Somatic mutation rates scale with time not growth rate in long-lived tropical trees
- Department of Biology, Faculty of Science, Kyushu University, Japan
- Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Japan
- Faculty of Forestry, Universitas Gadjah Mada, Indonesia
- PT. Sari Bumi Kusuma, Indonesia
- Gregor Mendel Institute of Molecular Plant Biology, Austrian Academy of Sciences, Austria
- Forestry Division, Japan International Research Center for Agricultural Sciences, Japan
- Faculty of Life and Environmental Sciences, University of Tsukuba, Japan
- Field Science Center, Graduate School of Agricultural Science, Tohoku University, Japan
Figures
Figure 1 with 3 supplements
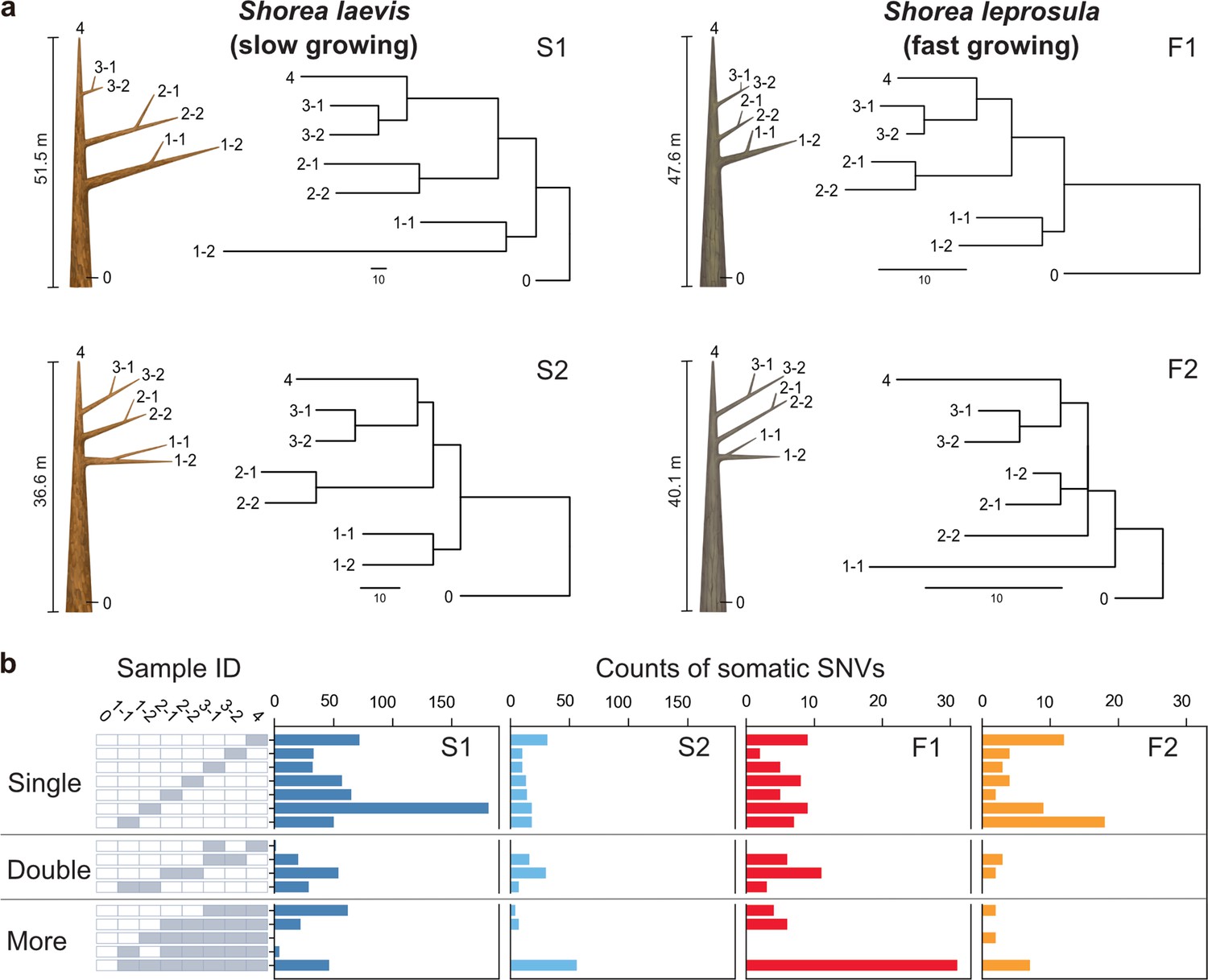
Physical tree structures and phylogenetic trees constructed from somatic mutations.
(a) Comparisons of physical tree structures (left, branch length in meters) and neighbor-joining (NJ) trees (right, branch length in the number of nucleotide substitutions) in two tropical tree species: S. laevis, a slow-growing species (S1 and S2), and S. leprosula, a fast-growing species (F1 and F2). IDs are assigned to each sample from which genome sequencing data were generated. Vertical lines represent tree heights. (b) Distribution of somatic mutations within tree architecture. A white and gray panel indicates the presence (gray) and absence (white) of somatic mutation in each of eight samples compared to the genotype of sample 0. Sample IDs are the same between panels (a) and (b). The distribution pattern of somatic mutations is categorized as Single, Double, and More depending on the number of samples possessing the focal somatic mutations. Among 27–1 possible distribution patterns, the patterns observed in at least one of the four individuals are shown.
Figure 1—figure supplement 1

Target tropical trees and location of study site.
(a) Images of S. laevis (S1), a slow-growing species, and S. leprosula (F1), a fast-growing species. (b), Location of the study site in central Borneo, Indonesia.
