Deep learning-driven insights into super protein complexes for outer membrane protein biogenesis in bacteria
- Center for the Study of Systems Biology, School of Biological Sciences, Georgia Institute of Technology, United States
- School of Computer Science, Georgia Institute of Technology, United States
Figures
Figure 1 with 1 supplement
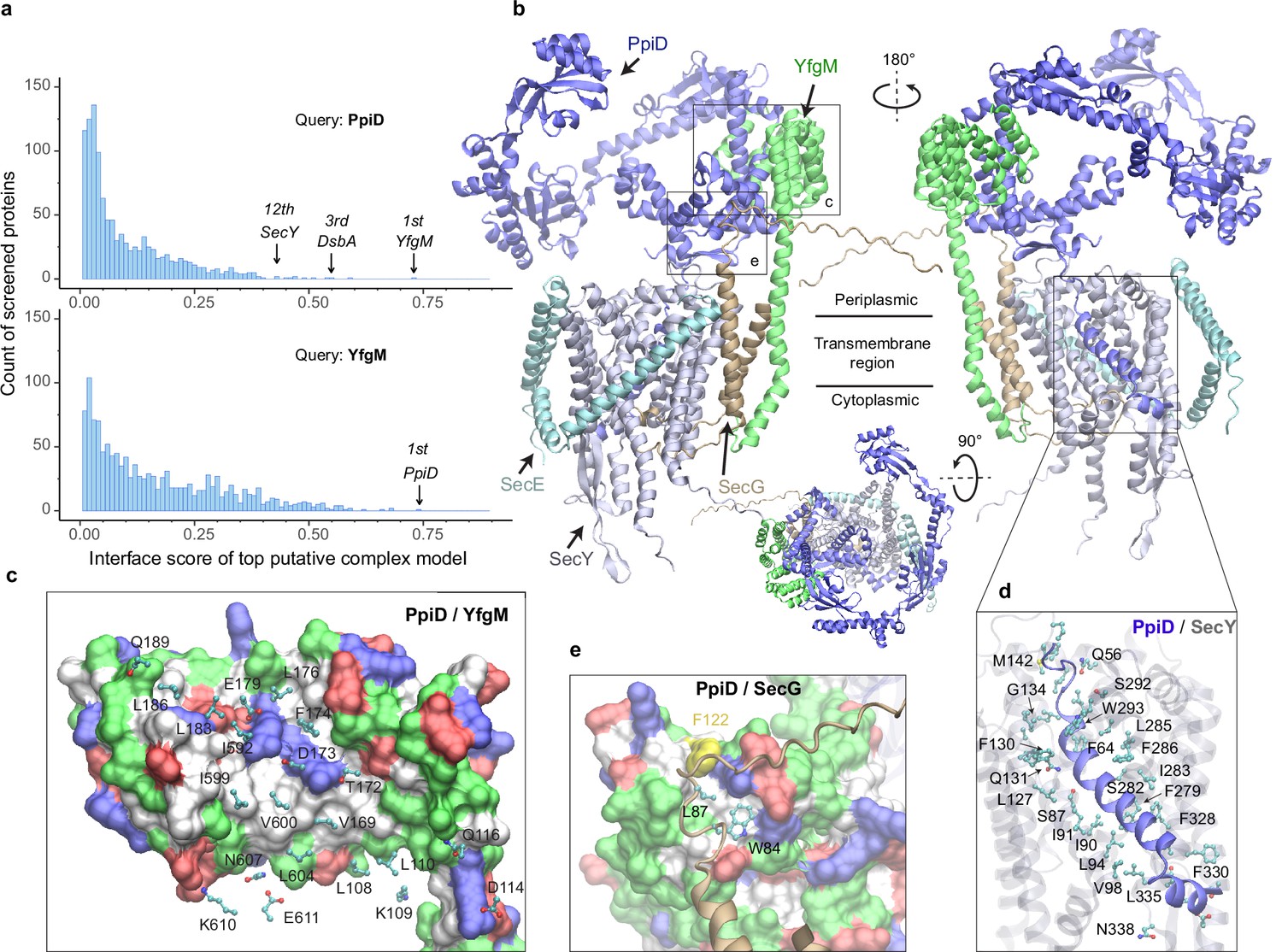
E. coli super-translocon SecYEG/PpiD/YfgM.
(a) Computational screening for protein-protein interaction partners of PpiD and YfgM within the E. coli envelopome, respectively. A histogram displays the distribution of the top interface scores (iScores) of all envelope proteins screened with each query. Black arrows mark the top hits that were further studied, along with their names and overall ranks. (b) The top AF2Complex model of a supercomplex made of PpiD (blue), YfgM (green), SecY (silver), SecE (cyan), and SecG (tan) in three different views. Proteins are shown in a cartoon representation. Viewpoint transition, from either left to right or top to bottom, is indicated by a rotation axis (dashed line) and the rotation angle in degrees (circled arrow). (c, d, and e) Predicted PPI sites. The corresponding locations in b are indicated by