Longitudinal map of transcriptome changes in the Lyme pathogen Borrelia burgdorferi during tick-borne transmission
- Department of Biochemistry & Biophysics, University of California, San Francisco, United States
- Department of Pharmaceutical Chemistry and Cardiovascular Research Institute, University of California, San Francisco, United States
- Division of Biological Sciences, University of Montana, United States
eLife assessment
In this Tools and Resources article, the authors overcome the challenge of low Borrelia burgdorferi numbers during infection for analyses such as RNA-sequencing or mass spectrometry. They do so by physically enriching for spirochetes, which is important, as it provides technical advances for the study of global transcriptomic changes of B. burgdorferi during tick feeding, helping to build on the knowledge already collected by the field. The evidence presented is compelling, and the strategy described here could benefit researchers in the field and possibly also support broader applications.
https://doi.org/10.7554/eLife.86636.3.sa0Significance of the findings:
Important: Findings that have theoretical or practical implications beyond a single subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Compelling: Evidence that features methods, data and analyses more rigorous than the current state-of-the-art
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
Borrelia burgdorferi (Bb), the causative agent of Lyme disease, adapts to vastly different environments as it cycles between tick vector and vertebrate host. During a tick bloodmeal, Bb alters its gene expression to prepare for vertebrate infection; however, the full range of transcriptional changes that occur over several days inside of the tick are technically challenging to capture. We developed an experimental approach to enrich Bb cells to longitudinally define their global transcriptomic landscape inside nymphal Ixodes scapularis ticks during a transmitting bloodmeal. We identified 192 Bb genes that substantially change expression over the course of the bloodmeal from 1 to 4 days after host attachment. The majority of upregulated genes encode proteins found at the cell envelope or proteins of unknown function, including 45 outer surface lipoproteins embedded in the unusual protein-rich coat of Bb. As these proteins may facilitate Bb interactions with the host, we utilized mass spectrometry to identify candidate tick proteins that physically associate with Bb. The Bb enrichment methodology along with the ex vivo Bb transcriptomes and candidate tick interacting proteins presented here provide a resource to facilitate investigations into key determinants of Bb priming and transmission during the tick stage of its unique transmission cycle.
Introduction
Vector-borne microbial pathogens are transmitted by the bite of arthropods and have evolved sophisticated ways to adapt to vastly different environments as they move between vector and host. Uncovering their adaptive mechanisms can not only open avenues for disrupting pathogen transmission but also provide fundamental insights into vector physiology and microbial symbioses (Shaw and Catteruccia, 2019). Lyme disease, the most reported vector-borne disease in North America, is caused by the bacterial pathogen Borrelia burgdorferi (Bb) (Rosenberg et al., 2018; Steere et al., 2016). Its primary vector, the blacklegged tick Ixodes scapularis, acquires and transmits Bb through two separate multi-day bloodmeals, one in which Bb is acquired from an infected vertebrate host and a second, during the subsequent life stage, in which Bb is transmitted to a new host (Tilly et al., 2008). During the transmission bloodmeal, Bb proliferates in the tick midgut before a subset of these cells disseminate to the salivary glands (Dunham-Ems et al., 2009). Bb is deposited into the new host via the tick saliva extruded into the bite site (Ribeiro et al., 1987). Given the prolonged nature of I. scapularis feeding, the high specificity of vector-pathogen relationships, and the complicated array of events needed for successful Bb transmission, the tick bloodmeal provides an opportune intervention point for preventing pathogen spread. However, we do not currently have a clear understanding of the molecular mechanisms involved in this process.
Bb must adapt to dramatically different environments as it cycles from tick to vertebrate host, and understanding the genes involved in this process will enable the identification of key interactions to target to prevent transmission. When an infected tick feeds, Bb responds to bloodmeal-induced environmental changes and undergoes cellular modifications driven by key transcriptional circuits, including the RpoN/RpoS sigma factor cascade and the Hk1/Rrp1 two-component system (Radolf et al., 2012). In vitro analyses of Bb cells cultured in tick- or mammal-like growth conditions have pointed to additional genetic determinants of tick-borne transmission by revealing widespread transcriptome remodeling during host switching (reviewed in Samuels et al., 2021). However, it is not fully clear how these in vitro expression changes correspond to complex in vivo changes over the course of a transmission bloodmeal. Capturing comprehensive, longitudinal data on Bb gene expression from inside its vector has been hampered by technical challenges due to the dynamic nature of the bloodmeal and the general low abundance of bacterial cells relative to the tick (Samuels et al., 2021). Some progress has been made in making transcriptome-wide measurements from the tick. Bb sequence enrichment coupled with microarrays identified large scale changes in Bb gene expression between the first and second tick bloodmeals (Iyer et al., 2015). More recently, enriching Bb sequences from infected tick RNA-seq libraries through TBDCapSeq has provided clearer resolution into differences in Bb gene expression as it cycles between the fed tick and mammalian host (Grassmann et al., 2023). Still, we have limited temporal resolution into the molecular transitions that happen across key steps of tick feeding. This problem necessitates novel approaches to capture the transcriptomic changes of Bb within the natural tick environment.
While the full landscape of Bb transmission determinants is not yet known, we do have a growing knowledge of functional processes that are critical during the tick stage, such as motility, metabolism, and immune evasion (Kurokawa et al., 2020; Phelan et al., 2019). These functions often rely on the unique protein-rich Bb outer surface. Notably, several specific tick–Bb protein–protein interactions are important for Bb survival, migration, or transmission to the next host. Bb encodes an extensive outer surface protein (Osp) family with members that are differentially expressed during host switching. One of these proteins, OspA, binds a tick cell surface protein, tick receptor for OspA (TROSPA), which is required for successful Bb colonization of the tick midgut during the first acquisition bloodmeal (Pal et al., 2004). Several other proteins have also been linked to Bb migration within the tick (Pal et al., 2021). For example, BBE31 binds a tick protein TRE31, and disruption of this interaction decreases the number of Bb cells that successfully migrate from the tick gut to salivary glands (Zhang et al., 2011). However, these interactions alone are not sufficient to block Bb growth or migration in ticks, suggesting there are likely additional molecular factors from Bb and ticks at play during tick-borne transmission.
To provide a more comprehensive set of Bb determinants driving tick-borne transmission of this important human pathogen, we developed a novel sequencing-based strategy for ex vivo transcriptomic profiling of Bb populations within infected nymphal I. scapularis ticks as they transmit Bb to a mouse host. We used this method to longitudinally map genome-wide Bb expression changes for bacterial cells isolated from ticks during the transmission bloodmeal from 1 to 4 days after attachment. We identified 192 highly differentially expressed genes, including genes previously implicated in Bb transmission as well as many others. Genes upregulated during tick transmission included many outer surface lipoproteins, suggesting Bb dramatically remodels its cell envelope as it migrates through the tick. Mass spectrometry analyses revealed dramatic changes in the tick environment over feeding, identifying new potential determinants of a more extensive and diverse set of tick–microbe molecular interactions than previously appreciated. The Bb enrichment method and resulting datasets serve as a community resource to facilitate further investigations into the key determinants of Bb transmission.
Results
A two-step enrichment process facilitates robust transcriptional profiling of Bb during the tick bloodmeal
To gain a more comprehensive understanding of Bb gene expression throughout the tick phase of the transmission cycle, we developed an experimental approach to characterize the Bb transcriptome of spirochetes isolated from nymphal I. scapularis ticks during a days-long bloodmeal in which Bb is transmitted to a vertebrate host. We aimed to establish a longitudinal transcriptional profile encompassing key pathogen transmission events each day of feeding after ticks attached to their mouse bloodmeal hosts (Figure 1A). We fed Bb-infected nymphal ticks on naive mice and collected the feeding ticks at daily intervals after the start of feeding until 4 days after attachment, at which time the ticks had fully engorged and detached from the mice. The major bottleneck for such an RNA sequencing (RNA-seq) approach is capturing sufficient quantities of Bb transcripts from complex multi-organism samples in which pathogen transcripts represent a very small minority. Our initial attempts to uncover Bb mRNA by simply removing tick mRNA with polyA-depletion and removing tick rRNA sequences using Depletion of Abundant Sequences by Hybridization (DASH; Dynerman et al., 2020; Gu et al., 2016) were unsuccessful. This approach resulted in an average of only 0.09% of RNA-seq reads mapping to Bb mRNA – approximately 10-fold less than we estimated would be needed to feasibly obtain robust transcriptome-wide differential gene expression analysis (Haas et al., 2012).
Figure 1 with 2 supplements see all
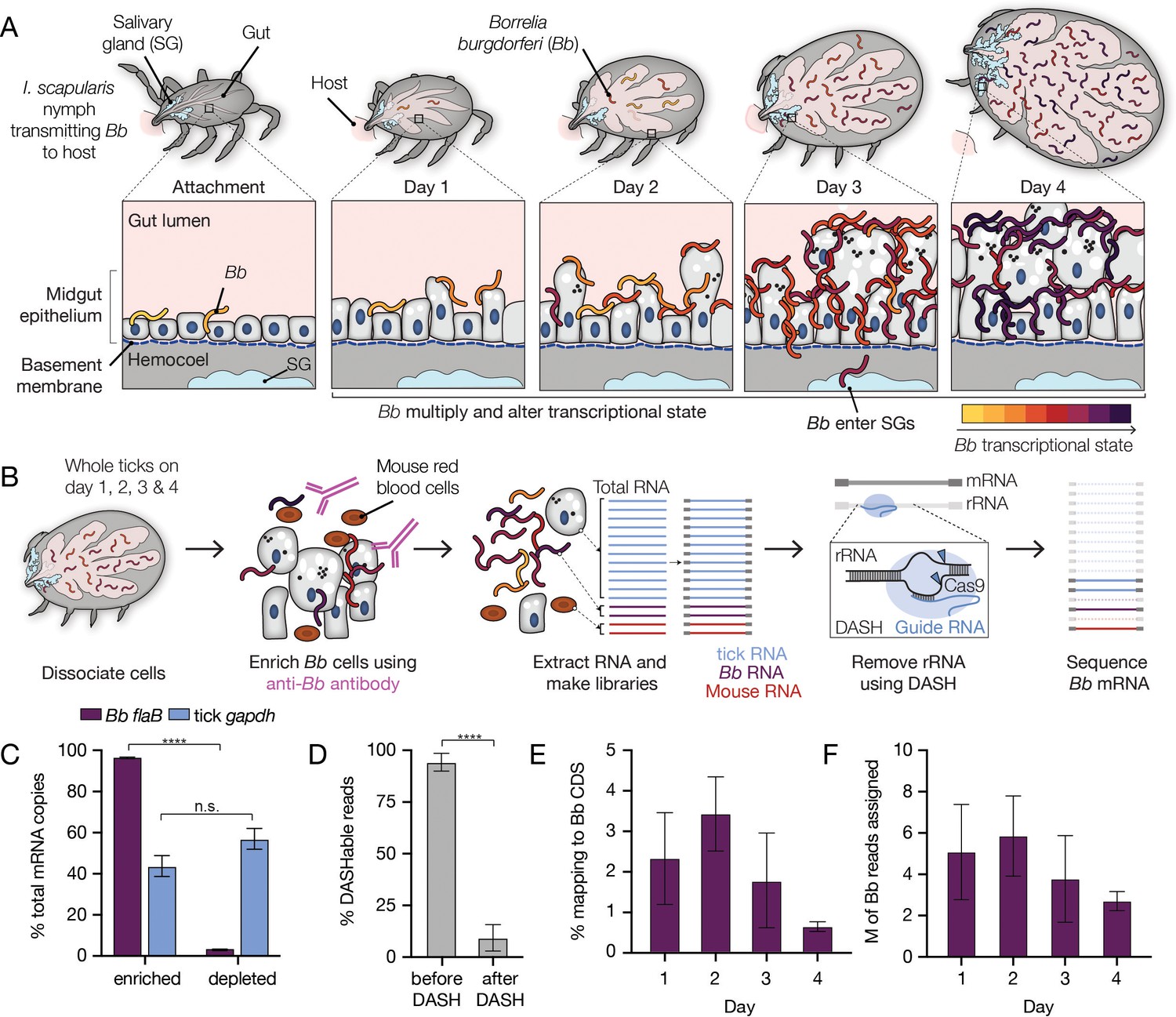
A two-step enrichment process facilitates robust transcriptional profiling of Bb during the tick bloodmeal.
