Flamingo participates in multiple models of cell competition
- Department of Pathology, Stanford University School of Medicine, United States
eLife Assessment
This study investigates the role of the Cadherin Flamingo (Fmi) in cell competition in developing tissues in Drosophila melanogaster. The findings are valuable in that they show that Fmi is required in winning cells in several competitive contexts. The evidence supporting the conclusions is solid, as the authors identify Fmi as a potential new regulator of cell competition, however, they don't delve into a mechanistic understanding of how this occurs.
https://doi.org/10.7554/eLife.98535.4.sa0Significance of the findings:
Valuable: Findings that have theoretical or practical implications for a subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Solid: Methods, data and analyses broadly support the claims with only minor weaknesses
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
The growth and survival of cells with different fitness, such as those with a proliferative advantage or a deleterious mutation, is controlled through cell competition. During development, cell competition enables healthy cells to eliminate less fit cells that could jeopardize tissue integrity, and facilitates the elimination of pre-malignant cells by healthy cells as a surveillance mechanism to prevent oncogenesis. Malignant cells also benefit from cell competition to promote their expansion. Despite its ubiquitous presence, the mechanisms governing cell competition, particularly those common to developmental competition and tumorigenesis, are poorly understood. Here, we show that in Drosophila, the planar cell polarity (PCP) protein Flamingo (Fmi) is required by winners to maintain their status during cell competition in malignant tumors to overtake healthy tissue, in early pre-malignant cells when they overproliferate among wildtype cells, in healthy cells when they later eliminate pre-malignant cells, and by supercompetitors as they compete to occupy excessive territory within wildtype tissues. ‘Would-be’ winners that lack Fmi are unable to overproliferate, and instead become losers. We demonstrate that the role of Fmi in cell competition is independent of PCP, and that it uses a distinct mechanism that may more closely resemble one used in other less well-defined functions of Fmi.
Introduction
Dividing cells in proliferative tissues must maintain tissue homeostasis and structural integrity as well as preserve their genetic integrity. One of the mechanisms operating in proliferative tissues to achieve these requirements is cell competition. Cell competition is a process in which cells of a higher fitness (‘winners’) eliminate less fit neighbors (‘losers’), by inducing cell death (Norman et al., 2012; Meyer et al., 2014; Di Giacomo et al., 2017; Watanabe et al., 2018) and/or extrusion from the epithelial layer (Norman et al., 2012; Kon et al., 2017; Kohashi et al., 2021). Cell competition was first recognized in 1975 (Morata and Ripoll, 1975), when Morata and Ripoll studied the growth of Drosophila carrying a mutation in the gene for the ribosomal protein RpS17 (termed minute). Animals heterozygous for this minute mutation are viable but develop slowly due to decreased proliferation rates. They further observed that clones of cells carrying this minute mutation in a wildtype background were eliminated from the Drosophila developing wing. Since then, extensive advances have been made in our understanding of the process (reviewed in Morata, 2021). We now know that cell competition can be triggered by mutations that cause a disadvantage, such as the aforementioned minute cells, and can also be triggered by mutations that confer a proliferative advantage, such as ectopic expression of proto-oncogenes such as Myc or Ras (Moreno and Basler, 2004; de la Cova et al., 2004; Hogan et al., 2009). In this case, the process is known as supercompetition, and wildtype cells behave as losers and are eliminated from the tissue by the mutant supercompetitors. The ‘survival of the fittest’ effect observed in cell competition has been shown to be critical during embryonic and larval development as well as for tissue homeostasis (Amoyel and Bach, 2014; Maruyama and Fujita, 2017; Morata, 2021). Much additional research has shown that cells and their neighbors are constantly evaluating their fitness, and that the active elimination of less fit cells is critical to maintain tissue integrity (Ellis et al., 2019), maintain chromosomal stability (Baillon et al., 2018; Ji et al., 2021), ameliorate effects of cellular aging (Merino et al., 2015), and suppress tumorigenesis (Kon et al., 2017; de Vreede et al., 2022).
While cell competition has been observed in many cell types, much attention has focused on studies in epithelia (Vincent et al., 2013). Polarity is a hallmark of epithelial tissues, and loss of polarity plays a prominent role in triggering cell competition and in the process of eliminating loser cells. Cells harboring mutations in apicobasal polarity genes have been shown to induce and/or regulate cell competition when surrounded by wildtype cells. In Drosophila, mutations in apicobasal polarity genes scribble (scrib), discs large (dlg), and lethal giant larva (lgl) all trigger cell competition when induced clonally (i.e. surrounded by wildtype cells), and the clones are eliminated from the epithelial tissue (Hariharan and Bilder, 2006; Morata and Calleja, 2020). However, clones that are deficient for any of these polarity genes can survive if they are competing against less fit cells, or if they carry additional mutations that confer a growth advantage. For example, when lgl clones, which would be eliminated when surrounded by wildtype cells, are made to express elevated levels of Myc, the competition is reversed, and they instead become winners (Froldi et al., 2010). This context dependence and fitness surveillance highlights the complexity of cell competition and its plasticity.
In epithelial tissues, cell competition is critical to suppress tumorigenesis, as polarity-deficient clones are prone to malignancy. When scrib mutant epithelial cells acquire elevated activity of a proto-oncogene such as the constitutively active RasV12, these clones acquire malignant properties of ectopic growth and invasiveness (Pagliarini and Xu, 2003; Brumby and Richardson, 2003). Surveillance and rapid elimination of cells that develop polarity defects therefore serves to protect epithelia from oncogenesis. This competitive mechanism is known as epithelial defense against cancer (EDAC) (Maruyama and Fujita, 2017; Kon and Fujita, 2021). Through EDAC, normal epithelial cells eliminate neighboring transformed cells by inducing apoptosis or by inducing their apical or basal extrusion from the epithelial layer (Watanabe et al., 2018; Kohashi et al., 2021). On the other hand, tumors that exhibit more aggressive properties and escape the defense mechanisms not only proliferate but actively engage in cell competition to promote their own growth and malignancy (Vishwakarma and Piddini, 2020; Madan et al., 2022). For example, growth of Drosophila APC-/- intestinal adenomas requires active elimination of wildtype cells, and inhibiting cell death in the wildtype tissue hampers adenoma growth (Suijkerbuijk et al., 2016).
In addition to apicobasal polarity, epithelial tissues are planar polarized. Planar cell polarity (PCP) signaling polarizes cells within the plane of the epithelium to orient cellular structures, cell divisions, and cell migration during development and homeostasis (Adler, 2002; Simons and Mlodzik, 2008; Vladar et al., 2009; Butler and Wallingford, 2017). Intercellular Fmi homodimers scaffold the assembly of a set of core PCP proteins: on one side of a cell, Frizzled (Fz), Dishevelled (Dsh), and Diego (Dgo) comprise a complex that interacts with another complex containing Van Gogh (Vang) and Prickle (Pk) on the opposite side of the adjacent cell. Fmi homodimers transmit differential signals in opposite directions to communicate the presence of either protein complex to the other, linking the proximal and distal complexes and mediating asymmetric signaling between them (Lawrence et al., 2004; Strutt and Strutt, 2007; Strutt and Strutt, 2008; Chen et al., 2008; Struhl et al., 2012). In addition to establishing planar polarity, numerous reports have suggested links between PCP signaling and cancer progression, promoting cell motility, invasiveness, and metastasis (Weeraratna et al., 2002; Katoh, 2005; Katoh and Katoh, 2007; Coyle et al., 2008; Gujral et al., 2014; VanderVorst et al., 2018; VanderVorst et al., 2023). Its roles, however, remain poorly characterized.
To probe a potential connection between PCP and cancer, we employed a Drosophila eye tumor model. We discovered a requirement for Fmi, but not other core PCP proteins, in tumor-associated cell competition. We found that aggressive tumors require Fmi to outcompete the neighboring wildtype tissue. We then found that this Fmi requirement is not unique to tumor competition; in several developmental cell competition scenarios, removing Fmi from winner cells prevents them from eliminating their neighbors and transforms them into losers. ‘Would-be’ winners that lack Fmi show both a decrease in proliferation and an increase in apoptosis. By several criteria, we show that this function for Fmi is independent of its role in PCP signaling.