black boxes. For clarity, the viewpoints and representations are adjusted. In the surface representations c and e, the color code is hydrophobic (white), polar (green), positive (blue), and negative (red), except for Phe122PpiD (yellow) in e. The same color code for the surface representation is employed below unless noted otherwise. PPI residues are shown in a ball-and-stick representation for PpiD in c, SecY in d, and SecG in e; the color scheme of atoms is carbon (cyan), oxygen (red), nitrogen (blue), and sulfur (yellow). The same scheme of atoms is adopted throughout this work.
Figure 1—figure supplement 1

Comparison of a computed structure of SecY (silver) and two experimental structures (magenta).
(a) Superposition of an X-ray crystal structure of SecY from G. thermodenitrificans (PDB code: 5EUL) onto SecY from the predicted structure of the E. coli SecYEG/PpiD/YfgM supercomplex. In the experimental structure, SecY was co-crystalized with the signal peptide (green) of a precursor OmpA. In the computed structure, the N-terminal transmembrane α-helix (blue) of PpiD is also displayed. All structures are shown in the cartoon representation. (b) Superposition of an EM structure of SecY from E. coli (PDB code: 5GAE, chain g) onto the computed model. For clarity, the α-helix of PpiD was omitted.
Figure 2
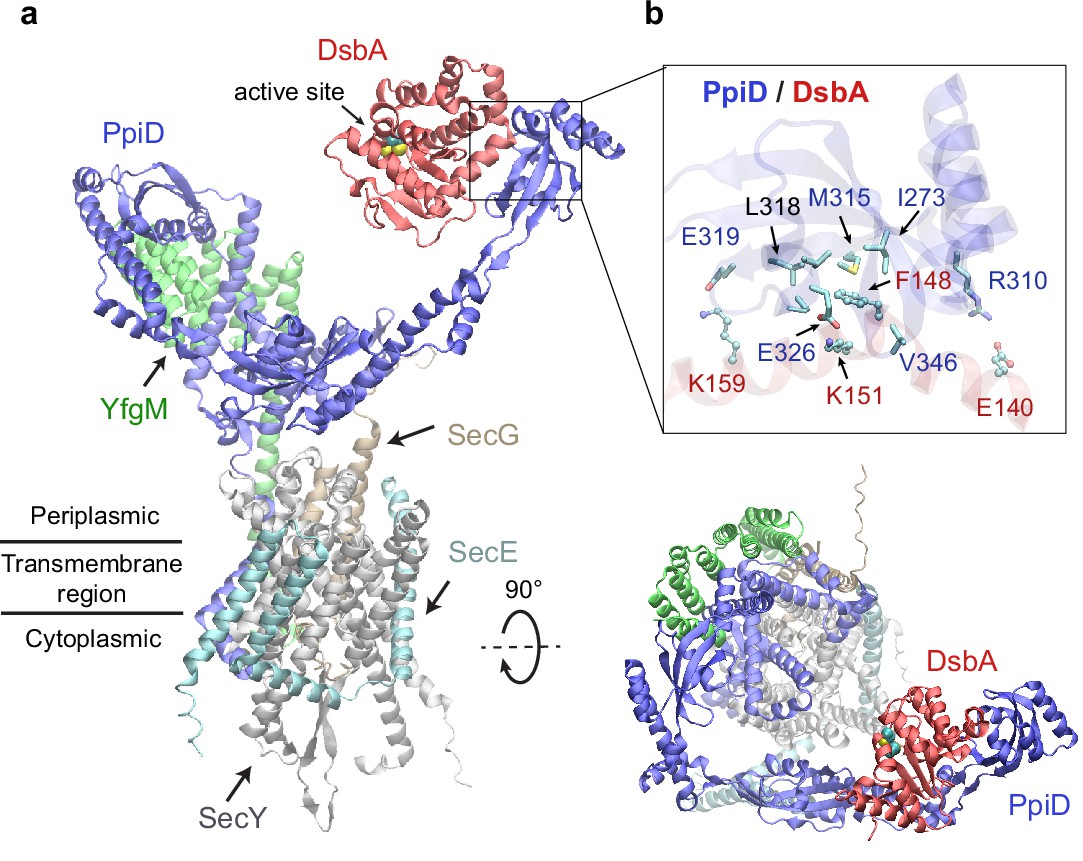
Structural model of the SecYEG/PpiD/YfgM/DsbA supercomplex.
(a) Two views of the predicted structure. DsbA is shown in red, while the other proteins are colored the same as in Figure 1. Two cysteines, Cys49 and Cys52, essential to the enzymatic function of DsbA, are shown as spheres. (b) Protein-protein interaction sites between PpiD and DsbA. For clarity, tertiary structures are transparent. Key interacting residues are shown in the licorice representation for PpiD and in the ball-and-stick representation for DsbA.
Figure 3

Predicted structure of the PpiD/YfgM/LepB/OmpA supercomplex.
(a) Two views in the cartoon representation are shown. Colors: PpiD (blue), YfgM (green), LepB (magenta), and OmpA (residue 1–87, yellow). For clarity, representations of PpiD and LepB are transparent. (b) Close-up view of the OmpA signal peptide in the active site of LepB. Essential catalytic residues, Ser89, Ser91, Lys146, and Ser279 of LepB are shown in a licorice representation, and the cleavage site Ala21 and Ala19 of OmpA is shown as spheres.
Figure 4 with 1 supplement
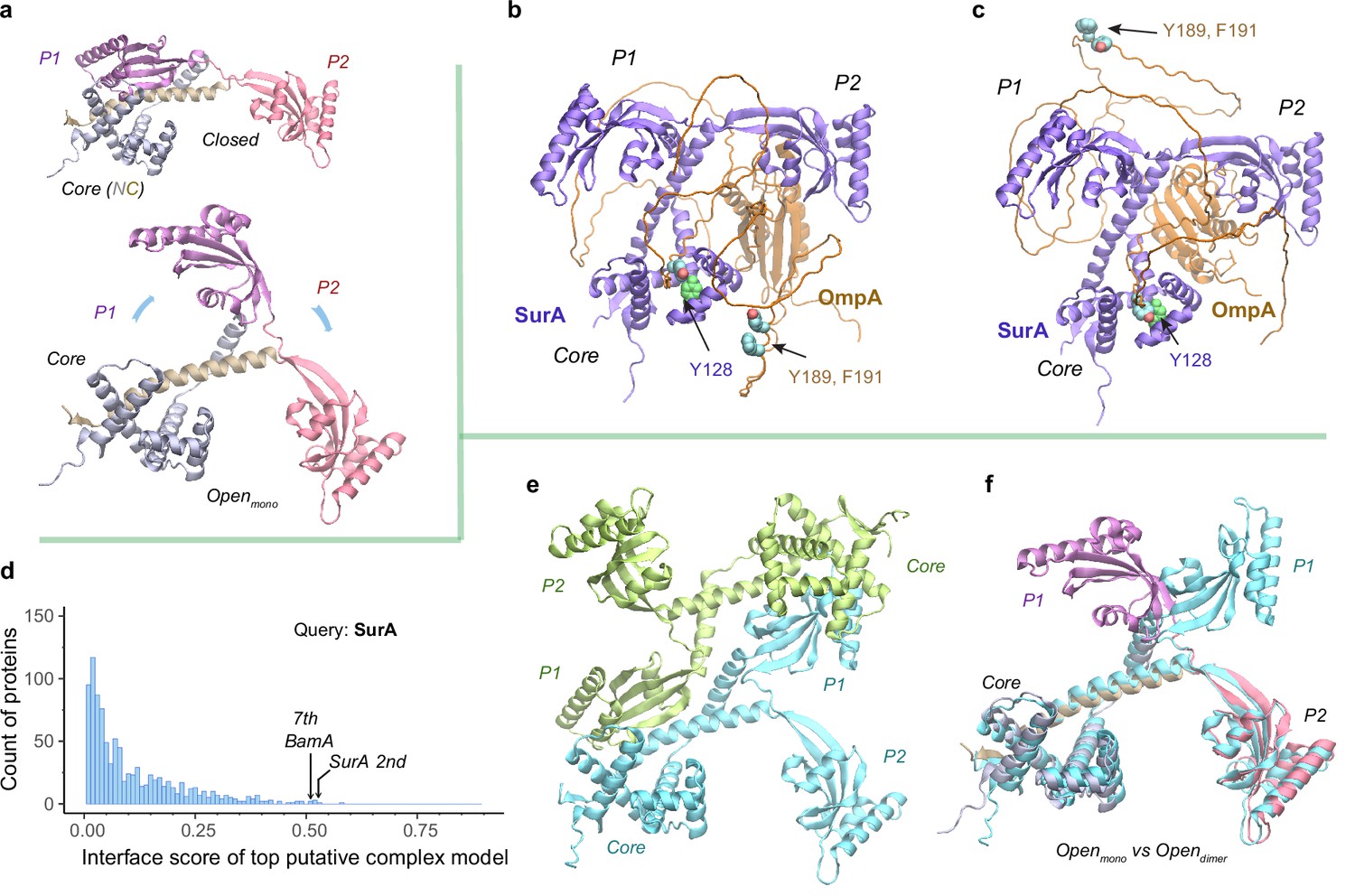
Structural models of SurA in the absence and presence of an OmpA substrate.
(a) Open and closed conformations of monomeric SurA, consisting of the core domain (N-terminal region in gray and C-terminal in tan), P1 (purple), P2 (red). (b and c) Two structures of SurA in the presence of an OmpA substrate from two separate modeling runs. In both, SurA is open as in a. The β-barrel domain of OmpA is completely unfolded and generally does not maintain the same residue-residue contacts with SurA, except that an OmpA aromatic residue consistently makes π-π interactions with Tyr128SurA located in the crevice of the SurA core domain. Two β-signal residues, Y189 and F191 of OmpA, are also shown as spheres. The folded periplasmic domain of OmpA is bound to the P2 domain of SurA. (d) Envelopome protein-protein interaction screening of SurA identifies itself and BamA among the top hits. (e) Two views of the top predicted structure of a SurA dimer (green and cyan). (f) Superimposition of two open conformations from the monomeric and dimeric SurA. Subscripts indicate the stoichiometry. The color schemes correspond to those used in a and e. Only a single SurA from the dimeric model is shown in the superposition.