(A) Schematic of Bb during nymphal I. scapularis feeding. Bb in the nymphal tick midgut respond to the nutrient-rich bloodmeal by multiplying and changing their transcriptional state (Ouyang et al., 2012; de Silva and Fikrig, 1995). At the same time, the tick gut undergoes numerous changes to digest the bloodmeal (Caimano et al., 2015; Sonenshine and Anderson, 2014). After two to three days of feeding, a small number of Bb leave the midgut and enter the salivary glands (blue), while the majority are left behind in the gut after engorgement (Dunham-Ems et al., 2009). (B) Schematic of Bb enrichment process from feeding ticks. Whole ticks are dissociated, αBb antibodies are added to lysates, and antibodies and Bb are captured magnetically. RNA is extracted and RNA-seq libraries are prepared. DASH is then used to remove rRNA before sequencing. This process increases Bb reads in the resulting sequencing data. (C) RT-qPCR results showing the percentage of Bb flaB and I. scapularis gapdh RNA in the enriched versus depleted fractions after the enrichment process. Data come from 4 replicates each from day 2, day 3, and day 4, mean +/-SE. ****p-value <0.0001, paired t test. Nearly all Bb flaB RNA was found in the enriched fraction. (D) The percentage of reads mapping to rRNA before and after DASH. n=4. Data are shown as mean +/-SD. ****p-value <0.0001, paired t test. rRNA reads are drastically reduced after DASH. (E) The percentage of reads in RNA-seq libraries mapping to Bb. Bb mRNA reads make up a larger proportion of libraries than without enrichment. n=4. Data are shown as mean +/-SD, see Figure 1—source data 1. (F) The number of reads in millions (M) mapped to Bb for each day. n=4. Data are shown as mean +/-SD. An average of 4.3 million reads per sample mapped to Bb genes, covering 92% of annotated genes with at least 10 reads.
-
Figure 1—source data 1
Overview of mapping statistics from 16 Bb sequencing samples.
- https://cdn.elifesciences.org/articles/86636/elife-86636-fig1-data1-v1.xlsx
To dramatically increase Bb transcript representation in our libraries, we physically enriched Bb cells from tick lysates prior to library preparation by adding an initial step of immunomagnetic separation (Figure 1B). We took advantage of a commercial antibody previously generated against whole Bb cells (αBb, RRID: AB_1016668). By western blot analysis, we confirmed that αBb specifically recognized several Bb proteins, including surface protein OspA (Figure 1—figure supplement 1A), which is highly prevalent on the Bb surface in the tick (Ohnishi et al., 2001). In addition, immunofluorescence microscopy with αBb showed clear recognition of Bb cells from within the tick at each day of feeding (Figure 1—figure supplement 1B). After collecting infected nymphal ticks from mice one, two, three, and four days post-attachment, we used αBb and magnetic beads to enrich Bb cells from the tick material in the lysates. We tracked relative Bb enrichment through RT-qPCR of Bb flaB RNA and tick gapdh RNA in the separated samples. Measuring Bb flaB RNA from both Bb-enriched samples and their matched Bb-depleted fractions, we found over 95% of total Bb flaB RNA was present in enriched fractions (Figure 1C), suggesting our approach captured the vast majority of Bb transcripts from the tick.
Total RNA recovered from the enrichment process was used to create RNA-seq libraries that subsequently underwent depletion of highly expressed rRNA sequences from tick, mouse, and Bb using DASH, which targets unwanted sequences for degradation by Cas9 (Gu et al., 2016; Ring et al., 2022). DASH reduced unwanted sequences from 94% to 9% of our total libraries, greatly increasing the relative abundance of Bb transcripts (Figure 1D). For resulting libraries generated across feeding, between 0.6% and 3.4% of reads mapped to annotated Bb genic regions (Figure 1E), which translated to an average of 4.3 million Bb genic reads per sample (Figure 1F and Figure 1—source data 1).
As expected, for samples pulled from the mice one, two, and three days after attachment, the majority of the remaining sequencing reads mapped to the I. scapularis genome (72–83%; Figure 1—source data 1). On day 4, when ticks were fully engorged and were recovered from mouse cages rather than pulled from the mice, we found that fewer reads mapped to the I. scapularis genome (24–44%). Only a small percentage of reads from all samples mapped to the host Mus musculus genome (1–3%). To identify the source of the remaining reads in the day 4 samples, we ran the data through a publicly available computational pipeline that identifies microbes in sequencing datasets, CZ ID (Kalantar et al., 2020). This analysis led us to discover that a large percentage of day 4 reads mapped to bacterial species Pseudomonas fulva (41–64%) (Figure 1—source data 1), which may have been present in our mouse cages. While these samples had a lower percentage of reads that mapped to Bb, broad transcriptome coverage was still obtained by increasing total sequencing depth. Across all samples from the 4 days, at least 10 reads mapped to 92% of annotated Bb protein coding genes and pseudogenes. The median number of reads per gene in each sample varied from 338 to 1167 reads (Figure 1—source data 1). This coverage was sufficient for statistically significant downstream differential expression analyses for the vast majority of Bb genes.
To evaluate whether our approach introduced any major artifacts in Bb expression, we sequenced and compared RNA-seq libraries from in vitro cultured Bb cells before and after immunomagnetic enrichment. We found minimal expression differences (29 genes with p<0.05, fold changes between 0.83 and 1.12; Figure 1—figure supplement 2 and Figure 1—figure supplement 2—source data 1), suggesting experimental enrichment did not significantly alter global transcriptome profiles for Bb. Thus, our enrichment approach enabled genome-wide analysis of Bb population-level expression changes that occur within the feeding nymph as Bb is transmitted to the host.
Global ex vivo profiling of Bb reveals extent and kinetics of transcriptional changes
To provide a broad overview of Bb expression changes in the tick during the nymphal I. scapularis transmission bloodmeal, we performed principal component analysis (PCA) on the Bb transcriptome data from one, two, three, and four days after attachment (n=4). We reasoned that if many longitudinal expression changes were occurring across Bb populations, we would observe greater data variability between time points than between biological replicates. Indeed, we found replicates from each day grouped together, whereas distinct time points were largely non-overlapping. The first principal component, which explained 64% of the variance in our data, correlated well with day of feeding (Figure 2A). The global pattern suggested that Bb gene expression changes generally trended in the same direction over the course of feeding with the most dramatic differences between flanking timepoints on day 1 and day 4.
Figure 2 with 2 supplements see all

Global ex vivo profiling of Bb reveals extent and kinetics of transcriptional changes.
(A) Principal component analysis of normalized read counts from samples from across feeding, see Figure 2—source data 1. PC1 correlates strongly with day of feeding. (B) Schematic depicting how data was analyzed, as pairwise comparisons between the first day after attachment and all other days. (C–E) Volcano plots of differentially expressed genes comparing day 2 versus day 1 (C), day 3 versus day 1 (D), and day 4 versus day 1 (E). The total number of upregulated genes is shown in the top right and the number of downregulated genes is shown in the top left. Yellow dots are genes that first change expression between day 1 and day 2, red dots are genes that first change expression between day 1 and day 3, and purple dots are genes that first change expression between day 1 and day 4. Two genes with log2 fold changes >4 are shown at x=4, and five genes with -log10(padj)>60 are shown at y=60. Only genes with p-value <0.05 from Wald tests and at least a twofold change are highlighted, see Figure 2—source data 2. n=4. By day 4 of feeding, 153 genes are upregulated and 33 genes are downregulated from day 1 baseline levels.
-
Figure 2—source data 1
DESeq2 normalized counts for all genes across all samples.
- https://cdn.elifesciences.org/articles/86636/elife-86636-fig2-data1-v1.xlsx
-
Figure 2—source data 2
Transcriptome-wide differential expression analysis results from Bb across tick feeding timepoints.
- https://cdn.elifesciences.org/articles/86636/elife-86636-fig2-data2-v1.xlsx
Using day 1 (early attachment) as a baseline, we performed differential expression analysis for all Bb genes at subsequent time points (day 2, day 3, day 4; Figure 2B and Figure 2—source data 2). We examined changes with p-values <0.05 when adjusted for multiple hypothesis testing and fold changes above a twofold threshold (listed in Figure 3—source data 1). These analyses mirrored the global longitudinal expression pattern predicted by the PCA. The total number of differentially expressed (DE) genes when compared to day 1 increased with each subsequent timepoint to day 4. By day 4, there were 186 DE genes, including 153 upregulated and 33 downregulated (Figure 2C–E). Across all later time point comparisons to day 1, DE genes were highly overlapping and largely changed in the same directions. For example, of the DE genes that increased on day 2, 29 of 30 were still increased on day 3, and 29 of 30 were still increased on day 4. In the day 2, day 3, and day 4 comparisons to the day 1 baseline, we found 192 DE genes in total (Figure 3—source data 1). We observed some differences between gene expression patterns, such as the overall timing and kinetics of expression changes. Transcript levels for some DE genes changed suddenly over the course of feeding, while others were more gradual. To our knowledge, this is the first comprehensive report of global Bb expression changes over multiple stages of a tick feeding.
To assess the integrity of our dataset, we first examined expression profiles of previously characterized targets of major transcriptional programs activated at the onset of the bloodmeal: the RpoN/RpoS sigma factor cascade and the Hk1/Rrp1 two-component system (Caimano et al., 2015; Grassmann et al., 2023). Between day 1 and day 4, the expression of rpoS increased (Figure 2—figure supplement 1A), as expected (Hübner et al., 2001), and the majority of genes activated by RpoS in the feeding tick (Grassmann et al., 2023), including canonical targets ospC and dbpA, were also significantly upregulated (79/89, p<0.05, Wald tests; Figure 2—figure supplement 1B). The majority of genes activated or repressed by Rrp1 in vitro (Caimano et al., 2015) also trended in the expected direction ex vivo between day 1 and day 4 (111/148 upregulated, 37/57 downregulated, p<0.05, Wald tests, Figure 2—figure supplement 1C). We also examined the expression trends of genes regulated by RelBbu as part of the stringent response, another major transcriptional program active in Bb in the tick during nutrient starvation (Drecktrah et al., 2015). About half of RelBbu-regulated genes changed in the direction expected if the stringent response was active during this time (129/251 upregulated, 111/226 downregulated, p<0.05, Wald tests) (Figure 2—figure supplement 1D); however, more of the genes that were twofold downregulated over feeding are regulated by RelBbu than either RpoS or Rrp1, suggesting it may play a role during this time frame.
