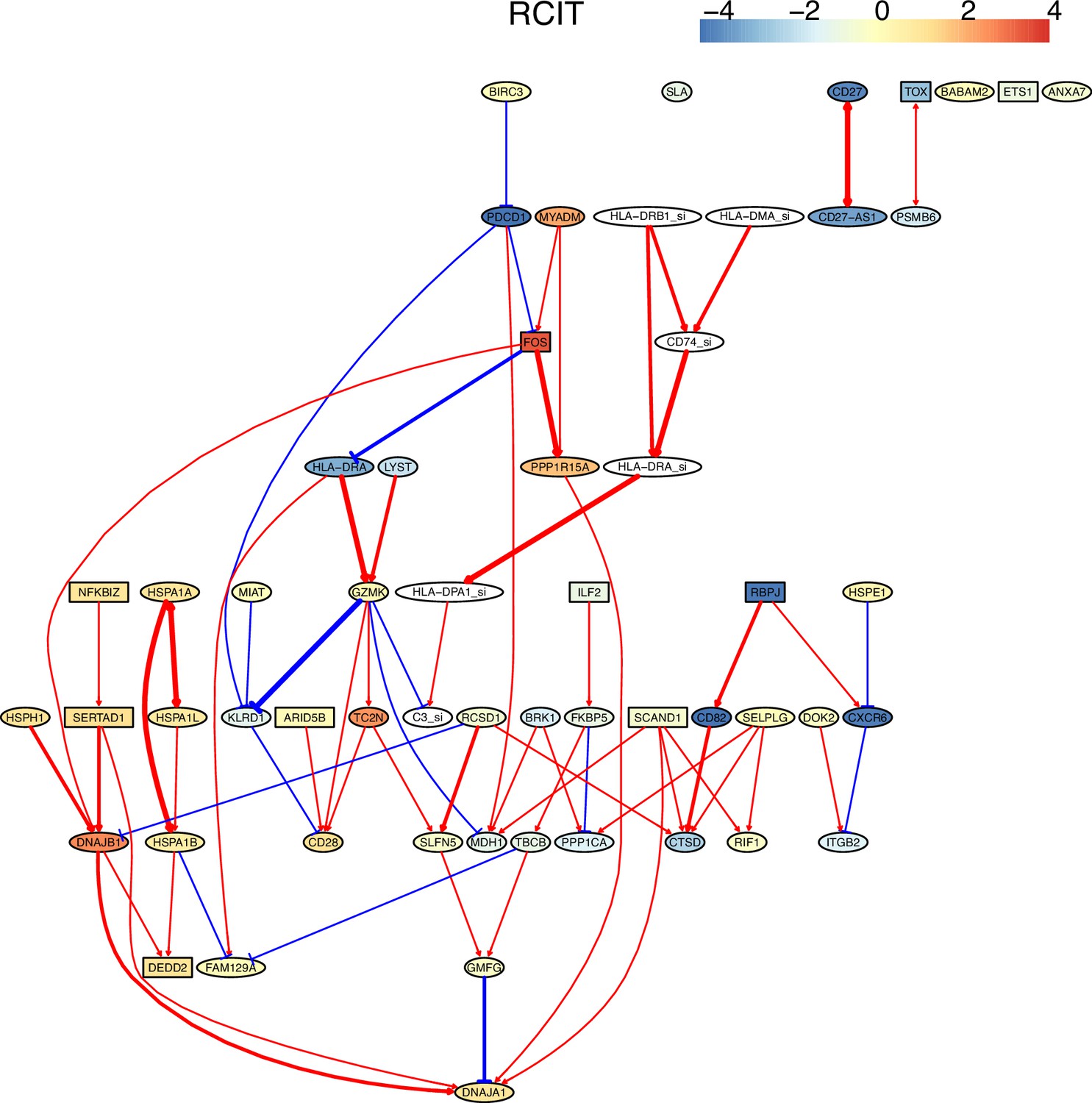
Multiple algorithms can identify genes and their relationships in the spike-in dataset from genes and their relationships in the primary dataset.
The six genes and their relationships were identified in the network of RCIT, but the network has multiple orphan nodes.
Appendix 4—figure 2
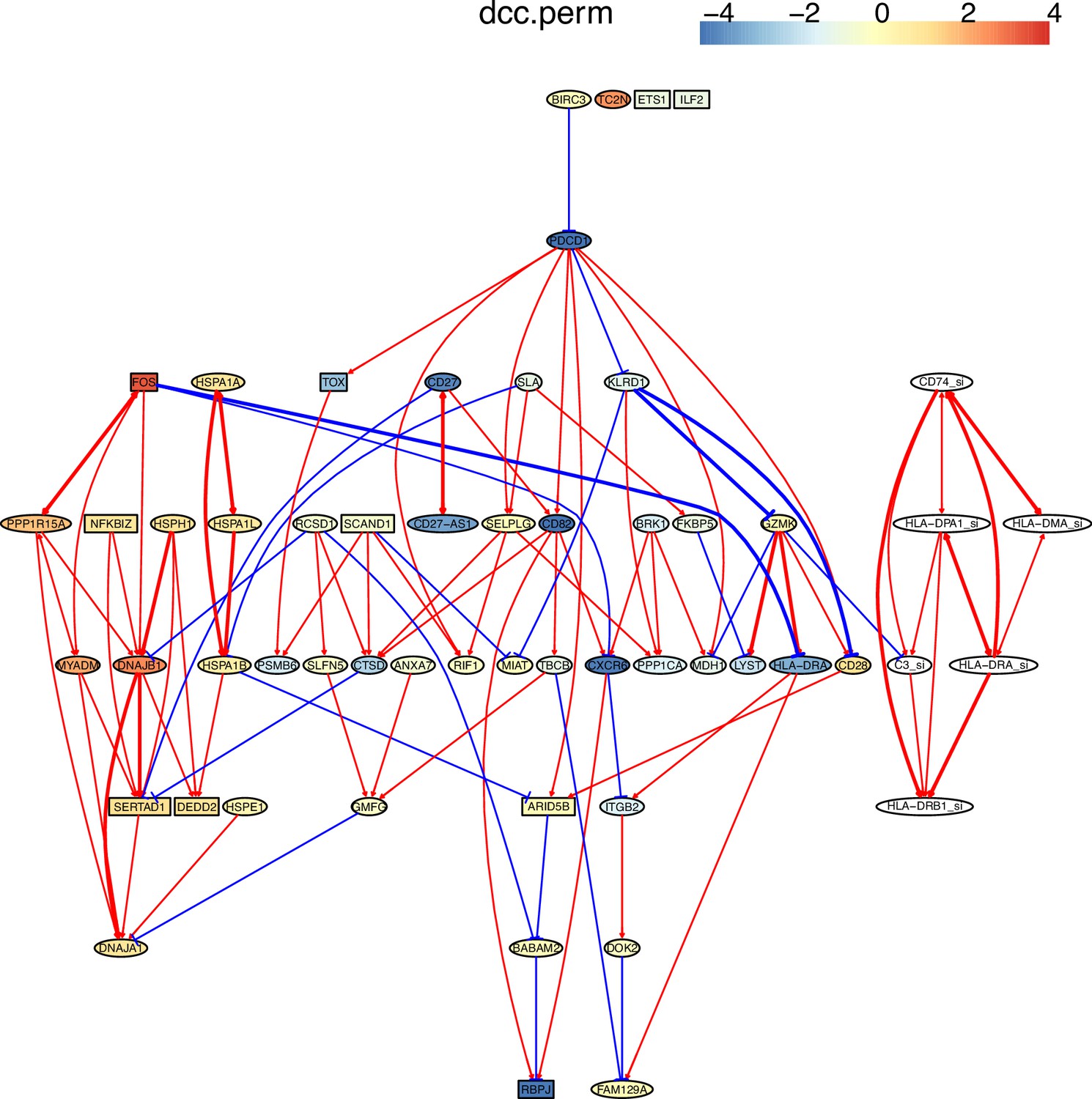
Multiple algorithms can identify genes and their relationships in the spike-in dataset from genes and their relationships in the primary dataset.
The six genes and their relationships were identified in the network of RCIT, but the network has multiple orphan nodes. GZMK-|C3_si is a wrong interaction in the networks of RCIT and DCC.perm.
Appendix 4—figure 3

Protein interactions in the STRING database (parameter settings: network type = full STRING network, meaning of network edges = confidence, active interaction sources = all, minimum required interaction score = medium confidence, 0.4).
(A) Interactions among MHC-II genes and CD74 and among CXCL and CXCR genes. (B) Interactions among MHC-I genes and B2M.
Appendix 4—figure 4
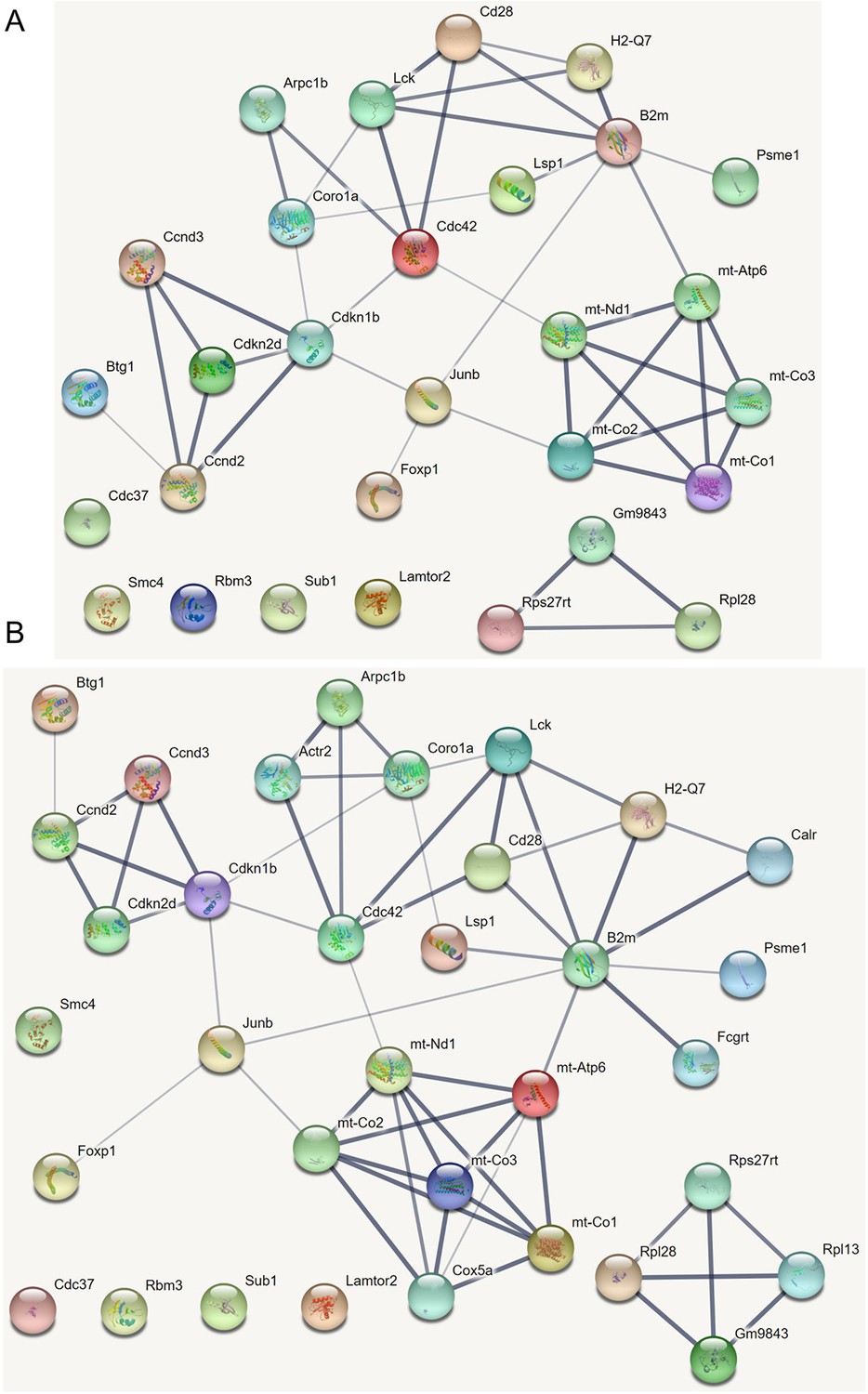
Interactions among the differentially expressed genes in CD4 T cells in the STRING database (https://string-db.org/) (parameter settings: network type = full STRING network, meaning of network edges = confidence, active interaction sources = all, minimum required interaction score = medium confidence, 0.4).
(A) The interactions. (B) The extended interactions (‘add more nodes to current network’ is chosen).
Appendix 4—figure 5
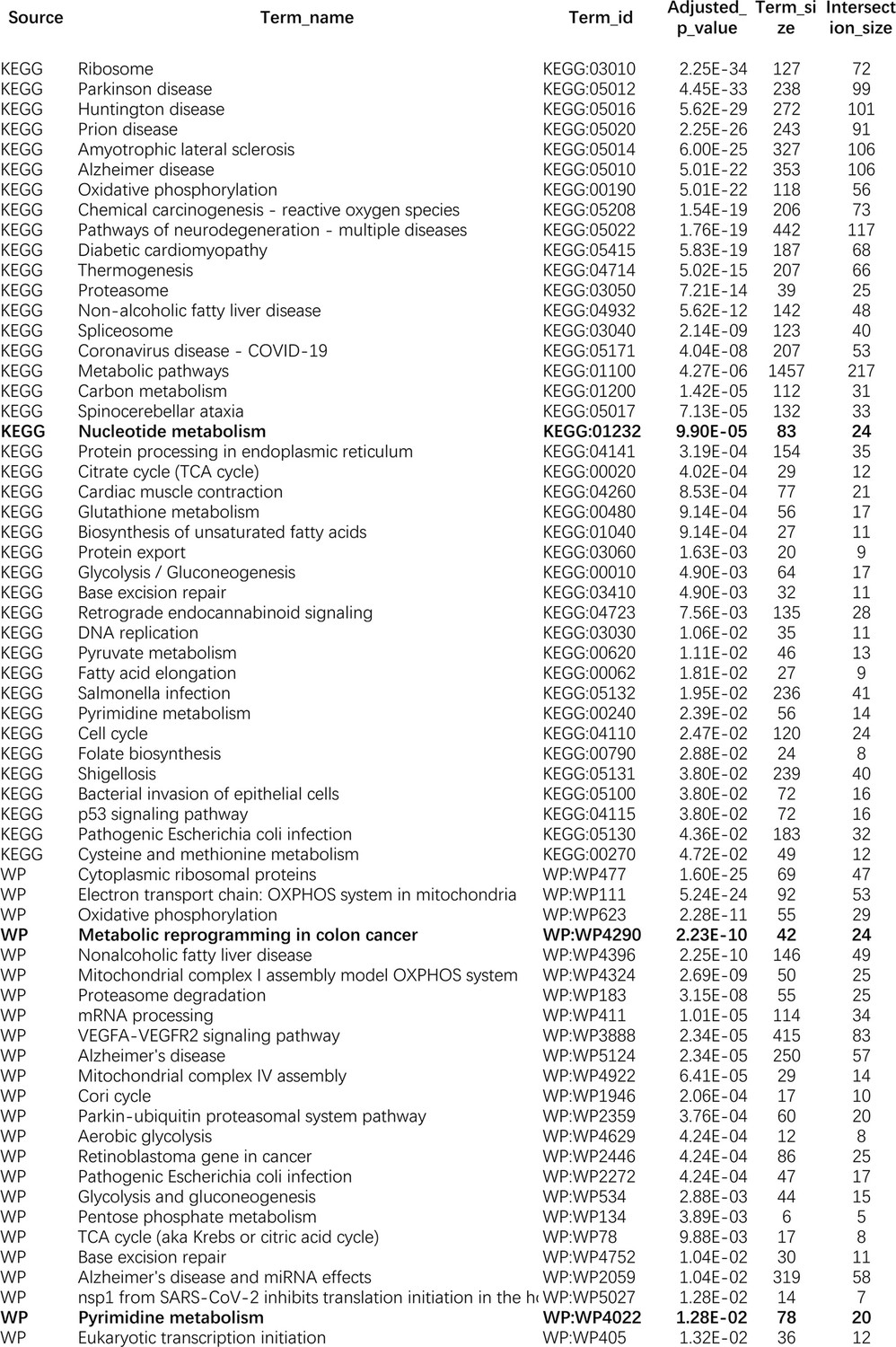
The enriched KEGG and WikiPathways pathways of differentially expressed genes in the A549 cell line.
KEGG:01232, WP4290, and WP4022 are enriched in all of the five lung cancer cell lines.
Appendix 4—figure 6
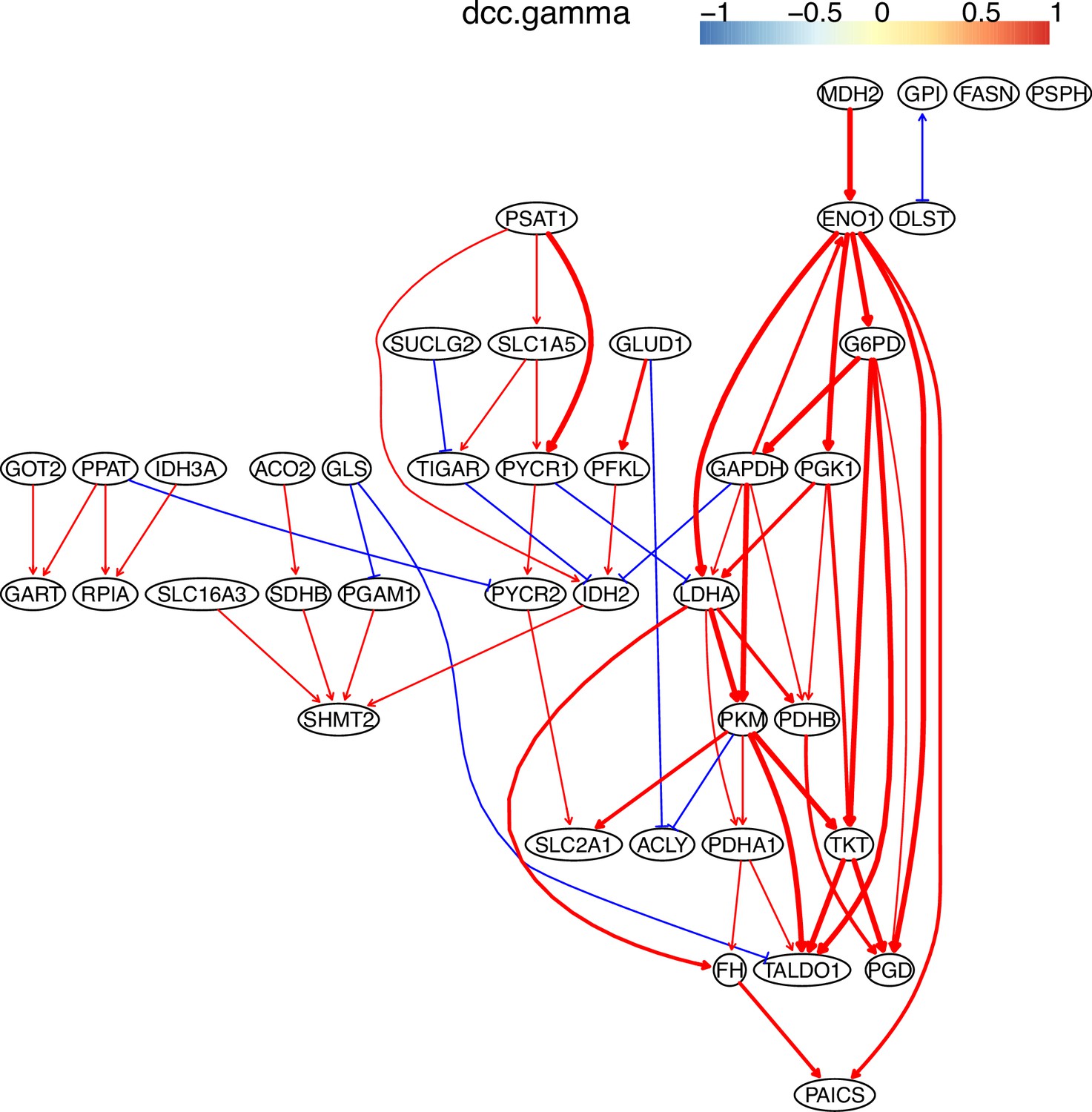
The causal relationships between genes in the WP4290 pathway in the cell line A549 inferred using PC+DCC.gamma.
The inference is flawed because the true network contains both gene products and metabolites but single-cell RNA-sequencing (scRNA-seq) data do not contain metabolites. Nevertheless, Appendix 4—figures 6–8 show that multiple inferred interactions reasonably reveal key features of reprogrammed glucose metabolism. First, shared interactions in ≥2 datasets are 21.12%, 58.82%, 50.51%, 40.82%, 53.09%, and 50.0% in alveolar cells, H838 cells, H2228 cells, HCC827 cells, H1975 cells, and A549 cells, indicating that causal inference differentiates glucose metabolism in cancer cells from in alveolar cells. Second, the inferred networks reflect key features of reprogrammed glucose metabolism, especially the activation of SLC2A1 (which encodes a major glucose transporter and controls glucose intake), PGD (which promotes glucose metabolism toward nucleotide synthesis), PSAT1 (which promotes glucose metabolism toward nucleotide synthesis), and LDHA (which promotes glucose metabolism toward lactate generation) in cancer cell lines.