Figure 4—figure supplement 1
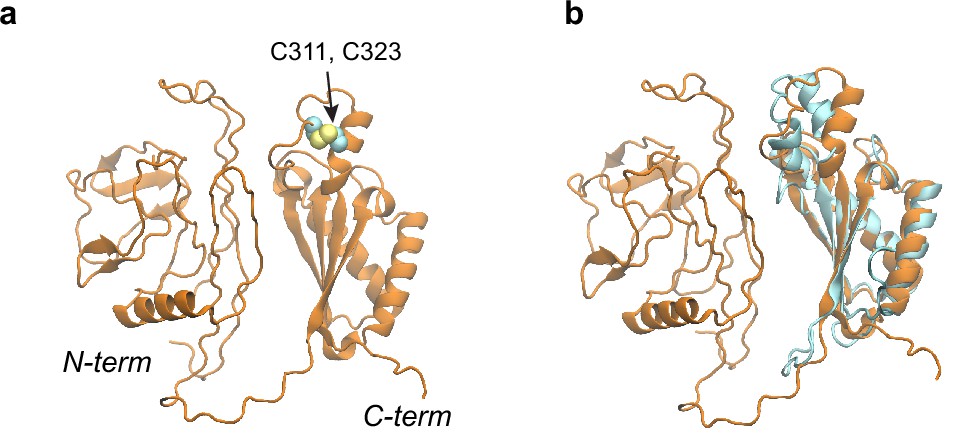
Structural models of the OmpA polypeptide in the absence of SurA.
(a) Predicted model of OmpA obtained with a shallow multiple sequence alignments (MSAs) and no structural templates. The N-terminal β-barrel domain is collapsed but not in its native fold; the C-terminal periplasmic domain is native-like. Two cysteines, Cys311 and Cys323, forming a disulfide bond are shown as spheres. (b) The predicted model aligned to an nuclear magnetic resonance (NMR) structure of the periplasmic domain (PDB code: 2MQE, TM-score [Zhang and Skolnick, 2004] ~0.75).
Figure 5
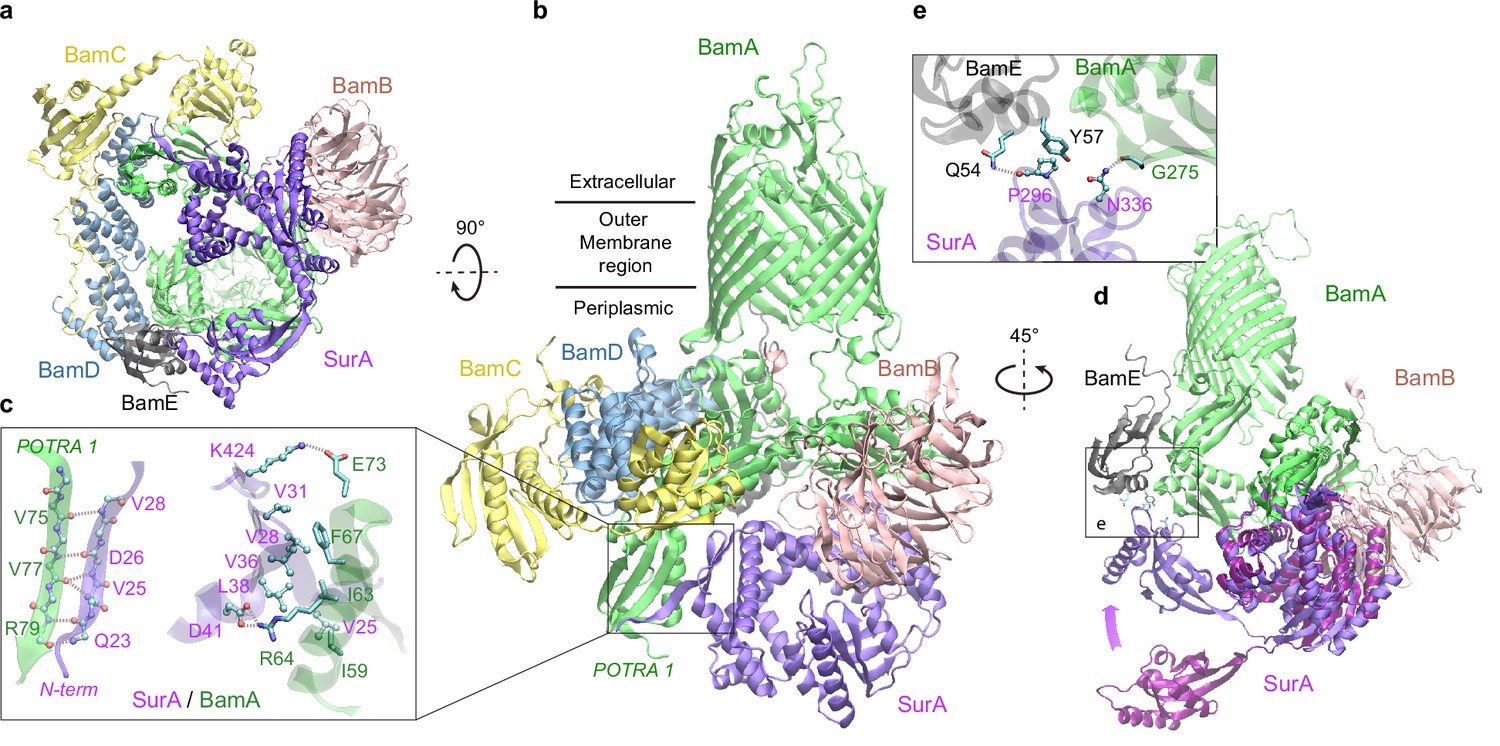
Structural model of SurA docked to β-barrel assembly machine (BAM).