Results
Flamingo is required for tumorigenesis in Drosophila RasV12 tumors
Planar polarity has been linked to tumorigenesis in several organisms, tumor models, and patient tumor samples (Kaucká et al., 2013; Asad et al., 2014; Puvirajesinghe et al., 2016; VanderVorst et al., 2018; Chen et al., 2021b; Li et al., 2021; Chen et al., 2021a). We explored how the core PCP complex might be involved in tumorigenesis. To do so, we used the versatile and widely used RasV12 tumor model in Drosophila eye imaginal discs. In this model, eye disc cells are transformed into highly tumorigenic cells by co-expressing the constitutively active RasV12 oncoprotein and RNAi against the tumor suppressor Scribble (Scrib) (Pagliarini and Xu, 2003). Cells expressing these genes quickly expand into massive tumors and invade the neighboring brain tissue (Figure 1A–C).
Figure 1 with 2 supplements see all
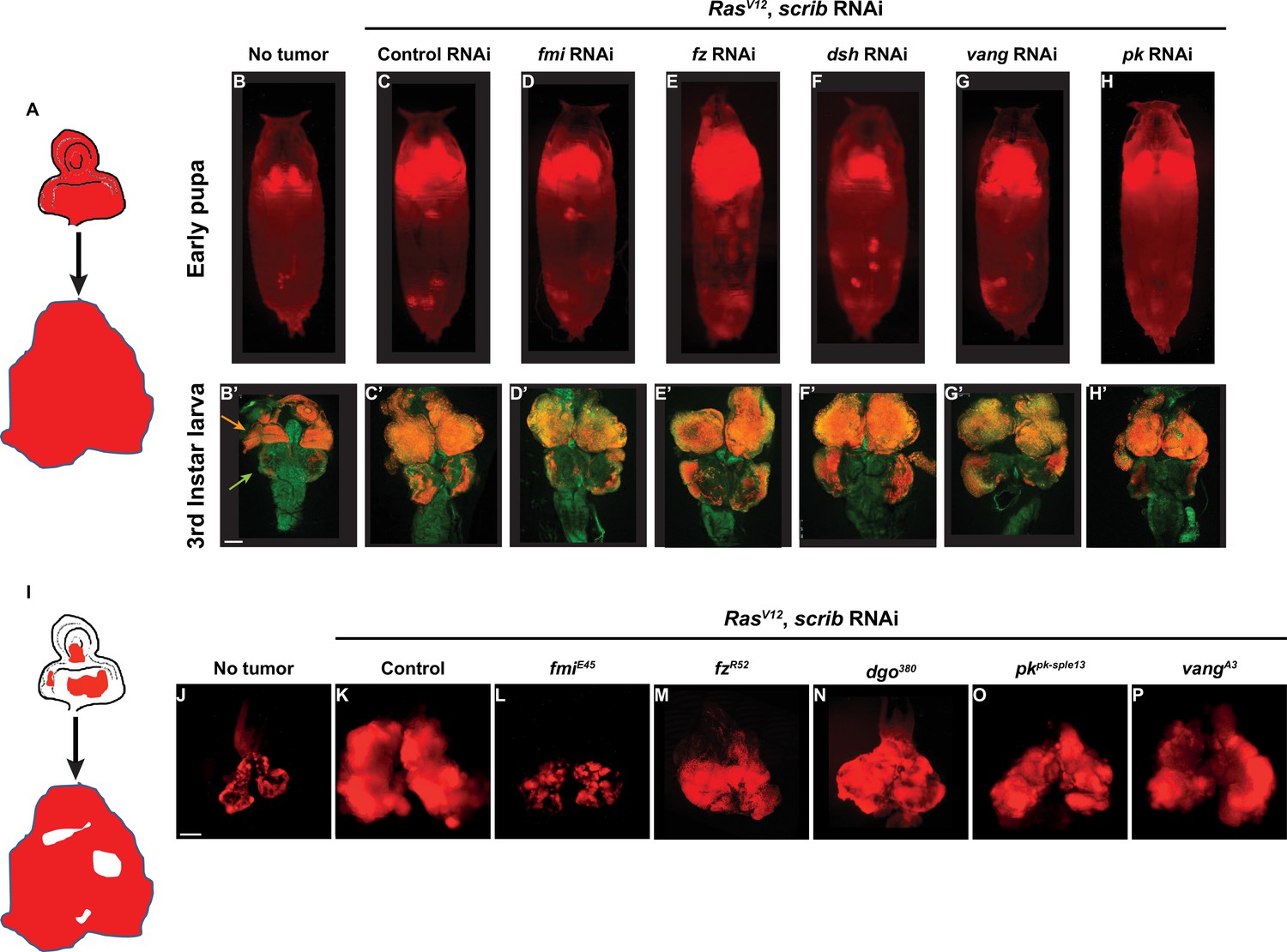
Fmi is required in clonal tumors to outcompete wildtype tissue.
(A) Schematic of the whole eye disc RasV12, scrib RNAi, RFP tumors. (B–H) Pupal RFP tumors imaged through the cuticle. All pupae are ey-Flp; act5C>CD2>Gal4, UAS-RFP with the indicated RNAi. N=5 (no tumor), 10 (control RNAi), 13 (fmi RNAi), 17 (fz RNAi), 19 (dsh RNAi), 17 (vang RNAi), and 8 (pk RNAi). (C) Control UAS-RasV12, UAS-scrib RNAi, UAS-w RNAi tumors. (D–H) UAS-RasV12, UAS-scrib RNAi tumors co-expressing UAS-RNAi against the planar cell polarity (PCP) genes indicated above each pupa. (B’–H’) Representative third instar larval brain and eye discs for each of the experimental groups from above. Arrowheads in B’ point to the eye-antenna imaginal disc (orange) and the brain lobes (green). N=3 (no tumor), 10 (control RNAi), 13 (fmi RNAi), 12 (fz RNAi), 15 (dsh RNAi), 20 (vang RNAi), and 10 (pk RNAi). (I) Schematic of eye disc RasV12, scrib RNAi, RFP clonal tumors. (J–P) Third instar larval eye discs RFP tumor clones generated via ey-Flp; FRT42D Gal80/FRT42D; act5C>CD2>Gal4, UAS-RFP. (J) Eye discs with non-tumor, control RFP clones. (K) Control RasV12, UAS-scrib RNAi, RFP clonal tumors. (L–P) RasV12, scrib RNAi, RFP clonal tumors carrying the PCP allele indicated above each panel. Scale bars: 100 μm.
To study the requirement for PCP signaling in tumors, we expressed RasV12 and scrib RNAi throughout the eye, and simultaneously downregulated PCP core proteins by also expressing validated RNAis against fmi, fz, dsh, vang, and pk. We then imaged tumor size through the pupal cuticle. We observed that tumor growth was unaffected by knockdown of any of the core PCP genes at third instar larval or at early pupal stages (Figure 1A–H). We then asked whether PCP signaling might be required by tumors when they’re confronted by wildtype cells. To test this hypothesis, we created clonal tumors in the eye disc by expressing RasV12 and scrib RNAi under control of ey-Flp. In this model, nearly every cell in the eye disc will perform chromosomal recombination (Figure 1—figure supplement 1), producing an eye disc composed of approximately 50% wildtype and 50% tumor cells. Under these conditions, tumor cells expand aggressively, displacing wildtype cells and causing massive overgrowth of the eye disc and metastasis to the larval brain (Figure 1I–K). We created clonal tumors that were also null for either fmi, fz, dgo, vang, or pk and evaluated them for their ability to grow and metastasize when confronted with wildtype cells (Figure 1I–P). Remarkably, only fmi interfered with tumorigenesis, restraining growth of tumors in the eye disc and preventing them from invading neighboring tissues (Figure 1L). To ensure that the growth inhibitory effect of loss of Fmi in clonal tumors but not in whole eye tumors was not a result from comparing a null allele in clonal tumors vs RNAi in whole eye tumors, we generated fmi RNAi clonal tumors and compared them to control RNAi clonal tumors. While the effect was not as dramatic as with null clones, the size of fmi RNAi clonal tumors was substantially reduced compared to that of clonal tumors expressing w RNAi (Figure 1—figure supplement 1A and B). Therefore, the same partial level of fmi knockdown impairs clonal tumor growth but not growth of whole eye tumors.