Appendix 4—figure 7
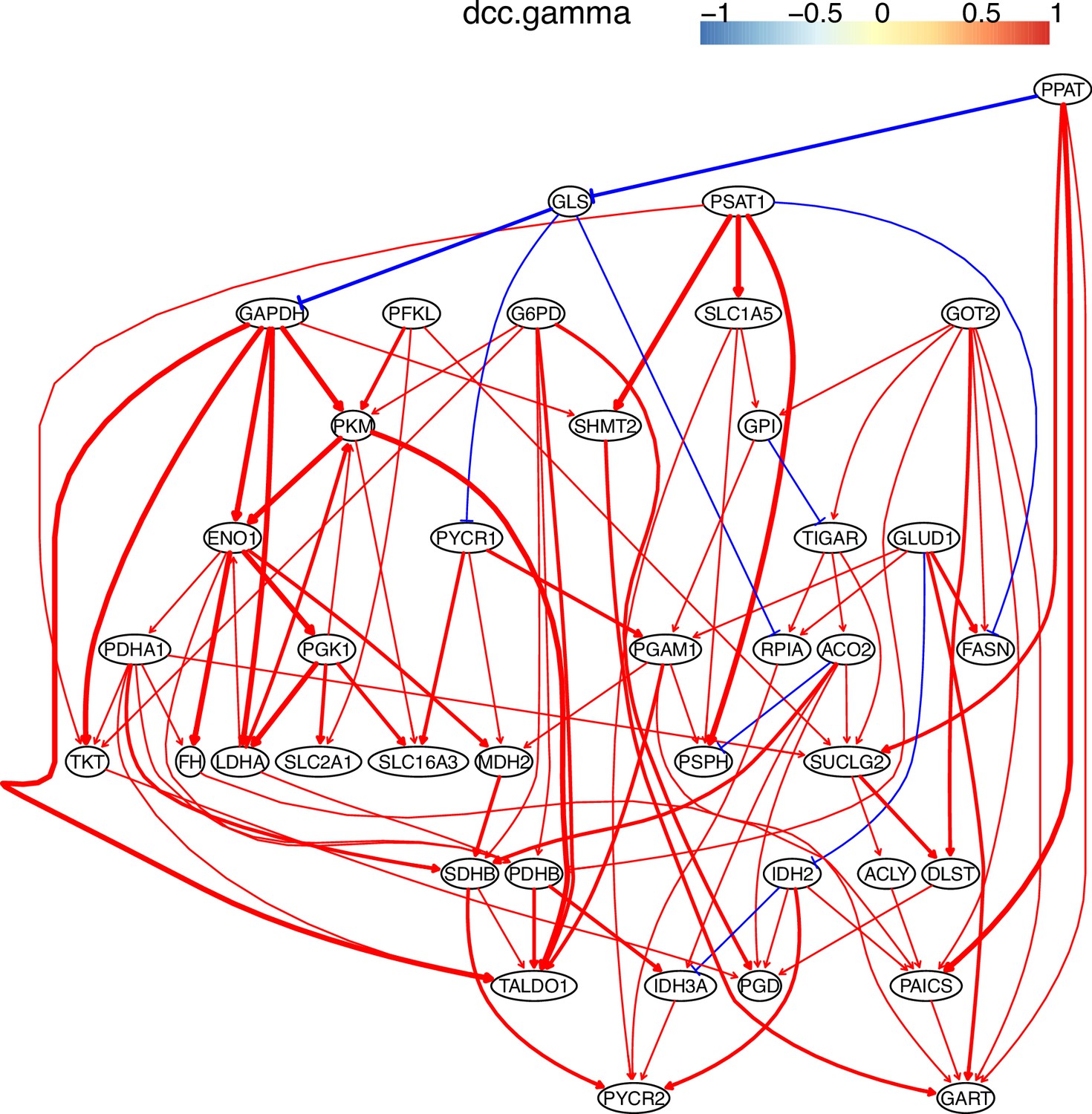
The causal relationships between genes in the WP4290 pathway in the cell line H2228 inferred using PC+DCC.gamma.
Appendix 4—figure 8
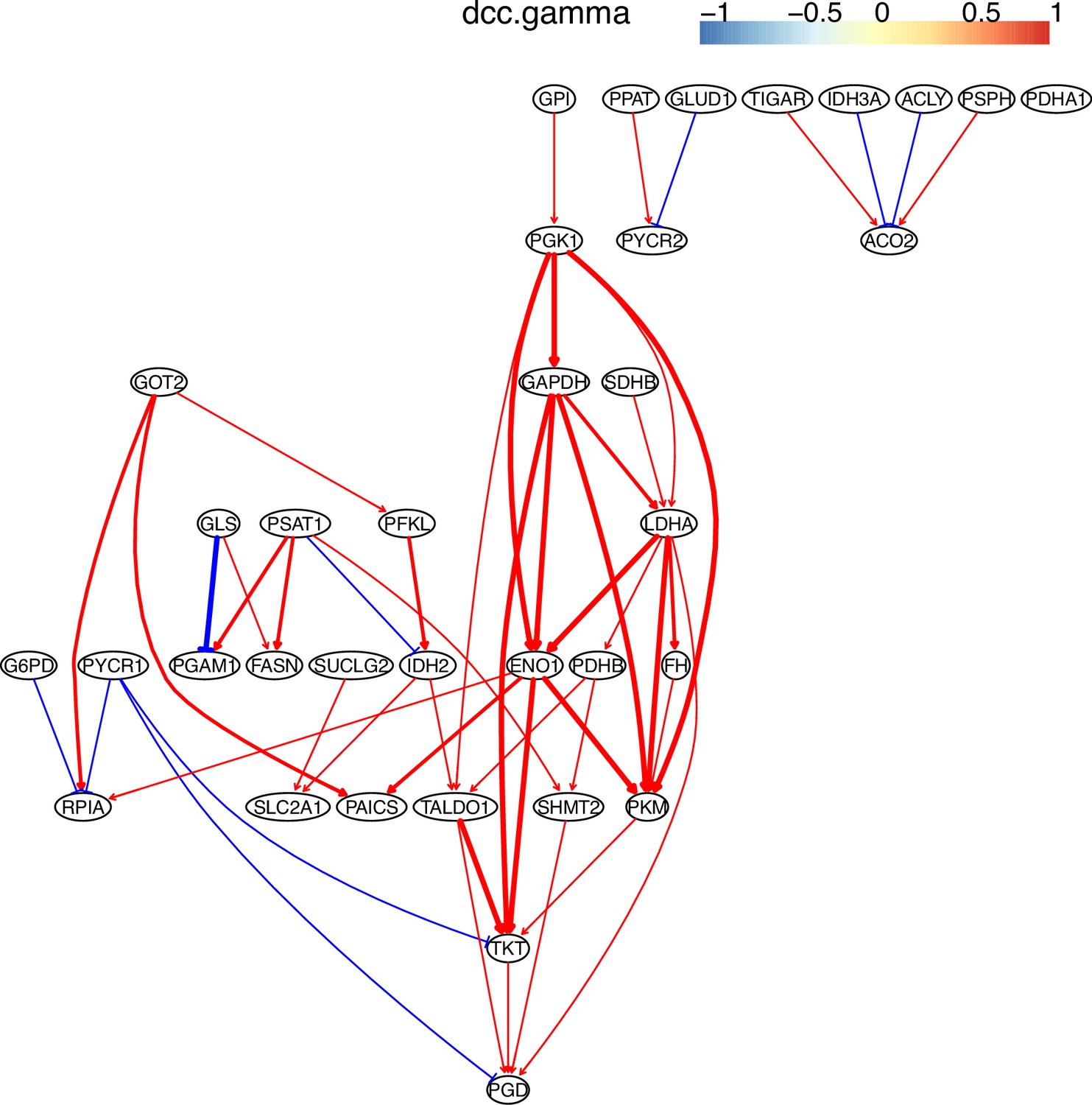
The causal relationships between genes in the WP4290 pathway in the cell line H838 inferred using PC+DCC.gamma.
Appendix 4—figure 9
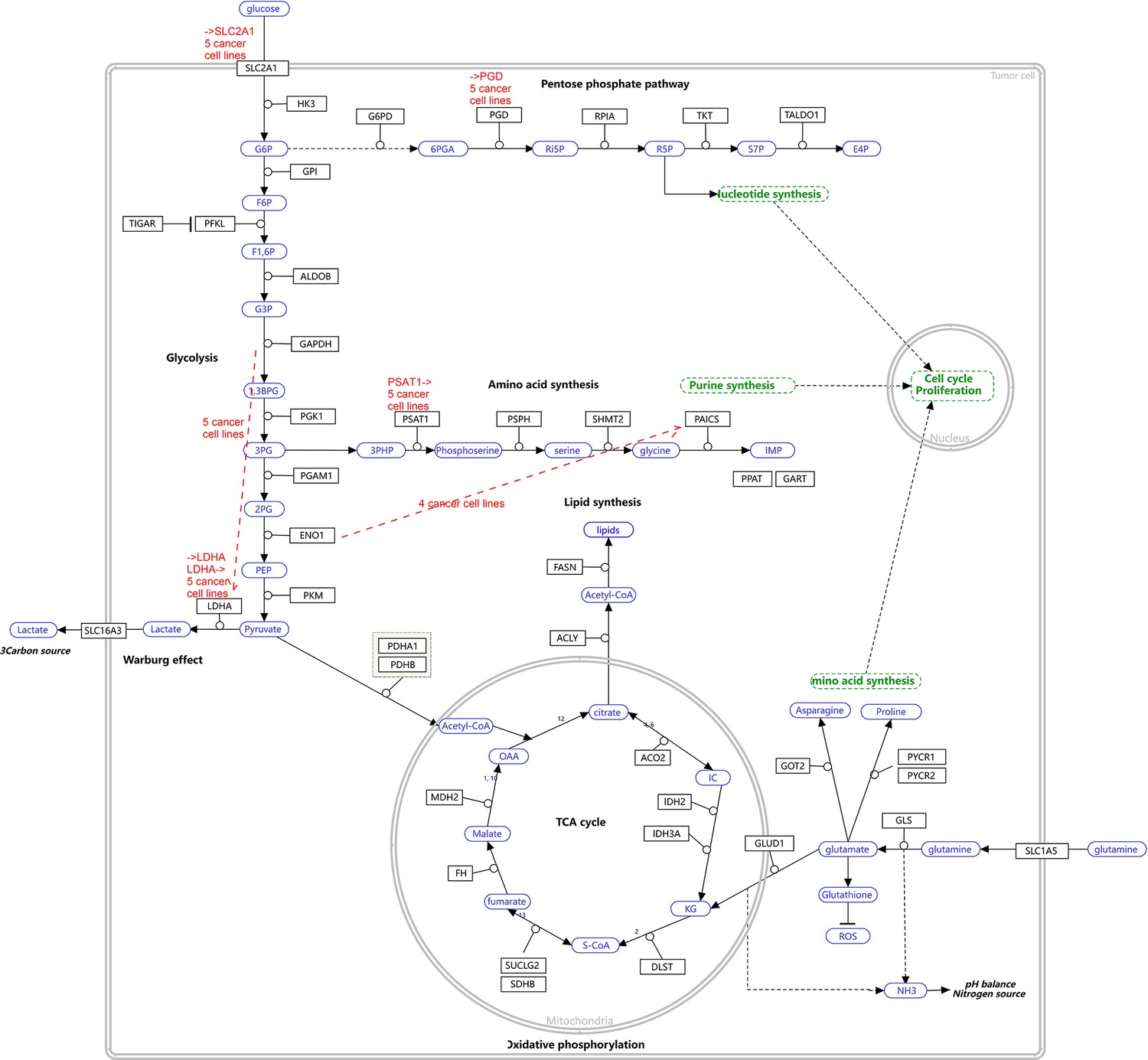
The WP4290 pathway and the key features of inferred interactions in lung cancer cell lines.
Glucose intake is greatly increased in cancer cells. The increased glucose consumption is used as a carbon source for anabolic processes and this excess carbon is also used for the de novo generation of nucleotides, lipids, and proteins. This so-called Warburg effect is proposed to be an adaptation mechanism to support these biosynthesis processes for uncontrolled proliferation of cancer cells. SLC2A1, PGD, PSAT1, and LDHA are critical genes controlling glucose intake and the generation of nucleotides and lactate. The added red notes indicate key inferred interactions. ‘→XXX’ and ‘XXX→’ indicate the activation of the gene XXX by others and the activation of others by the gene XXX, respectively. In the alveolar cells, SLC2A1, PGD, PSAT1, ENO1, and LDHA are not expressed and none of these interactions are inferred.
Appendix 4—figure 10
The causal interactions between genes in the hsa00240 pathway in the A549 cells.
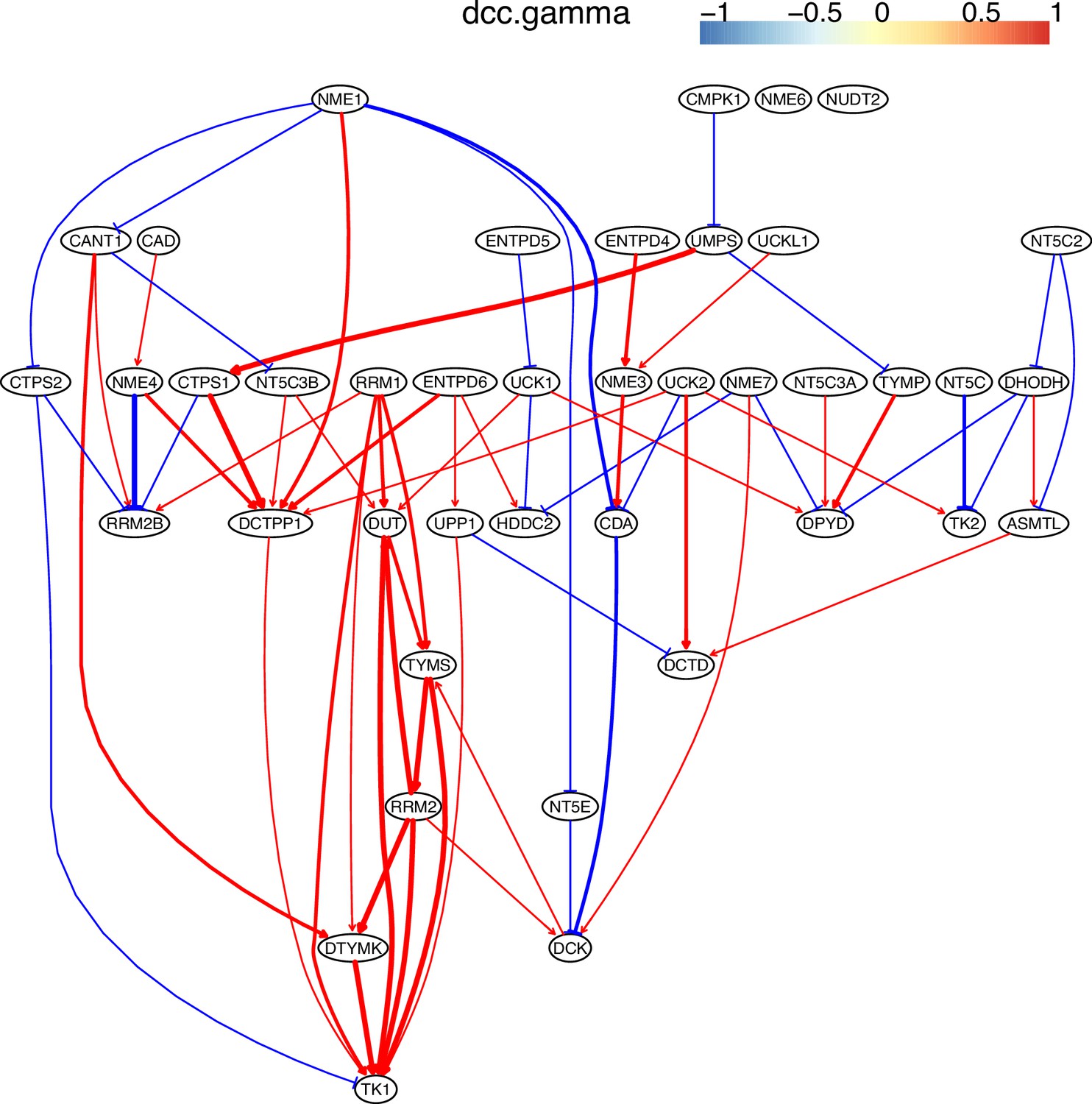
Since pyrimidine metabolism consists of many reversible reactions (Appendix 4—figure 13), inferred interactions are more varied than those of glucose metabolism. Appendix 4—figures 10–12 show that causal inference reasonably reveals the critical differences between cancer cells and alveolar cells, which include that percentages of interactions shared by ≥2 cell lines are 19.74%, 41.38%, 42.42%, 30.86%, 36.67%, and 32.99% in alveolar, H838, H2228, HCC827, H1975, and A549 cells. The regulations of important genes are notable (Appendix 4—figure 13). (1) TYMS is activated in five cancer cell lines but not in alveolar cells, and is not repressed in cancer cell lines. (2) Tk1/2 are activated in five cancer cells and alveolar cells. (3) DUT is not expressed in alveolar cells and is activated in the five cancer cell lines. (4) Multiple TYMP activations are inferred in alveolar cells. (5) ENTPD1/3 are activated only in alveolar. (6) NT5C/E/M are repressed in five cancer cell lines but are not expressed in alveolar cells. (7) There are many cases where downstream enzymes activate upstream enzymes, such as ENTPD3->CTPS2. Of note, DUT->Tk1 and DUT->TYMS in all five cancer cell lines indicate well-coordinated causal interactions for DNA synthesis in cancer cells.
Appendix 4—figure 11

The causal interactions between genes in the hsa00240 pathway in the HCC827 cells.
Appendix 4—figure 12
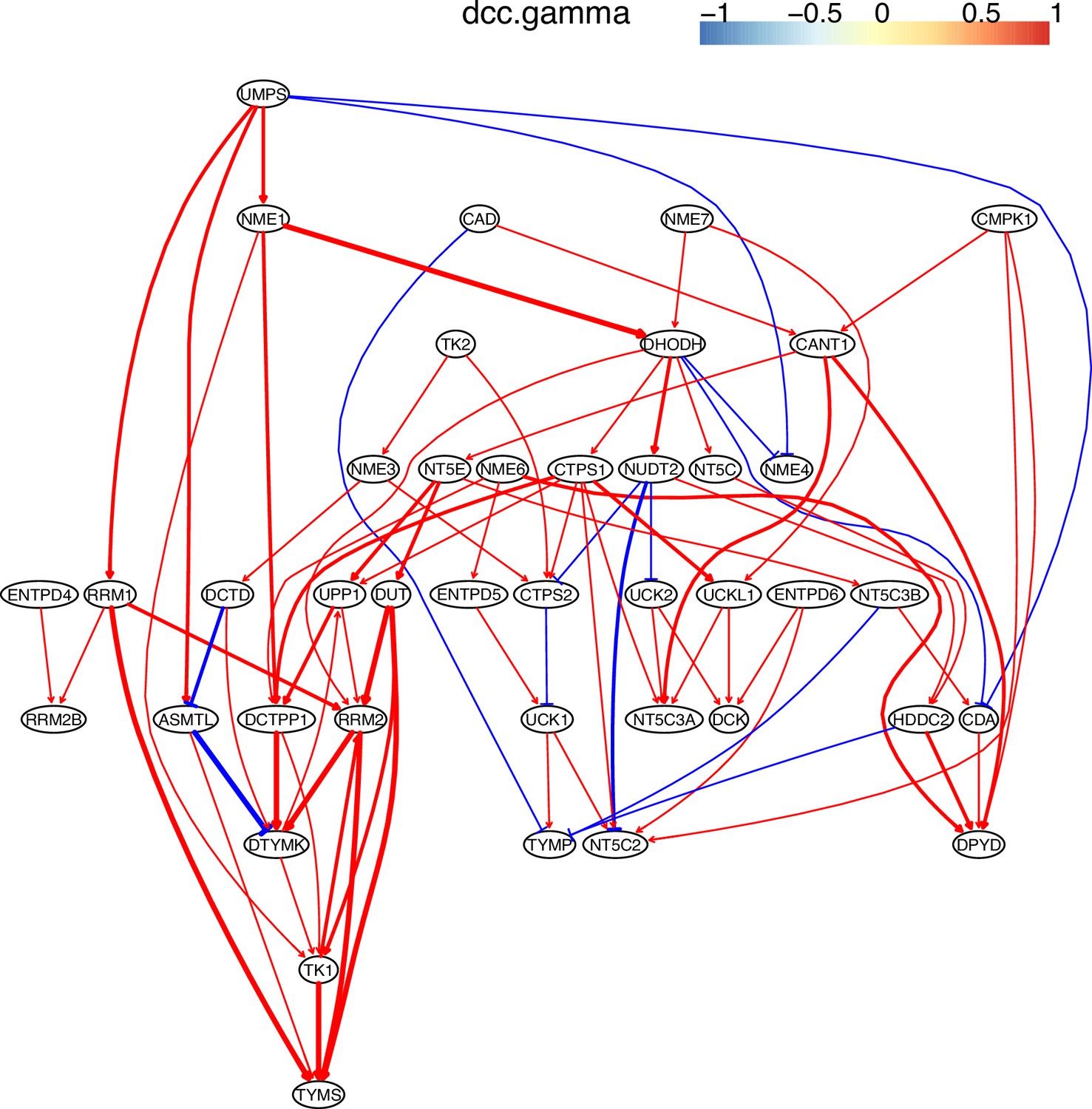
The causal interactions between genes in the hsa00240 pathway in the H1975 cells.