(a and b) Two views of the top ranked supercomplex model in the cartoon representation. The BAM constituents are BamA (green), BamB (pink), BamC (yellow), BamD (blue), and BamE (black). The N-terminal POTRA1 domain of BamA provides the main interaction sites for SurA (violet). (c) Close-up view of the interaction sites at POTRA1 and the core domain of SurA. Interacting residues are shown in the licorice (SurA) and ball-and-stick (BamA) representations. (d) Crystal structure of SurA (magenta, PDB 1M5Y) superimposed onto the computed structure of the supercomplex. The magenta arrow indicates the change of location in P2 between two structures. BamCD are omitted for clarity. (e) Protein-protein interaction between P2 of SurA and POTRA4 of BamA and BamE.
Figure 6 with 1 supplement

Structural models of β-barrel assembly machine (BAM) and BepA.
(a) Computational protein-protein interaction screening identifies BepA as a top hit to BamA. (b) Top structural model of the heterodimeric complex of BamA (green) and BepA (purple) in cartoon representation. The lid of BepA is colored red. The active sites with the protease domain of BepA are shown in a surface representation (orange). The five POTRA domains of BamA are labeled P1−P5. (c) Predicted structure of the BAM/BepA supercomplex. The lid of BepA extends to an open conformation. The image was created from the same viewpoint as b. (d) Specific