We also wished to directly compare growth of fmi-null clonal and fmi-null whole eye tumors. Because fmi alleles are embryonic lethal, we generated fmi-null tumors while eliminating WT cells from the eye disc using the GMR-Hid, l(2)CL system (Stowers and Schwarz, 1999). While fmi-null clonal tumors showed restrained growth (Figure 1L), elimination of WT cells in eye discs removed competition, and fmi-null tumors grew to a size comparable to control whole eye tumors (Figure 1—figure supplement 1C and D). Because the effect of fmi was only apparent when tumors were induced in juxtaposition to wildtype cells, we hypothesized that Fmi may only be required in tumors that undergo cell competition.
Flamingo is required in winner cells during cell competition
Molecular mechanisms used by tumors in cell competition are sometimes shared by developmental cell competition. Malignant cells have been shown to actively eliminate surrounding cells by mechanical cell competition, apoptosis, and engulfment (Levayer et al., 2016; Kohashi et al., 2021), similar to the cell death-mediated elimination of losers during developmental cell competition and EDAC (Maruyama and Fujita, 2017; Parker et al., 2021) Induction of JNK-mediated cell death is also common to developmental competition and tumor cell competition, as shown in intestinal adenomas (Suijkerbuijk et al., 2016). We hypothesized that the requirement of Fmi by competing tumors might also be shared in developmental cell competition. We therefore evaluated the role of Fmi in non-malignant, developmental cell competition models.
Cells expressing high levels of Myc behave as winners when growing among wildtype cells, proliferating faster than wildtype cells and inducing their elimination. Despite Myc being a proto-oncogene, in contrast to RasV12 scrib RNAi tumors, Myc overexpressing clones do not become tumors, but instead expand to occupy larger than normal domains of morphologically normal tissue (Moreno and Basler, 2004; de la Cova et al., 2004; Clavería et al., 2013). As for the RasV12 scrib RNAi tumor model, we used UAS-myc and ey-Flp to create eye discs comprising initially equal populations of Myc overexpressing (>>Myc) and wildtype cells. As expected, >>Myc cells behaved like supercompetitors, and the majority of cells (~65%) in late third instar discs were >>Myc cells (Figure 2A). However, when >>Myc cells were depleted of fmi, they failed to outcompete wildtype cells and behaved as one would expect of losers; the >>Myc, fmi-/- cells were outcompeted by wildtype cells such that late third instar eye discs were composed of only ~35% >>Myc, fmi-/- cells, suggesting that wildtype cells behave as winners against >>Myc, fmi-/- cells (Figure 2B and D). We then tested whether Fmi had an effect in the losers during >>Myc competition. Removing Fmi from the WT losers had no effect on this competition, as >>Myc clones did not compete more successfully than when confronting wildtype cells (Figure 3C and D). Importantly, loss of Fmi alone did not induce competition, as in eye discs comprising initially equal populations of fmi-/- and wildtype cells the fmi mutant cells grew equally well as the wildtype cells (Figure 2—figure supplement 1A–C).
Figure 2 with 1 supplement see all
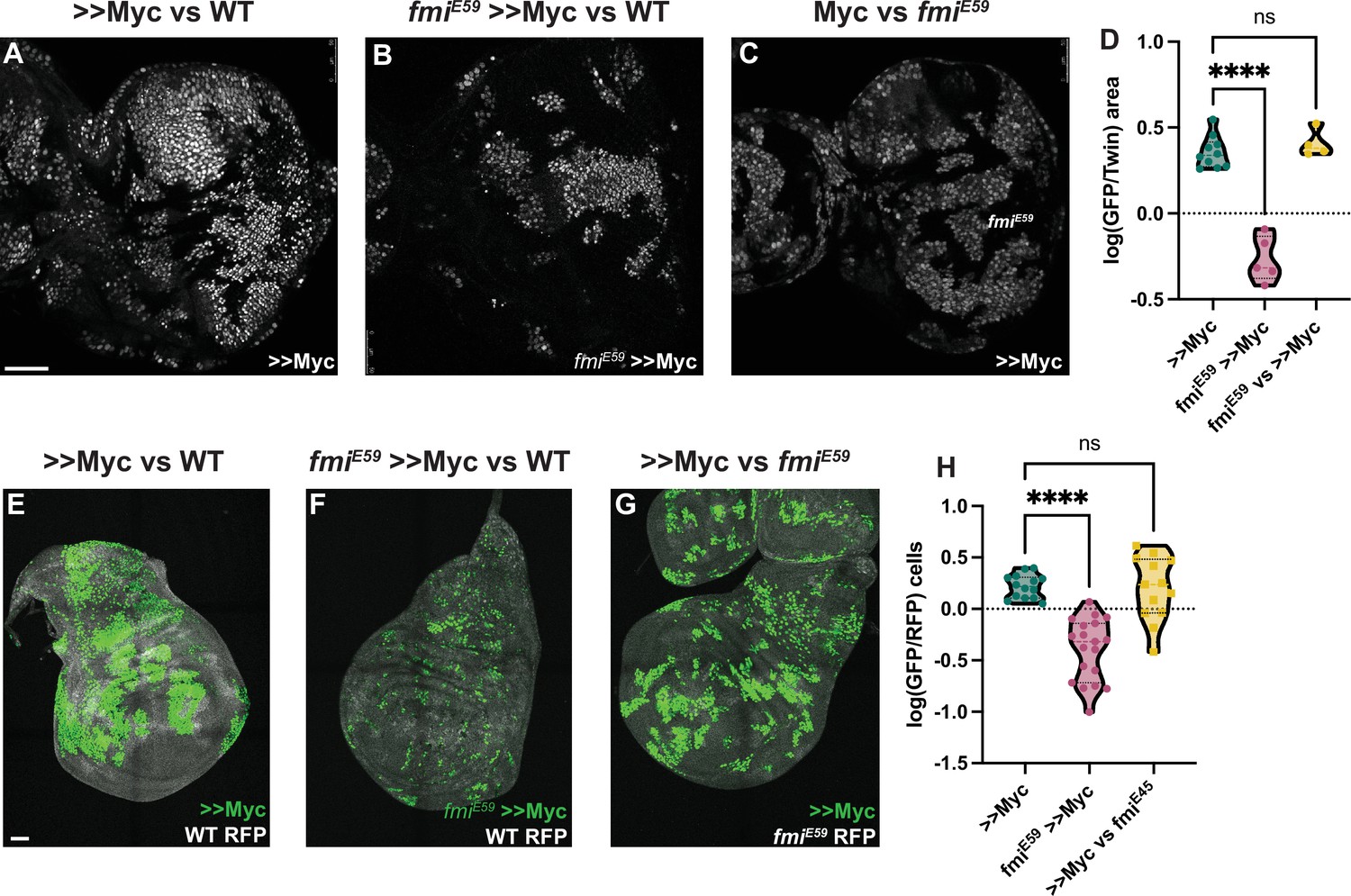
Winners require Fmi in developmental supercompetition.