Appendix 4—figure 13
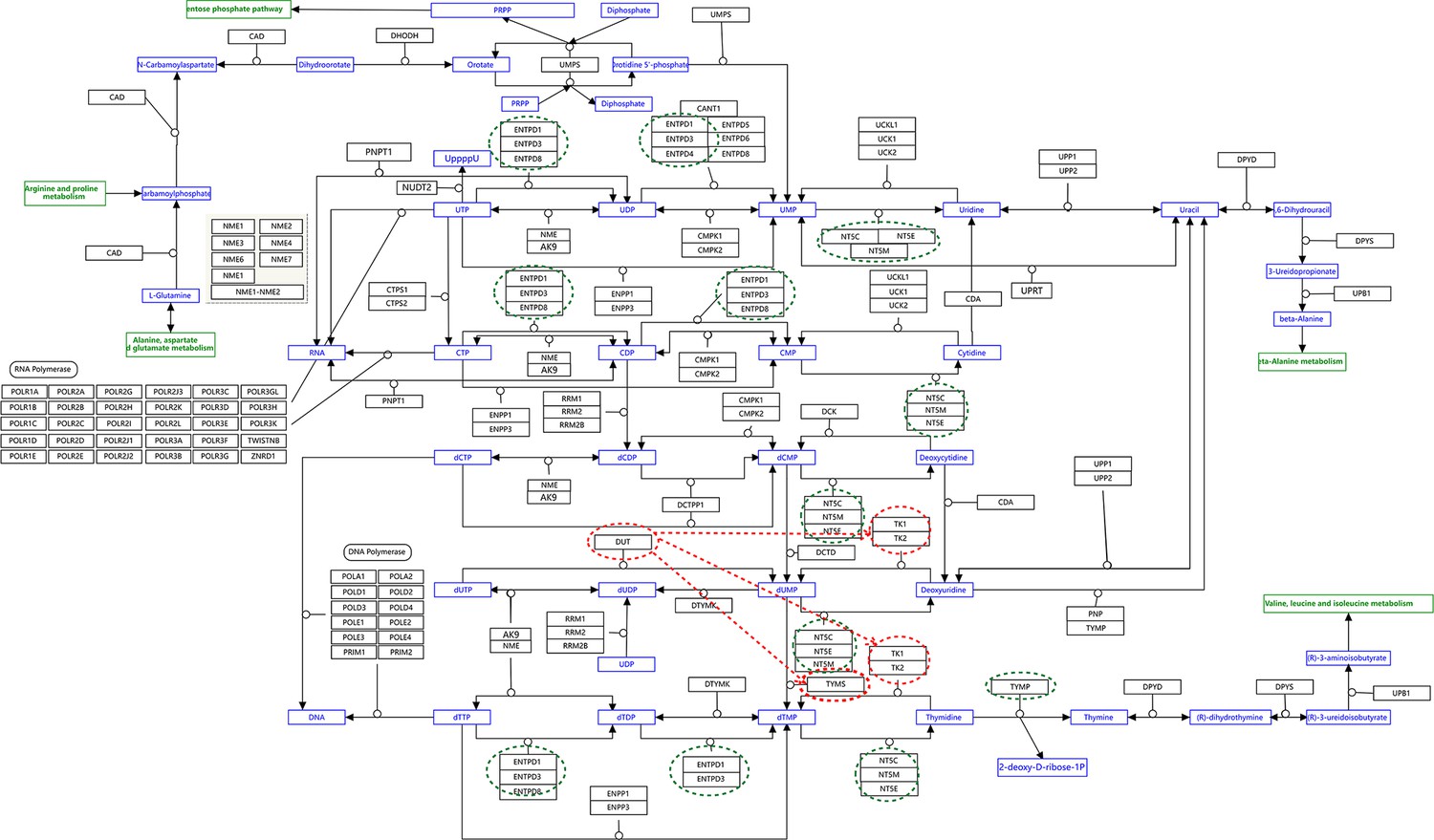
The pyrimidine metabolism pathway and key features of inferred causal networks in lung cancer cell lines and alveolar cells.
Genes in the KEGG ‘Pyrimidine metabolism’ (hsa00240) pathway were used to perform causal inference (because WP4022 contains too many POLR gene families) and the figure of WP4022 was used to illustrate the results (this figure is more readable). This figure indicates that pyrimidine metabolism has many reversible reactions, and these reactions somewhat blur the key features in cancer cell lines and alveolar cells. The following genes and reactions are notable. (1) TYMS turns dUMP->dTMP unidirectionally toward DNA synthesis. (2) Tk1/2 turn thymidine->dTMP and deoxyuridine->dUMP toward DNA synthesis (while NT5C/E/M do the opposite). (3) DUT turns dUTP->dUMP, and dUMP is the substrate for TYMS. (4) TYMP turns thymidine->thymine unidirectionally away from DNA synthesis. (5) ENTPD1/3 turn dTTP->dTDP->dTMP, UTP->UDP->UMP, and CTP->CDP->CMP away from DNA synthesis and RNA synthesis (but these reactions can be reversed by AK9/NME). (6) NT5C/E/M turn dCMP->deoxycytidine, dUMP->deoxyuridine, and dTMP->thymidine away from DNA synthesis. Red and green ellipses mark genes that promote DNA synthesis and genes that do not promote DNA synthesis. In the inferred causal networks, accordingly, there are following interactions. (1) TYMS is activated in five cancer cell lines but not in alveolar cells, and is not repressed in cancer cell lines. (2) Tk1/2 are activated in five cancer cells and alveolar cells. (3) DUT is not expressed in alveolar cells and is activated in the five cancer cell lines. (4) Multiple TYMP activations are inferred in alveolar cells. (5) ENTPD1/3 are activated only in alveolar. (6) NT5C/E/M are repressed in five cancer cell lines but are not expressed in alveolar cells. (7) There are many cases where downstream enzymes activate upstream enzymes, such as ENTPD3->CTPS2. Of note, there are DUT->Tk1 and DUT->TYMS in all five cancer cell lines, indicating coordinated molecular interaction and gene regulation for DNA synthesis in cancer cells.
Appendix 4—figure 14
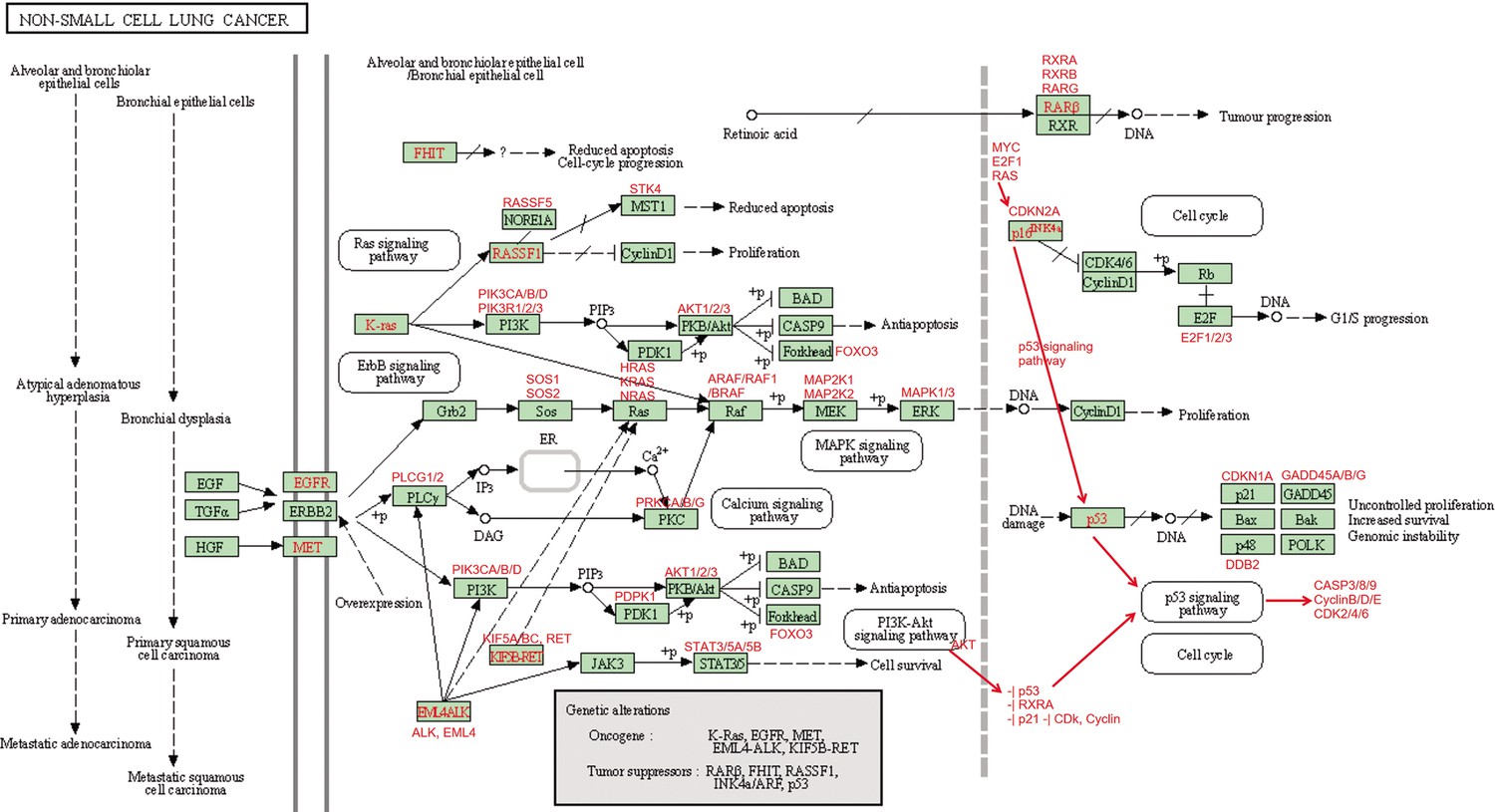
The ‘Non-small cell lung cancer’ (hsa05223) pathway.
Annotated sub-pathways, genes, and interactions are marked in red.
Appendix 5—figure 1
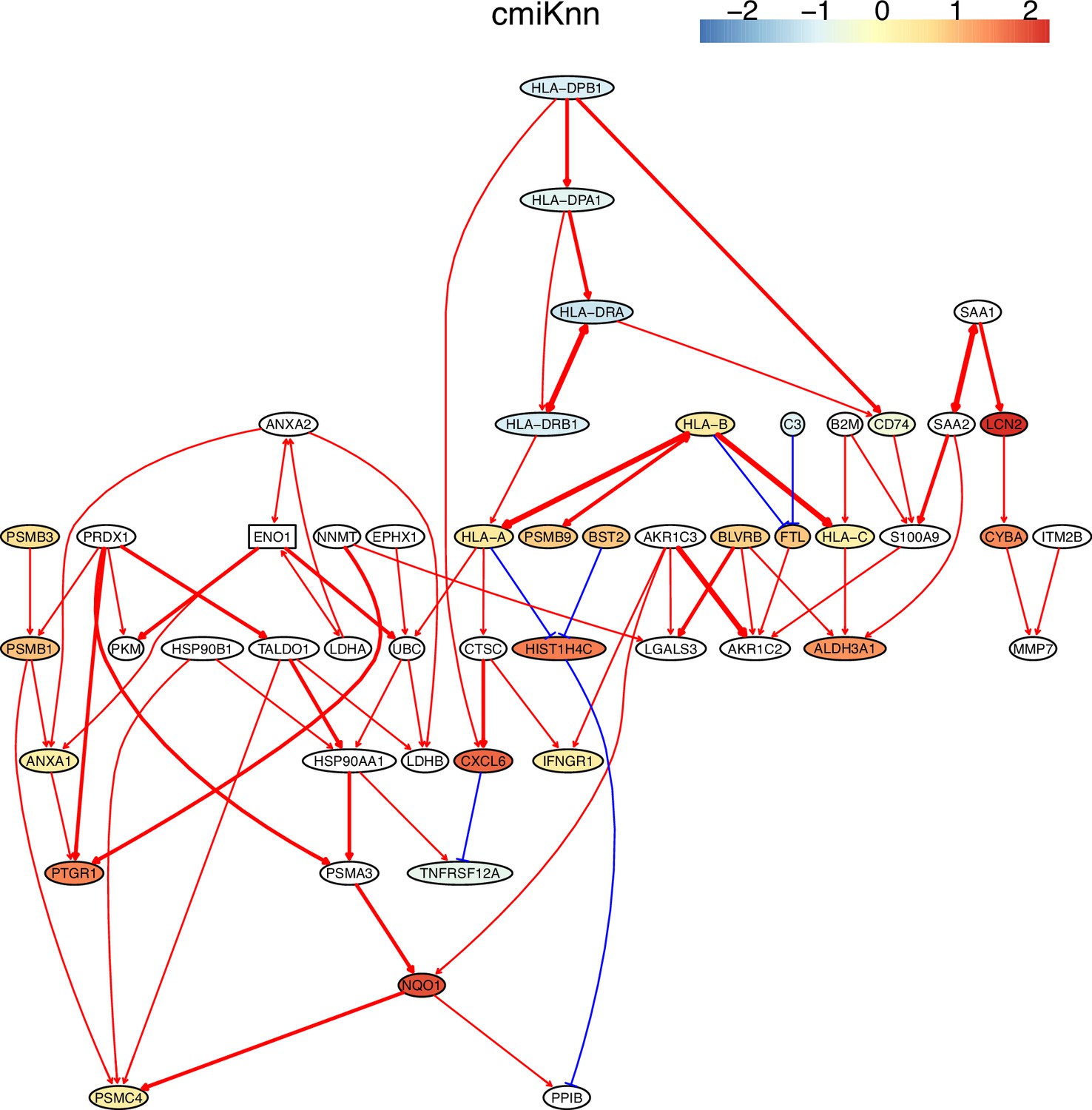
The causal network of the 50 feature genes inferred by PC+cmiKnn from the H2228 dataset (settings: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed in cells were used).
In these and the following figures, red and blue arrows indicate activation and inhibition, double arrows indicate undermined direction, arrows' thickness indicates the statistical significance of CI test, and node colors indicate fold changes of gene expression. MHC-II genes were significantly down-regulated compared with the control (the lung alveolar epithelial cells).
Appendix 5—figure 2
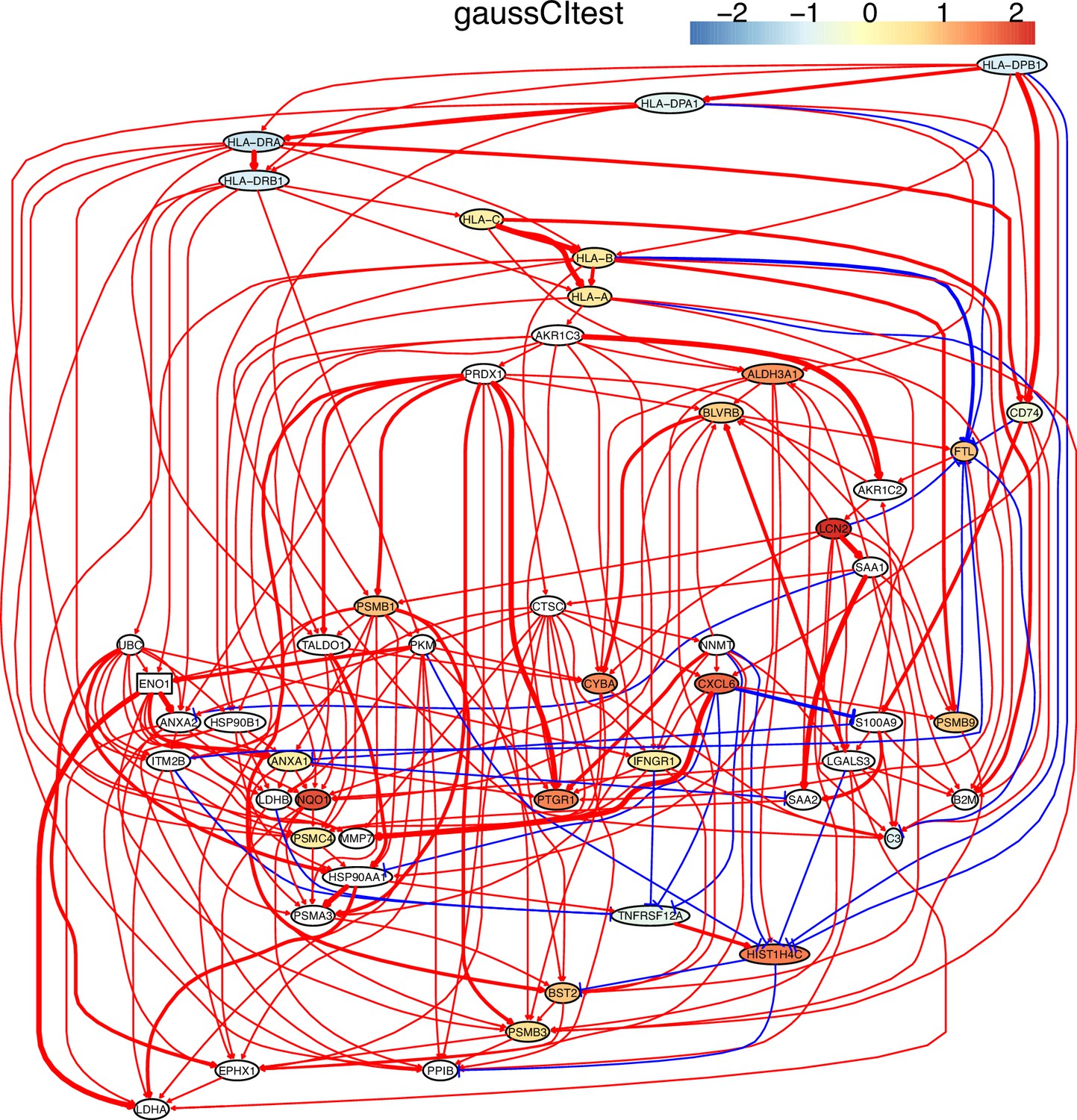
The causal network of the 50 feature genes inferred by PC+GaussCItest from the H2228 dataset (settings: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed in cells were used).
MHC-II genes were significantly down-regulated compared with the control (the lung alveolar epithelial cells).
Appendix 5—figure 3
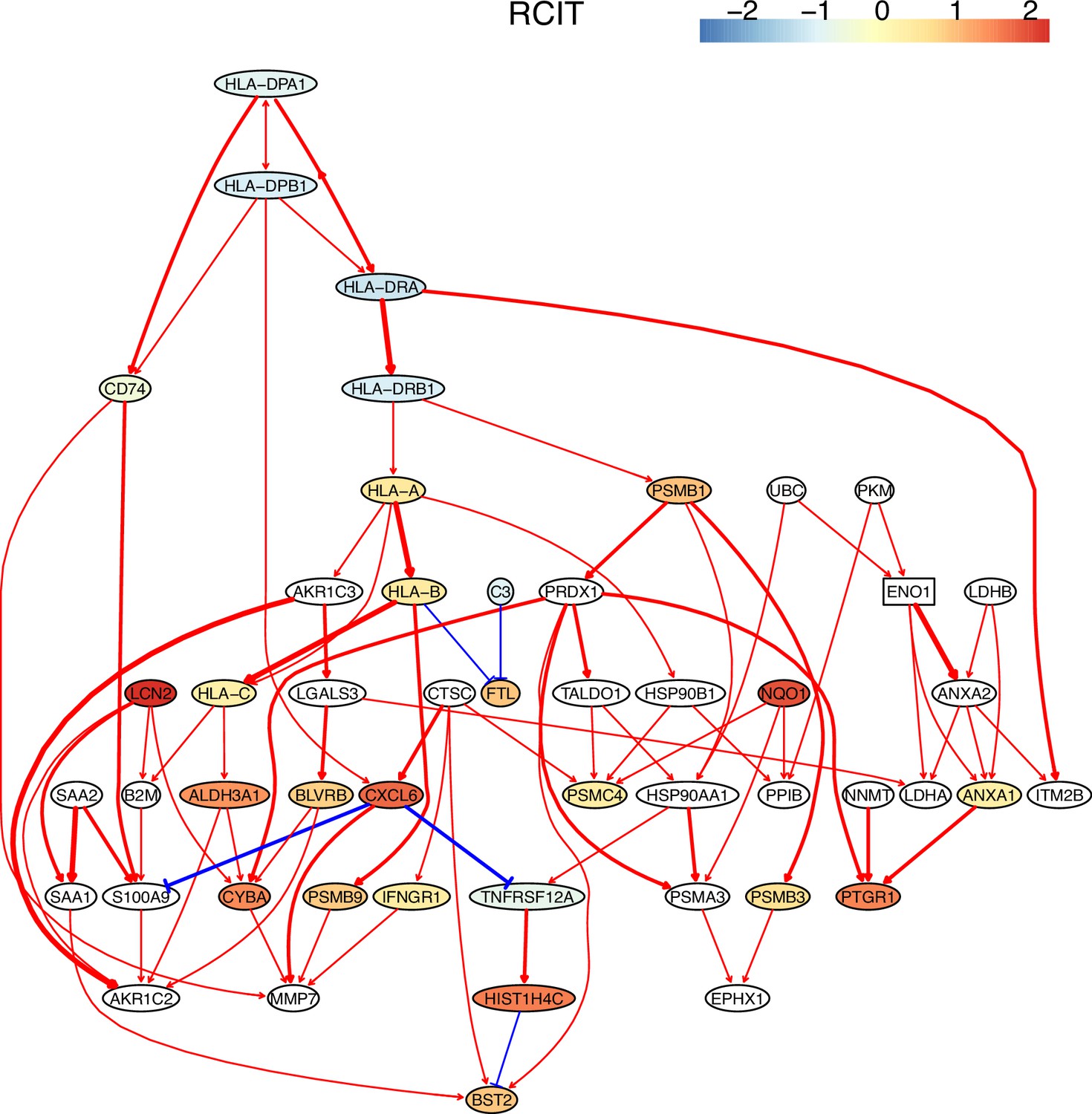
The causal network of the 50 feature genes inferred by PC+RCIT from the H2228 dataset (settings: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed in cells were used).
MHC-II genes were significantly down-regulated compared with the control (the lung alveolar epithelial cells).