Figure 1—figure supplement 2
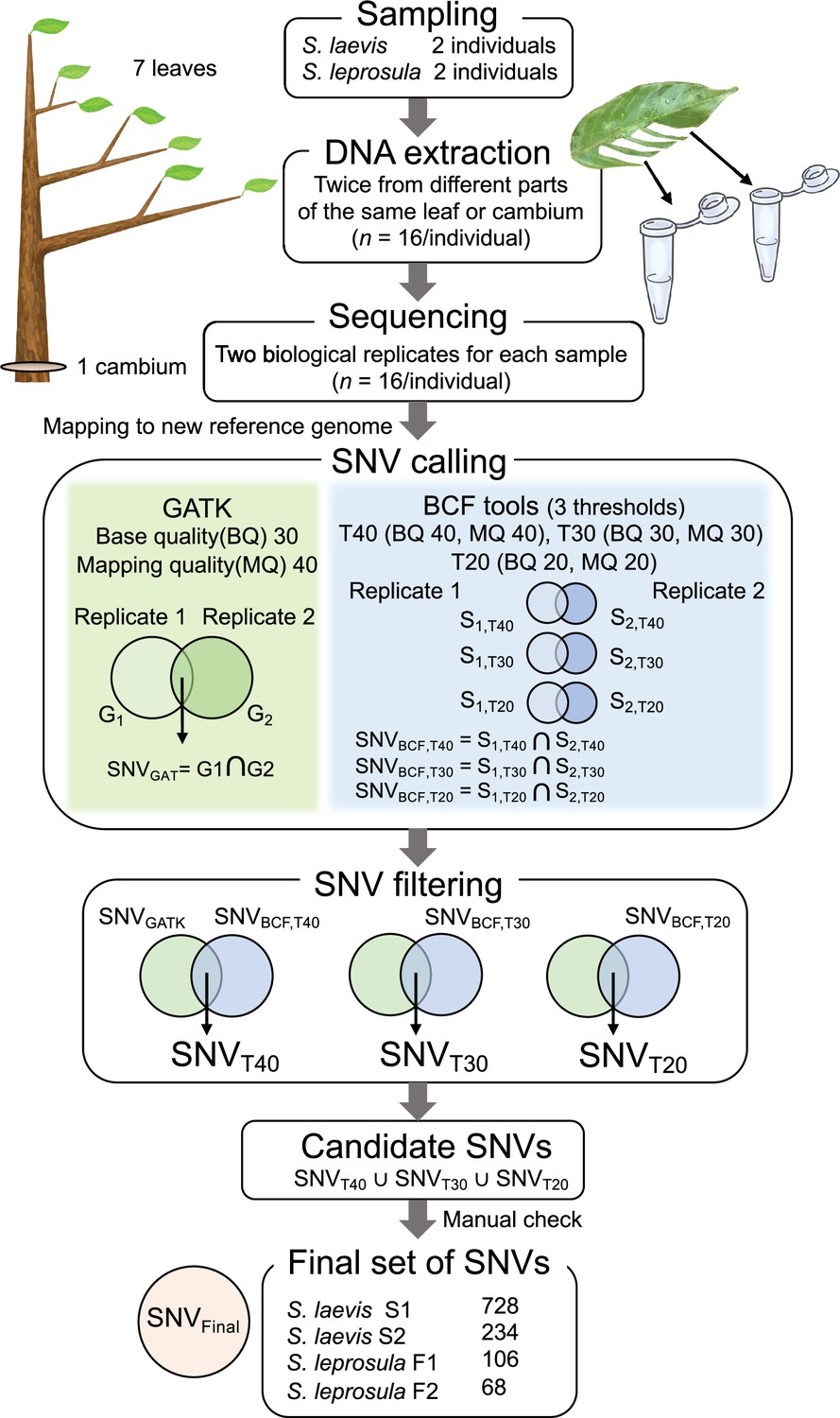
Workflow for identifying de novo somatic SNVs.
Eight samples (seven leaves and one cambium) were collected from four trees (two trees from each species). DNA was extracted twice independently from each sample and sequenced independently. Reads were mapped to the reference genome and used for SNV calling and filtering. SNVs over eight samples were called using GATK HaplotypeCaller (GATK) and Bcftools mpileup (BCF tools) for each set of biological replicates from seven branches and one cambium independently, generating potential SNVs for each set of replicates and for each SNP caller (G1 and G2 for GATK, B1 and B2 for BCF tools). For BCF tools, we set three thresholds (T40, T30, and T20) with different base quality (BQ) and mapping quality (MQ). SNVs detected in both replicates were extracted for each SNP callers and generated potential SNVs for each SNP caller, SNVGATK for GATK and SNVBCF for Bcftools with three thresholds. These SNVs were filtered by extracting SNVs detected in both SNP callers, generating potential SNVs for each threshold: SNVT40, SNVT30, and SNVT20. Finally, SNVs detected at any of the three thresholds were extracted to obtain candidate SNVs. We checked the candidate SNVs manually and obtained a final set of SNVs, SNVFinal.