residue-residue contacts between BamA and the tetratricopeptide repeat (TPR) and protease domains of BepA. (e) Close-up views of the lid and the BamA β-barrel in the surface (top) and cartoon representations (bottom). A hydrophobic contact between Ala180BepA and Leu780BamA is shown as spheres, and the lateral gate of BamA is between the β1 and β16 strands (dark blue).
Figure 6—figure supplement 1
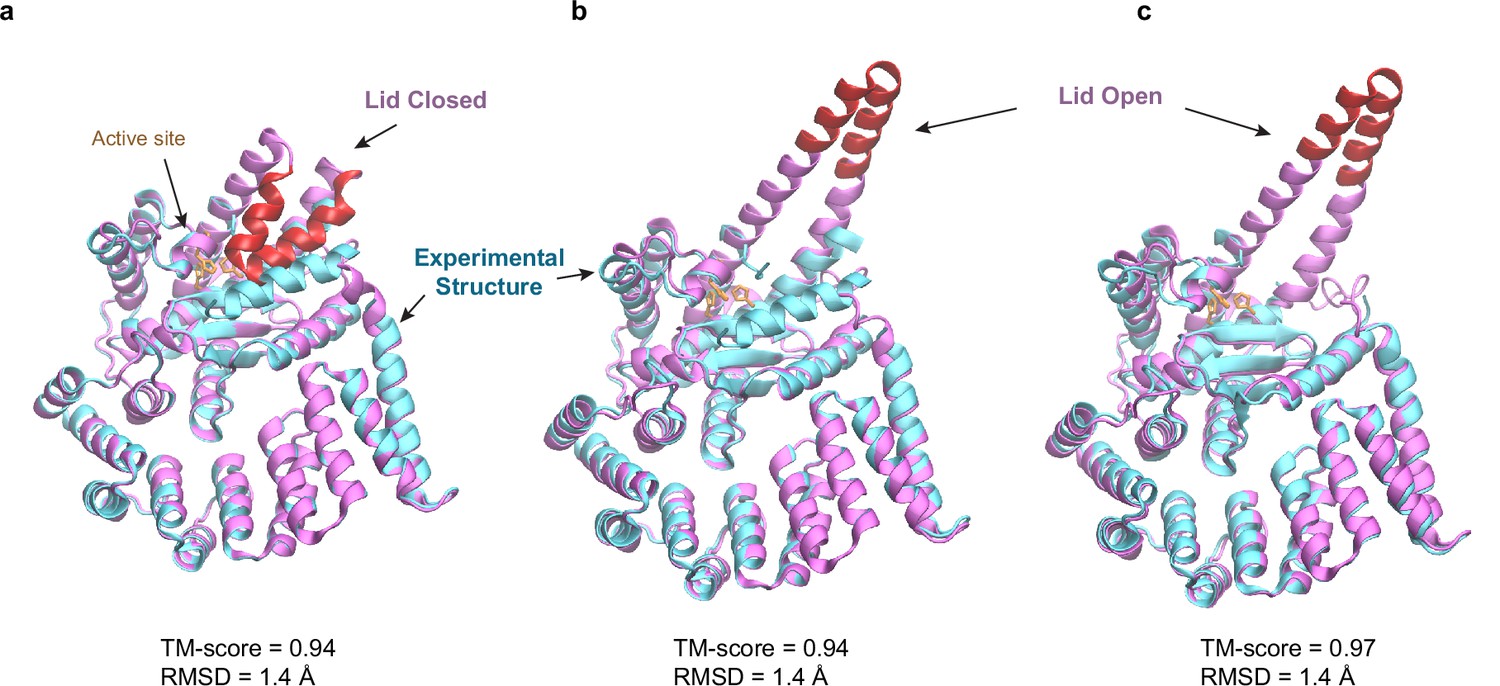
Predicted structures of BepA compared to two experimental structures.
(a and b) Superposition of structural models of the lid (red/magenta) in closed and open states, respectively, onto a crystal structure (PDB code: 6AIT, cyan). (c) Superposition onto another crystal structure (PDB code: 6ASR, cyan).
Figure 7
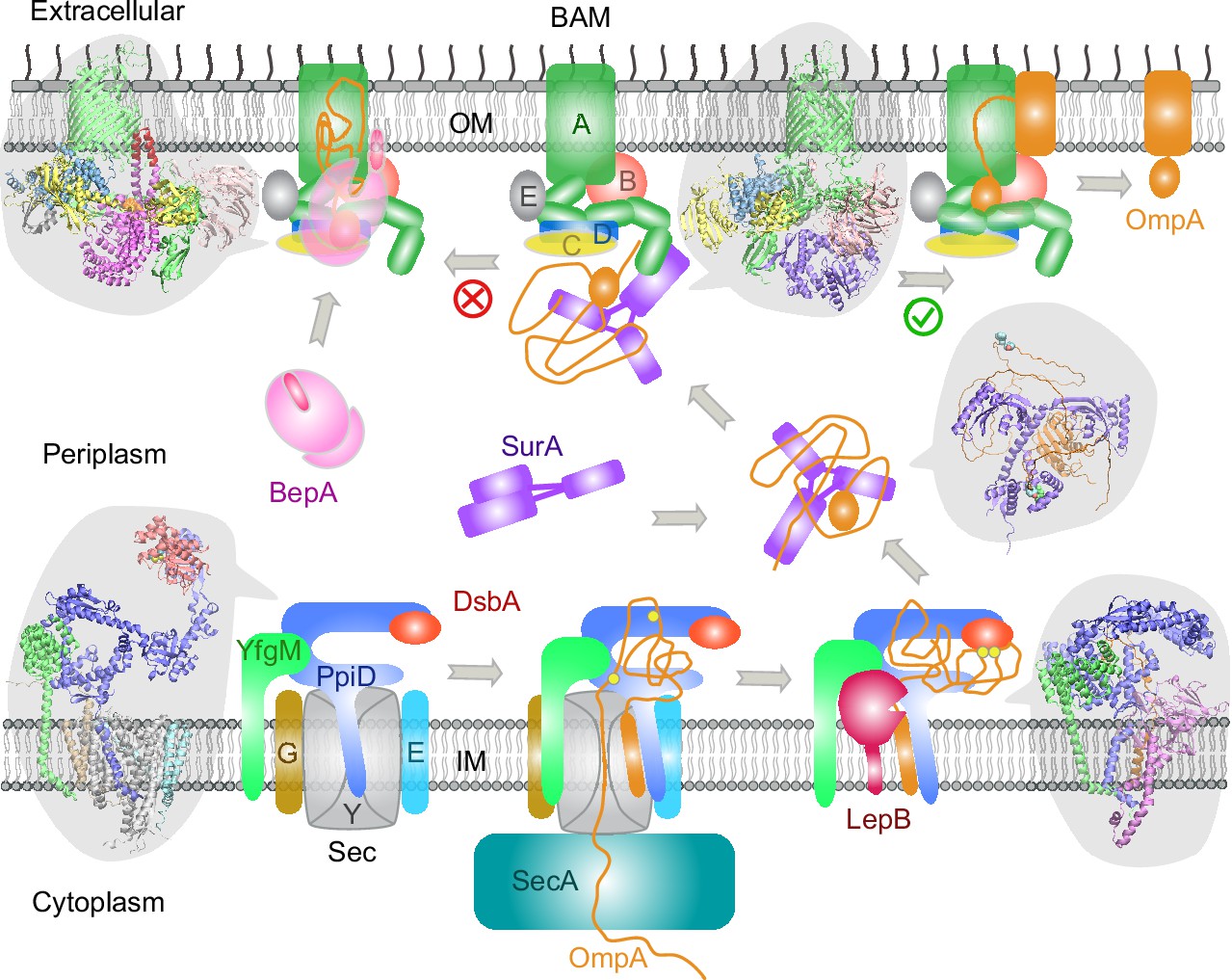
Proposed mechanisms involved in the outer membrane protein (OMP) biogenesis pathway in E. coli.
Complex structures resulting from this study accompany relevant cartoon diagrams. Powered by SecA, a precursor OmpA polypeptide (orange line) first passes through the SecYEG translocon in complex with PpiD and YfgM. PpiD, held in place by YfgM, senses the translocating substrate via its N-terminal α-helix bound to the lateral gate of SecY and temporarily dissociates from the translocon upon receiving the substrate OmpA. Protein disulfide isomerase DsbA is recruited by PpiD and promotes formation of a disulfide bond between