Appendix 5—figure 4
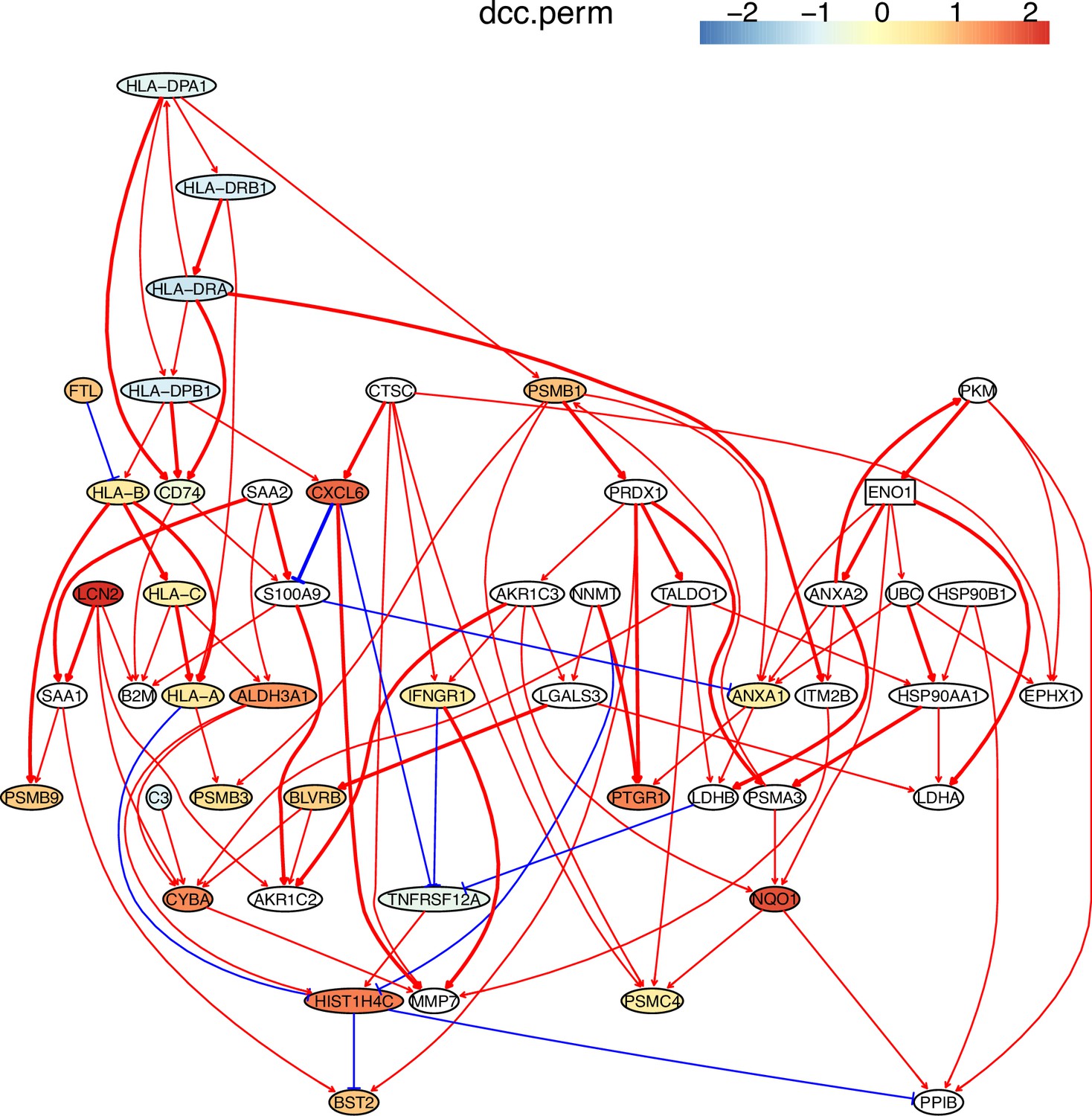
The causal network of the 50 feature genes inferred by PC+DCC.perm from the H2228 dataset (settings: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed in cells were used).
MHC-II genes were significantly down-regulated compared with the control (the lung alveolar epithelial cells).
Appendix 5—figure 5
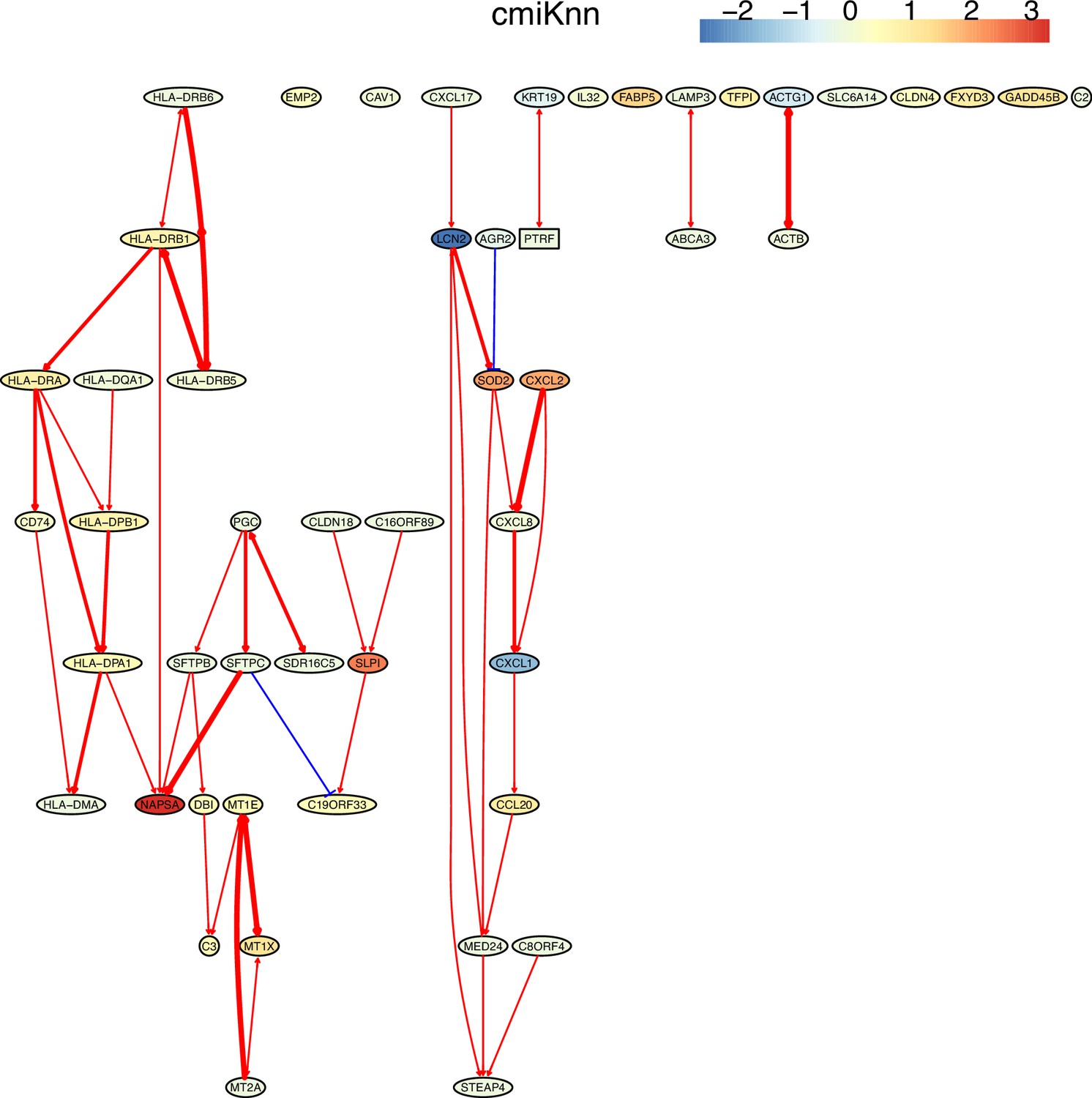
The causal network of the 50 feature genes inferred by PC+cmiKnn from the alveolar epithelial cell dataset (settings: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed the cells were used).
The control of the case was the H2228 cells. Compared with H2228 cells, MHC-II genes in alveolar epithelial cells were highly expressed. The relationships between MHC-II genes and the relationships between MHC-II genes and CD74 (which is a key regulator of MHC-II proteins) are supported by annotated interactions in the STRING database.
Appendix 5—figure 6
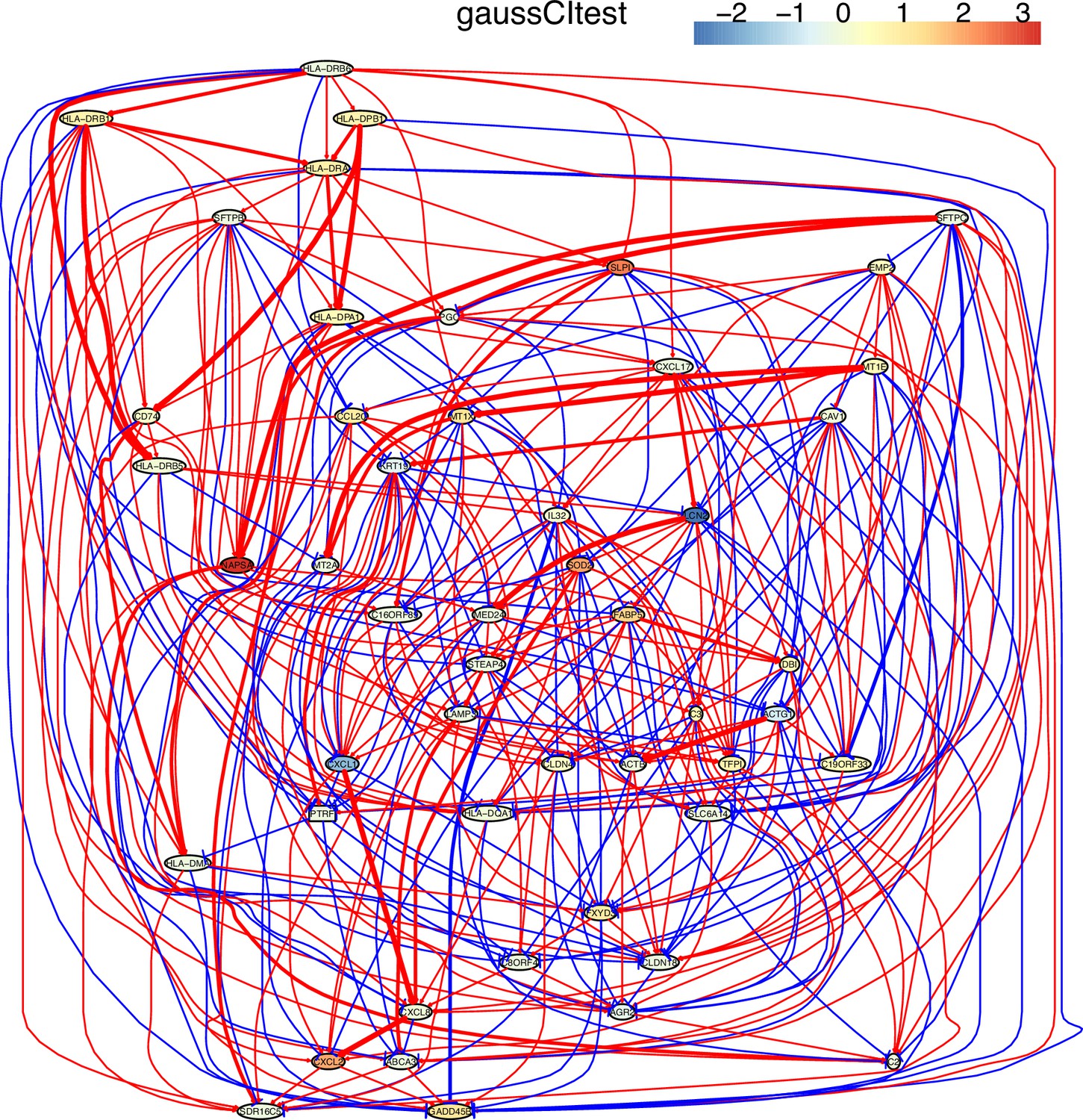
The causal network of the 50 feature genes inferred by PC+GaussCItest from the alveolar epithelial cell dataset (settings: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed the cells were used).
The control of the case was the H2228 cells. Compared with H2228 cells, MHC-II genes in alveolar epithelial cells were highly expressed. The relationships between MHC-II genes and between MHC-II genes and CD74 are much more dense than those inferred by PC+cmiknn.
Appendix 5—figure 7
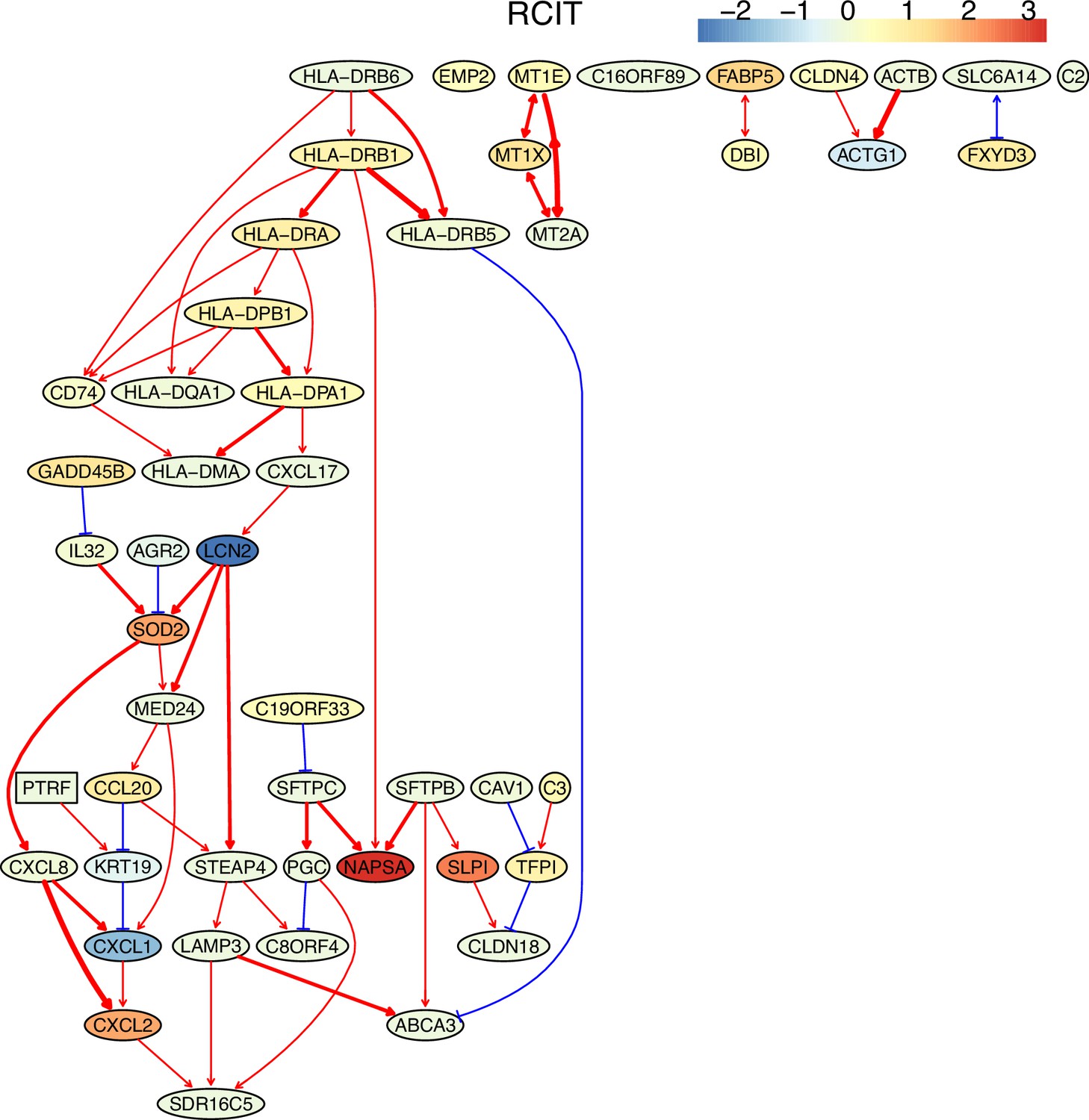
The causal network of the 50 feature genes inferred by PC+RCIT from the alveolar epithelial cell dataset (settings: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed the cells were used).
The control of the case was the H2228 cells. Compared with H2228 cells, MHC-II genes in alveolar epithelial cells were highly expressed.
Appendix 5—figure 8
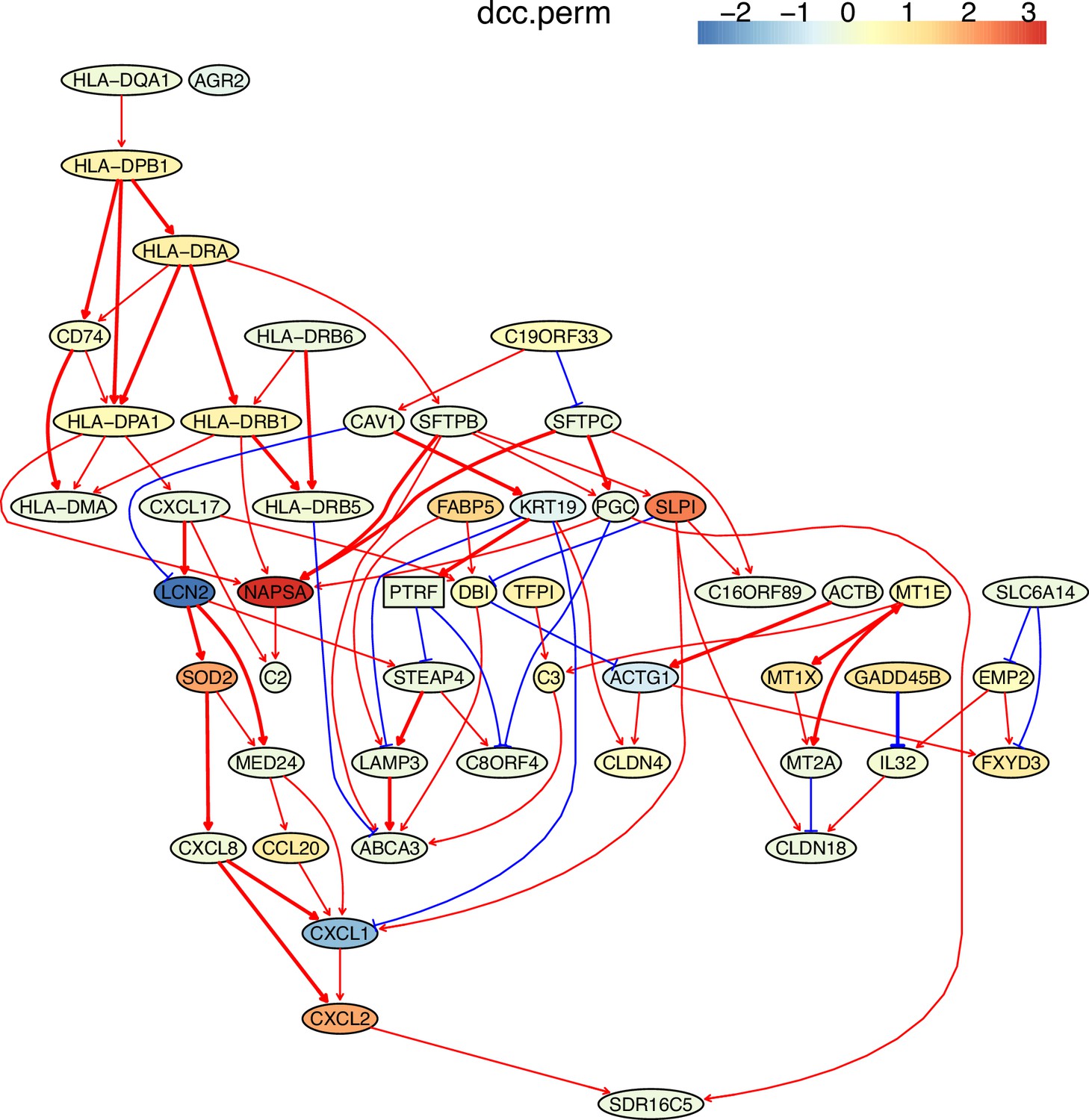
The causal network of the 50 feature genes inferred by PC+DCC.perm from the alveolar epithelial cell dataset (settings: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed the cells were used).
The control of the case was the H2228 cells. Compared with H2228 cells, MHC-II genes in alveolar epithelial cells were highly expressed.