Figure 1—figure supplement 3
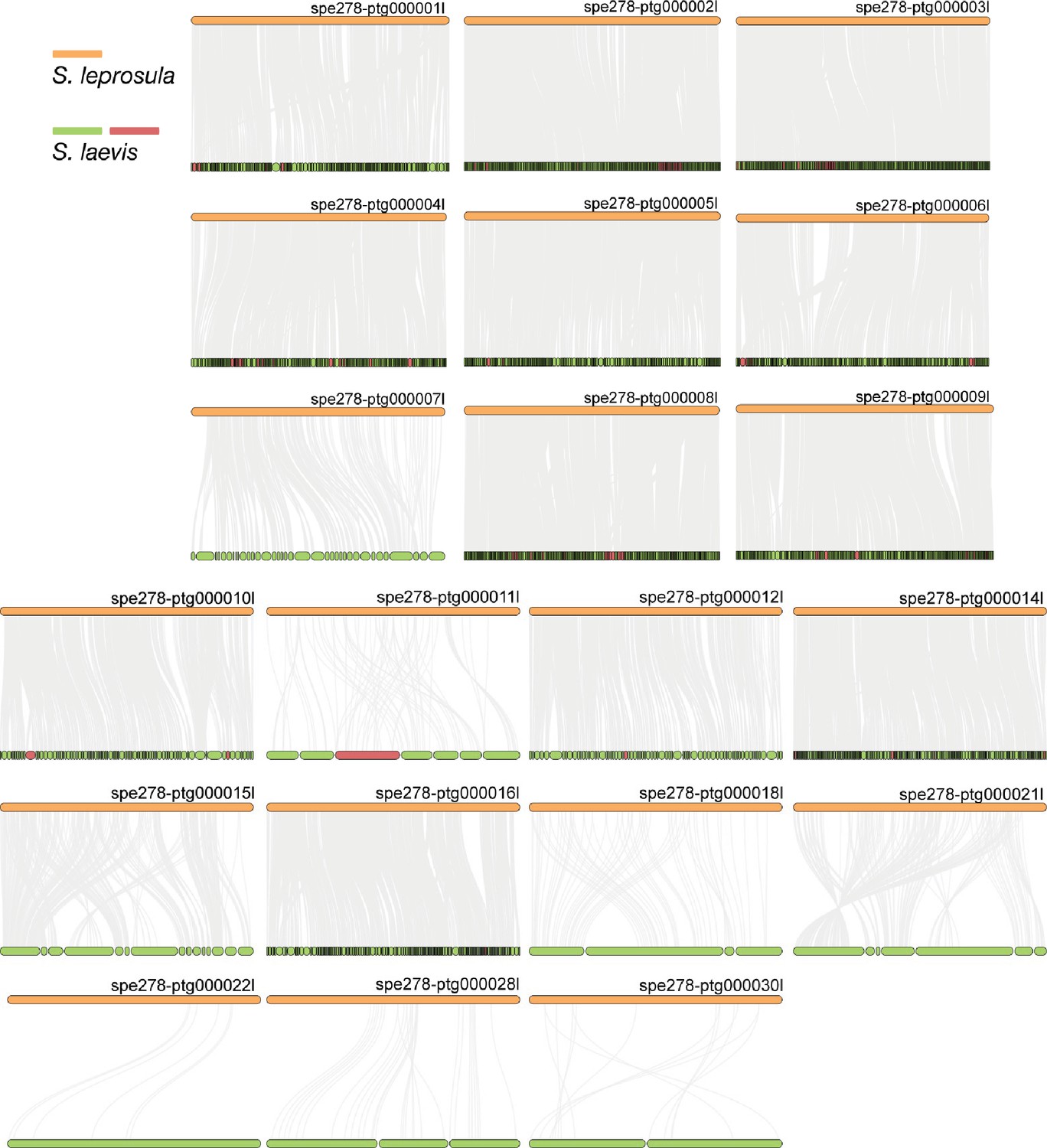
Synteny relationship between S. laevis and S. leprosula.
The collinear blocks within the genomes of S. leprosula and S. laevis were displayed by gray lines, with orange objects representing the contigs of the S. leprosula genome and green objects denoting the contigs of the S. laevis genome. In cases where the direction of a contig in S. laevis was partly different from that in S. leprosula, the contigs of the S. laevis genome were colored in red, otherwise it is indicated as green.
Figure 2
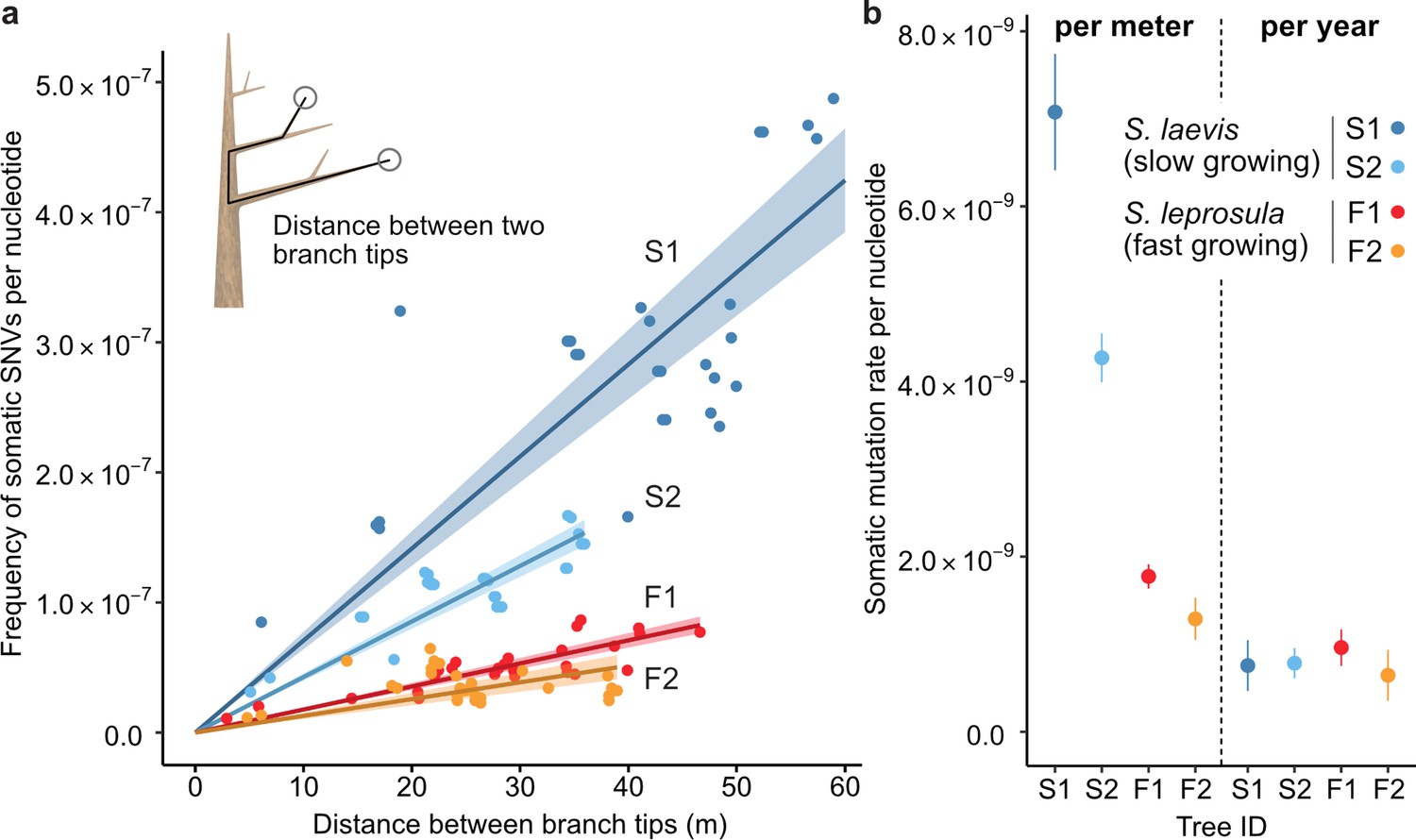
The relationship between the physical distance and the numbers of SNVs.