We then compared the 192 twofold DE genes in our longitudinal time course to those identified in two previous studies that measured Bb gene expression changes from culture conditions approximating the unfed tick and fed tick through modulation of temperature and/or pH (Ojaimi et al., 2003; Revel et al., 2002). 31% of the DE genes upregulated from day 1 (49/158) were more highly expressed in ‘fed tick’ conditions compared to ‘unfed tick’ conditions in one or both studies, while 24% of the DE genes downregulated from day 1 (8/24) were more highly expressed in ‘unfed tick’ conditions in one or both studies (Figure 2—figure supplement 2A and Figure 3—source data 1). The studies become more concordant when focusing on the DE genes that were upregulated on day 2, which were generally the genes that changed the most dramatically in the time course. 70% of DE genes upregulated on day 2 (21/30) were more highly expressed in ‘fed tick’ conditions in these previous studies, suggesting that the majority of the most dramatic gene expression changes we saw across feeding agree with what was observed in these previous studies.
We also compared the DE genes to two studies that assessed Bb gene expression differences in fed nymphs versus dialysis membrane chambers (DMCs), which mimic Bb conditions in the mammal (Grassmann et al., 2023; Iyer et al., 2015). 63% of all upregulated DE genes (100/158) were differentially expressed between fed nymphs and DMCs in one or both studies (Figure 2—figure supplement 2B and Figure 3—source data 1). The genes that were more highly expressed in nymphs were most concentrated amongst the day 2 DE genes (17/30, 57%), while the genes more highly expressed in DMCs were concentrated amongst the day 3 and day 4 DE genes (55/128, 43%). These comparisons suggested that the timing and magnitude of gene expression changes during feeding may indicate whether gene expression will peak in the tick or continue rising once Bb is transmitted to the host.
Through comparisons to these previous studies, we were able to verify that our data captured many expected transcriptional trends occurring during tick feeding. Nevertheless, 14% of the twofold DE genes were not previously found to change expression in these different tick-feeding contexts (Grassmann et al., 2023; Iyer et al., 2015; Ojaimi et al., 2003; Revel et al., 2002) or identified in these RNA-seq studies as dependent upon RpoS, Rrp1, or RelBbu (Caimano et al., 2015; Drecktrah et al., 2015; Grassmann et al., 2023), which are three known Bb regulatory programs active in the tick (Samuels et al., 2021). These additional genes highlight the necessity of measuring transcription in the tick environment and suggest we uncovered gene expression changes specific to the tick stage of the Bb enzootic cycle. The nature and dynamics of these changes provide insights into potential genetic determinants of Bb survival, proliferation, and dissemination in the tick during transmission.
Bb genes upregulated during feeding are found predominantly on plasmids
Bb has a complex, highly fragmented genome (Barbour, 1988; Figure 3A), including numerous plasmids that are necessary during specific stages of the enzootic cycle (Schwartz et al., 2021) suggesting they contain genes that are crucial for pathogen transmission and survival. In fact, many genes found on the plasmids have been previously shown to alter expression upon environmental changes or in different host environments (Iyer et al., 2015; Ojaimi et al., 2005; Revel et al., 2002; Tokarz et al., 2004). Thus, we reasoned that many of the 192 DE Bb genes that change expression from day 1 to any later feeding time point (Figure 3—source data 1) would reside on the plasmids, and we examined their distribution throughout the genome. Consistent with these previous reports, we found that most of the upregulated genes were located on the plasmids (143/158; 90%), while fewer were found on the chromosome (15/158; 10%; Figure 3B), which is home to the majority of metabolic and other housekeeping genes. In contrast, the majority of the downregulated genes were found on the chromosome (27/34, 79%; Figure 3C).
Figure 3
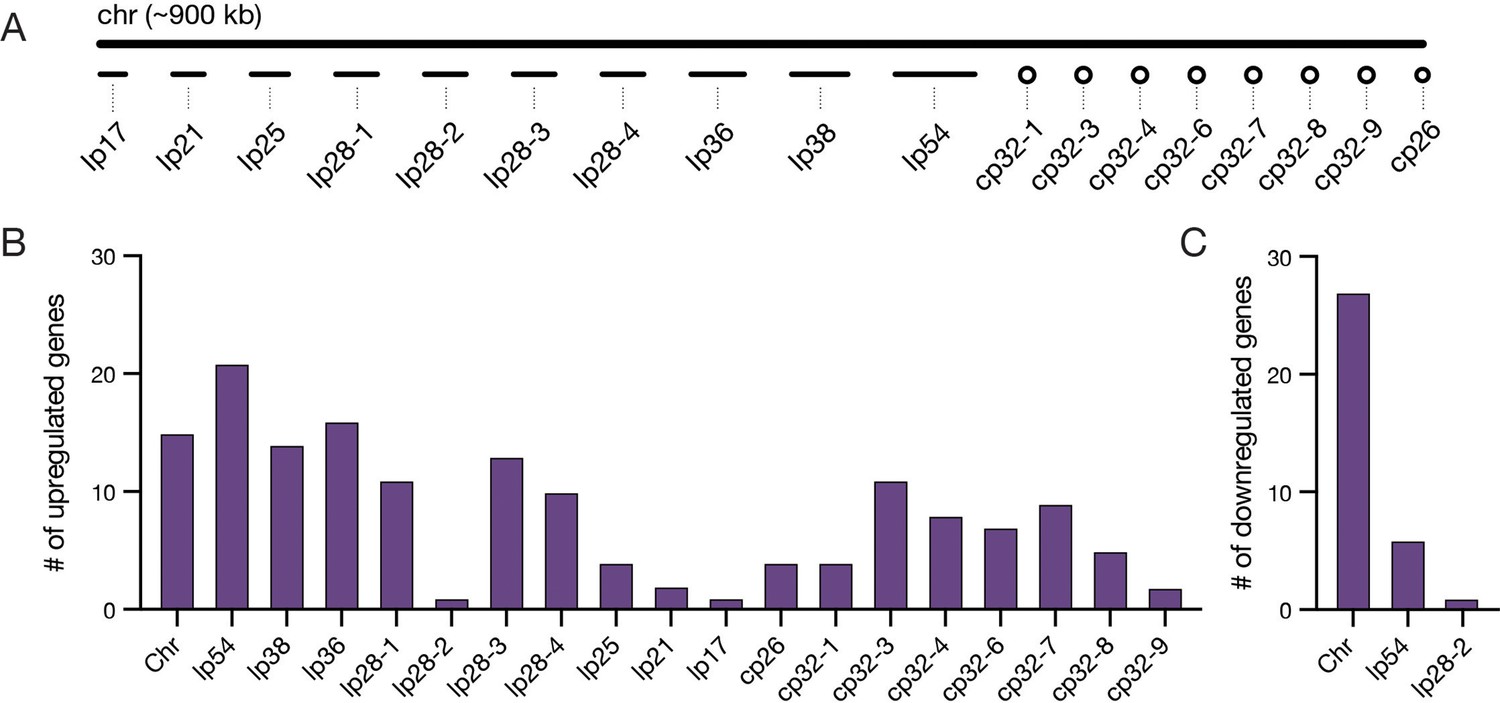
Bb genes upregulated during feeding are found predominantly on plasmids.
(A) Schematic of the chromosome and plasmids in the Bb B31-S9 genome. Plasmid names denote whether the plasmid is linear (lp) or circular (cp) and the length of plasmids in kilobases (kb). For example, lp17 is a 17 kb linear plasmid. Genome is shown approximately to scale. (B–C) The number of genes from each chromosome or plasmid that increased (B) or decreased (C) expression twofold during feeding, see Figure 3—source data 1 for gene information. Upregulated genes are distributed across plasmids, while most downregulated genes are found on the chromosome and lp54.
-
Figure 3—source data 1
Twofold differentially expressed Bb genes from across tick feeding timepoints.
- https://cdn.elifesciences.org/articles/86636/elife-86636-fig3-data1-v1.xlsx
Several plasmid-encoded genes that were longitudinally upregulated in our dataset have known roles during the tick bloodmeal or in mammalian infection. Linear plasmid 54 (lp54), which is an essential plasmid present in all Bb isolates (Casjens et al., 2012), contained the largest number of upregulated genes. Many of the genes on lp54 are regulated by RpoS during feeding, including those encoding adhesins DbpA and DbpB, which are important for infectivity in the host (Blevins et al., 2008). This set also included five members of a paralogous family of outer surface lipoproteins BBA64, BBA65, BBA66, BBA71, and BBA73. BBA64 and BBA66 are necessary for optimal transmission via the tick bite (Gilmore et al., 2010; Patton et al., 2013). These findings indicate our dataset captures key Bb transcriptional responses known to be important for survival inside the tick during a bloodmeal.
Many upregulated genes were also encoded by cp32 plasmid prophages. Bb strain B31-S9 harbors seven cp32 isoforms that are highly similar to each other (Casjens et al., 2012). When cp32 prophages are induced, phage virions called ϕBB1 are produced (Eggers and Samuels, 1999). In addition to phage structural genes, cp32 contain loci that encode various families of paralogous outer surface proteins (Stevenson et al., 2000). Amongst the cp32 genes that increased over feeding were members of the RevA, Erp, and Mlp families, which are known to increase expression during the bloodmeal (Gilmore et al., 2001). We also found several phage genes that were upregulated, including those encoding proteins annotated as phage terminases on cp32-3, cp32-4, and cp32-7 (BBS45, BBR45, and BBO44). Some cp32 genes have been shown to change expression in response to the presence of blood (Tokarz et al., 2004) and as a part of the stringent response regulated by RelBbu (Drecktrah et al., 2015), while BBD18 and RpoS regulate prophage production in the tick midgut after feeding (Wachter et al., 2023). Our data suggest that some prophage genes are upregulated over the course of tick feeding, raising the possibility that cp32 prophage are induced towards the end of feeding. Overall, our data support the long-held idea that the Bb plasmids, which house many genes encoding cell envelope proteins, proteins of unknown function, and prophage genes, play a critical role in the enzootic cycle during the key transition period of tick feeding.
Bb genes encoding outer surface proteins are highly prevalent among upregulated genes
To gain a better overall sense of the types of genes that changed over feeding and the timing of those changes, we grouped DE genes into functional categories. Since a high proportion of plasmid genes encode lipoproteins within the unique protein-rich outer surface of Bb, genes of unknown function, and predicted prophage genes (Casjens et al., 2000; Fraser et al., 1997), we expected that many of the DE genes would fall into these categories. We classified the genes as related to either: cell envelope, bacteriophage, cell division, DNA replication and repair, chemotaxis and motility, metabolism, transporter proteins, transcription, translation, stress response, protein degradation, or unknown (Drecktrah et al., 2015; see Figure 3—source data 1).
Of the genes that increased twofold over feeding, the clear majority each day of feeding fell into two broad categories, cell envelope (55 of 158 total genes) and proteins of unknown function (69 of 158 total genes), with fewer genes related to metabolism, chemotaxis and motility, transporters, bacteriophage, cell division, and transcription (Figure 4A). In contrast to the upregulated genes, genes downregulated during feeding were more evenly distributed among the functional categories – including translation, protein degradation, transcription, and metabolism – consistent with many of them being located on the chromosome.