Appendix 5—figure 9
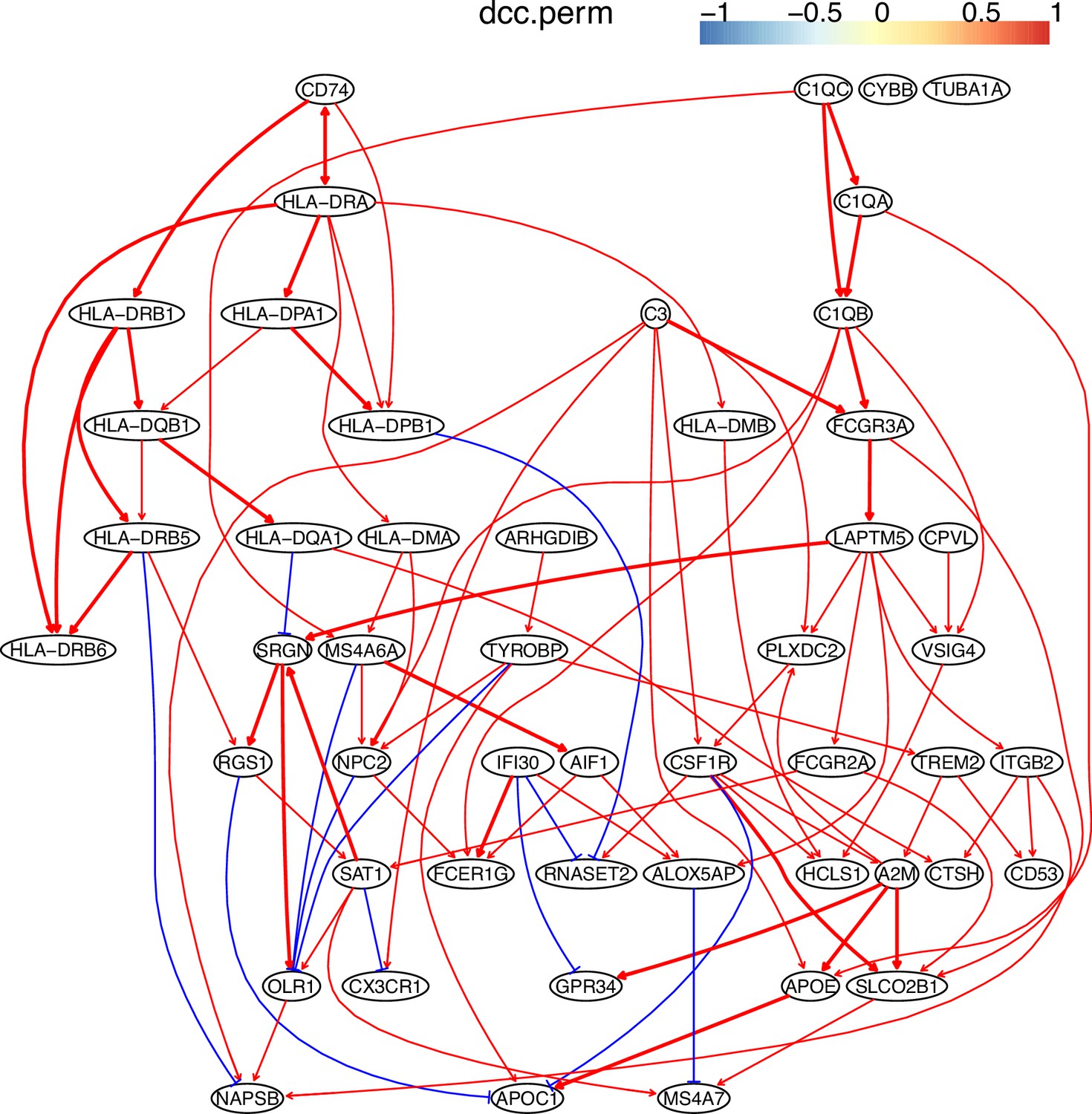
The causal network of the 50 feature genes inferred by PC+DCC.perm from the macrophage dataset (macrophages isolated from human glioblastoma) (setting: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed were used).
Because no control data was used, the differential expression of genes was not computed. Note the interactions between MHC-II genes and CD74, between C1QA/B/C, and the TYROBP→TREM2→A2M→ APOE→APOC1 cascade.
Appendix 5—figure 10
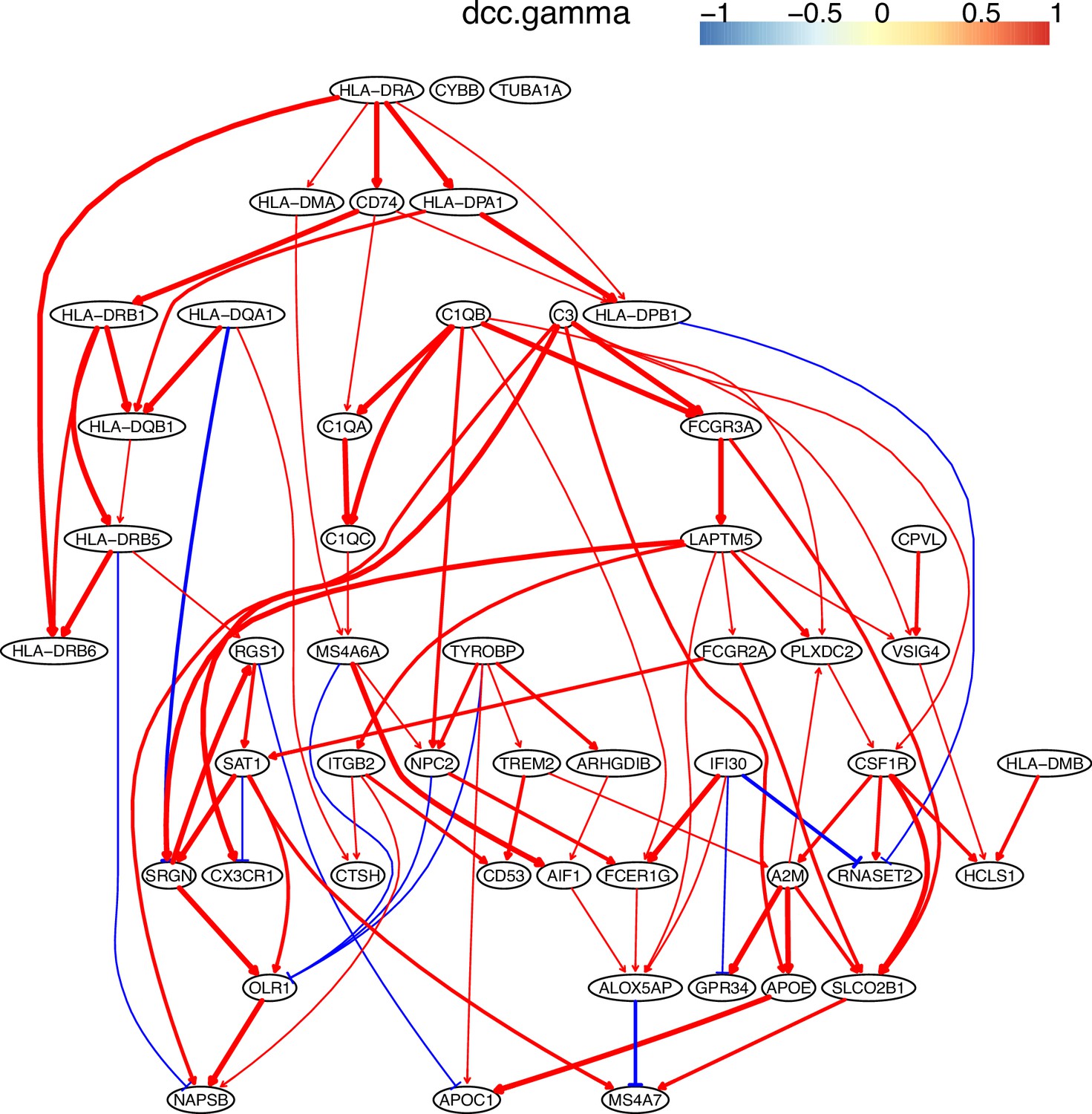
The causal network of the 50 feature genes inferred by PC+DCC.gamma from the macrophage dataset (macrophages isolated from human glioblastoma) (setting: feature genes were expressed in >50% cells, the alpha level for CI test was 0.1, and the 300 cells with more feature genes expressed were used).
Because no control data was used, the differential expression of genes was not computed. Note the interactions between MHC-II genes and CD74, between C1QA/B/C, and the TYROBP→TREM2→A2M→ APOE→APOC1 cascade.
Appendix 5—figure 11
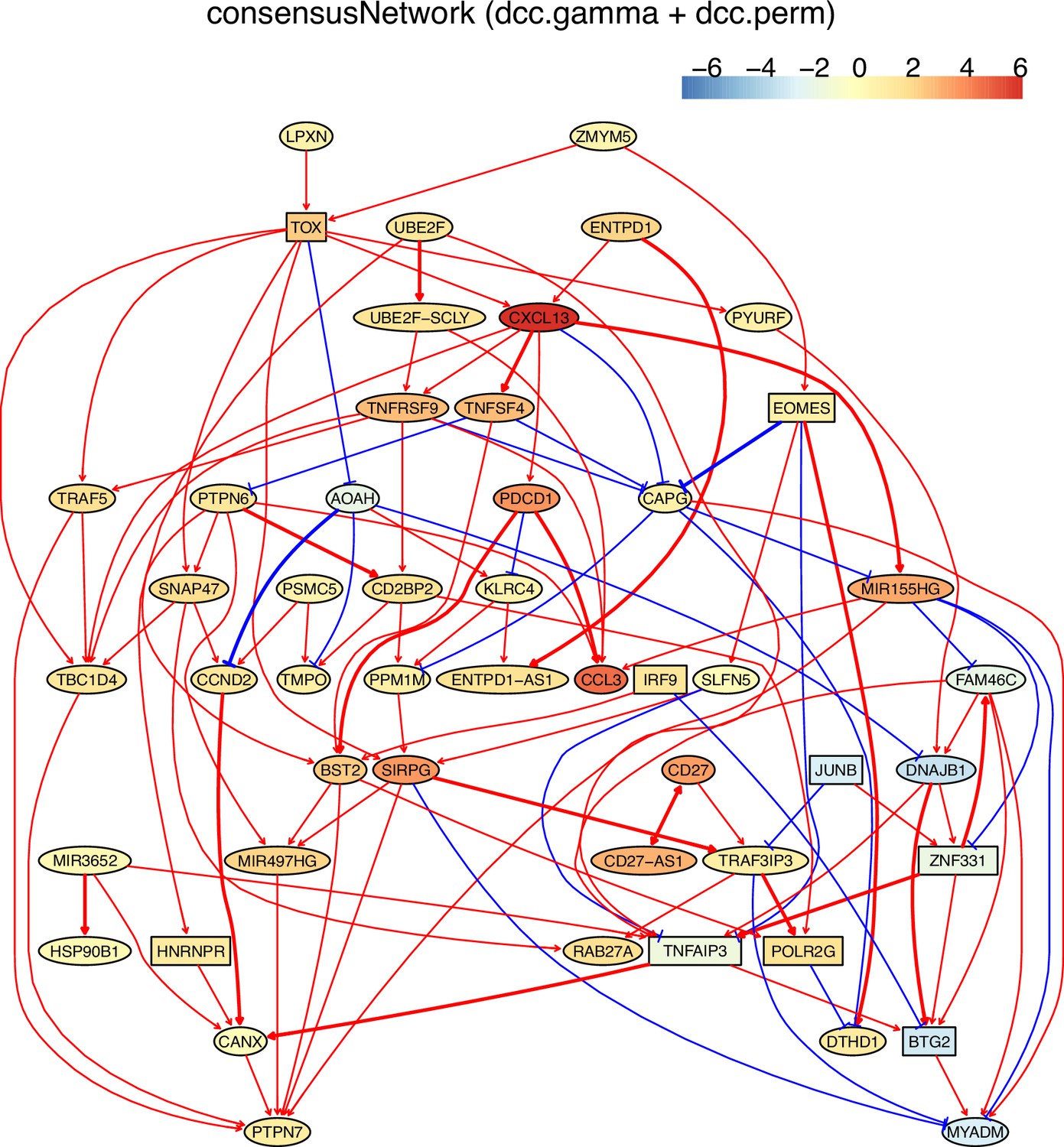
The networks of 50 genes in exhausted CD8 T cells and non-exhausted CD8 T cells from colorectal and lung cancers and the normal tissues neighboring the cancers.
(A) The TOX-network inferred from exhausted CD8 T cells from colorectal cancer. TOX→→PDCD1 (TOX→CXCL13→PDCD1), TNFRSF9→TRAF5, and TOX→→MIR155HG have related reports including (1) TOX up-regulates PDCD1 expression (Khan et al., 2019), (2) TNF receptors bind to TRAF2/5 to activate NF-kB signaling, (3) in mice up-regulated miR-155 represses Fosl2 by inhibiting Fosb and causes long-term persistence of exhausted CD8 T cells during chronic infection (Stelekati et al., 2018). We found that if more feature genes were selected (to include FOSB), the MIR155HG-|YPEL5→DNAJB1→FOSB were inferred, agreeing with inhibited FOXB by up-regulated MIR155.
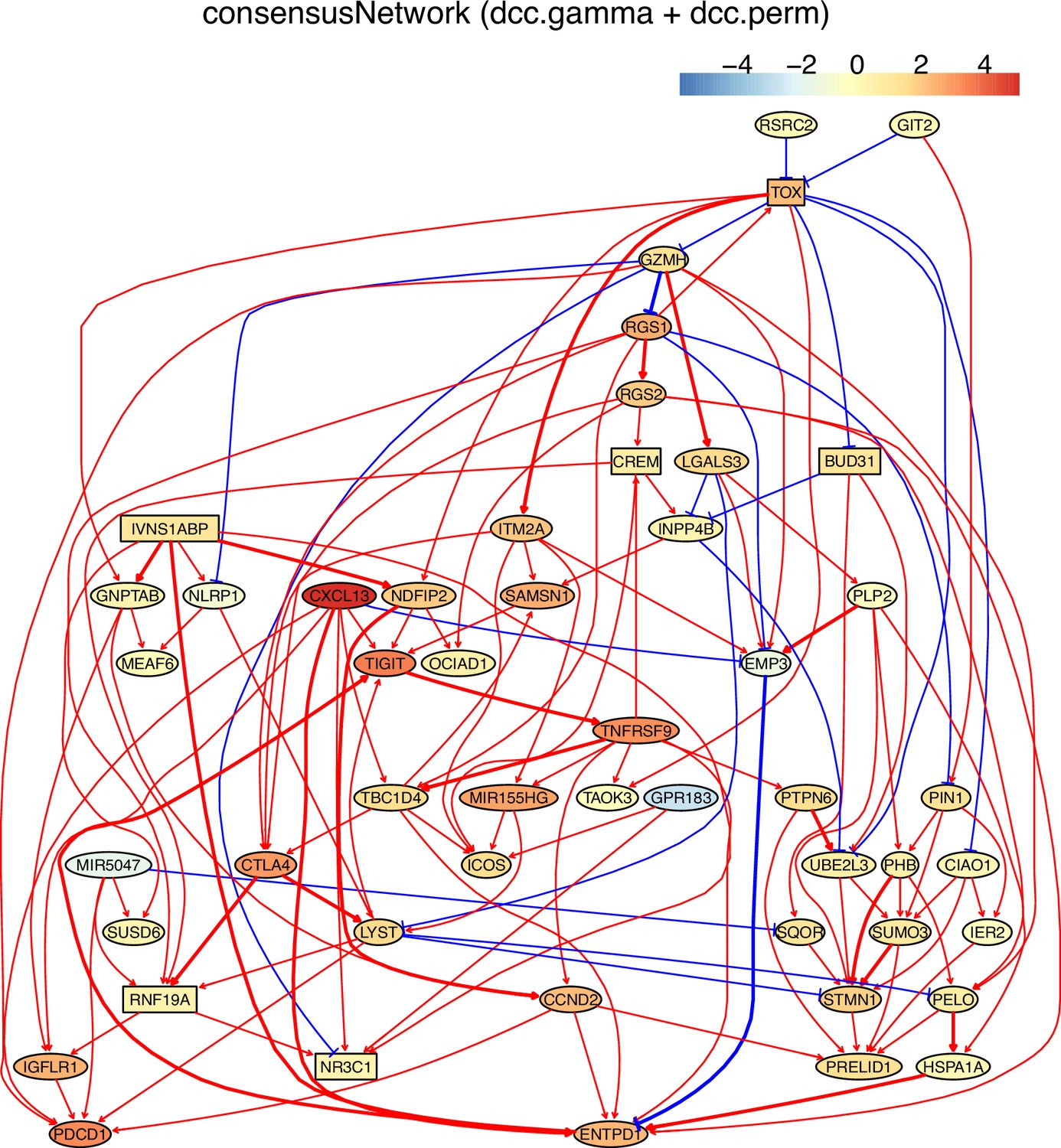
Appendix 5—figure 12
The networks of 50 genes in exhausted CD8 T cells and non-exhausted CD8 T cells from colorectal and lung cancers and the normal tissues neighboring the cancers.
(B) The TOX-network inferred from exhausted CD8 T cells from lung cancer. Different route of TOX→→PDCD1 and TOX→→MIR155HG were inferred (i.e. TOX→GNPTAB→IGFLR1→PDCD1, TOX→ITM2A→MIR155HG).
Appendix 5—figure 13
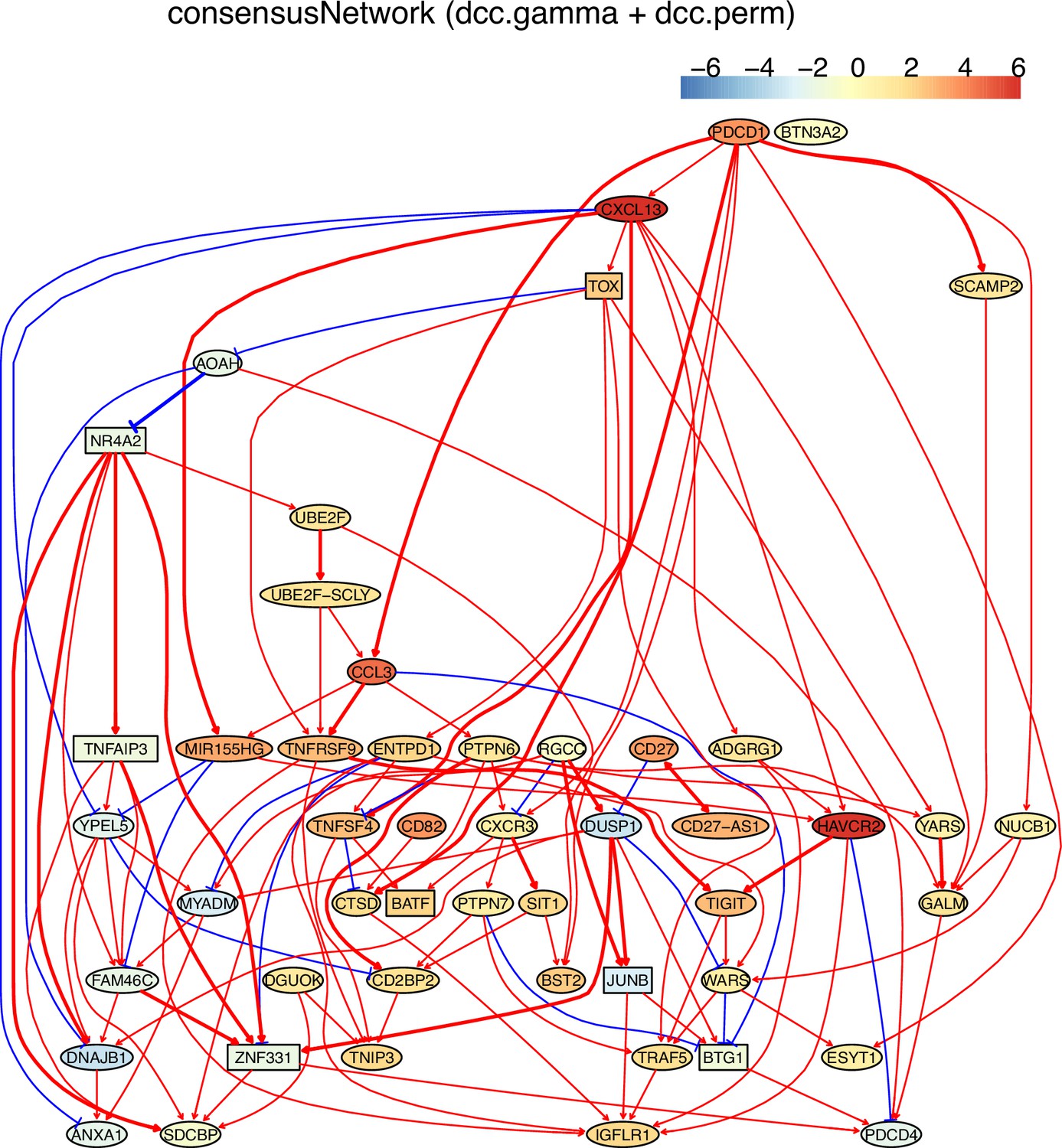
The networks of 50 genes in exhausted CD8 T cells and non-exhausted CD8 T cells from colorectal and lung cancers and the normal tissues neighboring the cancers.
(C) The PDCD1-network inferred from exhausted CD8 T cells from colorectal cancer. PDCD1→→TOX (PDCD1→CXCL13→TOX), PDCD1→→MIR155HG (there were two routes: PDCD1→CXCL13→MIR155HG, PDCD1→CCL3→MIR155HG), MIR155HG-|YPEL5→DNAJB1, and HAVCR2-|PDCD4 were inferred. Related reports of these interactions include (1) TOX transcription factors cooperate with NR4A transcription factors to impose CD8+ T cell exhaustion (Seo et al., 2019), (2) CCL3 is one of the up-regulated chemokine genes in exhausted CD8 T cells (Wherry et al., 2007).
Appendix 5—figure 14

The networks of 50 genes in exhausted CD8 T cells and non-exhausted CD8 T cells from colorectal and lung cancers and the normal tissues neighboring the cancers.
(D) The TOX-network inferred from non-exhausted CD8 T cells from the normal tissue neighboring colorectal cancer. Direct TOX→PDCD1 was inferred, and MIR155HG was not associated with TOX.