(a) Linear regression of the number of SNVs against the pair-wise distance between branch tipcs with an intercept of 0 for each tree (S1: blue, S2: right blue, F1: red, and F2: orange). Shaded areas represent 95% confidence intervals of regression lines. Regression coefficients are listed in Supplementary file 1h. (b) Comparison of somatic mutation rates per nucleotide per growth and per year across four tropical trees. Bars indicate 95% confidence intervals.
Figure 3 with 4 supplements
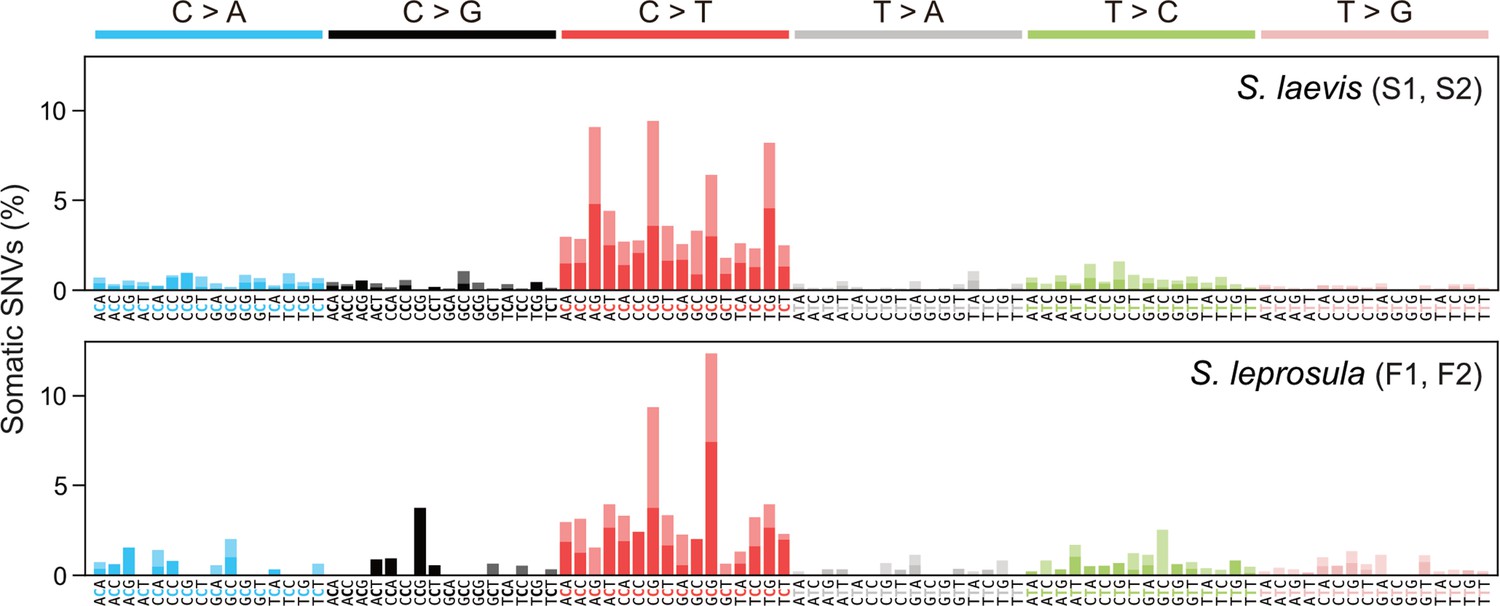
Mutational spectra of somatic SNVs.
Somatic mutation spectra in S. laevis (upper panel) and S. leprosula (lower panel). The horizontal axis shows 96 mutation types on a trinucleotide context, coloured by base substitution type. Different colours within each bar indicate complementary bases. For each species, the data from two trees (S1 and S2 for S. laevis and F1 and F2 for S. leprosula) were pooled to calculate the fraction of each mutated triplet.