Figure 4
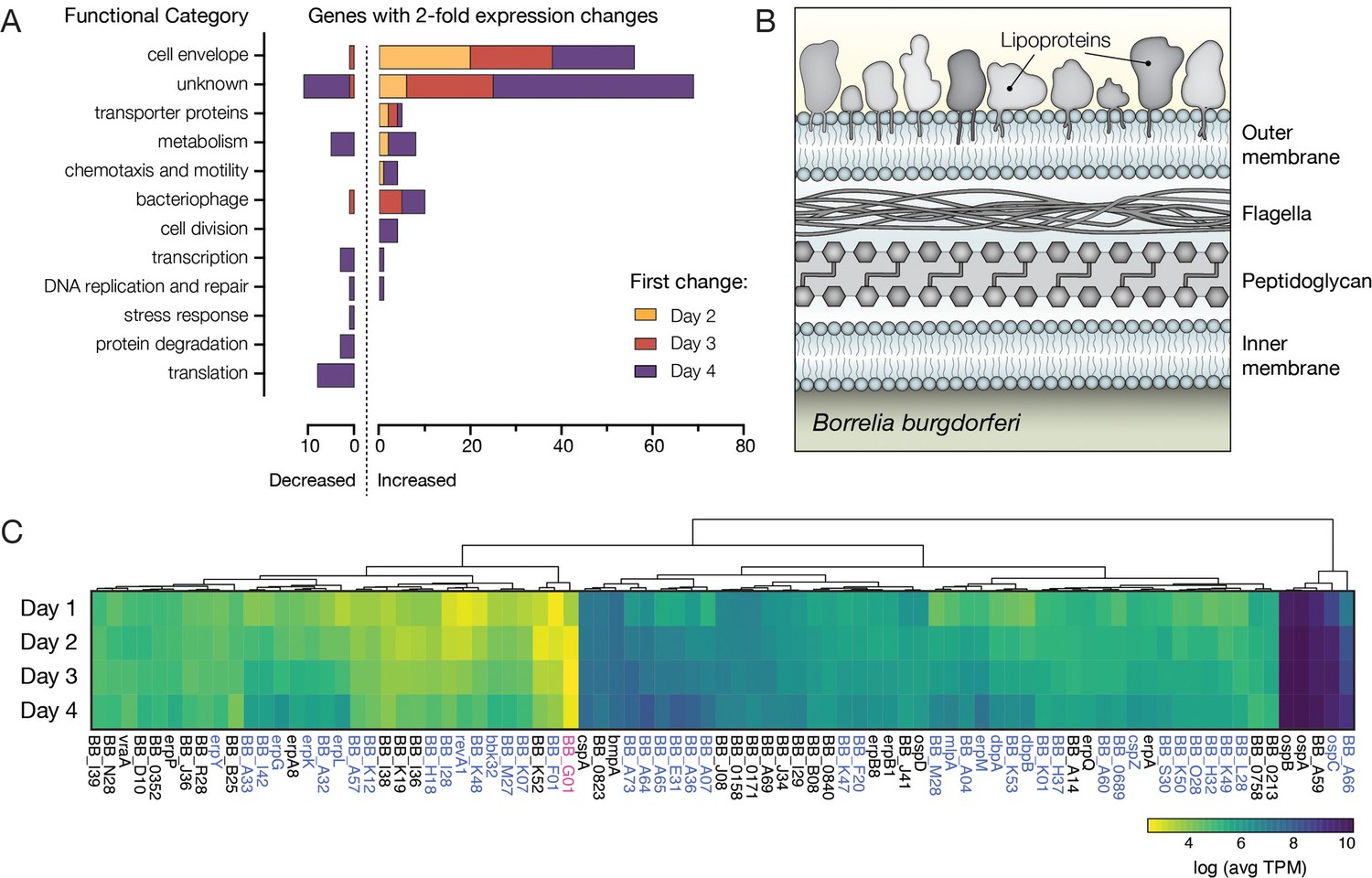
Bb genes encoding outer surface proteins are highly prevalent among upregulated genes.
(A) The number of Bb genes that change over the course of tick feeding sorted into functional categories. Genes that first change 2 days after attachment are shown in yellow, 3 days after attachment in red, and 4 days after attachment in purple. A majority of upregulated genes fall into cell envelope and unknown categories. (B) Schematic of the outer membrane of Bb showing outer surface lipoproteins. Lipoproteins can also reside in the periplasmic space. (C) Heat map of expression levels of all genes encoding outer surface lipoproteins as average Transcripts Per Million (TPM) across the 4 days of tick feeding, see Figure 4—source data 1. Gene names highlighted in blue were twofold upregulated and genes in pink twofold downregulated over feeding (see Figure 2). A majority of genes encoding outer surface proteins increased in expression throughout feeding, while having different magnitudes of expression.
-
Figure 4—source data 1
Transcripts per million (TPM) for all Bb genes across feeding timepoints.
- https://cdn.elifesciences.org/articles/86636/elife-86636-fig4-data1-v1.xlsx
When looking at changes across these functional categories, the overrepresentation of cell envelope proteins was striking, while not unexpected. The Bb outer surface is covered with lipoproteins (Figure 4B), and these proteins are critical determinants in Bb interactions with the various environments encountered during the enzootic cycle (Kurokawa et al., 2020). We found that more than half (46 of 83) of annotated outer surface lipoproteins (Dowdell et al., 2017; Iyer et al., 2015) changed expression twofold over the time course (Figure 4C). These data suggested widespread changes may be occurring on the Bb outer surface during feeding.
To understand the functional implications of expression changes in a majority of outer surface lipoproteins, we also compared their relative expression. The magnitude of expression varied greatly, with ospA, ospB, ospC, and bba59 being the most highly expressed outer surface protein genes. Many of the outer surface protein genes that we found had increased expression over feeding were much less abundantly expressed (Figure 4C and Figure 4—source data 1). However, even the genes that appeared to have low expression in these population level measurements could play important roles in transmission if they are highly expressed in a small number of crucial cells, such as those that ultimately escape the midgut. While bulk RNA-seq cannot distinguish what is happening at the single-cell level, our data suggest that during the bloodmeal, Bb are undergoing a complex outer surface transformation driven by increases in transcription of a majority of the genes encoding these lipoproteins.
Identification of candidate tick interaction partners of Bb cells ex vivo
Our RNA-seq data suggested that the outer surface of Bb transforms over the course of feeding as Bb are primed for transmission to a vertebrate host. At the same time, the tick midgut environment is changing as the tick begins to digest its bloodmeal (Sonenshine and Anderson, 2014). Tick-Bb interactions are likely crucial at the beginning of the tick bloodmeal, as Bb adhere to the tick gut epithelium before becoming motile and migrating out of the midgut and into the salivary glands (Dunham-Ems et al., 2009). Some Bb outer surface proteins, such as BBE31 and BBA52 play key roles in pathogen migration through interactions with the tick environment (reviewed in Kurokawa et al., 2020). We wanted to explore the changing tick environment to identify tick proteins with which Bb could interact throughout the tick bloodmeal. Since our Bb enrichment process retained some tick material, we reasoned that tick proteins that interact with Bb would be present in these samples.
To outline the changes occurring in the tick during feeding and to identify candidate Bb-interacting tick proteins, we used mass spectrometry to survey the content of the tick material that was enriched along with the Bb cells we sequenced at early and late stages during feeding. We purified proteins from the αBb-enriched fraction of crushed infected ticks one day after attachment and four days after attachment in triplicate. As controls for each day, we also performed the Bb enrichment process on lysate from uninfected ticks mixed with in vitro cultured Bb to help rule out proteins that were not pulled down through in vivo tick-Bb interactions (Figure 5A). When querying against the Bb and I. scapularis proteomes, we identified between 414 and 2240 protein groups per sample replicate. The vast majority of all detected proteins were from ticks (2801/2858, 98%). To identify proteins of interest, we looked for those that were detected in at least two of three replicates within infected ticks and had a mean average coverage twice that of uninfected ticks mixed with cultured Bb. We found 256 proteins (251 from I. scapularis and 5 from Bb) that were enriched with Bb from infected ticks one day after attachment and 226 proteins (220 from I. scapularis and 6 from Bb) that were enriched with Bb from infected ticks four days after attachment (Figure 5—source data 1). Of these proteins, only 27 (24 from I. scapularis and 3 from Bb) were detectable on both days, suggesting the tick proteins present upon Bb enrichment change dramatically over the course of feeding (Figure 5A). Amongst the small number of Bb proteins, we identified OspC in the samples from day 4 after attachment but not day 1 after attachment, confirming the expression change we saw in our RNA-seq. The distinct sets of tick proteins we identified at each timepoint suggest dramatic changes occur in the Bb-infected tick midgut environment during feeding that may alter the landscape of tick-Bb interactions.
Figure 5
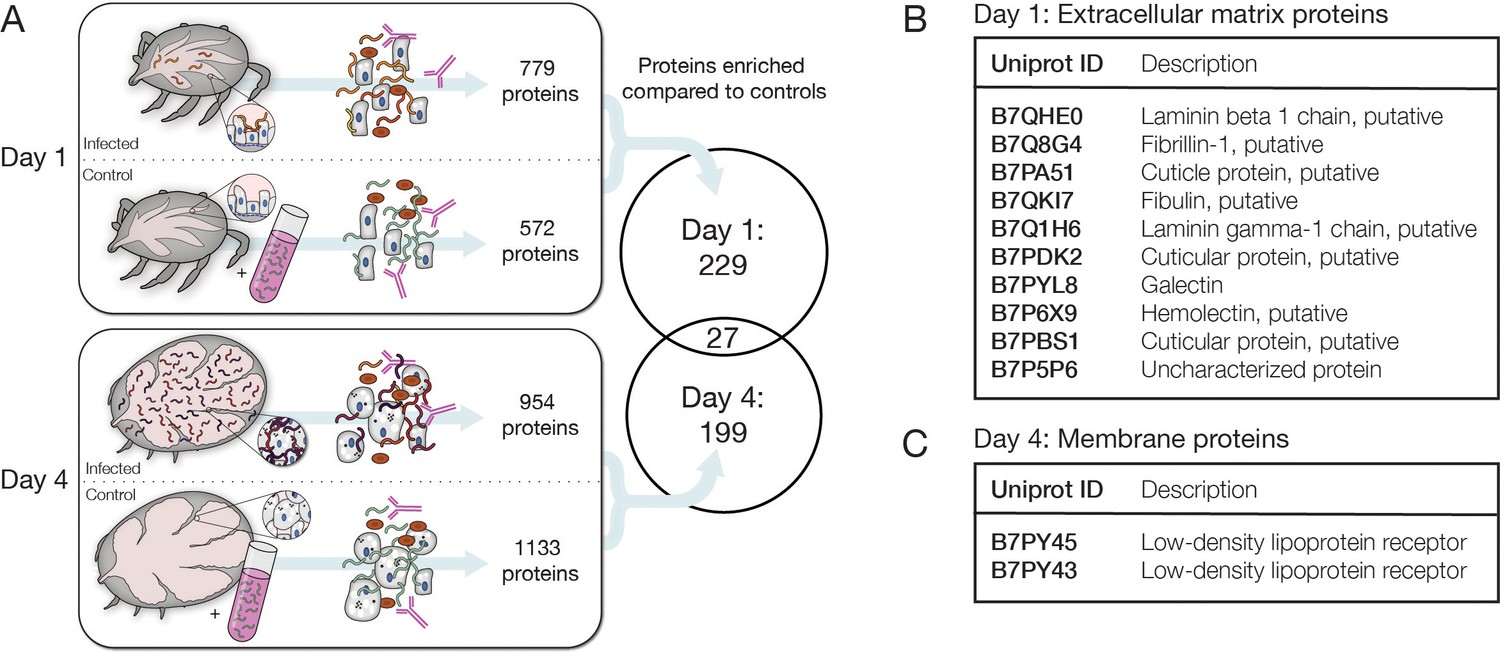
Identification of candidate tick interaction partners of Bb cells ex vivo.