Appendix 5—figure 15
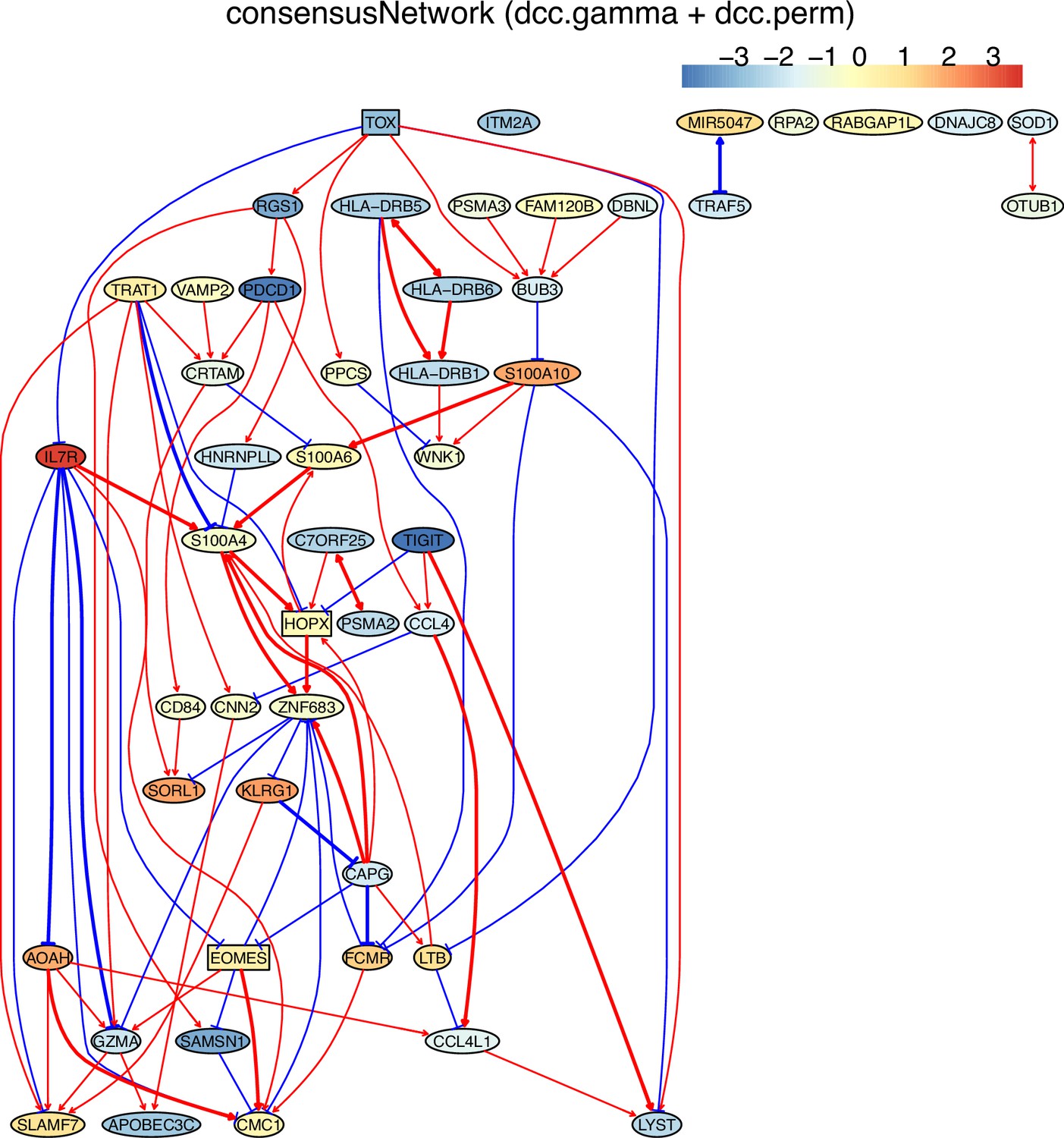
The networks of 50 genes in exhausted CD8 T cells and non-exhausted CD8 T cells from colorectal and lung cancers and the normal tissues neighboring the cancers.
(E) The TOX-network inferred from non-exhausted CD8 T cells from the normal tissue neighboring lung cancer. TOX→RGS1→PDCD1 and interactions between HLA-DRB2, HLA-DRB6, and HLA-DRB1 were inferred.
Appendix 5—figure 16
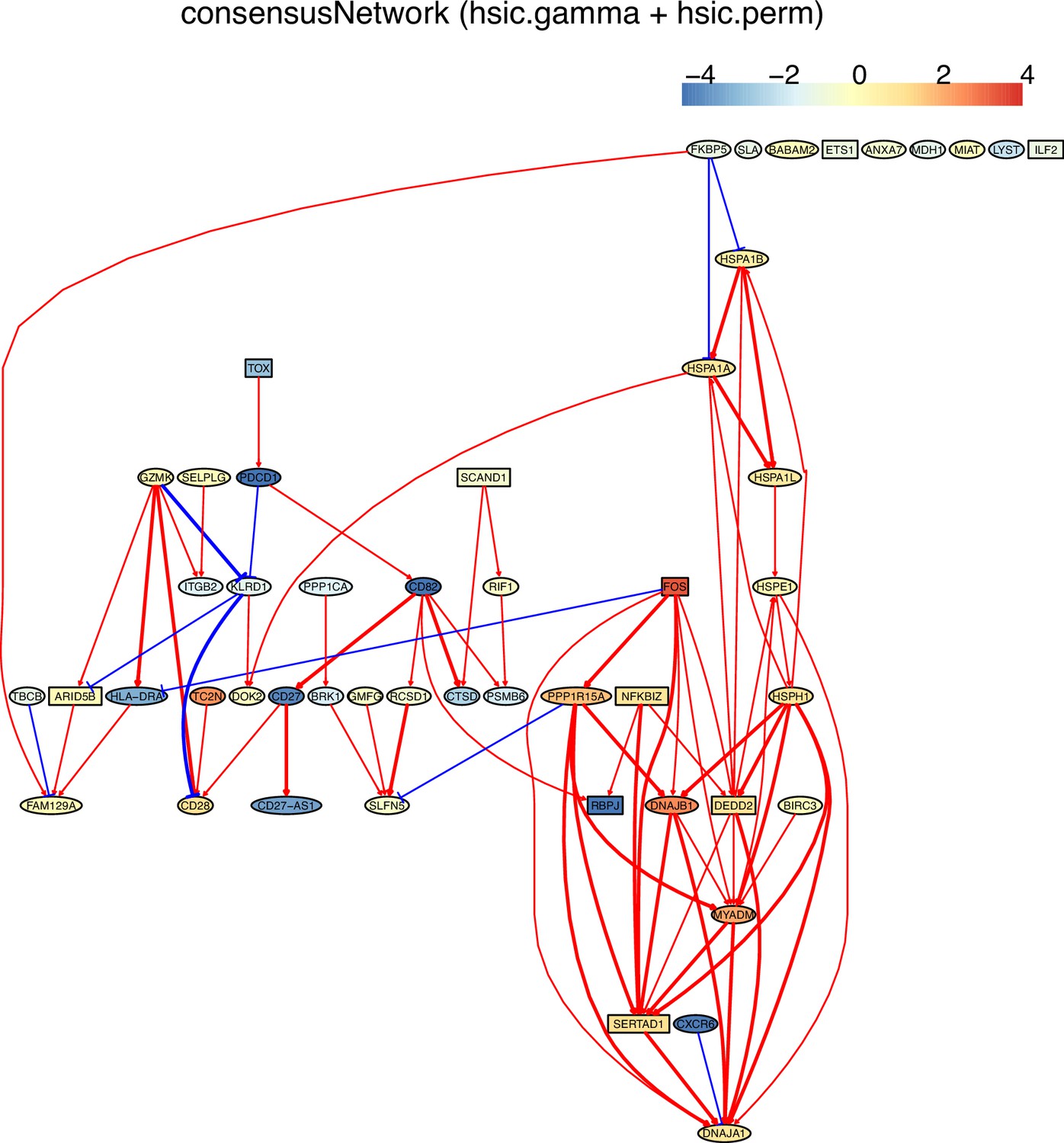
The networks of 50 genes in exhausted CD8 T cells and non-exhausted CD8 T cells from colorectal and lung cancers and the normal tissues neighboring the cancers.
(F) The PDCD1-network of HSIC algorithms inferred from non-exhausted CD8 T cells from the normal tissue neighboring colorectal cancer.
Appendix 5—figure 17
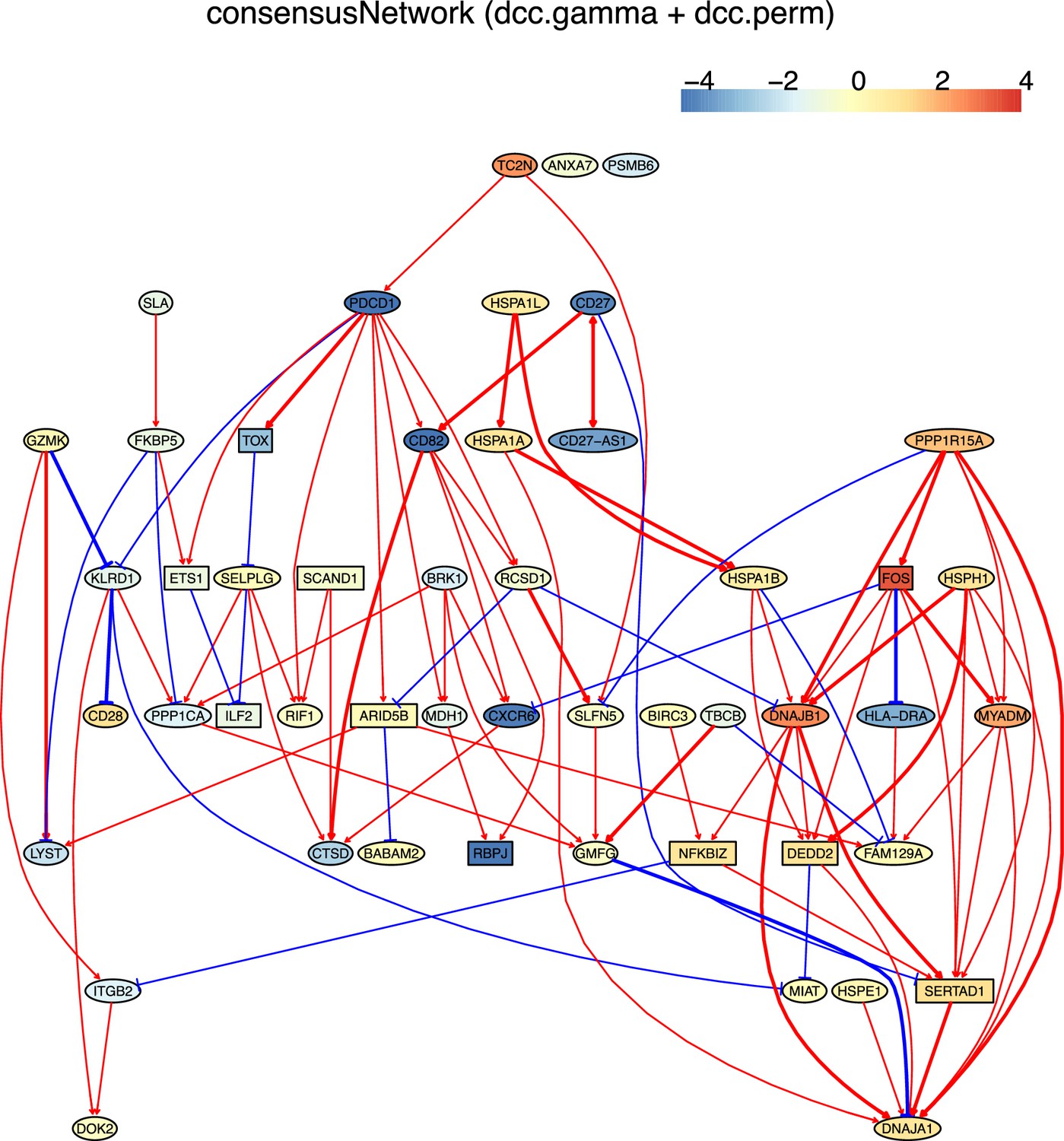
The networks of 50 genes in exhausted CD8 T cells and non-exhausted CD8 T cells from colorectal and lung cancers and the normal tissues neighboring the cancers.
(G) The PDCD1-network inferred from non-exhausted CD8 T cells from the normal tissue neighboring colorectal cancer.
Appendix 5—figure 18
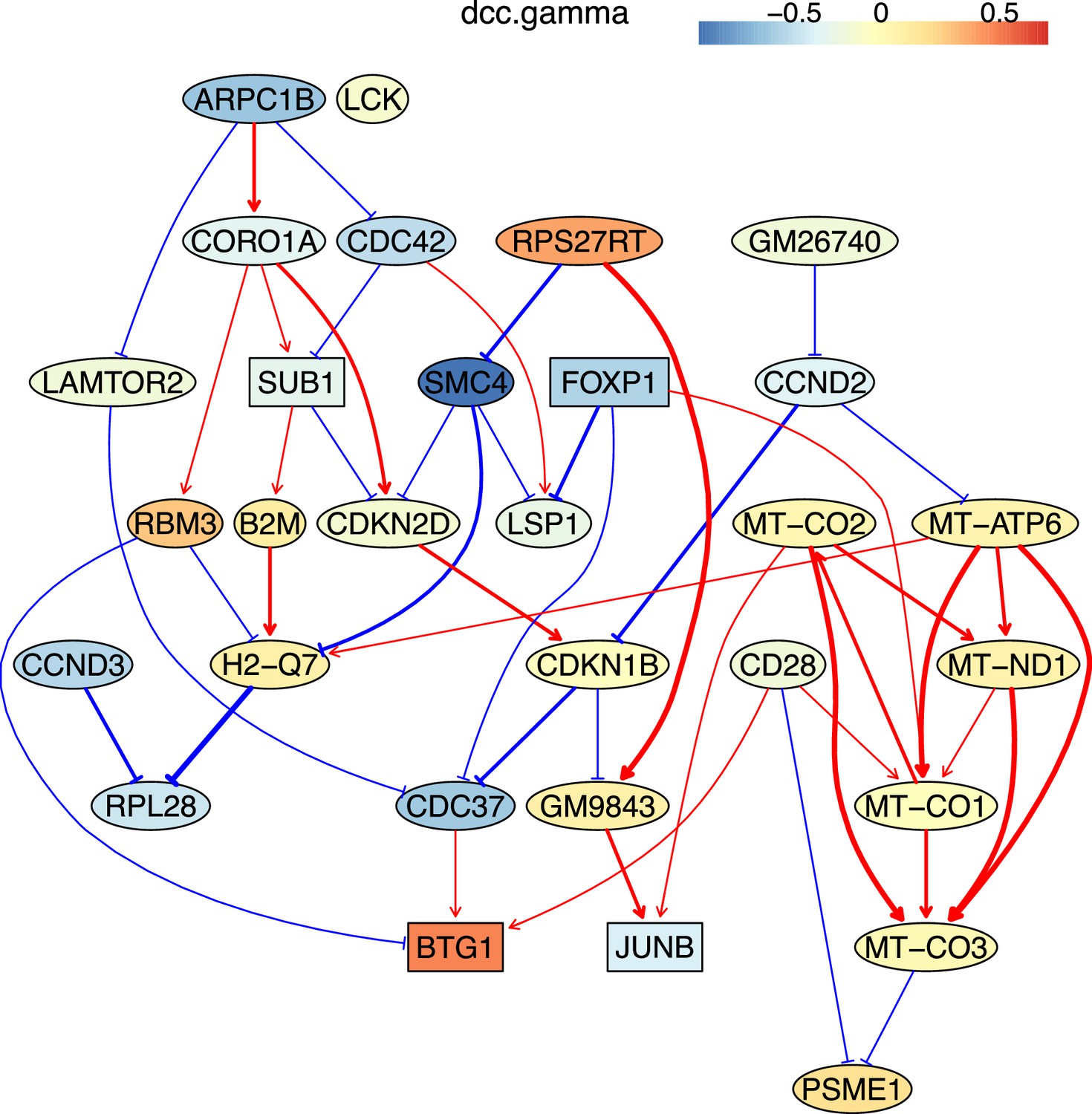
The causal relationships between genes that were differentially expressed in old cytotoxic CD4 T cells.
The network was inferred from 600 cells.
Appendix 5—figure 19
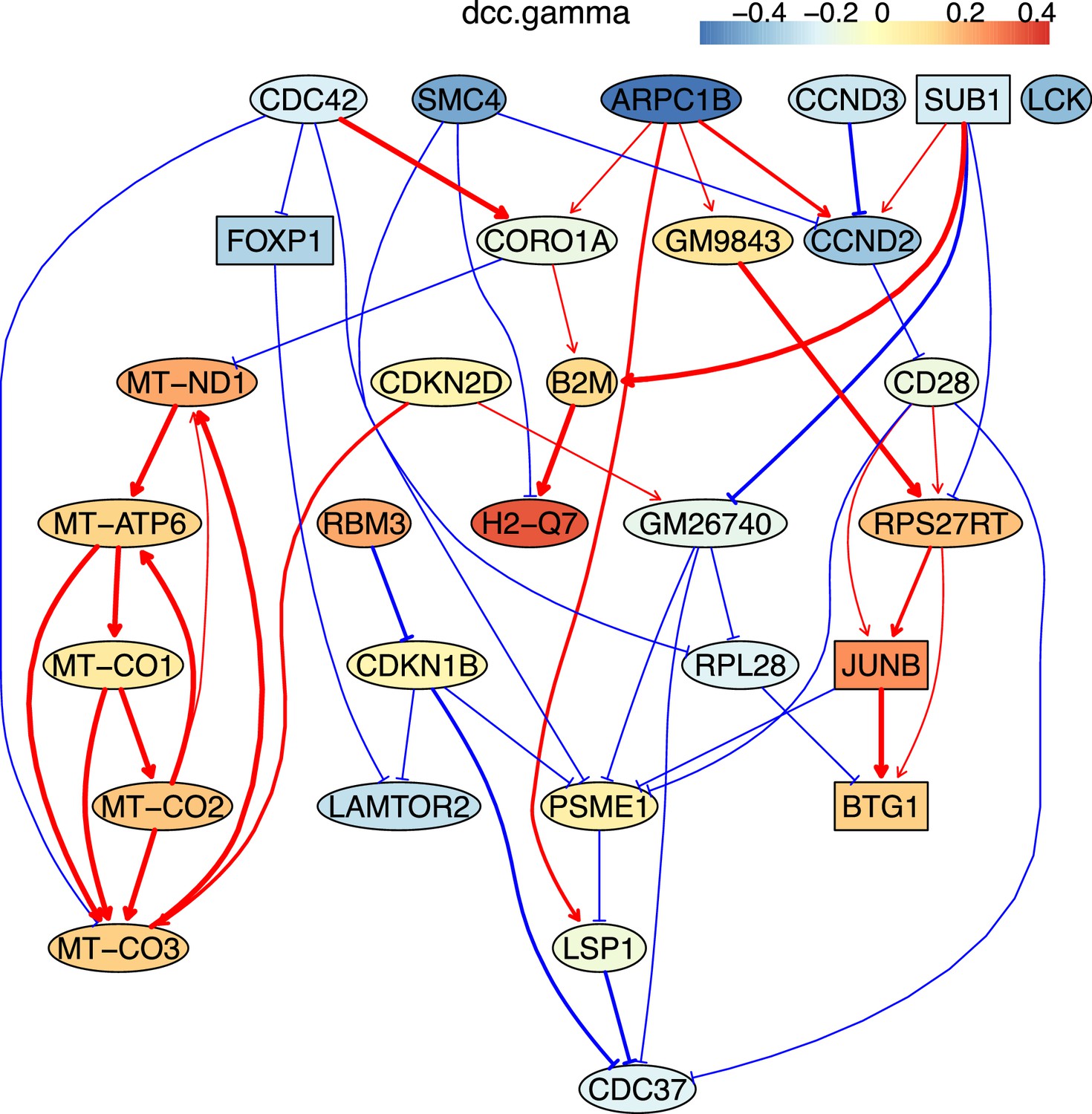
The causal relationships between genes that were differentially expressed in old exhausted CD4 T cells.
The network was inferred from 600 cells.
Appendix 5—figure 20
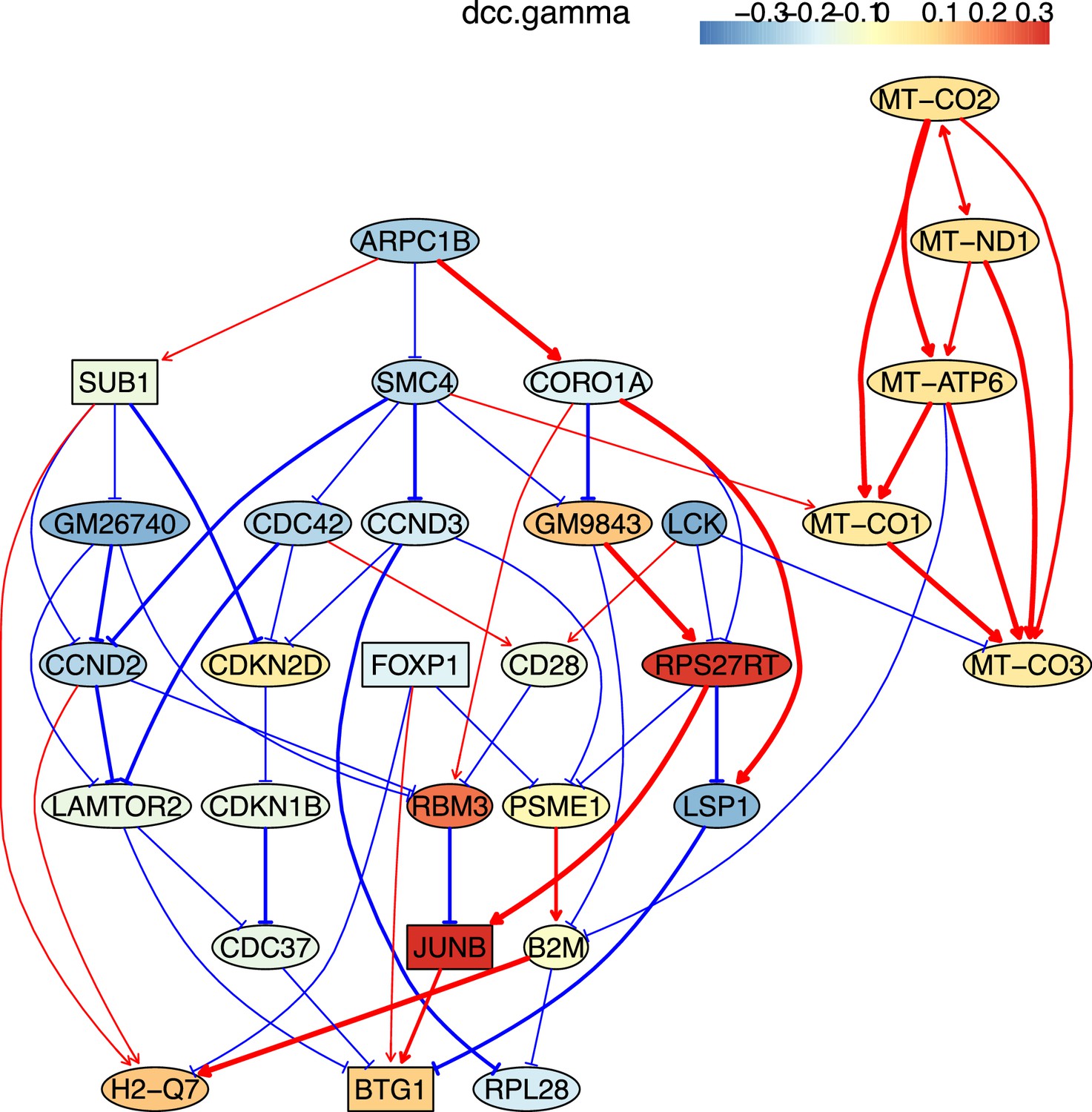
The causal relationships between genes that were differentially expressed in young TEM CD4 T cells.