(A) Eye imaginal disc clones overexpressing UAS-Myc (>>Myc) and UAS-nGFP under control of the tub >CD2>GAL4 driver. Clones were generated with ey-Flp, so half of the cells are clones and the other half are wildtype twins. (B) Eye imaginal disc >>Myc, UAS-nGFP, fmiE59 clones, competing against wildtype twins. (C) Eye imaginal disc >>Myc, UAS-nGFP clones, competing against fmiE59 twins. (D) Ratio of RFP-labeled clone area vs unlabeled wildtype area. The unlabeled wildtype area was obtained by subtracting the RFP-labeled area from the total eye disc area. A ratio over 1 implies RFP-positive clones are over-represented, likely behaving as supercompetitors, while a ratio below 1 means the RFP cells are under-represented, likely losers. N=9 discs (>>Myc), 5 discs (fmiE59,>>Myc), 4 discs (fmiE59 vs >>Myc), groups were analyzed using multiple unpaired, two-tailed t-test; p-values:<0.0001 (****), 0.6340 (ns). (E) Representative wing imaginal disc overexpressing >>Myc and UAS-nGFP under control of the tub-Gal4 driver. Clones were generated using hsp70-Flp, with a 15 min 37°C heat-shock. Wildtype cells are labeled with tub-nRFP. Non-recombinant cells are heterozygous for nRFP, while twin spots are homozygous nRFP. n=14 discs. (F) Representative wing imaginal disc with >>Myc, UAS-nGFP, fmiE59 clones, over a wildtype background. Clones were generated in the same fashion as D. n=19 discs. (G) Representative wing imaginal disc with >>Myc, UAS-nGFP clones, competing against fmiE59 twin spots. n=13 discs. (H) log10 of the >>Myc/Twin spot cell ratio. nGFP total cells were divided by the number of twin spot homozygous nRFP cells, and then log10-transformed. A ratio over 0 indicates over-representation (likely supercompetition) of nGFP+ clones, and below 0 indicates nGFP+ cells are under-represented (likely behaving as losers). The difference between groups was analyzed using a one-way ANOVA, with Dunnett correction for multiple comparisons; p-value: >>Myc vs fmiE59 >>Myc < 0.0001 (****); >>Myc vs fmiE59=0.9705 (ns). Scale bars: 50 μm.
Figure 3 with 1 supplement see all
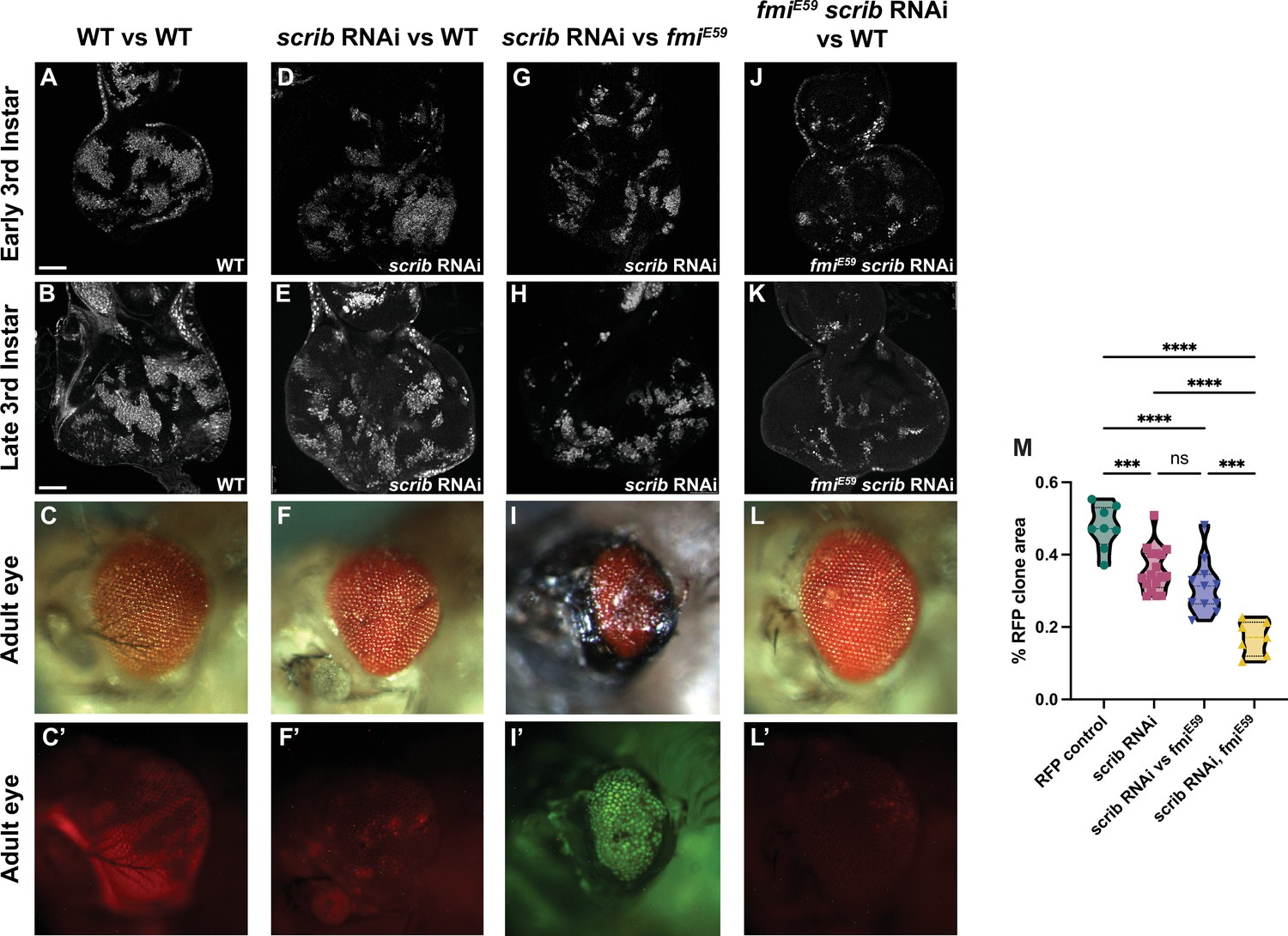
Winners require fmi in scribble cell competition.
(A–L’) scrib RNAi clone analysis in the prospective eye. UAS-scrib RNAi was expressed with the act5C>CD2>Gal4 driver and clones were generated by ey-Flp. Clones are marked both with UAS-nGFP and tub-nRFP. (A) Representative early third instar disc with control UAS-nGFP clones. (B) Representative late third instar disc with control UAS-nGFP clones. (C, C’) Representative adult eye with control UAS-nGFP clones. C’ shows fluorescently labeled control clone cells. (D) Representative early third instar disc with UAS-scrib RNAi clones vs wildtype twin clones. (E) Representative late third instar disc with UAS-scrib RNAi clones vs wildtype twin clones. (F, F’) Representative adult eye with UAS-scrib RNAi clones vs wildtype twin clones. F’ shows fluorescently labeled surviving UAS-scrib RNAi cells. (G) Representative early third instar disc with UAS-scrib RNAi clones vs fmiE59 twin clones. (H) Representative late third instar disc with UAS-scrib RNAi clones vs fmiE59 twin clones. (I, I’) Representative adult eye with UAS-scrib RNAi clones vs fmiE59 twin clones. This phenotype was lethal. The adult eye shown was from an escaper. Escapers had trouble eclosing and died within hours. I’ shows fluorescently labeled surviving scrib RNAi cells. tub-nRFP fluorescence was barely visible, so the stronger UAS-nGFP is shown. (J) Representative early third instar disc with fmiE59, UAS-scrib RNAi clones vs wildtype twin clones. (K) Representative late third instar disc with fmiE59, UAS-scrib RNAi clones vs wildtype twin clones. (L–L’) Representative adult eye with fmiE59, UAS-scrib RNAi clones vs wildtype twin clones. L’ shows fluorescently labeled surviving fmiE59, UAS-scrib RNAi cells. (M) Quantification of fluorescently tagged clone area for the phenotypes mentioned above. Each dot in the violin plot displays the ratio of RFP+ area vs total disc area. Of special significance is the fact that scrib RNAi clones competing with wildtype clones survive similarly to scrib RNAi clones competing with fmiE59. p-Values are as follows: ****<0.0001; ***<0.0005; ns = 0.2634. Scale bars: 50 μm.
If the requirement of Fmi is a general feature of cell competition, it should be observable in other tissues. We therefore assessed whether depleting Fmi in wing disc >>Myc winner clones would also reverse the competition outcome. We used hsp-Flp to generate twin spot clones expressing UAS-Myc, and as in eye discs, those clones behaved like supercompetitors. Myc clones were on average 1.7 times larger than the homozygous RFP+ (hRFP) twin spots (Figure 2E). However, when >>Myc clones lacked Fmi, their ability to compete was severely impaired, producing much smaller clones, being on average half as large as their hRFP twin spots, and thus, like in eye discs, behaved as one would expect of losers when competing against wildtype cells (Figure 2F–H). Interestingly, fmiE59 Myc clones were also highly fragmented in the wing disc, a phenomenon we did not observe in eye discs. As observed in eye discs, removing Fmi from the wildtype losers had no effect on the ability of >>Myc clones to compete (Figure 3G and H). Similarly, making clones mutant for fmi alone had no effect on their growth in wing discs (Figure 2—figure supplement 1D–F).