(A) Schematic of experiment to determine candidate tick proteins interacting with Bb over the course of feeding. Ticks were collected 1 day and 4 days after placement on mice. Uninfected ticks at the same time points were mixed with cultured Bb as controls. Bb was enriched with αBb antibody as in RNA-seq experiments and then subjected to mass spectrometry to identify tick proteins present in the samples. Venn diagram depicts the proteins enriched in day 1 and day 4 samples over controls in at least two of three replicates, see Figure 5—source data 1 for all proteins. Tick proteins that are enriched with Bb vary greatly over the course of feeding. (B) Tick proteins uniquely identified one day after placement that are annotated as extracellular matrix (ECM) proteins, see Figure 5—source data 2. (C) Tick proteins uniquely identified four days after attachment that are annotated as low-density lipoprotein receptors, see Figure 5—source data 3. ECM and membrane proteins may be good candidates for Bb-interacting proteins.
-
Figure 5—source data 1
Mass spectrometry analysis for Bb-enriched samples.
Sheet 1, all proteins; sheet 2, proteins enriched on day 1 only; sheet 3, proteins enriched on day 4 only; sheet 4, proteins enriched on day 1 and day 4.
- https://cdn.elifesciences.org/articles/86636/elife-86636-fig5-data1-v1.xlsx
-
Figure 5—source data 2
Annotation and GO term enrichment for tick proteins enriched on feeding day 1.
- https://cdn.elifesciences.org/articles/86636/elife-86636-fig5-data2-v1.xlsx
-
Figure 5—source data 3
Annotation and GO term enrichment for tick proteins enriched on feeding day 4.
- https://cdn.elifesciences.org/articles/86636/elife-86636-fig5-data3-v1.xlsx
Some of the proteins enriched with Bb from the tick may be good candidates for key Bb-interacting partners during feeding, especially if they are localized to the surface of tick cells where they may encounter Bb. The scarcity of both predicted and experimentally validated functions and localizations for tick proteins makes it difficult to fully assess the potential for tick protein interactions with extracellular Bb. Nevertheless, of the proteins found exclusively one day after attachment, 10 were categorized as extracellular matrix proteins using the PANTHER gene database (Thomas et al., 2022), and this category was statistically enriched (Fisher’s exact test, FDR = 0.000093; Figure 5B and Figure 5—source data 2). 30 additional proteins were annotated with a cellular component as plasma membrane. Four days after attachment, we did not detect any annotated extracellular matrix proteins; however, we identified 31 proteins that are likely to be found at the membrane, including two proteins annotated as putative low-density lipoprotein receptors (Figure 5C and Figure 5—source data 3). These extracellular matrix and membrane proteins may be the most likely to directly interact with Bb during this timeframe and are candidates for tick proteins important in the Bb dissemination process. The proteins present in the changing tick environment may be key determinants of pathogen transmission as Bb remodels its outer surface while preparing to migrate through the tick to a new host.
Discussion
Vector-borne pathogens must adapt to distinct environments as they are transmitted by arthropods to colonize new bloodmeal hosts. The tick-borne Lyme disease pathogen Bb undergoes transcriptional changes inside its vector over the course of a single bloodmeal that are important for a number of key transmission events. For example, expression changes enable survival in the feeding tick (He et al., 2011), facilitate transmission across several internal compartments into the bloodmeal host (Kurokawa et al., 2020), and prime bacteria for successful infection of the next host (Kasumba et al., 2016). Capturing these changes as they occur in vivo has been challenging due to the low relative abundance of Bb material inside of the rapidly growing, blood-filled tick. To date, many advances in our understanding of Bb expression during transmission from tick to host have come from tracking changes in small subsets of genes during tick feeding (Bykowski et al., 2007; Gilmore et al., 2001; Narasimhan et al., 2002), leveraging Bb culture conditions that approximate environmental changes across its lifecycle (Ojaimi et al., 2005; Revel et al., 2002; Tokarz et al., 2004), and defining transcriptional regulons outside of the tick (Caimano et al., 2019; Caimano et al., 2015; Drecktrah et al., 2015). Here, we developed an experimental RNA-seq-based strategy to more directly and longitudinally profile gene expression for Bb populations isolated from ticks over the course of a transmitting bloodmeal.
Our longitudinal collection of transcriptomes for Bb cells isolated from ticks serves as a resource and starting point for delineating the functional determinants of Bb adaptation and transmission. We focused our analysis on 192 genes that changed twofold between one day after attachment and later days in feeding. While these genes were highly concordant with those found in previous studies probing transcriptional changes throughout the Bb enzootic cycle (Caimano et al., 2019; Caimano et al., 2015; Drecktrah et al., 2015; Grassmann et al., 2023; Iyer et al., 2015; Ojaimi et al., 2003; Revel et al., 2002), our transcriptome profiles revealed changes in expression of 26 genes not observed in these previous studies. These novel gene expression changes, like all of the 192 DE genes, were concentrated in genes of unknown function and those encoding outer surface proteins. Although challenging to address, efforts focused on uncovering the contribution of the genes of unknown function to the Bb life cycle could be groundbreaking for understanding the unique aspects of tick-borne transmission. Our data reveal more extensive expression changes for Bb outer surface proteins than previously appreciated, with nine additional genes encoding cell envelope proteins upregulated over feeding that were not found to change expression in the previous studies examining conditions mimicking unfed ticks versus fed ticks or in nymphs versus DMCs. The extensive changes in genes encoding outer surface proteins suggest Bb is actively remodeling its outer coat to navigate the dynamic tick environment during feeding, akin to wardrobe changes across seasons. These outer surface changes may play roles in the persistence of Bb in the tick, cell adhesion, or in immune evasion, either inside of the tick or later in the vertebrate host (Kenedy et al., 2012). It has long been observed that molecular interactions between pathogens and the midgut of their vectors are key determinants of transmission (Barillas-Mury et al., 2022). Modifications to the Bb surface could facilitate different tick–pathogen interactions that are critical for its physical movement through tick compartments.
More comprehensive knowledge about Bb outer surface proteins and their functional consequences could lead to new avenues for curtailing the spread of Lyme disease. Protective vaccines against vector-borne pathogens have often targeted surface proteins encoded by pathogens (Kovacs-Simon et al., 2011). While previous Lyme disease vaccine efforts effectively targeted the highly expressed Bb outer surface protein OspA (Steere et al., 1998), more targets could increase the likelihood of a successful vaccine. Our study provides a catalog of new Bb candidates that could be explored. In addition, our biochemical pull-downs using tick-isolated Bb cells as bait unearthed a preliminary list of potential tick proteins that could be involved in tick–Bb interactions. Blocking molecular interactions that are functionally critical for transmission could also be explored as a therapeutic strategy (Barillas-Mury et al., 2022; Manning and Cantaert, 2019).
To identify the genes changing expression during tick feeding, we used an antibody targeting Bb to overcome the low abundance of Bb in the tick. This method produced broad coverage of the transcriptome over our time course, but there are caveats. While we confirmed that the hour-long enrichment procedure does not cause significant gene expression changes in cultured Bb, this may still influence gene expression in our ex vivo samples. The αBb antibody, which targets OspA and a number of other proteins, captured the vast majority, but not all, Bb flaB transcripts from inside of the tick. This loss may introduce some degree of bias. Further, a large number of day 4 reads mapping to P. fulva suggests that this particular antibody may enrich other bacterial species. It is also important to note that our method was unable to produce consistent expression data from Bb from unfed ticks, limiting the full longitudinal scope of the study and potentially overlooking key changes occurring in the first 24 hours of feeding. We predict this difficulty may be due to low numbers of Bb in flat nymphs (de Silva and Fikrig, 1995) or the stress of the prolonged nutrient-deprived period between tick feedings (Kung et al., 2013). The transcriptional landscape of unfed ticks may be better probed through enrichment after RNA-seq library preparation such as TBDCapSeq (Grassmann et al., 2023) or perhaps a combination of the two enrichment strategies. Despite these caveats, this type of antibody-based enrichment strategy is a simple and flexible technique that can probe gene expression from Bb from multiple timepoints in feeding ticks. With small modifications, this protocol may be able to assist in facilitating sequencing of Bb or other Borrelia species from other milieu or enrich other tick-borne bacterial species.
Our method has produced a transcriptomic resource providing critical insights into Bb population-level changes during the vector stage of its lifecycle, which serves as a starting point for understanding the primary drivers of tick-borne transmission. Strikingly few Bb cells out of the total pathogen population in ticks are ultimately transmitted to the next host during feeding (Dunham-Ems et al., 2009; Rego et al., 2014). There may be important molecular variations across Bb cells within the population residing in the tick that contribute to these differential outcomes including heterogeneously expressed proteins across cells within the feeding tick midgut (Ohnishi et al., 2001). Genes that appear unaltered or show low expression levels across the bulk population could still play an outsized role in infection for a minority of the cells, and conducting transcriptomic analyses at the single-cell level will be key. Our work provides a foundational methodology that can be leveraged to greatly improve the resolution of tick–microbe studies, which may unearth surprising mechanistic insights into the unique lifestyles of tick-borne pathogens.
Methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain (Borrelia burgdorferi) | B31-S9 | Sourced from Dr. Patricia Rosa | Streptomycin resistant strain | |
| Strain (Ixodes scapularis) | Ixodes scapularis nymphal ticks | Sourced from Tick Lab at Oklahoma State University (OSU) (RNA-seq) and BEI Resources (mass spec) | BEI Cat# NR-44115 | Ticks were sourced as larvae; fed on mice and allowed to molt to nymphs in lab |
| Strain (Mus musculus) | C3H/HeJ | Jackson Laboratories | Strain#:00659 | Tick hosts were 4–6 week old female mice |
| Strain (Borrelia burgdorferi) | B31-A3 | Sourced from Dr. Patricia Rosa | Used for anti-Bb western only | |
| Strain (Borrelia burgdorferi) | ospA1 | Sourced from Dr. Patricia Rosa; Battisti et al., 2008 | ospA mutant; Used for anti-Bb western only | |
| Strain (Borrelia burgdorferi) | ospA+B1 | Sourced from Dr. Patricia Rosa; Battisti et al., 2008 | ospA restored; Used for anti-Bb western only | |
| Antibody | anti-Borrelia burgdorferi (Rabbit polyclonal) | Thermo Fisher Scientific | Invitrogen: PA1-73004; RRID: AB_1016668 | 10 mg added to immunomagnetic enrichment; IF(1:100), WB (1:10,000) |
| Antibody | anti-Rabbit HRP secondary (goat polyclonal) | Advansta | Advansta: R-05072–500; RRID: AB_10719218 | WB (1:5000) |
| Antibody | Anti-Rabbit IgG (H+L) Alexa 488 (goat polyclonal) | Thermo Fisher Scientific | Invitrogen: A-11008; RRID: AB_143165 | IF (1:100) |
| Commercial assay or kit | Dynabeads Protein G | Thermo Fisher Scientific | Invitrogen 10003D | |
| Commercial assay or kit | Zymo Direct-zol RNA Microprep Kit | Zymo Research | R2062 | With on column DNase |
| Commercial assay or kit | NEBNext Ultra II Directional RNA Library Prep Kit for Illumina | New England Biolabs | E7760L | |
| Commercial assay or kit | NEBNext Multiplex Oligos for Illumina Dual Index | New England Biolabs | E7600S | |
| Commercial assay or kit | Kapa HiFi Real-Time Library Amplification Kit | Roche | Kapa KK2702 | |
| Commercial assay or kit | Cas9 | New England Biolabs | M0386S | |
| Commercial assay or kit | Taqman Universal PCR Master Mix | Thermo Fisher Scientific | Applied Biosystems 4304437 | |
| Commercial assay or kit | PowerUp SYBR Green Master Mix | Thermo Fisher Scientific | Applied Biosystems A25741 | |
| Sequence-based reagent | crRNAs targeting tick, mouse, Bb rRNA | Dynerman et al.; Ring et al., 2022 | See Supplementary file 1 | |
| Sequence-based reagent | flaB F | Jewett et al., 2007 | qPCR primers | 5’- TCTTTTCTCTGGTGAGGGAGCT |
| Sequence-based reagent | flaB R | Jewett et al., 2007 | qPCR primers | 5’-TCCTTCCTGTTGAACACCCTCT |
| Sequence-based reagent | flaB probe | Jewett et al., 2007 | qPCR probe | /56-FAM/AAACTGCTCAGGCTGCACCGGTTC/36-TAMSp |
| Sequence-based reagent | gapdh F | This paper | qPCR primer | 5’-TTCATTGGAGACACCCACAG |
| Sequence-based reagent | gapdh R | This paper | qPCR primer | 5’-CGTTGTCGTACCACGAGATAA |
| Chemical compound | Propidium iodide | Thermo Fisher Scientific | Invitrogen P3566 | |
| Chemical compound | TRIzol | Thermo Fisher Scientific | Invitrogen 15596018 | |
| Software, algorithm | DASHit | Dynerman et al., 2020 | http://dashit.czbiohub.org/ | |
| Software, algorithm | Salmon | Patro et al., 2017 | RRID:SCR_017036 | |
| Software, algorithm | STAR | Dobin et al., 2013 | RRID:SCR_004463 | |
| Software, algorithm | Bowtie2 | Langmead and Salzberg, 2012 | RRID:SCR_016368 | |
| Software, algorithm | DESeq2 | Love et al., 2014 | RRID:SCR_015687 | |
| Software, algorithm | GraphPad Prism v9.5.1 | GraphPad Software | RRID:SCR_002798 | |
| Software, algorithm | PEAKS Online Xpro 1.6 | Bioinformatics Solutions Inc. | RRID:SCR_022841 | |
| Other | BSK II | Rosa Lab Recipe | Media used to grow Bb; https://www.niaid.nih.gov/sites/default/files/lzp_recipes.pdf |
B. burgdorferi culture
Bb strain B31-S9 (Rego et al., 2011) was provided by Dr. Patricia Rosa (NIAID, NIH, RML) and cultured in BSK II media at 35 °C, 2.5% CO2. B31-S9 was used for all RNA-seq and Bb enrichment experiments. Wildtype Bb strain B31-A3, ospA1-mutants (ospA1) and ospA-restored Bb (ospA+B1) Battisti et al., 2008 used in αBb western blot were also provided by Dr. Rosa.