The network was inferred from 600 cells.
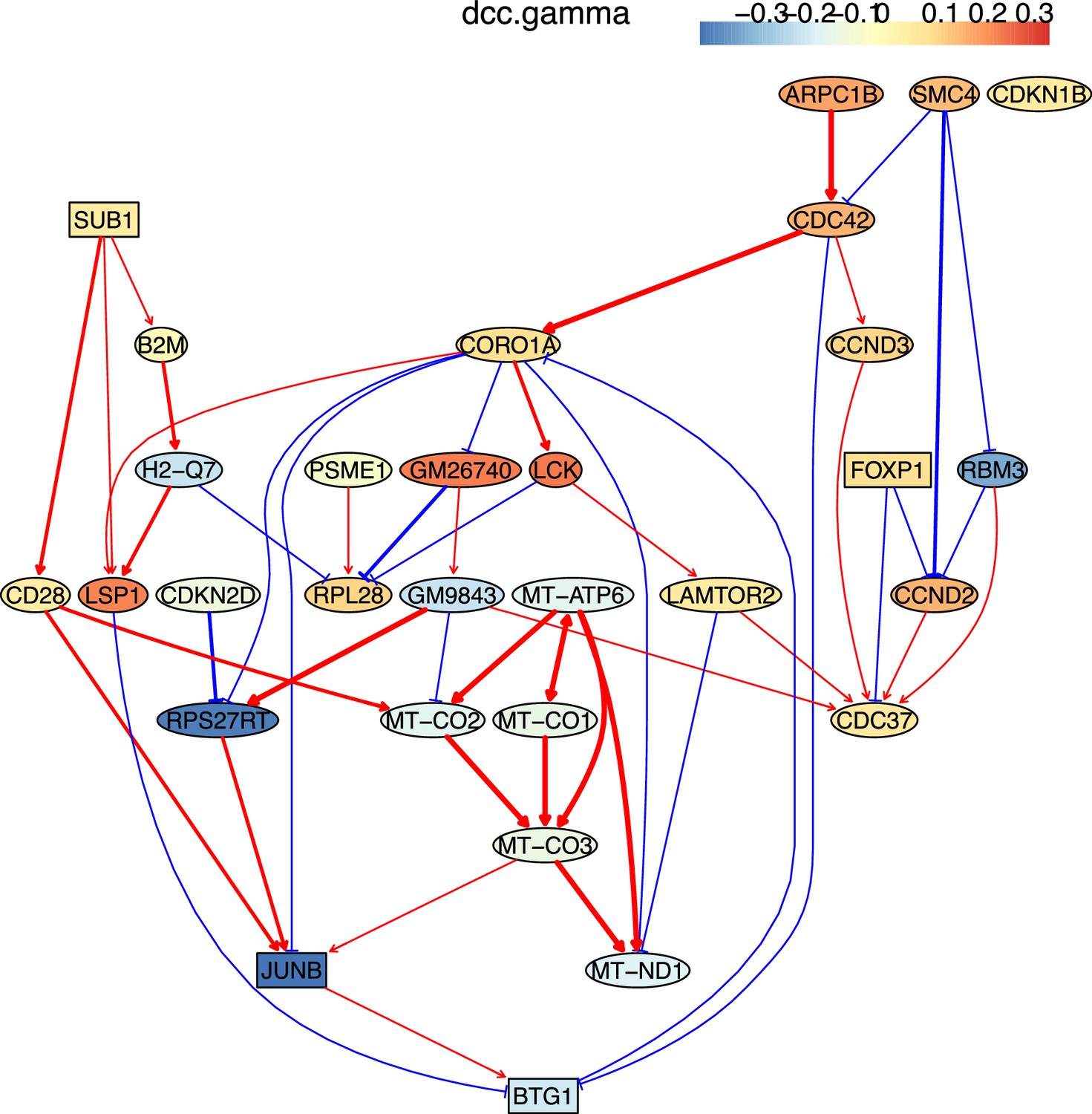
Appendix 5—figure 21
The causal relationships between genes that were differentially expressed in old TEM CD4 T cells.
The network was inferred from 600 cells.
Appendix 5—figure 22
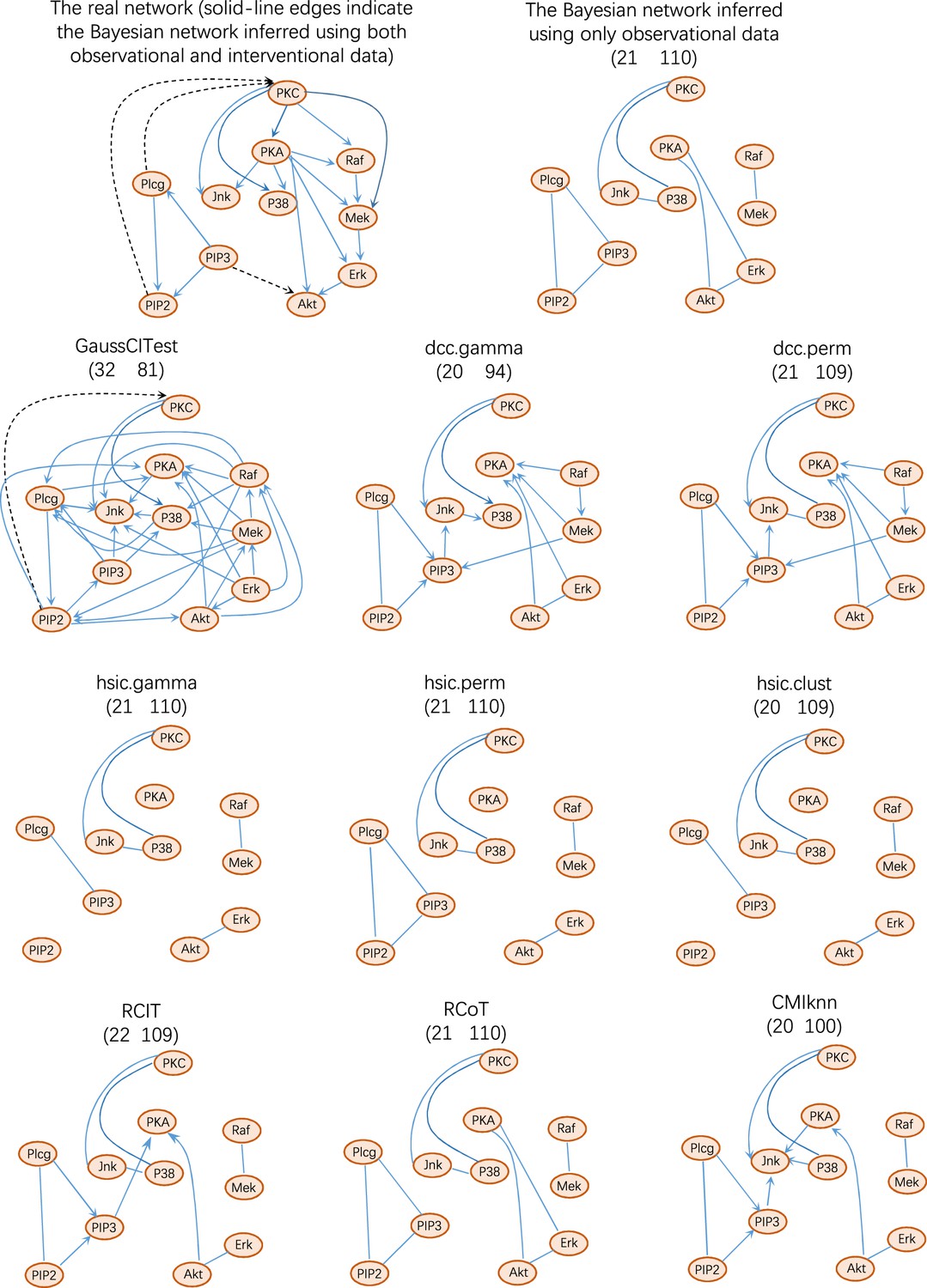
The performance of causal discovery algorithms upon the Sachs dataset (Sachs et al., 2005).
Structural intervention distance (SID) is another important measure for evaluating causal graphs. The numbers in the bracket are structural Hamming distance (SHD) and SID values (the smaller, the better). These values indicate that DCC.gamma and DCC.perm outperform others. The network inferred by Bayesian inference contains only undirected edges.
Author response image 1
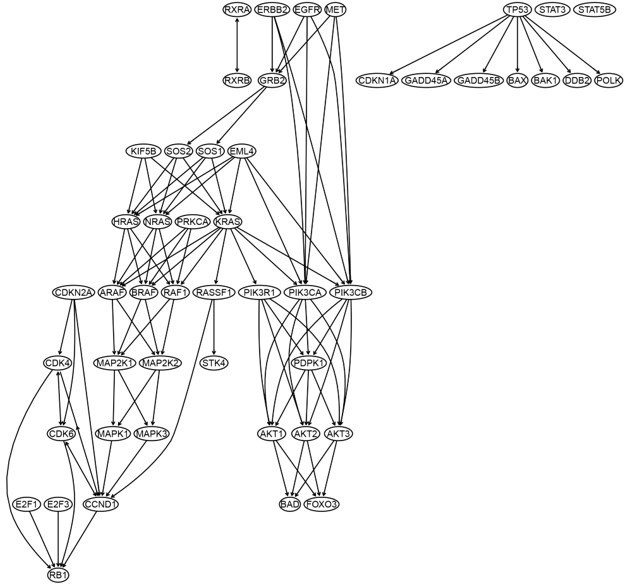
The DAG of the hsa05223 "non-small cell lung cancer" pathway.
Author response image 2
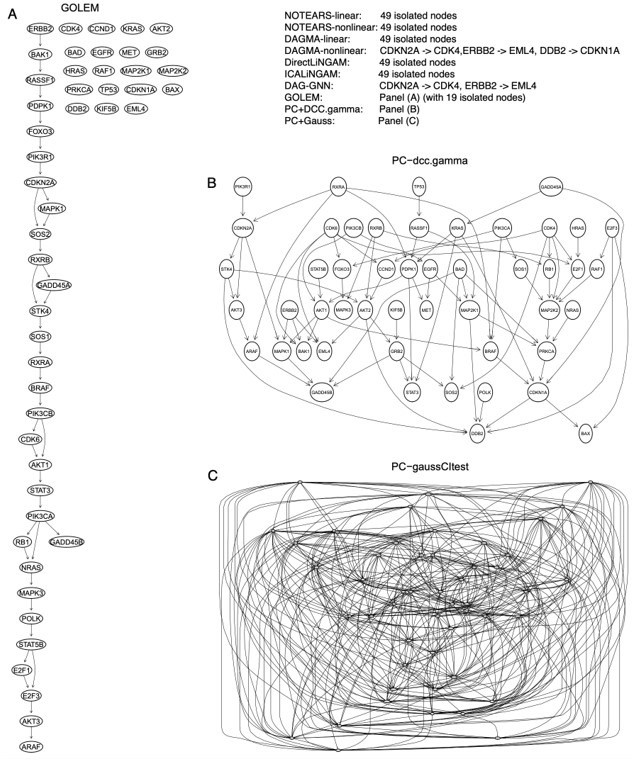
The inferred networks of the 49 genes in the hsa05223 pathway by the ten methods in A549 cells.
(A) The network inferred by GOLEM. (B) The network inferred by PC+DCC.γ. (C) The network inferred by PC+GaussCItest.
Author response image 3
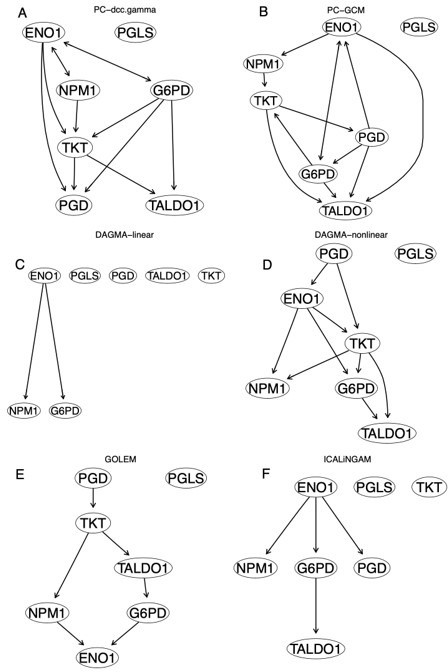
Some inferred networks of the WP134 pathway in the A549 cells.
(A) The network of PC+DCC.γ. (B) The network of PC+GCM. (C) The network of DAGMA-linear. (D) The network of DAGMA-nonlinear. (E) The network of GOLEM, (F) The network of ICLiNGAM.
Author response image 4
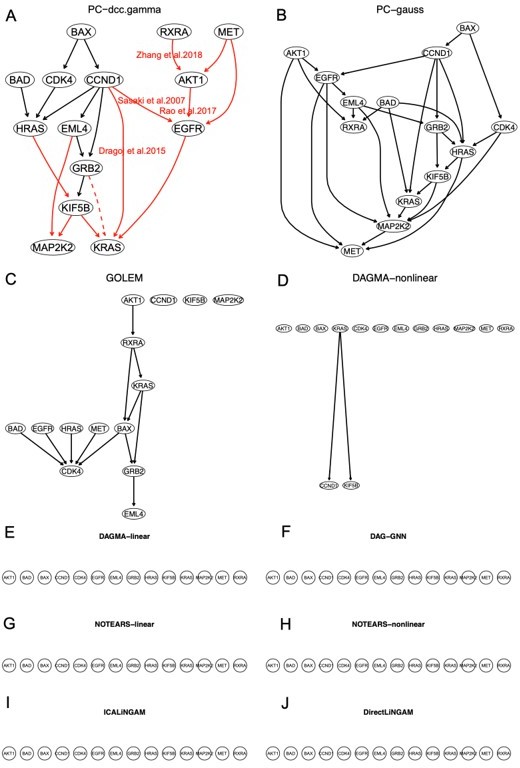
The networks of the 14 enriched hsa05223 genes inferred by the ten methods.
Most continuous optimization-based methods do not infer relationships between genes. In the network of PC+DCC.γ, solid and dashed red edges map direct and indirect interactions in the hsa05223 pathway and red edges with paper citations mark relationships supported by experimental findings. Edges between BAX, CDK3, and CCND1 (functioning for uncontrolled proliferation and driving cell division) are unannotated in hsa05223, but some of these relationships may be true because they reflect feedback regulations in cancer cells.
Author response image 5
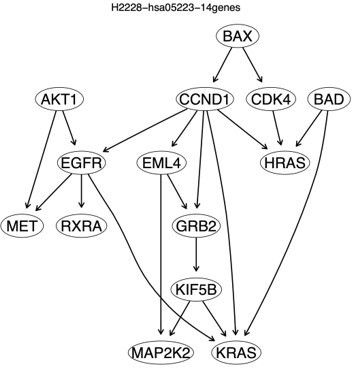
An approximate ground truth for the 14 enriched hsa05223 genes in H2228 cells built upon causal hypothesis testing using gene expression data in H2228 cells.
Author response image 6
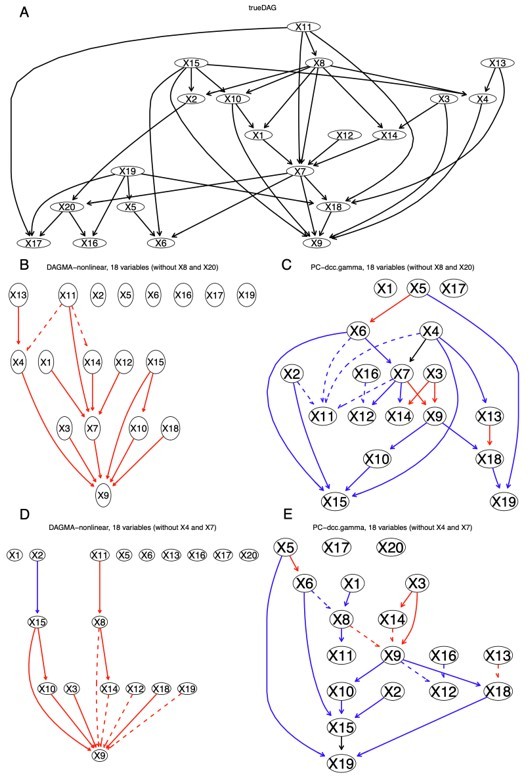
DAGMA-nonlinear and PC+DCC.
γ show different ability to infer indirect causal relationships from simulated data. Red and blue edges indicate mapped direct relationships between variables with different direction, dashed edges indicate mapped indirect relationships between variables (e.g., X11X4 in panel (B) maps X11X8X4 in the true DAG). When an edge maps both direct and indirect relationships, it is assumed to map the direct one. We do not care about the edges' orientation when checking whether it maps any edge in the true DAG. (A) The true DAG of the nonlinear relationships between the 20 variables. (B) The network of DAGMA-nonlinear without X8 and X20, in which two edges map indirect relationships. (C) The network of PC+DCC.γ without X8 and X20, in which five edges map indirect relationships. (D) The network of DAGMA-nonlinear without X4 and X7, in which four edges map indirect relationships. (E) The network of PC+DCC.γ without X4 and X7, in which six edges map indirect relationships.
Author response image 7
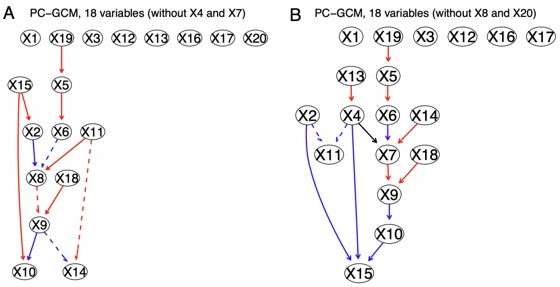
The network inferred by PC+GCM without X4 and X7 (left) and X8 and X20 (right).
Compared with the network of PC+DCC.γ (Author response image 6CE), PC+GCM infers fewer edges.
Tables
Table 1
Performance of feature selection methods.
| Algorithm | Category | Time consumption | Accuracy | Scalability | Advantage/disadvantage |
|---|---|---|---|---|---|
| RandomForest | Ensemble learning-based methods use many trees of a random forest to calculate the importance of features, then perform regression based on the response variable(s) to identify the most relevant features. | + | ++ | ++ | These algorithms are indeterministic (the same input may generate slightly different outputs). ExtraTrees and RandomForest perform better than XGBoost. |
| ExtraTrees | + | ++ | ++ | ||
| XGBoost | ++ | + | + | ||
| BAHSIC | The three are Hilbert-Schmidt independence criterion (HSIC)-based algorithms. HSIC is used as the measure of dependency between the response variable and features. | + | +++ | + | BAHSIC and SHS are the best and second best. |
| SHS | + | +++ | + | ||
| HSIC Lasso | ++ | ++ | ++ | Inferior to BAHSIC and SHS. | |
| Lasso | Lasso is a regression analysis method that performs both variable selection and regularization (which adds additional constraints or penalties to a regression model). Lasso, RidgeRegression, and ElasticNet are three regulation terms. | +++ | + | +++ | Inferior to BAHSIC and SHS. Accuracy is not high and scalability is poor. |
| RidgeRegression | +++ | + | +++ | ||
| ElasticNet | +++ | + | +++ |
-
# Time consumption is estimated upon simulated data (Appendix 2—figure 1). Accuracy is estimated upon simulated and real data (Appendix 2—figures 2–7). Scalability is estimated upon simulated data (Appendix 2—figure 2). Advantage/disadvantage is made upon accuracy together with algorithms’ other properties.
Table 2
Performance of causal discovery methods.
| Methods | CI tests | Category | Time consumption | Accuracy | Stability | Features |
|---|---|---|---|---|---|---|
| PC GSP | GaussCItest | GaussCItest assumes all variables are multivariate Gaussian, which impairs GussCItest’s performance when data are complex. | +++ | + | +++ | Fast and inaccurate |
| CMIknn | Conditional mutual information (CMI) is based on mutual information. | +++ | ++ | + | Fast and inaccurate | |
| RCIT | Two approximation methods of KCIT (the Kernel conditional independence test). | ++ | ++ | ++ | Fast and moderately accurate | |
| RCoT | ++ | ++ | ++ | |||
| HSIC.clust | Extra transformations make HSIC determine if X and Y are conditionally independent given a conditioning set. HSIC.gamma and HSIC.perm employ gamma test and permutation test to estimate a p-value. | + | ++ | + | Slow and accurate | |
| HSIC.gamma | + | +++ | ++ | |||
| HSIC.perm | + | +++ | + | |||
| DCC.gamma | Distance covariance is an alternative to HSIC for measuring independence. DCC.gamma and DCC.perm employ gamma test and permutation test to estimate a p-value. | + | +++ | ++ | Slow and accurate | |
| DCC.perm | + | +++ | + | |||
| GCM | The generalized covariance measure-based (also classified as regression-based). | + | ++ | +++ | Slower than DCC.gamma | |
| GES | Score-based causal. | ++ | ++ | ++ | Fast and moderately accurate | |
| DAGMA-nonlinear | Continuous optimization-based. | + | ++ | +++ | Performs well with complete models | |
-
# Time consumption is estimated upon simulated data (Appendix 3—figure 1). Accuracy is estimated upon the lung cancer cell lines (Figure 2; Appendix 3—figure 2). Stability is estimated upon the relative structural Hamming distance (SHD, a standard distance to compare graphs by their adjacency matrix), which is used to measure the extent an algorithm produces the same results when running multiple times (Appendix 3—table 1). Advantage/disadvantage is made upon accuracy.