In the setting of tumorigenesis, either tumor cells or wildtype cells may emerge as winners of cell competition. Tumors outcompete wildtype cells in the processes of invasion and metastasis, whereas transformed pre-malignant cells are often eliminated by wildtype cells through competition-dependent defense mechanisms such as EDAC (Kon et al., 2017; Kon and Fujita, 2021). We therefore further explored the potential requirement for Fmi in a system in which both outcomes occur at different times. In eye discs, cells lacking Scrib display a proto-oncogenic behavior, losing polarity and overproliferating in the developing eye disc. However, as eye development progresses, they subsequently become losers, and are eliminated via JNK-mediated apoptosis during late third instar and pupal development so that they are virtually absent from the tissue before the fly ecloses (Brumby and Richardson, 2003).
Using the ey-Flp system to activate RNAi against scrib in eye discs, we confirmed that downregulation of scrib generated clones that perdure through larval development (Figure 3D , E, and M) but were eliminated from the tissue by the end of pupal development and were not detected in the adult eye (Figure 3F and F’). Compared to control RFP clones, which represented around 40% of the eye cell population in early third instar larva (Figure 3A, B, and M, Figure 3—figure supplement 1A) and remained at roughly that proportion in the adult eye (Figure 3C and C’), scrib RNAi clones began to be eliminated at or before the time the morphogenetic furrow progresses (Figure 3E and M, Figure 3—figure supplement 1B), and were almost completely absent in adult eyes (Figure 3F and F’). Their elimination left scars in the adult eye, likely because the structure of the eye is established with the passing of the morphogenetic furrow, and the compound eyes were smaller compared to wildtype eyes (Figure 3C and F). Wildtype cells therefore behave as winners when confronted with scrib RNAi clones during late larval and pupal development.
We then evaluated the effect of removing Fmi from the wildtype winner clones in scrib cell competition. Loss of fmi in wildtype cells had little impact on the size of scrib RNAi clones in third instar larval discs (Figure 3G, H, and M, Figure 3—figure supplement 1C), but as development progressed, wildtype winner cells depleted of fmi lost their ability to compete with and eliminate scrib RNAi clones; the scrib RNAi clones survived during pupal development and indeed constituted the majority of the adult eye (Figure 3I and I’), showing a reversal in the competition outcome much as the outcome of >>Myc competition is reversed when the winners lack Fmi. The surviving scrib RNAi cells failed to differentiate, and instead produced large scars in the adult eye (Figure 3I and I’). We also observed considerable lethality in this population of flies, presumably due to the uncontrolled proliferation of scrib RNAi cells that could not be eliminated.
During >>Myc supercompetition, removing Fmi from the loser wildtype cells showed no effect in cell competition in eye and wing discs (Figure 2C and G). We therefore considered what might happen if scrib RNAi loser clones are made to lack Fmi. However, this situation is more complex than the >>Myc competition, since scrib RNAi clones have been previously documented to initially overproliferate, perhaps behaving as winners, before ultimately becoming losers (Brumby and Richardson, 2003). We were unable to quantify this early proliferation of scrib RNAi eye clones before they are eliminated by their wildtype neighbors, as the discs are too small for us to dissect and count. However, assuming that they are behaving as winners in early larval development, one might expect that removing Fmi from scrib RNAi clones would impair their ability to overproliferate early, such that by third instar, they would be smaller than scrib RNAi clones with intact Fmi. Indeed, this is what we observed (Figure 3J, K, and M, Figure 3—figure supplement 1). While some portion of their smaller size relative to control clones is attributed to their elimination by wildtype winners as described above, the remainder may be a result of the loss of their ability to overproliferate earlier in larval development.
The small population of fmiE59 scrib RNAi clones remaining at third instar was almost completely eliminated by the end of larval development, such that the adult eyes were indistinguishable from wildtype eyes (Figure 3L and L’). We hypothesize that the lack of fmi in scrib clones hinders their ability to overproliferate in early larval stages, resulting in smaller clones that are quickly eliminated by their wildtype neighbors, allowing compensatory differentiation to generate a morphologically normal eye.
Loss of fmi triggers cell death and reduces proliferation in ‘would-be’ winner clones
Our observations suggest that lack of fmi renders winner clones and tumors incapable of outcompeting the neighboring tissue in multiple cell competition scenarios. Cell competition relies mainly on two mechanisms to allow winner cells to take over the tissue when confronting less fit cells: faster proliferation than losers and elimination of loser cells by induced apoptosis or extrusion (Amoyel and Bach, 2014; Maruyama and Fujita, 2017; Morata, 2021). We therefore explored how the lack of fmi in tumors and winner >>Myc cells during competition affected both mechanisms.
Drosophila control RasV12, scrib RNAi eye tumors trigger apoptosis in the neighboring cells, as detected by Dcp1 staining (Figure 4A–D), consistent with previous reports (Karim and Rubin, 1998; Pérez et al., 2017) and with their highly invasive behavior when surrounded by wildtype cells. However, when fmi was removed from the clonal tumors, the outcome of this competition was reversed, as described above, and instead, fmi-/-, RasV12, scrib RNAi tumors displayed excess apoptosis compared to the surrounding wildtype tissue (Figure 4E–H). Moreover, we found cell debris from fmi-deficient tumor cells inside lysosomal vesicles in wildtype cells, suggesting that wildtype cells are clearing neighboring loser tumor cells through engulfment (Figure 4—figure supplement 1). We then tested whether this reversal of apoptosis burden was specific to the tumor model, or if it is a mechanism shared in other cell competition models. We counted apoptotic cells in WT and >>Myc clones in the eye disc only when these cells were located near to the clone boundary (1–3 cells from the boundary, see Materials and methods for details). We observed that control >>Myc clones showed low levels of apoptosis and similar levels in their neighboring WT cells (Figure 4I–L; p-value 0.6049), in line with previous results (de la Cova et al., 2004), whereas, consistent with the tumor model, apoptosis was significantly increased in fmiE59, >>Myc clones but not in their WT neighbors (Figure 4M–P, p-value 0.0006), likely contributing to the reversal in competition we previously observed (Figure 2A–D).