Figure 3—figure supplement 1
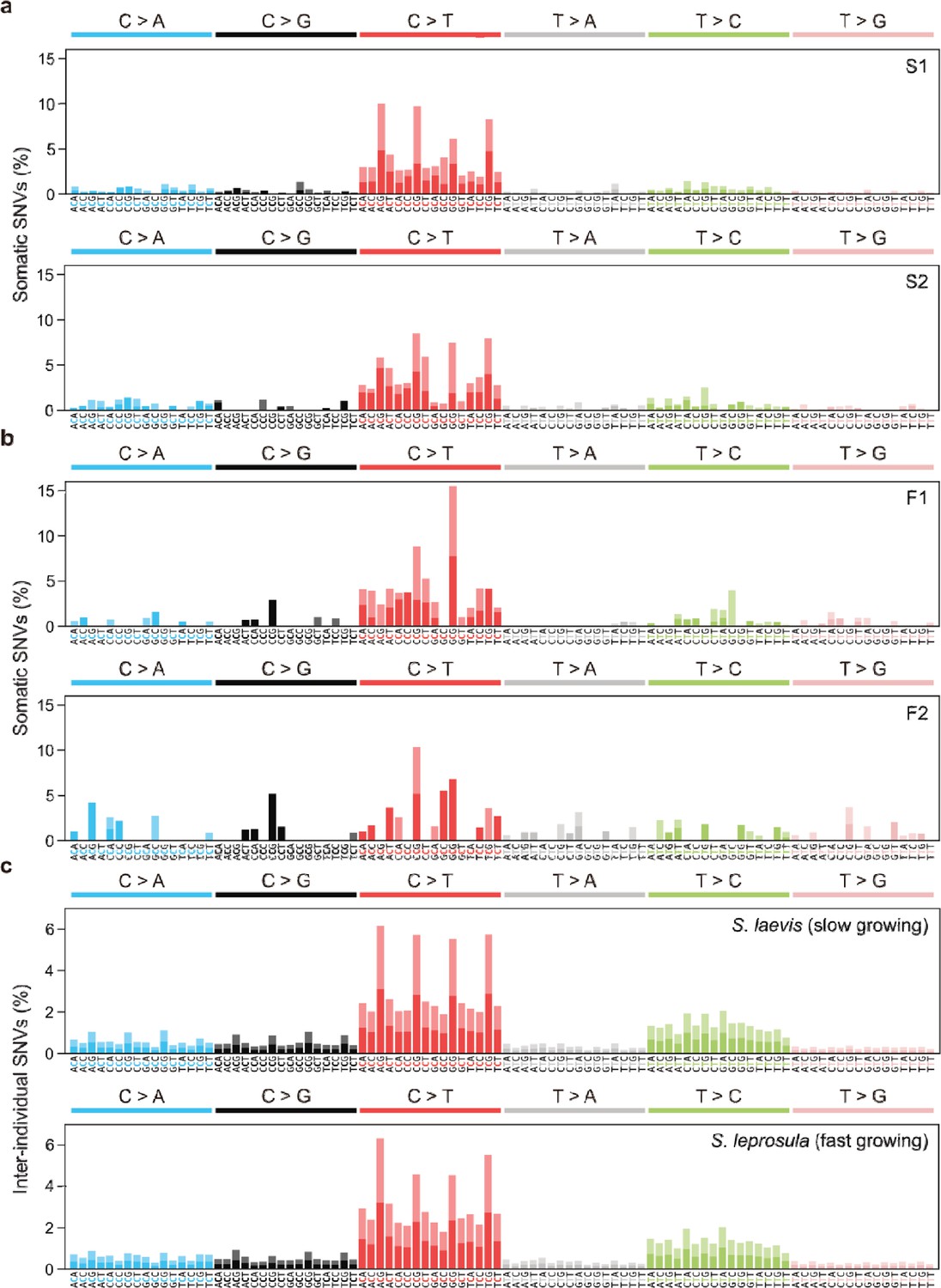
Mutational spectra of somatic and inter-individual substitutions.
(a) Somatic mutation spectra for S1 and S2 individuals in S. laevis. (b), Somatic mutation spectra for F1 and F2 individuals in S. leprosula. (c), Inter-individual SNVs between S1 and S2 (upper panel) and between F1 and F2 (lower panel). The horizontal axis shows 96 mutation types on a trinucleotide context, coloured by base substitution type. Different colours in each bar indicate complementary bases.
Figure 3—figure supplement 2
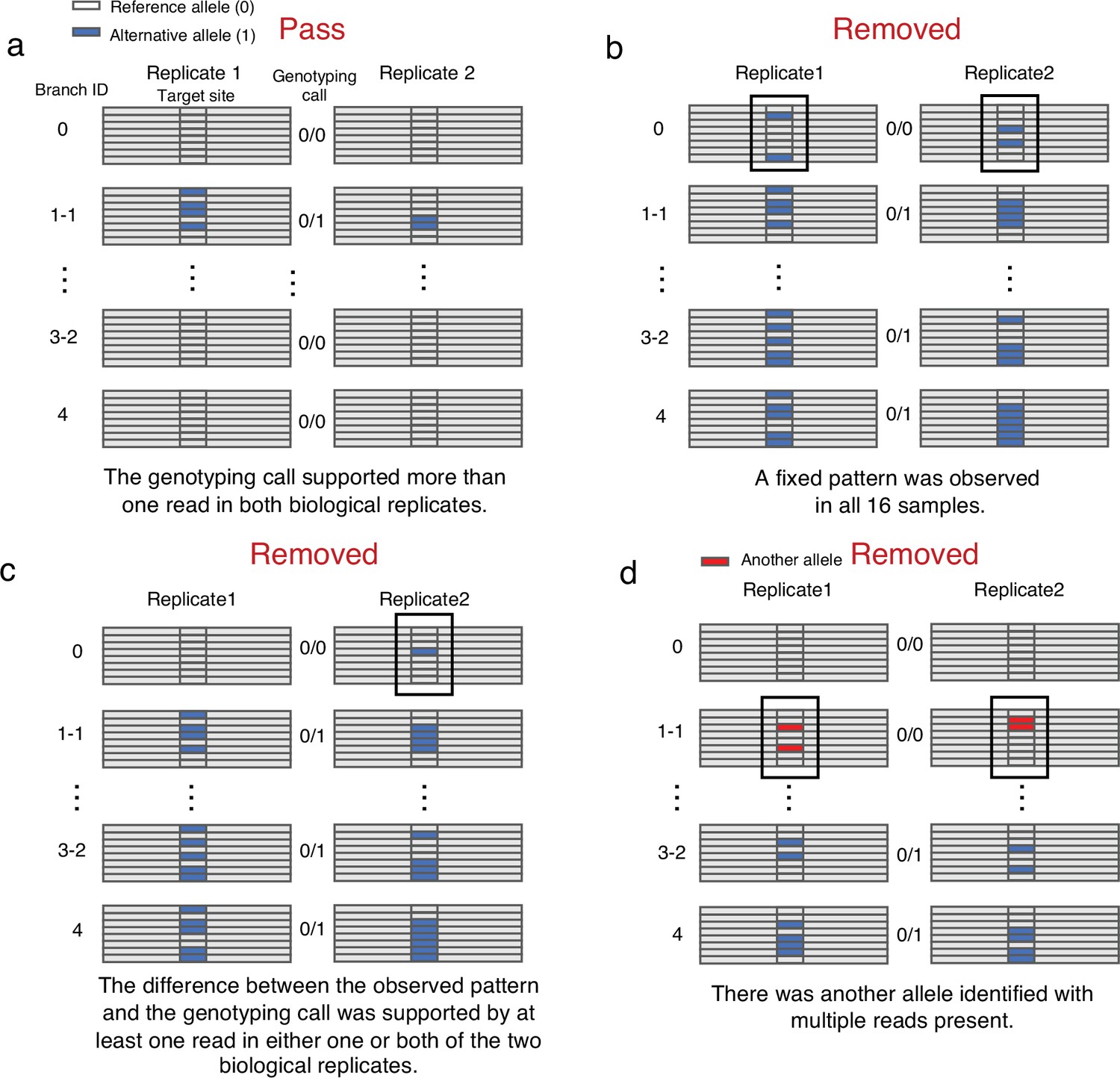
Manual confirmation of candidate SNVs.