Tick feeding experiments
I. scapularis larvae were purchased from the Tick Lab at Oklahoma State University (OSU) for RNA-seq experiments or provided by BEI Resources, a division of the Center for Disease Control, for mass spectrometry experiments. Before and after feeding, ticks were maintained in glass jars with a relative humidity of 95% (saturated solution of potassium nitrate) in a sealed incubator at 22 °C with a light cycle of 16 hr/8 hr (light/dark). Animal experiments were conducted in accordance with the approval of the Institutional Animal Care and Use Committee (IACUC) at UCSF, Project Number AN183452. Ticks were fed on young (4–6 week-old) female C3H/HeJ mice acquired from Jackson Laboratories. Mice were anesthetized with ketamine/xylazine before placement of ≤100 larval or ≤30 nymphal ticks. Replete larval ticks were placed in the incubator to molt before being used as nymphs in experiments. Nymphal ticks were either pulled off isoflurane anesthetized mice at various times during feeding (1–3 days after placement) or allowed to feed to repletion and collected from mouse cages (4 days after placement).
Western blot with αBb antibody
To determine whether the αBb antibody targeted ospA, wildtype Bb (B31-A3), ospA1-mutants (ospA1) and ospA-restored Bb (ospA+B1) (Battisti et al., 2008) were cultured to approximately 5x107 Bb/mL. 3 mLs of culture were centrifuged for 7 min at 8000 x g, washing twice with PBS. Pelleted cells were lysed in 50 µL of water, and 25 µg of protein per sample were mixed with 5 X loading dye (0.25% Bromophenol Blue, 50% Glycerol, 10% Sodium Dodecyl Sulfate, 0.25 M Tris-Cl pH 6.8, 10% B-Mercaptoethanol), run on a Mini-PROTEAN TGX 4–15% gel (Bio-Rad, Hercules, CA), and transferred using the Trans-Blot Turbo Transfer System (Bio-Rad). After transfer, the blot was blocked for 30 minutes at 4 °C in TBST (Tris buffered saline with 0.1% tween) with 5% milk, then treated with αBb antibody (Invitrogen, Waltham, MA: PA1-73004; RRID: AB_1016668) diluted 1:10,000 for 1 hr at room temperature, followed by anti-rabbit HRP secondary antibody (Advansta, San Jose, CA: R-05072–500; RRID: AB_10719218) diluted 1:5000 for 45 min at room temperature with three short PBST washes between each step. Blots were exposed using Clarity Western ECL Substrate (Bio-Rad) and imaged using the Azure C400 imaging system (Azure Biosystems, Dublin, CA). This experiment was repeated three times.
Enrichment of Bb from feeding ticks
To sequence RNA from Bb inside of feeding ticks, Bb were enriched to increase the ratio of Bb to tick material. Larval ticks were fed to repletion on three mice that were infected with Bb through intraperitoneal and subcutaneous injection with 104 total Bb. Approximately 5 months later, the molted nymphal ticks were fed on eight mice, which were housed individually during the feeding. We estimated that 83% of the ticks were infected with Bb by crushing 12 unfed nymphs in BSK II media and checking for viable Bb days later. Ticks were pulled from all mice and pooled into four biological replicates 1 day after placement (14 ticks per replicate), 2 days after placement (12 ticks per replicate), and 3 days after placement (6 ticks per replicate) and collected from cages 4 days after placement (7 ticks per replicate). Shortly after collection, ticks were washed with water and placed in a 2 mL glass dounce grinder (Kimble, DWK Life Sciences, Millville, NJ) in 500 µL of phosphate-buffered saline (PBS). Ticks were homogenized first with the large clearance pestle and then the small clearance pestle. The homogenate was transferred to a 1.5 mL Eppendorf tube and 500 µL of PBS was added to total 1 mL. At this stage, 50 µL of homogenate was removed as an input sample and mixed with 500 µL of TRIzol (Invitrogen) for RNA extraction. Two µL of αBb antibody (Invitrogen: PA1-73004; RRID: AB_1016668) was added to the homogenate, which was then placed on a nutator at 4 °C for 30 min. During incubation, 50 µL of Dynabeads Protein G (Invitrogen) per sample were washed twice in PBS. After incubation with the antibody, the homogenate and antibody mixture were added to the beads. This mixture was placed on a nutator at 4 °C for 30 min. Tubes were then placed on a magnet to secure beads, and the homogenate was removed and saved to create depleted samples. The depleted homogenate was centrifuged at 8000 x g for 7 min, 900 µL of supernatant was removed, and 500 µL of TRIzol was added to the pellet to create depleted samples. The beads were washed twice with 1 mL of PBS, resuspending the beads each time. The second wash was removed and 500 µL of TRIzol was added to the beads to create enriched samples. RNA was extracted from all input, enriched, and depleted samples using the Zymo Direct-zol RNA Microprep Kit with on-column DNase treatment (Zymo Research, Irvine, CA). The step-by-step Bb enrichment protocol is available at: https://dx.doi.org/10.17504/protocols.io.36wgqjrbovk5/v1.
Enrichment of Bb from culture
To test whether the Bb enrichment process altered gene expression levels, we performed the enrichment protocol on cultured Bb. Tubes of Bb in BSK II media were grown to 9x104 Bb/mL at 35 °C. 1 mL of culture was spun down at 8000 x g for 7 min, media was removed, Bb were washed in 1 mL of PBS and spun again. Pelleted Bb were resuspended in 1 mL of fresh PBS. These samples were used as starting homogenate for the Bb enrichment protocol and input, enriched, and depleted fractions were collected as above. RNA-seq libraries from these samples were prepared and sequenced as below.
RNA-seq library preparation and sequencing
To make RNA-seq libraries from enriched Bb RNA, 50 ng of total RNA was used as input into the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA). Libraries were prepared following the manufacturer’s protocol for use of the kit with purified mRNA or rRNA-depleted RNA, despite starting with total RNA. Libraries were barcoded using NEBNext Multiplex Oligos for Illumina Dual Index (New England Biolabs).
To deplete reads from the libraries that were from highly expressed tick, Bb, or mouse rRNA sequences, we used Depletion of Abundant Sequences by Hybridization (DASH Gu et al., 2016), which targets Cas9 to unwanted reads in RNA-seq libraries using custom dual-guide RNAs (dgRNAs). The dgRNAs targeted short sequences within tick rRNA, mouse rRNA, and Bb rRNA that were designed using DASHit software (Dynerman et al., 2020). We ordered crRNA oligos (Supplementary file 1) that targeted these sequences (Ring et al., 2022). To transcribe the dgRNAs, we followed the protocol for In Vitro Transcription for dgRNA V2 (Lyden et al., 2019b) as follows. Both tracrRNA and pooled crRNA DNA templates were annealed to equimolar amounts of T7 primer by heating to 95 °C for 2 min and slowly cooling to room temperature. Annealed templates were used in 1 mL in vitro transcription reactions with: 120 μL 10X T7 buffer (400 mM Tris pH 7.9, 200 mM MgCl2, 50 mM DTT, 20 mM spermidine (Sigma-Aldrich, St. Louis, MO)), 100 μL of T7 enzyme (custom prepped enzyme from E. Crawford, diluted 1:100 in T7 buffer, final concentration: 100 μg/mL), 300 μL NTPs (25 mM each, Thermo Fisher Scientific, Waltham, MA), 4 μg of annealed crRNA template or 8 μg of annealed tracrRNA template, and water to 1 mL. In vitro transcription was performed for 2 hr at 37 °C. Reactions were purified twice with the Zymo RNA Clean & Concentrator-5 Kit (Zymo Research). To form the dgRNA complex for DASH, crRNA and tracrRNAs were diluted to 80 μM, mixed in equimolar amounts and annealed by heating to 95 °C for 30 s and cooling slowly to room temperature.
After transcription of dgRNAs, we performed DASH Protocol Version 4 (Lyden et al., 2019a) on each library individually as follows. Cas9 and transcribed dgRNAs were prepped by mixing: 2.5 μL 10 X Cas9 buffer, 5 μL 20 μM Cas9 (New England Biolabs), and 5 μL of 40 μM transcribed dgRNAs. The mixture was incubated at 37 °C for 5 min before 7.5 μL of RNA-seq library (2.8 nM) was added. The mixture was incubated at 37 °C for 1 hr and then purified with Zymo DNA Clean & Concentrator-5 (Zymo Research) following the PCR product protocol and eluting DNA into 10.5 μL of water. During cleanup, Cas9 was again mixed with buffer and dgRNAs and incubated at 37 °C for 5 min. Following cleanup, eluted DNA was added to the second Cas9-dgRNA mixture and incubated at 37 °C for 1 hr for a second time. Then, 1 μL of proteinase K (New England Biolabs) was added, and the mixture was incubated at 50 °C for 15 min. The libraries were then purified with 0.9 X volume of sparQ PureMag Beads (QuantaBio, Beverly, MA) following the standard protocol, eluting in 24 μL of water. rRNA-depleted RNA-seq libraries were then amplified in a Bio-Rad CFX96 using the Kapa HiFi Real-Time Amplification Kit (Roche, Basel, Switzerland) in a 50 μL reaction with 25 μL master mix, 23 μL of the DASHed library pool, and 2 μL of a 25 μM mix of Illumina P5 (5’-AATGATACGGCGACCACCGAGATCT) and P7 (5’- CAAGCAGAAGACGGCATACGAGAT) primers. The qPCR program for amplification was as follows: 98 °C for 45 sec (1 cycle), (98 °C for 15 s, 63 °C for 30 s, 72 °C for 45 s, plate read, 72 °C for 20 s) for 10 cycles (day 3 and day 4 samples) or 11 cycles (day 1 and day 2 samples). The libraries were removed from cycling conditions before leaving the exponential phase of amplification and then purified with 0.9 X volume of sparQ PureMag Beads according to the standard protocol.