Figure 4 with 2 supplements see all
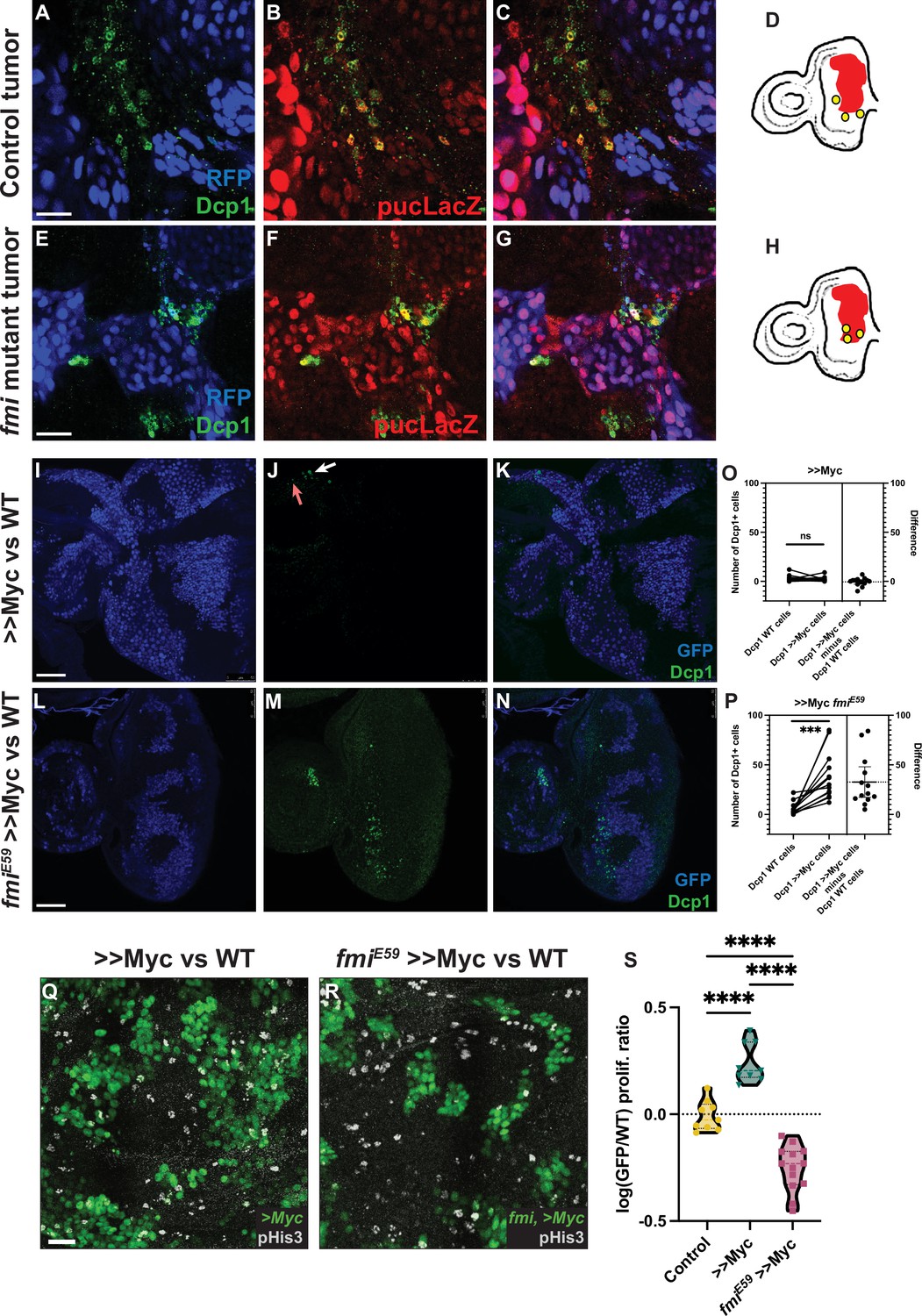
Lack of Fmi increases cell death and reduces proliferation in would-be winners.
(A–C) RasV12, scrib RNAi tumors stained for DAPI, Dcp1+ (A), and puc-lacZ (B). (C) Merged channels. (D) Representation of how Dcp1+ staining localizes in the wildtype (WT) cells at the boundary with the tumor. (E–G) RasV12, scrib RNAi tumors mutant for Fmi, stained with DAPI, Dcp1+ (E), and puc-LacZ (F). (G) Merged channels. (H) Representation of how Dcp1+ staining localizes in the tumor cells in contact with the surrounding WT tissue. Scale bar for A–G: 25 μm. (I–K) Dcp1+ staining in >>Myc clones in eye discs. >>Myc clones are marked by GFP (I) and were stained against Dcp1+ (J). The arrows show apoptotic WT (red arrow) and >>Myc (white arrow) cells at the clone boundary. (K) Merged channels, showing apoptotic cells evenly distributed between WT and >>Myc cells. (L–N) Dcp1+ staining in >>Myc clones lacking Fmi in the eye disc. Eye disc >>Myc clones are marked by GFP (L) and were stained for Dcp1+ (M, N) Merged channels, showing apoptotic cells localized mainly in the >>Myc, fmiE59 clones. Scale bar: 50 μm. (O) Quantification of apoptotic cells in WT vs >>Myc clones in eye discs. Apoptosis occurs similarly in WT and >>Myc cells (two-tailed paired t-test; p-value = 0.6049). The left side of the graph shows the number of apoptotic WT and >>Myc cells. Each imaginal disc is displayed as a pair of dots, linked by a line, to easily visualize the Dcp1+ apoptotic cells in WT vs >Myc cells. Dots represent the number of apoptotic WT (left) or >>Myc (right) cells per disc. The right side of the graph displays the difference (>>Myc minus WT apoptotic cells). The dashed line indicates the mean difference between those values for all samples. N=14 discs. (P) Quantification of apoptotic cells in WT vs >>Myc, fmiE59 clones in eye discs. Apoptosis is found mainly in >>Myc, fmiE59 cells (two-tailed paired t-test; p-value = 0.0006). The left side of the graph shows the number of apoptotic WT and >>Myc, fmiE59 cells, side by side. Dots represent the number of apoptotic >>WT (left) or >>Myc, fmiE59 (right) cells per disc. The right side of the graph displays the difference (>>Myc, fmiE59 minus WT apoptotic cells). The dashed line indicates the mean difference between those values for all samples. N=14 discs. (Q–R) Proliferation analysis performed by pHis3 staining in wing discs with either >>Myc clones (Q) or >>Myc, fmiE59 clones (R). Scale bar: 20 μm. (S) Proliferative ratio of GFP cells in a non-competition Control (n=9 discs), >>Myc (n=9 discs), or >>Myc, fmiE59 (n=13 discs) clones. The proliferative ratio for each group was calculated as the ratio of pHis3 cells within the GFP+ clone vs the non-GFP WT tissue and the differences were analyzed as an ordinary ANOVA with a Tukey’s test for multiple comparisons, with all p-values<0.0001 (****).
JNK signaling plays a prominent role during cell competition. Previous reports have shown that activation of JNK mediates engulfment and elimination of scribble clones during cell competition (Ohsawa et al., 2011; Yamamoto et al., 2017). However, JNK signaling is also responsible for increased tumor growth and invasion in Ras-activated cells or scrib mutants (Igaki et al., 2006; Uhlirova and Bohmann, 2006; Leong et al., 2009). We therefore asked whether the role of Fmi in tumorigenesis and cell competition could be related to the regulation of JNK signaling. Confirming previous observations, we detect activation of JNK signaling via the puckered lacZ reporter pucE69 both in RasV12, scrib RNAi tumors (Figure 4A–C) and scrib RNAi clones in eye discs (Figure 4—figure supplement 2A–C). However, RasV12 tumors lacking fmi, despite displaying cell death in tumor cells competing with wildtype neighbors, show no change in Puc activation (Figure 4E–G). Similarly, scrib RNAi clones activate JNK signaling independently of fmi (Figure 4—figure supplement 2D–F). We therefore conclude that Fmi acts independently of JNK signaling.
Cell competition does not rely solely on apoptosis to eliminate loser cells. An increased proliferation rate of winners is also a hallmark of cell competition (Morata, 2021; Madan et al., 2022). To evaluate whether Fmi is also required to maintain a higher proliferative ratio in winners, we quantified mitotic cells in winner clones with and without Fmi. The wing disc displays distributed cell divisions throughout larval development, in contrast to eye discs, where passing of the morphogenetic furrow limits proliferation as cells start differentiating into compound eye cells (Wolff and Ready, 1991; Tsachaki and Sprecher, 2012). Therefore, for the quantification of proliferation, the wing is a better model than the eye disc. When wing disc >>Myc clones were depleted of Fmi, cell proliferation, as measured by pHis3 staining, was significantly reduced (Figure 4Q–S). Taken together, these observations directly link the need for Fmi to proliferation and induced apoptosis, the two key events of cell competition, in several models of cell competition.
Fmi-mediated cell-cell communication is not required for competition between winners and losers
Previously, some key players involved in cell competition, including Flower (Rhiner et al., 2010) and Xrp-1 (Baillon et al., 2018), were shown to be either up- or downregulated during competition. We evaluated whether Fmi protein levels might also be affected during competition. Fmi is ubiquitously expressed at low levels in Drosophila larva, pupa, and adult (Brown et al., 2014). We stained third instar larval wing discs with >>Myc clones to determine whether >>Myc supercompetition affects the levels of Fmi protein at the membrane, either inside the clone or near the boundary where cell competition occurs. We observed no change in Fmi protein levels (Figure 5—figure supplement 1), suggesting that Fmi is involved in competition through a mechanism that does not rely on altering protein levels.