Appendix 1—table 1
Real single-cell RNA-sequencing (scRNA-seq) data.
| Dataset | Cell type | Species | Protocols | Dataset URL | Database and Identifier | References |
|---|---|---|---|---|---|---|
| 1 | Microglia from humans and mice | Human and mouse | MARS-seq | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE134705 | NCBI Gene Expression Omnibus, GSE134705 | Geirsdottir et al., 2019 |
| 2 | Five lung cancer cell lines (A549, H1975, H2228, H838, HCC827) from the CellBench benchmarking dataset | Human | 10x Genomics | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126906 | NCBI Gene Expression Omnibus, GSE126906 | Tian et al., 2019 |
| 3 | Lung alveolar epithelial cells | Human | 10x Genomics | https://www.synapse.org/#!Synapse:syn21041850 | Synapase, syn21041850 | Travaglini et al., 2020 |
| 4 | Six types of CD4 T cells (naïve, TEM, rTregs, naïve_Isg15, cytotoxic, exhausted) from young and old mice | Mouse | 10x Genomics | https://singlecell.broadinstitute.org/single_cell/study/SCP490/aging-promotes-reorganization-of-the-cd4-t-cell-landscape-toward-extreme-regulatory-and-effector-phenotypes | Single Cell Portal, SCP490 | Elyahu et al., 2019 |
| 5 | Macrophages isolated from glioblastomas | Human | Smart-seq2 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE131928 | NCBI Gene Expression Omnibus, GSE131928 | Neftel et al., 2019 |
| 6 | Exhausted CD8 T cells isolated from liver cancer, lung cancer, and CRC | Human | Smart-seq2 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99254. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108989. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98638. | NCBI Gene Expression Omnibus, GSE99254. NCBI Gene Expression Omnibus, GSE108989. NCBI Gene Expression Omnibus, GSE98638. | Guo et al., 2018; Zhang et al., 2018; Zheng et al., 2017 |
| 7 | Non-exhausted CD8 T cells isolated from the normal liver, lung, and colorectal tissues | Human | Smart-seq2 | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE99254. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108989. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE98638. | NCBI Gene Expression Omnibus, GSE99254. NCBI Gene Expression Omnibus, GSE108989. NCBI Gene Expression Omnibus, GSE98638. | Guo et al., 2018; Zhang et al., 2018; Zheng et al., 2017 |
| 8 | CD4 T cell | Human | Flow cytometry | https://www.science.org/doi/10.1126/science.1105809 | Science Supplementary Materials, doi: 10.1126/science.1105809 | Sachs et al., 2005 |
Appendix 1—table 2
Feature selection and causal discovery algorithms.
| Feature selection | Category | Causal discovery | Category |
|---|---|---|---|
| Random forests | Ensemble learning-based | GaussCItest | Test for CI between Gaussian random variables upon partial correlation |
| Extremely randomized trees | DCC.perm | Test for CI using a distance covariance-based kernel | |
| XGBoost | DCC.gamma | ||
| SHS | HSIC-based | HSIC.perm | Test for CI using a HSIC-based kernel |
| BAHSIC | HSIC.gamma | ||
| Block HSIC Lasso | HSIC.clust | ||
| Lasso | Regularization-based | RCIT | Test for CI using an approximate KCIT kernel |
| Ridge regression | RCoT | ||
| Elastic net | CMIknn | Test for CI based on conditional mutual information |
Appendix 3—table 1
Performance of the nine causal discovery algorithms (‘+++’ and ‘+’ indicate the best and worst, respectively).
| Algorithm | Time complexity* | Time consumption† | Accuracy ‡ | Sample size | Stability (mean of rSHD) § |
|---|---|---|---|---|---|
| GaussCItest | +++ | + | +++ | 0.075 (+++) | |
| CMIknn | + | ++ | + | 0.25 (+) | |
| RCIT | ++ | ++ | ++ | 0.12 (++) | |
| RCoT | ++ | ++ | ++ | 0.12 (++) | |
| HSIC.clust | + | +++ | ++ | 0.26 (+) | |
| HSIC.gamma | + | +++ | ++ | 0.12 (++) | |
| HSIC.perm | + | +++ | ++ | 0.28 (+) | |
| DCC.gamma | + | +++ | +++ | 0.14 (++) | |
| DCC.perm | + | +++ | +++ | 0.24 (+) | |
| GCM | Depending on regression methods | + | ++ | ++ |
-
*
(a) Assuming a dataset has n samples and the total dimension of X,Y,Z is q. Generally, q<<n. (b) r is the time of permutation. (c) d is the number of random Fourier features, generally d<n. (d) The time complexity of the PC algorithm is where N is the number of nodes and deg is the maximal degree.
-
†
Time-consuming levels are estimated upon simulated data (Appendix 3—figure 1).
-
‡
Accuracy is estimated upon the lung cancer cell lines (Main text-Figure 2; Appendix 3—figure 2). We performed causal discovery using the nine algorithms five times for the H2228 cell line and obtained 9*5=45 causal networks.
-
§
We estimated the stability of each algorithm’s performance by computing the mean relative SHD for the five causal networks the algorithm generated using the equation: In this equation, SHD(Gi, Gj) is the structural Hamming distance between causal network Gi and Gj, #edges(Gi) is the number of edges in Gi, and NG = 5 because each algorithm generates five causal networks.
Appendix 4—table 1
The percentages of mapped edges between inferred networks and the hsa05223 pathway.
| Cell lines | Inferred interactions | Num of ‘forward’ | Num of ‘reverse’ | Num of ‘undirected’ | Num of ‘epistatic’ and ‘synergistic’ | All |
|---|---|---|---|---|---|---|
| A549 | 82 | 10 (12.2%) | 12 (14.63%) | 2 (2.44%) | 17 (20.73%) | 50% |
| H838 | 77 | 9 (11.69%) | 10 (12.99%) | 1 (1.3%) | 21 (27.27%) | 53.25% |
| H1975 | 102 | 13 (12.75%) | 16 (15.69%) | 1 (0.98%) | 17 (16.67%) | 46.09% |
| H2228 | 106 | 13 (12.26%) | 20 (18.87%) | 2 (1.89%) | 25 (23.58%) | 56.60% |
| HCC827 | 122 | 13 (10.66%) | 26 (21.31%) | NA (NA%) | 28 (22.95%) | 54.92% |
Appendix 5—table 1
Up- and down-regulated genes in CD4 T cells from young and old mice.
| Gene | FC >0 cases | Annotation and evidence | References |
|---|---|---|---|
| Rpl28 | 24 | Ribosomal proteins influence aging. | Kirkland et al., 1993; Steffen and Dillin, 2016 |
| Arpc1b | 24 | Arpc1b may induce senescence in a p53-independent manner. | Li et al., 2022; Yun et al., 2011 |
| Smc4 | 24 | Goronzy and Weyand, 2019; McCartney et al., 2021 | |
| Sub1 | 23 | Sub1 is increased and becomes activated with age, and transgenic expression of PC4 disturbs mTOR-regulated proteostasis and causes global accelerated aging. | Chen et al., 2021 |
| Cdc37 | 23 | ||
| Lck | 23 | Lck is a positive regulator of inflammatory signaling and a potential treatment target for age-related diseases. | Garcia and Miller, 2009; Kim et al., 2019 |
| Cdc42 | 23 | Mouse model studies have found that aging is associated with elevated activity of the Rho GTPase Cdc42 in hematopoietic stem cells. In humans, CDC42 and CORO1A exhibited strong associations with age. | Amoah et al., 2021; Geiger and Zheng, 2013; Kerber et al., 2009 |
| Ccnd2 | 22 | Ccnd2 is an aging marker | Goronzy and Weyand, 2019; McCartney et al., 2021 |
| Ccnd3 | 22 | Ccnd2 is an aging marker | Goronzy and Weyand, 2019; Li et al., 2020; McCartney et al., 2021 |
| Foxp1 | 22 | FOXP1 controls mesenchymal stem cell commitment and senescence during skeletal aging. | Li et al., 2017 |
| Coro1a | 21 | CORO1A is a senescence-related gene. | Avelar et al., 2020; Kerber et al., 2009 |
| Gm26740 | 21 | A mouse-specific gene without annotation. | |
| Lamtor2 | 20 | MAPK and MTOR activator 2. It is involved in the activation of mTORC1. | Morita et al., 2017; Walters and Cox, 2021 |
| Lsp1 | 20 | Lymphocyte-specific protein 1; may play a role in mediating neutrophil activation and chemotaxis. | |
| Gene | FC <0 cases | Annotation and evidence | References |
| Rbm3 | 24 | Muscle from aged rats exhibited an increase in heat shock protein (HSP) 25 and HSP70 and in the cold shock protein RNA-binding motif 3 (RBM3). | Dupont-Versteegden et al., 2008; Van Pelt et al., 2019 |
| H2-Q7 | 23 | A strong increase of the MHC class I genes (including H2-Q7) and B2m is observed in the aging lung. | Angelidis et al., 2019 |
| Btg1 | 23 | Btg1 is involved in neural aging. | Micheli et al., 2021 |
| Gm9843 | 23 | A mouse-specific gene without annotation. | |
| Rps27rt (Gm9846) | 23 | Ribosomal protein S27 retrogene, mouse-specific. | |
| mt-Atp6 | 22 | Mitochondrial proteins involved in the electron transport chain are overrepresented in cells from older participants, with prevalent dysregulation of oxidative phosphorylation and energy metabolism molecular pathways. | Bektas et al., 2019; Goronzy and Weyand, 2019; Haas, 2019 |
| mt-Co1 | 22 | ||
| mt-Co2 | 22 | ||
| mt-Co3 | 22 | ||
| mt-Nd1 | 21 | ||
| Junb | 21 | JUNB is increased in all human immune cells during aging. | Maity et al., 2021; Zheng et al., 2020 |
| Psme1 | 21 | Proteasome activator subunit 1. It is implicated in immuno-proteasome assembly and required for efficient antigen processing. | Hwang et al., 2007 |
| B2m | 20 | B2m is in GO:0007568, a mouse aging GO term. B2M is elevated in the blood of aging humans and mice. | Smith et al., 2015 |
| Gene | Other cases | Annotation and evidence | References |
| Cd28 | 12 | Cd28 is an aging biomarker of T cells. | Le Page et al., 2018; Zhang et al., 2021a |
| Cdkn2d | 6 | Cdkn2d is an aging biomarker. | Goronzy and Weyand, 2019 |
| Cdkn1b | 5 | Cdkn1b is an aging biomarker. | Goronzy and Weyand, 2019 |
-
#1Genes and numbers in red indicate fold change (FC)>0, genes and numbers in blue indicate FC <0, genes and numbers in black do not show clear differential expression in a majority of cell groups.
Author response table 1
The results of 600 samples of 10 variables in linear relationships (* Dual Intel(R) Xeon(R) CPU E7-4820 v4 @ 2.00GHz,256G RAM).
| Methods | FDR | TPR | FPR | SHD | Precision | Recall | F1 | #Edges | Time* |
|---|---|---|---|---|---|---|---|---|---|
| NOTEARS_linear | 0.00 | 0.95 | 0.00 | 1 | 1.00 | 0.95 | 0.97 | 19 | 13.93 |
| NOTEARS_nonlinear | 0.17 | 0.75 | 0.12 | 6 | 0.83 | 0.75 | 0.79 | 18 | 505.72 |
| GOLEM | 0.00 | 1.00 | 0.00 | 0 | 1.00 | 1.00 | 1.00 | 20 | 21.89 |
| ICALiNGAM | 0.16 | 0.80 | 0.12 | 5 | 0.84 | 0.80 | 0.82 | 19 | 0.74 |
| DirectLiNGAM | 0.52 | 0.55 | 0.48 | 19 | 0.48 | 0.55 | 0.51 | 23 | 0.38 |
| DAGMA_linear | 0.00 | 1.00 | 0.00 | 0 | 1.00 | 1.00 | 1.00 | 20 | 4.59 |
| DAGMA_nonlinear | 0.15 | 0.55 | 0.08 | 10 | 0.85 | 0.55 | 0.67 | 13 | 508.95 |
| DAG_GNN | 0.05 | 1.00 | 0.04 | 1 | 0.95 | 1.00 | 0.98 | 21 | 522.86 |
| PC+DCC.γ | 0.17 | 0.50 | 0.08 | 10 | 0.71 | 0.50 | 0.59 | 12 | 296.28 |
| PC+Gauss | 0.13 | 0.65 | 0.08 | 8 | 0.72 | 0.65 | 0.68 | 15 | 0.20 |
| PC+GCM | 0.00 | 0.70 | 0.00 | 6 | 0.70 | 0.70 | 0.70 | 14 | 1515.40 |
| PC+KRESIT | 0.36 | 0.35 | 0.16 | 13 | 0.54 | 0.35 | 0.42 | 11 | 20432.41 |
Author response table 2
The results of 600 samples of 10 variables in nonlinear relationships.
| Methods | FDR | TPR | FPR | SHD | Precision | Recall | F1 | #Edges | Time |
|---|---|---|---|---|---|---|---|---|---|
| NOTEARS_linear | 0.00 | 0.30 | 0.00 | 14 | 1.00 | 0.30 | 0.46 | 6 | 3.18 |
| NOTEARS_nonlinear | 0.00 | 0.80 | 0.00 | 4 | 1.00 | 0.80 | 0.89 | 16 | 90.91 |
| GOLEM | 0.55 | 0.45 | 0.44 | 21 | 0.45 | 0.45 | 0.45 | 20 | 21.83 |
| ICALiNGAM | 0.33 | 0.30 | 0.12 | 17 | 0.67 | 0.30 | 0.41 | 9 | 0.76 |
| DirectLiNGAM | 0.14 | 0.30 | 0.04 | 14 | 0.86 | 0.30 | 0.44 | 7 | 0.40 |
| DAGMA_linear | 0.00 | 0.30 | 0.00 | 14 | 1.00 | 0.30 | 0.46 | 6 | 3.53 |
| DAGMA_nonlinear | 0.00 | 0.80 | 0.00 | 4 | 1.00 | 0.80 | 0.89 | 16 | 257.68 |
| DAG_GNN | 0.33 | 0.30 | 0.12 | 16 | 0.67 | 0.30 | 0.41 | 9 | 485.38 |
| PC+DCC.γ | 0.28 | 0.65 | 0.20 | 9 | 0.684 | 0.65 | 0.67 | 18 | 369.64 |
| PC+Gauss | 0.31 | 0.55 | 0.20 | 11 | 0.65 | 0.55 | 0.60 | 16 | 0.17 |
| PC+GCM | 0.14 | 0.60 | 0.08 | 9 | 0.86 | 0.60 | 0.70 | 14 | 1036.18 |
Author response table 3
The results of 600 samples of 10 variables in nonlinear relationships.
| Methods | FDR | TPR | FPR | SHD | Precision | Recall | F1 |
|---|---|---|---|---|---|---|---|
| NOTEARS_linear | - | - | - | - | - | - | - |
| NOTEARS_nonlinear | - | - | - | - | - | - | - |
| GOLEM | 0.97 | 0.09 | 0.03 | 127 | 0.029 | 0.010 | 0.015 |
| ICALiNGAM | - | - | - | - | - | - | - |
| DirectLiNGAM | - | - | - | - | - | - | - |
| DAGMA_linear | - | - | - | - | - | - | - |
| DAGMA_nonlinear | - | - | - | - | - | - | - |
| DAG_GNN | - | - | - | - | - | - | - |
| PC+DCC.γ | 0.92 | 0.15 | 0.07 | 162 | 0.085 | 0.071 | 0.077 |
| PC+Gauss | 0.94 | 0.31 | 0.36 | 451 | 0.057 | 0.232 | 0.091 |
Author response table 4
The results of 600 A549 cells, 5 enriched WP134 genes, and 2 of their transcription factors.
| Methods | FDR | TPR | FPR | SHD | Precision | Recall | F1 | #Edge | Time |
|---|---|---|---|---|---|---|---|---|---|
| NOTEARS_linear | - | - | - | - | - | - | - | - | 0.01 |
| NOTEARS_nonlinear | - | - | - | - | - | - | - | - | 0.61 |
| GOLEM | 0.83 | 0.08 | 0.56 | 11 | 0.17 | 0.08 | 0.11 | 6 | 19.53 |
| ICALiNGAM | 0.00 | 0.33 | 0.00 | 8 | 1.00 | 0.33 | 0.50 | 4 | 0.58 |
| DirectLiNGAM | 0.50 | 0.08 | 0.11 | 11 | 0.50 | 0.08 | 0.14 | 2 | 0.14 |
| DAGMA_linear | 0.00 | 0.17 | 0.00 | 10 | 1.00 | 0.17 | 0.29 | 2 | 3.17 |
| DAGMA_nonlinear | 0.56 | 0.33 | 0.56 | 10 | 0.44 | 0.33 | 0.38 | 9 | 374.06 |
| DAG_GNN | - | - | - | - | - | - | - | - | 546.36 |
| PC+DCC.γ | 0.40 | 0.50 | 0.44 | 8 | 0.50 | 0.50 | 0.50 | 10 | 234.71 |
| PC+Gauss | 0.38 | 0.42 | 0.33 | 8 | 0.63 | 0.42 | 0.50 | 8 | 0.14 |
| PC+GCM | 0.45 | 0.50 | 0.56 | 7 | 0.50 | 0.50 | 0.50 | 11 | 1056.60 |
Author response table 6
The results of 600 samples of 10 variables in nonlinear relationships, with 20% missing values.
| Methods | FDR | TPR | FPR | SHD | Precision | Recall | F1 | #Edges | Time |
|---|---|---|---|---|---|---|---|---|---|
| NOTEARS_linear | 0.20 | 0.2 | 0.04 | 17 | 0.80 | 0.2 | 0.32 | 5 | 1.66 |
| NOTEARS_nonlinear | 0.60 | 0.8 | 0.96 | 25 | 0.40 | 0.8 | 0.53 | 40 | 77.95 |
| GOLEM | 0.71 | 0.3 | 0.6 | 24 | 0.29 | 0.3 | 0.29 | 21 | 14.98 |
| ICALiNGAM | 0.50 | 0.1 | 0.08 | 20 | 0.50 | 0.1 | 0.17 | 4 | 0.07 |
| DirectLiNGAM | 0.25 | 0.15 | 0.04 | 18 | 0.75 | 0.15 | 0.25 | 4 | 0.23 |
| DAGMA_linear | 0.43 | 0.2 | 0.12 | 19 | 0.57 | 0.2 | 0.30 | 7 | 1.91 |
| DAGMA_nonlinear | - | - | - | - | - | - | - | 0 | 266.44 |
| DAG_GNN | 0.60 | 0.1 | 0.12 | 20 | 0.40 | 0.1 | 0.16 | 5 | 182.71 |
| PC_dcc_γ | 0.44 | 0.25 | 0.16 | 18 | 0.45 | 0.25 | 0.32 | 11 | 49.63 |
| PC_gauss | 0.70 | 0.15 | 0.28 | 19 | 0.27 | 0.15 | 0.19 | 11 | 0.07 |
| PC_GCM | 0.00 | 0.25 | 0 | 15 | 0.71 | 0.25 | 0.37 | 7 | 48.18 |
Author response table 5
The results of 600 samples of 10 variables in nonlinear relationships, with 10% missing values.
| Methods | FDR | TPR | FPR | SHD | Precision | Recall | F1 | #Edges | Time |
|---|---|---|---|---|---|---|---|---|---|
| NOTEARS_linear | 0.4 | 0.15 | 0.08 | 19 | 0.6 | 0.15 | 0.24 | 5 | 1.60 |
| NOTEARS_nonlinear | 0.56 | 0.8 | 0.8 | 21 | 0.44 | 0.8 | 0.57 | 36 | 97.66 |
| GOLEM | 0.52 | 0.55 | 0.48 | 19 | 0.48 | 0.55 | 0.51 | 23 | 14.75 |
| ICALiNGAM | 0.25 | 0.15 | 0.04 | 18 | 0.75 | 0.15 | 0.25 | 4 | 0.04 |
| DirectLiNGAM | 0.4 | 0.15 | 0.08 | 19 | 0.6 | 0.15 | 0.24 | 5 | 0.23 |
| DAGMA_linear | 0.43 | 0.2 | 0.12 | 19 | 0.57 | 0.2 | 0.30 | 7 | 1.88 |
| DAGMA_nonlinear | 0 | 0.15 | 0 | 17 | 1 | 0.15 | 0.26 | 3 | 277.04 |
| DAG_GNN | 0.67 | 0.15 | 0.24 | 23 | 0.33 | 0.15 | 0.21 | 9 | 215.29 |
| PC_dcc_γ | 0.22 | 0.35 | 0.08 | 14 | 0.64 | 0.35 | 0.45 | 11 | 82.42 |
| PC_gauss | 0.5 | 0.3 | 0.24 | 18 | 0.5 | 0.3 | 0.38 | 12 | 0.08 |
| PC_GCM | 0.29 | 0.25 | 0.08 | 17 | 0.5 | 0.25 | 0.33 | 10 | 68.43 |
Additional files
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Applying causal discovery to single-cell analyses using CausalCell
eLife 12:e81464.
https://doi.org/10.7554/eLife.81464




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}