Following DASH, RNA-seq libraries were sequenced on an Illumina NovaSeq S2 (2 lanes) with paired-end 100 base pair reads. Libraries from in vitro cultured control experiment were sequenced on an Illumina NextSeq with paired-end 75 base pair reads. FASTQ files and raw Bb read counts for in vitro control experiment (GSE217146) and ex vivo experiment (GSE216261) have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) under SuperSeries accession number GSE217236.
RNA-seq data analysis
DASH
To measure the success of our rRNA depletion through DASH, we used DASHit software (Dynerman et al., 2020) to determine the percentage of reads that would be DASHable by our guide RNAs (Ring et al., 2022). For pre-DASH data, we sequenced the input of each of our RNA-seq libraries before performing DASH on a MiSeq V2 Micro (Illumina, San Diego, CA). We tested DASHability on a random subset of 200,000 paired-end reads chosen by seqtk v1.3 (RRID:SCR_018927) from each pre- and post-DASH library. Paired t test comparing DASHable reads before and after DASH was performed using GraphPad Prism v9.5.1 (GraphPad Software, San Diego, CA).
Differential expression analysis
To map our RNA-seq data to Bb, we wanted to optimize for mapping reads that came from the many paralogous gene families found across the plasmids of the genome. We used the pseudoalignment tool Salmon v1.2.1 (Patro et al., 2017), which is used to accurately map reads coming from different isoforms of the same gene, for this reason. While using Salmon to map to gene sequences may improve mapping to paralogous genes, it may also have a tradeoff of reduced mapping of reads that fall on the ends of genes that reside in operons. Nevertheless, all samples should be similarly affected, and any undercounting should not change differential expression results. Reads were first trimmed of bases with quality scores less than 20 using Cutadapt (Martin, 2011) via Trim Galore v0.6.5 (RRID:SCR_011847). Reads were mapped to Bb gene sequences (for all protein coding and pseudogenes in the Genbank feature table) as a reference transcriptome from NCBI Genbank GCA_000008685.2 ASM868v2 (with plasmids lp5, cp9, and lp56 removed as they are not present in B31-S9) using Salmon with the following parameters: --validateMappings --seqBias --gcBias. Before mapping, the transcriptome was indexed using the Salmon index command with the whole genome as decoys and the parameter --keepDuplicates to keep all duplicate genes.
Read counts from Salmon were used as input into DESeq2 v1.24.0 (Love et al., 2014) for differential expression analysis in R version 3.6.1. DESeq2 function PlotPCA() was used to create a PCA plot from read counts after running the varianceStabilizingTransformation() function. For differential expression analysis between days, a DESeq object was created from count data using the DESeq() function. The lfcShrink() function with the apeglm method (Zhu et al., 2019) was used to calculate fold changes between days. DESeq2 uses a Benjamini-Hochberg multiple testing correction, and we focused the majority of our analysis on genes that had an adjusted p-value <0.05 and used an additional cutoff requiring genes to change twofold between conditions. Code used for differential expression analysis is available at: https://github.com/annesapiro/Bb-tick-feeding (copy archived at Sapiro et al., 2023).
Mapping to other species
To determine the source of non-Bb reads in our RNA-seq libraries, trimmed reads were mapped to tick and mouse genomes using STAR v2.7.3a (Dobin et al., 2013). The I. scapularis ISE6 genome (RefSeq assembly GCF_002892825.2, ISE6_asm2.2_dedeplicated) (Miller et al., 2018) was indexed using STAR run mode genomeGenerate with option --genomeChrBinNbits 18. The Mus musculus genome GRCm39 (RefSeq assembly GCF_000001635.27) was indexed using STAR run mode genomeGenerate with basic options. Reads were mapped using STAR to each genome using basic options. The percentage of reads that mapped to these genomes was determined by adding the percentage of uniquely mapped reads, reads mapped to multiple loci, and reads mapped to too many loci. To identify the potential source of reads that did not map to tick, mouse, or Bb in day 4 samples, one million reads from day 1 and day 4 libraries were used as input into CZ ID (Kalantar et al., 2020), which determined that a large number of reads mapped to bacterial species Pseudomonas fulva. Full RNA-seq libraries were then mapped to the P. fulva genome (NCBI GenBank GCF_001186195.1 ASM118619v1), using the standard options of Bowtie2 (Langmead and Salzberg, 2012) to calculate the overall alignment rate.
Comparisons to other studies
Genes identified in previous studies were compared to time course expression changes. Here, we considered RpoS-regulated genes as those found in Grassmann et al., 2023 that were upregulated by RpoS in both fed nymphs and DMCs and those upregulated by RpoS only during tick transmission (Grassmann et al. Supplemental Tables 5 and 6). RpoS did not suppress the expression of any genes in fed nymphs in the study. Genes up- and down-regulated by RpoS in DMCs only (Grassmann et al. Supplemental Tables 7 and 8) are noted in Figure 2—source data 2 and Figure 3—source data 1 for reference along with genes found to be regulated by RpoS in DMCs in Caimano et al., 2019 (Tables 2 and 3), which were used for RpoS comparisons in previous versions of this study. Rrp1 up- and down-regulated genes were those identified in vitro in Caimano et al., 2015, Table S2. RelBbu up- and down-regulated genes were examined by Drecktrah et al., 2015 in three different in vitro conditions: starvation (Tables S6 and S9), recovery (Tables S7 and S10), and stationary phase (Tables S5 and S8). For simplicity, we considered genes as RelBbu-regulated if they were up- or down-regulated in one or more of these conditions (Figure 2—source data 2 and Figure 3—source data 1). One gene was regulated in opposing directions across conditions and is noted in our tables as ‘both’ and was excluded from the comparison analysis. Genes changing between ‘unfed tick’ and ‘fed tick’ culture conditions in Revel et al., 2002 were those found in Table 3. Genes from Ojaimi et al., 2003 Table 4 with increased expression in vitro at 35 °C relative to 25 °C were considered higher in ‘fed tick’ while those in Ojaimi et al., 2003 Table 5 with increased expression at 25 °C relative to 35 °C were considered higher in ‘unfed tick’. Genes more highly expressed in nymphs than DMCs from Iyer et al., 2015 were found in Table S4, and genes more highly expressed in DMCs than nymphs were found in Table S8. Genes differentially expressed between nymphs and DMCs in Grassmann et al., 2023 were determined from the DESeq2 comparison between WT DMC vs Fed Nymphs found in Supplemental Table 3, in accordance with author cutoffs of at least a threefold difference and q-value<0.05. As many of these studies used different strains of Bb and different genome annotations, some genes were not examined here as they were not present in the B31-S9 strain used.
Gene classification
To classify genes into functional groups, functional categories were sourced from Drecktrah et al., 2015 where available. Other gene functions were sourced from Fraser et al., 1997. Genes found within the co-transcribed ‘late’ bacteriophage operon (Zhang and Marconi, 2005) were considered ‘bacteriophage’ even if their function is unknown. Outer surface proteins were those found in Dowdell et al., 2017 plus additional outer surface proteins listed in Iyer et al., 2015 that were also found in Dowdell et al., 2017 Supporting Table S2 categories SpII and SpI as evidence of outer surface localization. Outer surface and periplasmic lipoproteins were classified as ‘cell envelope’ in the absence of other classifications. Gene family information from Casjens et al., 2000 was considered to aid in classification. Figure 3—source data 1 contains the classification source for each gene.
RT-qPCR measuring Bb enrichment
To test the efficacy of the Bb enrichment protocol, RT-qPCR was used to quantify Bb flaB and I. scapularis gapdh transcript levels in enriched and depleted fractions. cDNA was synthesized from 8 μL of RNA extracted from Bb enrichment samples and their matched depleted samples from day 2, day 3, and day 4 post-attachment using the qScript cDNA Synthesis Kit (Quantabio). cDNA was diluted 2 X before use in qPCR. To measure flaB copies, standards of known concentration were created from purified PCR products. These standards were made from PCR with primers with the following sequences: 5’-CACATATTCAGATGCAGACAGAGGTTCTA and 5’-GAAGGTGCTGTAGCAGGTGCTGGCTGT. A dilution series with 10-fold dilutions between 106 copies and 101 copies of this PCR template was run alongside enriched and depleted samples. qPCR was performed using Taqman Universal PCR Master Mix (Applied Biosystems, Waltham, MA). The primers used to amplify flaB were: 5’- TCTTTTCTCTGGTGAGGGAGCT and 5’-TCCTTCCTGTTGAACACCCTCT (used at 900 nM) and the probe was /56-FAM/AAACTGCTCAGGCTGCACCGGTTC/36-TAMSp (used at 250 nM) (Jewett et al., 2007). For tick gapdh RT-qPCR, the cDNA samples were diluted an additional 2 X. Standards of known concentration were created using the qPCR primer sequences: 5’-TTCATTGGAGACACCCACAG and 5’-CGTTGTCGTACCACGAGATAA (used at 900 nM). qPCR was performed using PowerUp SYBR Green Master Mix (Applied Biosystems). For both flaB and gapdh, the number of copies in each sample was calculated based on the standards of known concentration. Three technical replicates were averaged from each of four biological replicates at each time point tested. We totaled the number of copies in each matched enriched and depleted fraction to calculate the percentage of flaB or gapdh that was found in either sample. All qPCR was performed on the QuantStudio3 Real-Time PCR System (Applied Biosystems). Paired t tests were performed using GraphPad Prism v9.5.1.
Immunofluorescence microscopy
To test whether the αBb antibody recognized Bb inside of the tick, ticks at each day of feeding were crushed in 50 µL of PBS. Ten µL of lysate was spotted onto slides and allowed to air dry before slides were heated briefly three times over a flame. Heat fixed slides were then treated with acetone for 1 hr. Slides were incubated with αBb primary antibody (1:100 diluted in PBS +0.75% BSA) for 30 min at 37 °C in a humid chamber. A control without primary antibody was also used for each day. Slides were washed once in PBS for 15 min at room temperature, then rinsed in distilled water and air dried. Anti-rabbit IgG Alexa 488 (Invitrogen: A-11008; RRID: AB_143165) diluted 1:100 in PBS +0.75% BSA was added for 30 min at 37 °C in a humid slide chamber. Slides were washed in PBS for 15 min at room temperature three times, adding 1:100 Propidium Iodide (Invitrogen) during the second wash. Slides were then rinsed with distilled water and air dried before the addition of mounting media (Fluoromount-G, SouthernBiotech, Birmingham, AL) and cover slips. Fluorescence imaging was performed on a Nikon Ti2 inverted microscope for widefield epifluorescence using a 100 X/1.40 objective. Images were captured with NIS-Elements AR View 5.20 and then processed with ImageJ software (Schneider et al., 2012). No strong florescence signal was observed on the control slides without primary antibody.