Cell competition is thought to rely on intercellular communication to compare fitness and determine the outcome between prospective winner and loser cells (Kon et al., 2017; Baker, 2020; Ogawa et al., 2021). If Fmi is involved in these determinative intercellular communication events by signaling as a trans-homodimer, it should be required in both prospective winner and loser cells. Our previous eye and wing disc >>Myc supercompetition results already suggested this is not the case. While the removal of fmi caused winner clones to effectively become losers during >>Myc competition (Figure 2), removing fmi from the losers had no effect on the losers’ outcome, either in eye discs (Figure 2C and D) or wing discs (Figure 2G and H). In both tissues, winner >>Myc clones showed no change in relative size to their twin spot counterparts, nor were loser clones eliminated more effectively (Figure 2D and H).
Our results so far suggested that the effect Fmi exerts on cell competition does not operate through bidirectional intercellular PCP communication. To further consolidate this hypothesis, we examined the effect of removing a dedicated core PCP protein other than Fmi from >>Myc supercompetitors. Generating vangA3 >>Myc clones in the wing disc, we observed that the >>Myc clones were unaffected and remained winners (Figure 5). vangA3, >>Myc clones were on average 2.2±0.82 times larger than their hRFP twins, not significantly different from the 1.7±0.45-fold value for >>Myc clones (Figure 2H; Figure 5C).
Figure 5 with 1 supplement see all
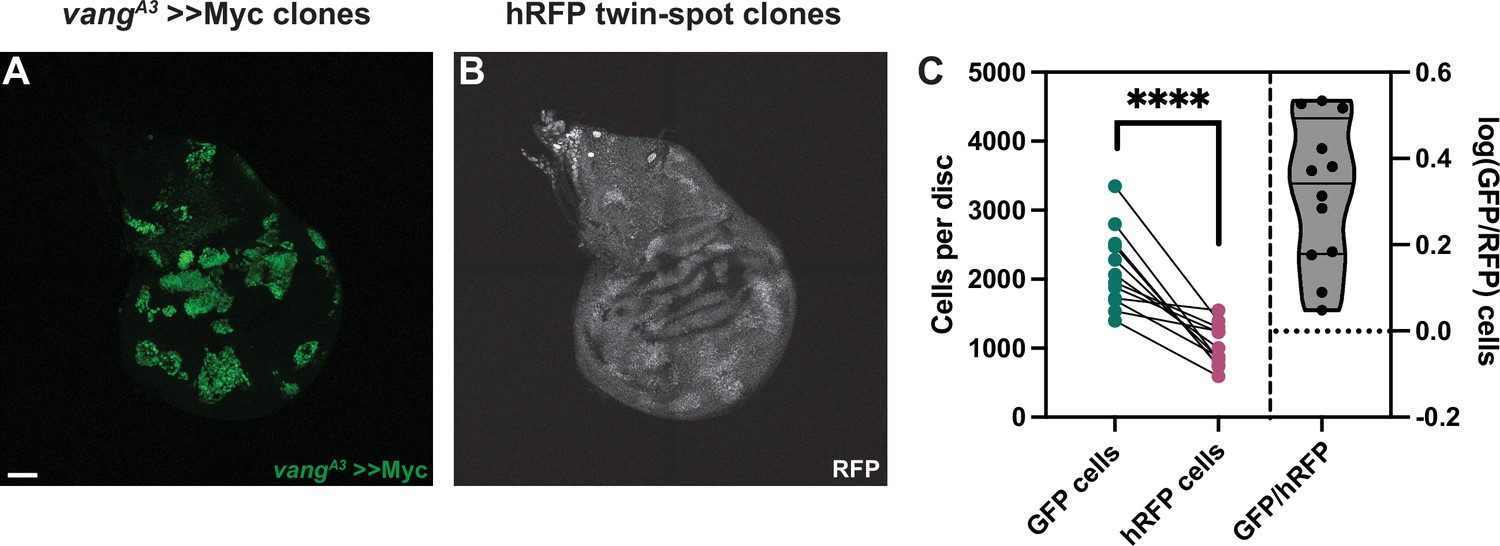
Vang is not required for >>Myc supercompetition.
(A) Representative wing imaginal disc showing the GFP-tagged, vangA3 >>Myc clones. Clones were generated using hsp70-Flp. (B) RFP-labeled twin spots for the clones shown in A. Twin spot homozygous for RFP can be observed adjacent to the supercompetitor clones. (C) Graph showing the total GFP and homozygous RFP counts per disc, with each disc counts linked by a line and the log10 ratio of GFP/hRFP cells per disc (n=12 discs). Differences between GFP/hRFP clone size were analyzed using a two-tailed, ratio-paired t-test. p-Value<0.0001. When the ratio was compared with a two-tailed t-test to >>Myc, the difference was not significant, with a p-value of 0.078. Scale bar: 50 μm.
The activity of Fmi in cell competition does not require its cadherin domains
Our results have so far demonstrated that Fmi is required only in winner cells and that its function in cell competition is independent of PCP. Furthermore, our results rule out a role for Fmi homodimers in directly communicating fitness information between prospective winner and loser cells. However, the possibility remained that Fmi might function through trans-homodimerization between prospective winner cells to sustain winner status via a signal, via adhesion, or both. To ask if Fmi fulfills its role by mediating adhesion, we tested whether we could restore winner status to >>Myc clones that lack Fmi by providing a transgenic fmi that lacks the nine cadherin domains of Fmi (arm-fmiΔCad).
Co-expression of arm-fmiΔCad in fmiE59, >>Myc clones re-established the ability of these clones to outcompete their neighbors, and they again became supercompetitors (Figure 6). When comparing the ratio of >>Myc clone cell number to twin spot cell number, the presence of arm-fmiΔcad restores competition to a level comparable to >>Myc supercompetitors (1.43±0.48 GFP/hRFP cells vs 1.71±0.45 GFP/hRFP cells respectively), far above the 0.49±0.29 value for fmiE59 >>Myc clones compared to twin spots.
Figure 6
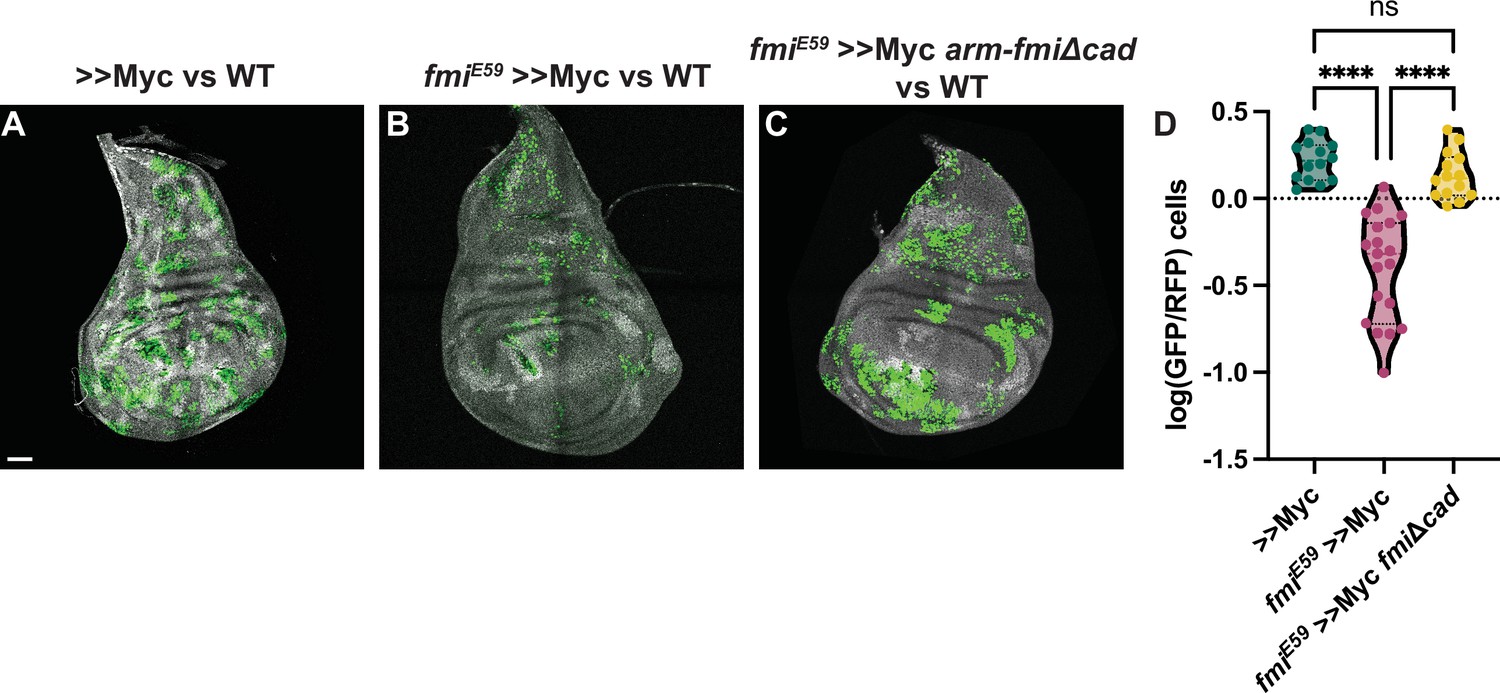
The Fmi cadherin repeats are not required for cell competition.