(a) SNVs that passed manual confirmation. (b) SNVs that were removed due to their fixed heterozygote pattern. (c) SNVs that have been removed due to the difference between the observed pattern and the genotyping call. (d) SNVs that were removed due to the presence of another allele with multiple reads.
Figure 3—figure supplement 3

Proportion of potential false positive SNVs for S. laevis (S1, S2) and S. leprosula (F1, F2).
Potential false positive SNVs was identified as the subset of candidate SNVs that were not included in the final set for each threshold (T40, T30, and T20). This subset was then divided by the total number of potential SNVs at each threshold to determine the proportion.
Figure 3—figure supplement 4
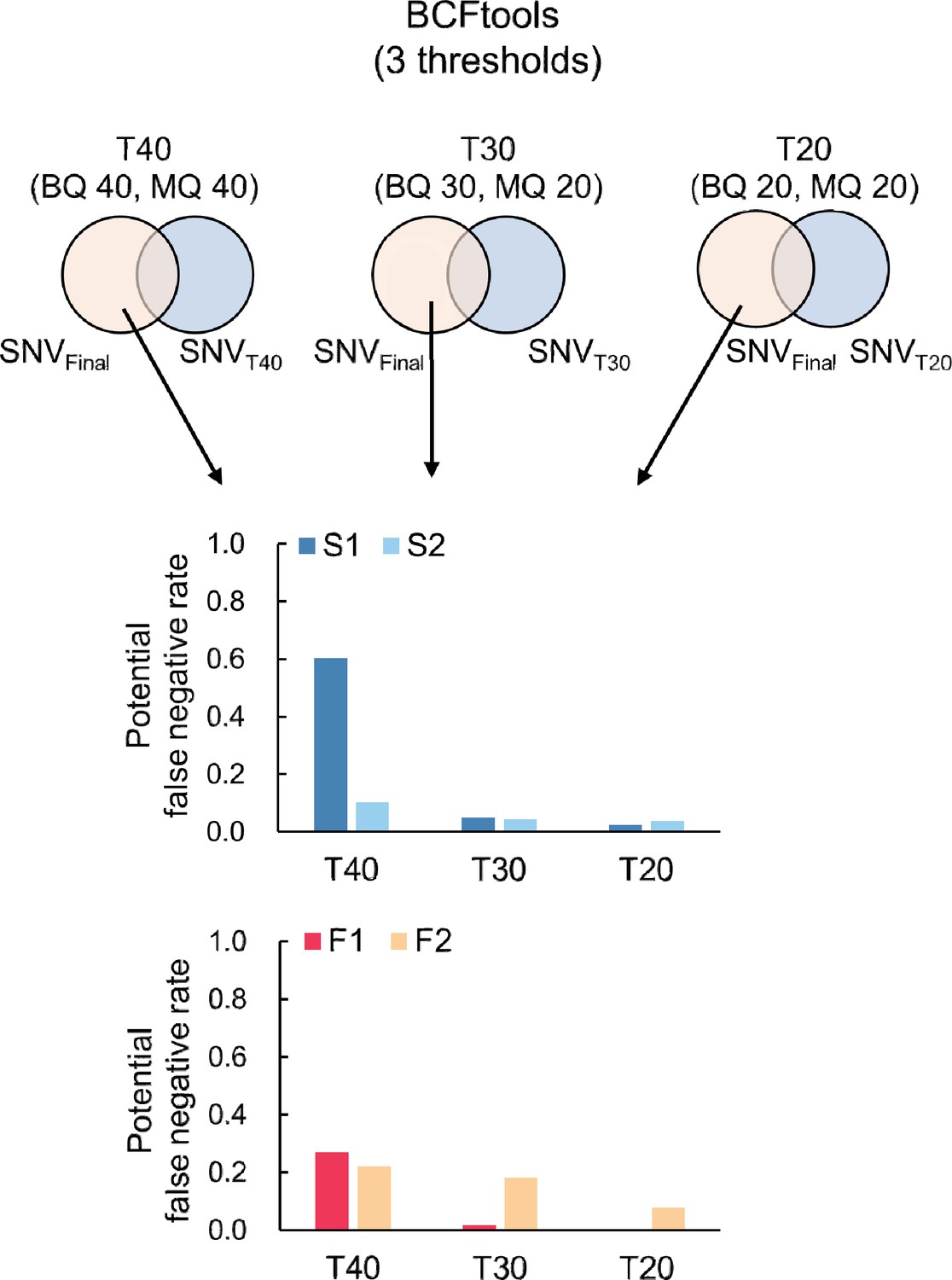
Proportion of potential false negative SNVs for S. laevis (S1, S2) and S. leprosula (F1, F2).