Mass spectrometry of Bb-enriched samples
To identify which tick proteins were found in samples after Bb enrichment across feeding, both uninfected and infected ticks were fed on mice. Three biological replicates of uninfected ticks one day after placement (11 ticks per replicate), infected ticks 1 day after placement (27 ticks per replicate), uninfected ticks 4 days after placement (8 ticks per replicate), and infected ticks four days after placement (16 ticks per replicate) were collected. Before αBb enrichment, the uninfected tick samples were mixed with Bb grown in culture that was washed with PBS (3x104 Bb one day after placement and 3x106 Bb 4 days after placement) and mixed lysates were rotated at room temperature for 30 min. Infected tick samples underwent the Bb enrichment process immediately. The enrichment process followed the same protocol used for RNA-seq. Sample volumes were increased to 1 mL as needed, and then 2 µL of αBb antibody (Invitrogen: PA1-73004; RRID: AB_1016668) was added, and samples were rotated at 4 °C for 30 min. Fifty µL of Dynabeads Protein G per sample were washed in PBS during this incubation and added to the lysates, which were rotated at 4 °C for 30 min. The beads were washed twice with 1 mL of PBS, and then placed into 50 µL of lysis buffer (iST LYSE, PreOmics, Martinsried, Bayern, Germany). Samples were boiled at 95 °C for 5 min, and lysates were removed from beads and frozen for mass spectrometry preparation.
For mass spectrometry, a nanoElute was attached in line to a timsTOF Pro equipped with a CaptiveSpray Source (Bruker, Billerica, MA). Chromatography was conducted at 40 °C through a 25 cm reversed-phase C18 column (PepSep) at a constant flowrate of 0.5 μL min−1. Mobile phase A was 98/2/0.1% water/MeCN/formic acid (v/v/v) and phase B was MeCN with 0.1% formic acid (v/v). During a 108 min method, peptides were separated by a 3-step linear gradient (5%–30% B over 90 min, 30% to 35% B over 10 min, 35% to 95% B over 4 min) followed by a 4 min isocratic flush at 95% for 4 min before washing and a return to low organic conditions. Experiments were run as data-dependent acquisitions with ion mobility activated in PASEF mode. MS and MS/MS spectra were collected with m/z 100–1700 and ions with z = +1 were excluded.
Raw data files were searched using PEAKS Online Xpro 1.6 (Bioinformatics Solutions Inc). The precursor mass error tolerance and fragment mass error tolerance were set to 20 ppm and 0.03 respectively. The trypsin digest mode was set to semi-specific and missed cleavages was set to 2. The I. scapularis reference proteome (Proteome ID UP000001555, taxon 6945) and Bb reference proteome (Proteome ID UP000001807, strain ATCC 35210/B31) were downloaded from Uniprot, totaling 21,774 entries. The I. scapularis proteome was the primary search reference and the Bb was used as a secondary to identify any bacterial proteins present. Carbamidomethylation was selected as a fixed modification. Oxidation (M) and Deamidation (NQ) was selected as a variable modification.
Experiments were performed in biological triplicate, with samples being a single run on the instrument. Proteins present in a database search (−10 log(p-value)≥20, 1% peptide and protein FDR) were subjected to the following filtration process. Proteins were filtered to include only those found in two out of three biological replicates, within each respective day (one or four days after placement). The mean area of proteins found in uninfected and infected samples was calculated. Proteins with missing values (i.e. not identified in a sample) were set to 1. The ratio of mean area for each protein was calculated as infected/uninfected, and enriched proteins were identified by having an infected to uninfected ratio greater than 2 within their respective feeding day (1 or 4 days after attachment).
To classify the identified proteins into functional groups, a PANTHER Overrepresentation Test (released 07/12/2022) was used (Mi et al., 2019) with PANTHER version 17.0 (released 02/22/2022) (Thomas et al., 2022). All I. scapularis genes in the database were used as the reference list and all proteins enriched on each day were analyzed for their PANTHER Protein Class. The test type used was Fisher’s Exact, calculating a false discovery rate as the correction.
Raw data files and searched datasets are available on the Mass Spectrometry Interactive Virtual Environment (MassIVE), a full member of the Proteome Xchange consortium under the identifier: MSV000090560.
Data availability
For sequencing data, FASTQ files and raw Bb read counts for in vitro control experiment (GSE217146) and ex vivo experiment (GSE216261) have been deposited in NCBI's GEO database under SuperSeries accession number GSE217236. For mass spectrometry data, raw data files and searched datasets are available on the Mass Spectrometry Interactive Virtual Environment (MassIVE) under the identifier: MSV000090560. Code used for data analysis is available at on GitHub (copy archived at Sapiro et al., 2023). Source data files contain the numerical data used to generate the figures.
-
NCBI Gene Expression OmnibusID GSE217236. Longitudinal map of transcriptome changes in the Lyme pathogen Borrelia burgdorferi during tick-borne transmission.
-
MassIVEID MSV000090560. Tick proteins interacting with borrelia burgdorferi Sapiro et al 2022.
References
-
Plasmid analysis of Borrelia burgdorferi, the Lyme disease agentJournal of Clinical Microbiology 26:475–478.https://doi.org/10.1128/jcm.26.3.475-478.1988
-
Outer surface protein A protects Lyme disease Spirochetes from acquired host immunity in the tick vectorInfection and Immunity 76:5228–5237.https://doi.org/10.1128/IAI.00410-08
-
Growth and migration of Borrelia burgdorferi in Ixodes ticks during blood feedingThe American Journal of Tropical Medicine and Hygiene 53:397–404.https://doi.org/10.4269/ajtmh.1995.53.397
-
STAR: Ultrafast universal RNA-Seq AlignerBioinformatics 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Live imaging reveals a biphasic mode of dissemination of Borrelia burgdorferi within ticksThe Journal of Clinical Investigation 119:3652–3665.https://doi.org/10.1172/JCI39401
-
Gene expression omnibus: NCBI gene expression and hybridization array data repositoryNucleic Acids Research 30:207–210.https://doi.org/10.1093/nar/30.1.207
-
Molecular evidence for a new Bacteriophage of Borrelia burgdorferiJournal of Bacteriology 181:7308–7313.https://doi.org/10.1128/JB.181.23.7308-7313.1999
-
BosR and PlzA reciprocally regulate RpoS function to sustain Borrelia burgdorferi in ticks and mammalsThe Journal of Clinical Investigation 133:e166710.https://doi.org/10.1172/JCI166710
-
The critical role of the linear plasmid Lp36 in the infectious cycle of Borrelia burgdorferiMolecular Microbiology 64:1358–1374.https://doi.org/10.1111/j.1365-2958.2007.05746.x
-
The role of Borrelia burgdorferi outer surface proteinsFEMS Immunology and Medical Microbiology 66:1–19.https://doi.org/10.1111/j.1574-695X.2012.00980.x
-
Lipoproteins of bacterial pathogensInfection and Immunity 79:548–561.https://doi.org/10.1128/IAI.00682-10
-
Interactions between Borrelia burgdorferi and ticksNature Reviews. Microbiology 18:587–600.https://doi.org/10.1038/s41579-020-0400-5
-
Fast gapped-read alignment with bowtie 2Nature Methods 9:357–359.https://doi.org/10.1038/nmeth.1923
-
Examination of the Borrelia burgdorferi transcriptome in Ixodes scapularis during feedingJournal of Bacteriology 184:3122–3125.https://doi.org/10.1128/JB.184.11.3122-3125.2002
-
Interactions between ticks and lyme disease spirochetesCurrent Issues in Molecular Biology 42:113–144.https://doi.org/10.21775/cimb.042.113
-
Borrelia burgdorferi Bba66 gene inactivation results in attenuated mouse infection by tick transmissionInfection and Immunity 81:2488–2498.https://doi.org/10.1128/IAI.00140-13
-
Of ticks, mice and men: understanding the dual-host lifestyle of Lyme disease SpirochaetesNature Reviews. Microbiology 10:87–99.https://doi.org/10.1038/nrmicro2714
-
Defining the plasmid-borne restriction-modification systems of the Lyme disease Spirochete Borrelia burgdorferiJournal of Bacteriology 193:1161–1171.https://doi.org/10.1128/JB.01176-10
-
Dissemination and salivary delivery of Lyme disease Spirochetes in vector ticks (Acari: Ixodidae)Journal of Medical Entomology 24:201–205.https://doi.org/10.1093/jmedent/24.2.201
-
Host blood meal identity modifies vector gene expression and competencyMolecular Ecology 31:2698–2711.https://doi.org/10.1111/mec.16413
-
Vital signs: trends in reported Vectorborne disease cases — United States and territories, 2004–2016MMWR. Morbidity and Mortality Weekly Report 67:496–501.https://doi.org/10.15585/mmwr.mm6717e1
-
Gene regulation and TranscriptomicsCurrent Issues in Molecular Biology 42:223–266.https://doi.org/10.21775/cimb.042.223
-
SoftwareBb-tick-feeding, version swh:1:rev:fb50ad41c831198d69757e07ba73306e8e913eeaSoftware Heritage.
-
NIH image to Imagej: 25 years of image analysisNature Methods 9:671–675.https://doi.org/10.1038/nmeth.2089
-
Multipartite genome of Lyme disease Borrelia: structure, variation and prophagesCurrent Issues in Molecular Biology 42:409–454.https://doi.org/10.21775/cimb.042.409
-
BookMouthparts and digestive system inIn: Sonenshine DE, Roe RM, editors. Biology of Ticks. Oxford University Press. pp. 122–162.
-
Vaccination against Lyme disease with recombinant Borrelia burgdorferi outer-surface lipoprotein A with adjuvantThe New England Journal of Medicine 339:209–215.https://doi.org/10.1056/NEJM199807233390401
-
Repetition, conservation, and variation: the multiple Cp32 plasmids of Borrelia speciesJ Mol Microb Biotech 2:411–422.
-
PANTHER: making Genome‐Scale Phylogenetics accessible to allProtein Science 31:8–22.https://doi.org/10.1002/pro.4218
-
Biology of infection with Borrelia burgdorferiInfectious Disease Clinics of North America 22:217–234.https://doi.org/10.1016/j.idc.2007.12.013
Article and author information
Author details
Funding
Life Sciences Research Foundation
- Anne L Sapiro
Arnold and Mabel Beckman Foundation
- Balyn W Zaro
Montana INBRE (P20GM103474)
- Margie Kinnersley
- Patrick R Secor
Chan Zuckerberg Initiative
- Seemay Chou
Pew Charitable Trusts
- Seemay Chou
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We are grateful to all members of the Chou lab for their feedback throughout the project, specifically to Fauna Yarza and Patrick Rockefeller Grimes for assistance with tick feeding and Ethel Enoex-Godonoo for administrative assistance. We thank Amy Lyden and Emily Crawford for assistance with and reagents for DASH along with Olga Botvinnik, Michelle Tan, and The Chan Zuckerberg Biohub sequencing team for their input and sequencing assistance. We thank Patricia Rosa, Jenny Wachter, Scott Samuels, and Meghan Lybecker for their feedback on the project. We also thank William Hatleberg for assistance with figure schematics.
Ethics
Animal experiments were conducted in accordance with the guidelines and approval of the Institutional Animal Care and Use Committee (IACUC) at UCSF, Project Number AN183452.
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.86636. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2023, Sapiro et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,047
- views
-
- 294
- downloads
-
- 19
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 17
- citations for umbrella DOI https://doi.org/10.7554/eLife.86636
-
- 1
- citation for Reviewed Preprint v1 https://doi.org/10.7554/eLife.86636.1
-
- 1
- citation for Version of Record https://doi.org/10.7554/eLife.86636.3
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Longitudinal map of transcriptome changes in the Lyme pathogen Borrelia burgdorferi during tick-borne transmission
eLife 12:RP86636.
https://doi.org/10.7554/eLife.86636.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}