two cysteine residues (yellow spheres) of OmpA. Meanwhile, peptidase LepB fills the vacancy left by SecYEG and cleaves the transmembrane signal peptide from OmpA, which is then handed over to chaperone SurA. At this point, the periplasmic domain of OmpA is folded, but the unfolded β-barrel region wraps around SurA, which carries OmpA to BAM. Lastly, SurA docks to POTRA1, the N-terminal domain of BamA, where the β-barrel domain of OmpA is folded and released from the lateral gate of BamA. If this folding and assembly process are stalled for some reason, metalloprotease BepA senses the failure with its flexible lid and cleans up by cleaving a stuck substrate. For clarity, the peptidoglycan layer in the periplasm is not shown, and the schematic drawings are not to scale.
Additional files
-
Supplementary file 1
Top hits from the protein-protein interaction (PPI) screening over the E. coli envelopome with AF2Complex for query proteins: PpiD, YfgM, SurA, and BamA.
- https://cdn.elifesciences.org/articles/82885/elife-82885-supp1-v1.xlsx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/82885/elife-82885-mdarchecklist1-v1.pdf
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Deep learning-driven insights into super protein complexes for outer membrane protein biogenesis in bacteria
eLife 11:e82885.
https://doi.org/10.7554/eLife.82885




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}