Potential false negative SNVs was identified as the subset of potential SNVs present in the final set but excluded from the candidate SNVs for each threshold (T40, T30, and T20). This subset was then divided by the total number of potential SNVs at each threshold to calculate the proportion.
Figure 4 with 1 supplement

Detecting selection on somatic and inter-individual SNVs.
(a) An illustration of somatic and inter-individual SNVs. Different colours indicate different genotypes. (b) Expected (Exp.) and observed (Obs.) rates of somatic non-synonymous substitutions. (c) Expected (Exp.) and observed (Obs.) rates of inter-individual non-synonymous substitutions. (d) The difference between the fractions of inter-individual and somatic substitutions spectra in S. laevis (upper panel) and S. leprosula (lower panel). The positive and negative values are plotted in different colours. The horizontal axis shows 96 mutation types on a trinucleotide context, coloured by base substitution type.
Figure 4—figure supplement 1
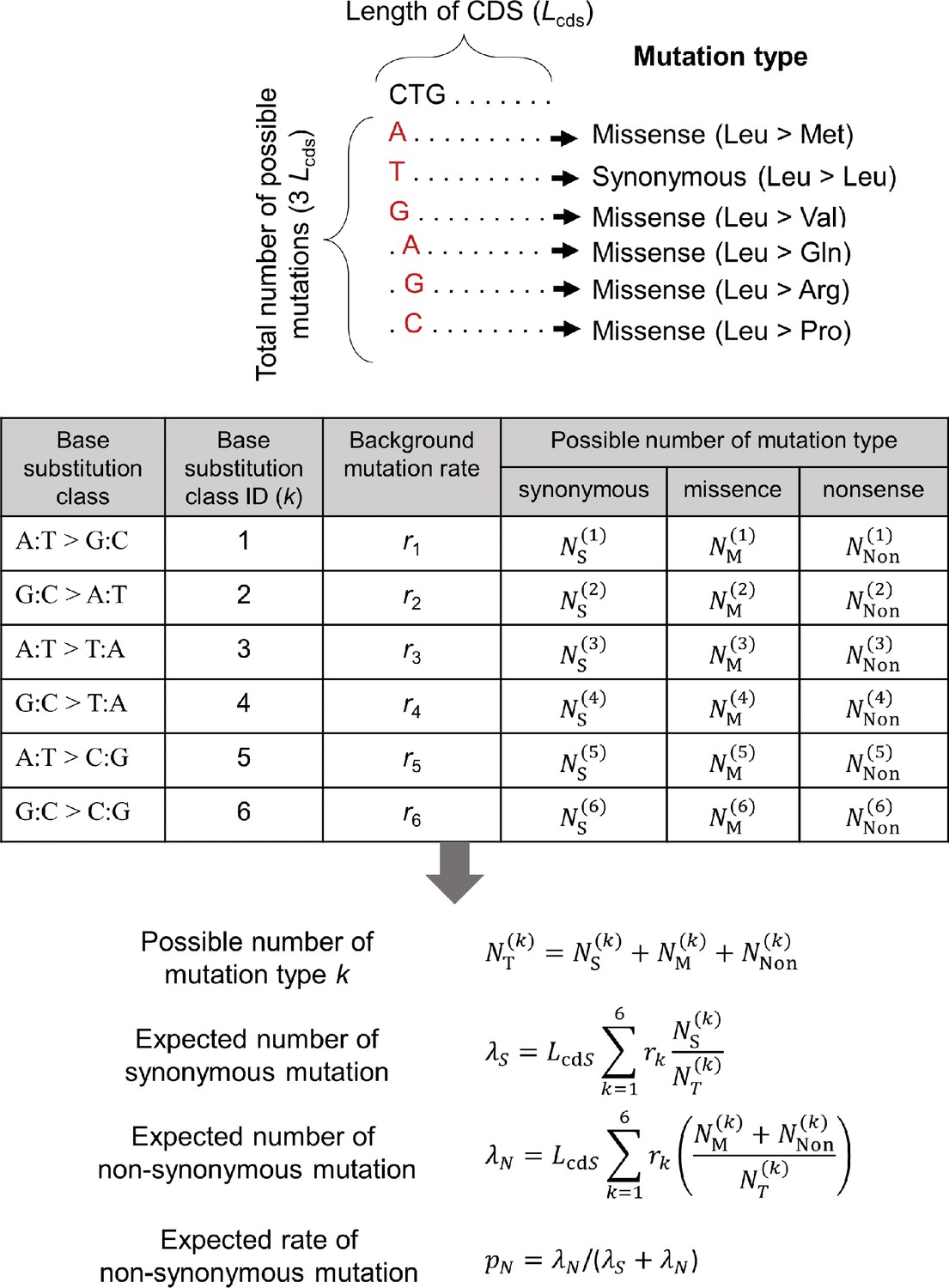
A calculation scheme for the expected rate of non-synonymous mutation.
The possible numbers of synonymous (, missense (, and nonsense ( mutations were counted for each of six base substitution classes from all possible mutations in CDS of length Lcds and used for the calculation of expected rate of non-synonymous mutation. For non-synonymous mutation, we pooled the number for missense and nonsense mutations. The background mutation rate for each substitution class i () is calculated from the observed somatic substitutions in intergenic regions.
Tables
Author response table 1
The branch-level increment of SNVs per meter.
| Branch ID | 0 | 1_1 | 12 | 2.1 | 22 | 3_1 | 32 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1-1 | 3.23 | ||||||||||||
| S. laevis | 1.2 | 5.18 | 13.32 | ||||||||||
| 2.1 | 4.01 | 4.67 | 6.42 | ||||||||||
| S1 | 2.2 | 3.78 | 4.42 | 6.19 | 7.18 | ||||||||
| 3.1 | 4.33 | 5.06 | 6.89 | 6.81 | 6.43 | ||||||||
| 3.2 | 4.30 | 5.02 | 6.85 | 6.75 | 6.38 | 10.82 | |||||||
| 4 | 4.14 | 4.77 | 6.43 | 5.95 | 5.86 | 7.43 | 7.41 | ||||||
| 1.1 | 2.37 | ||||||||||||
| 1.2 | 2.36 | 1.96 | |||||||||||
| 2-1 | 3.11 | 2.86 | 2.84 | ||||||||||
| latevis | 2.2 | 3.05 | 2.79 | 2.77 | 3.91 | ||||||||
| S2 | 3.1 | 2.61 | 2.23 | 1.86 | 3.45 | 3.36 | |||||||
| 3.2 | 2.59 | 2.20 | 1.84 | 3.40 | 3.31 | 3.92 | |||||||
| 4 | 2.77 | 2.43 | 2.41 | 3.73 | 3.63 | 3.74 | 3.67 | ||||||
| 1.1 | 1.21 | ||||||||||||
| 1.2 | 1.11 | 1.18 | |||||||||||
| - leprosula | 2-1 | 1.50 | 1.35 | 1.19 | |||||||||
| F1 | _(2)^(2)2 | 1.57 | 1.46 | 1.28 | 2.22 | ||||||||
| 3=1 | 1.27 | 1.05 | 0.96 | 1.41 | 1.52 | ||||||||
| 32 | 1.19 | 0.95 | 0.87 | 1.27 | 1.38 | 2.41 | |||||||
| 4 | 1.07 | 0.83 | 0.78 | 1.05 | 1.14 | 0.97 | 0.82 | ||||||
| 1.1 | 0.83 | ||||||||||||
| 12 | 0.55 | 2.07 | |||||||||||
| 2-1 | 0.34 | 1.10 | 0.54 | ||||||||||
| S. leprosula | 2.2 | 0.39 | 1.19 | 0.62 | 1.25 | ||||||||
| F? | 3+1 | 0.44 | 1.24 | 0.68 | 0.46 | 0.53 | |||||||
| 3.2 | 0.47 | 1.32 | 0.74 | 0.50 | 0.58 | 1.15 | |||||||
| 4 | 0.60 | 1.57 | 0.95 | 0.71 | 0.78 | 0.97 | 1.05 |
Additional files
-
Supplementary file 1
Supplementary tables.
(a) Mean annual increment (MAI) of diameter at breast height (DBH). (b) Matrix of physical distances (m) between sampling positions and the number of SNVs indicated in parentheses. (c) Summary statistics of the studied trees. Height and DBH were directly measured for two individuals of S. laevis and S. leprosula. Age was estimated as DBH divided by a mean annual increment (MAI). (d) Summary statistics of genome assemblies for S. laevis and S. leprosula. We assembled the genome using DNA extracted from the apical leaf at branch 1–1 of the tallest individual of each species (S1 and F1). Summary statistics of genome assemblies are listed here. (e) Summary statistics of whole genome sequencing. (f) The number of candidate SNVs during each step of the filtering process. (g) Assessment of candidate SNVs using amplicon sequencing. (h) Somatic mutation rates. The somatic mutation rate per nucleotide per meter was estimated as , where b indicates the slope of linear regression. The somatic mutation per nucleotide per year () was estimated as , where M indicates the total number of SNVs accumulated from the base to the branch tip and A represents tree age, respectively. R denotes the number of callable sites. (i) Cosine similarity of mutation spectra between Shorea trees and humans. (j) Results of the binomial test for selection on somatic and inter-individual SNVs. To test whether somatic and inter-individual SNVs are subject to selection, we calculated the expected rate of non-synonymous mutation. Given the observed number of non-synonymous and synonymous mutations, we rejected the null hypothesis of neutral selection using a binomial test with the significance level of 5%. pN_expected and pN_observed represent the expected and observed rate of non-synonymous substitutions. (k) The final set of SNVs. (l) Fractions of synonymous, missense, and nonsense substitutions. (m) Somatic mutation rates for six substitution classes. Somatic mutation rates for six substitution classes were calculated based on the observed number of SNVs both from the intergenic region and the whole genome. S1 +S2 and F1 +F2 represent the use of pooled data from two individuals for each species: S. laevis (S1, S2) and S. leprosula (F1, F2). The values based on the pooled data (indicated in bold type) were used to calculate the expected rate of non-synonymous mutation. (n) List of genes with somatic SNVs.
- https://cdn.elifesciences.org/articles/88456/elife-88456-supp1-v1.xlsx
-
MDAR checklist
- https://cdn.elifesciences.org/articles/88456/elife-88456-mdarchecklist1-v1.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Somatic mutation rates scale with time not growth rate in long-lived tropical trees
eLife 12:RP88456.
https://doi.org/10.7554/eLife.88456.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}