(A) Representative disc with nGFP-labeled, >>Myc (A), >>Myc, fmiE59 (B) or >>Myc, fmiE59, arm-fmiΔcad (C) clones competing against wildtype twin spots in the wing disc. Twin spot clones are labeled with homozygous nRFP and clones were generated with hsp70-Flp. (D) Ratio of GFP vs RFP cells in the three groups, represented as the log10(GFP/hRFP) cell ratios. To evaluate the effect of the arm-fmiΔcad rescue (n=14 discs), the GFP/hRFP cell ratio was directly compared against the two other groups, already quantified and shown in Figure 2F. Differences between the groups were analyzed using an unpaired, ordinary one-way ANOVA, which found a p-value<0.0001. Inter-group differences were analyzed with a Tukey’s multiple comparisons test, which found no differences between >>Myc and >>Myc, fmiE59, arm-fmiΔcad clones, whereas both groups strongly differed from >>Myc, fmiE59 clones, both returning a p-value<0.0001 (****) when compared directly against >>Myc, fmiE59. Scale bar: 50 μm.
Supercompetitor clones mutant for fmi consistently show clone fragmentation (Figures 2F, 4R, and 6B). Despite lacking the cadherin repeats, rescued clones not only restored their supercompetitor status, but fully recovered the ability of >>Myc clones to remain cohesive (Figure 6C). This suggests that the clone fragmentation we observed is unlikely due to the ability of Fmi to contribute to adhesion and more likely caused by a feature of the competition between wildtype cells and fmi-/->>Myc loser clones that causes their elimination.
Taken together, these results demonstrate that, while Fmi is essential in winner cells to eliminate less fit neighbors, this effect is independent of PCP or other homodimer-mediated signaling, and independent of Fmi-mediated cell adhesion, suggesting instead an as yet uncharacterized function for Fmi.
Discussion
We have identified a requirement for Fmi in winner cells in both tumorigenic and developmental cell competition models. Cells that would otherwise behave as winners instead behave as losers when they lack Fmi. Fmi is notable in that it is required in cell competition in each of four distinct competition scenarios examined: RasV12 scrib RNAi tumors, Myc supercompetitor clones in eye and wing discs, wildtype cells vs scrib RNAi loser clones in pupal eyes, and likely scrib RNAi clones in larval eye discs. Just how universal this requirement is in other cell competition scenarios in Drosophila and perhaps in competition in other organisms remains to be determined.
Fmi acts in cell competition independently of PCP
Several arguments support the conclusion that the role for Fmi in cell competition is distinct from its role in PCP signaling. First, Fmi is the only core PCP component among the six that were surveyed to inhibit the ability of RasV12 scrib RNAi tumor clones to compete in the eye. If Fmi’s role in cell competition were to signal winner fate to losers and vice versa by mirroring its role in PCP signaling, where it signals the presence of proximal (Vang, Pk) components in one direction and the presence of distal components (Fz, Dsh, Dgo) in the other direction between adjacent cells (Lawrence et al., 2004; Strutt and Strutt, 2007; Strutt and Strutt, 2008; Chen et al., 2008; Struhl et al., 2012), then one might expect either the proximal or distal components to also be required in winner clones. No such requirement was observed. Second, in PCP signaling, Fmi functions as a trans-homodimer to transmit those signals and requires the cadherin repeats and other extracellular domains, implying its function as a trans-homodimer (Kimura et al., 2006). In PCP signaling, removing Fmi from either of two adjacent cells completely blocks PCP signaling. In contrast, in cell competition, while Fmi is required in winners, removing Fmi from losers has no effect on competition. Thus, the model of bidirectional signaling via Fmi is not supported by our results.
These observations, however, do not rule out the possibility that Fmi trans-homodimers contribute to intercellular signaling among winner cells. Nonetheless, as discussed above, adhesion seems not to be a meaningful part of the function for Fmi in winners, as an adhesion-deficient Fmi construct (fmiΔCad) fully rescues both competition and clone adhesion. The potential contributions of adhesion vs other possible signaling mechanisms are discussed at more length below.
Unlike the other core PCP genes, fmi-/- mutations are lethal due to requirements in the nervous system (Usui et al., 1999). Though not fully characterized, its roles in the nervous system appear to be distinct from PCP signaling. Fmi is required for outgrowth and guidance of the R8 axon of the eye to the M3 layer of the medulla via a mechanism that appears independent of other components of the core PCP signaling pathway (Gao et al., 2000; Lee et al., 2003; Senti et al., 2003), but does interact with Golden goal, a transmembrane phosphoprotein that is not associated with PCP signaling (Takechi et al., 2021). Growth of the dendrites of dorsal da neurons is also regulated by Fmi. During embryogenesis, da dendrites in fmi mutant embryos emerge precociously and overgrow as they approach the dorsal midline, and later, during larval growth, dendrites from opposite sides fail to avoid each other (tile) and instead overlap (Gao et al., 2000; Sweeney et al., 2002; Kimura et al., 2006).
Fmi is classified as an atypical cadherin and a Class-B adhesion G protein-coupled receptor (AGPCR), as it contains in its extracellular domain several conserved functional domains including cadherin repeats, epidermal growth factor-like repeats, laminin A G-type repeats, and a GPCR autoproteolytic inducing (GAIN) domain that contains within it a GPCR proteolytic site (GPS) (Rosa et al., 2021; Einspahr and Tilley, 2022; Sreepada et al., 2022). These extracellular domains are followed by seven transmembrane domains and an intracellular C-terminal domain. Although much remains to be learned about this large subfamily of GPCRs, a general model has emerged in which activation by membrane bound or extracellular protein, peptide, proteoglycan or small molecule ligand, or mechanical force exposes a tethered ligand at the N-terminus of the C-terminal fragment in the GAIN domain that, upon exposure, interacts with the transmembrane portion to activate a G-protein signaling cascade. In many but not all AGPCRs, the tethered ligand is exposed by GAIN domain-mediated autoprotolysis of its GPS. Non-cleaved AGPCRs are hypothesized to expose the tethered ligand by an allosteric conformational change. Some de-orphanized AGPCRs interact with multiple ligands, and ligand binding can result in partial or full activation. In some cases, it appears that engineered truncation of portions of the extracellular domain can produce some level of ligand-independent activation (Rosa et al., 2021).
When expressed in da neurons, an Fmi construct lacking the cadherin, laminin G, and EGF-like repeats partially rescued the embryonic dendritic overgrowth phenotype (Kimura et al., 2006), suggesting a function independent of homodimerization. The G protein Gαq (Gq) has been proposed to function downstream of Fmi to mediate this repressive function (Wang et al., 2016). A recent preprint reports the resolved structure of the CELSR1 extracellular domain, showing that the protein has two distinct domains: an adhesion domain comprised of the first eight cadherin repeats, and a compact domain that extends from the ninth cadherin repeat to the transmembrane domains that is involved in GPCR signaling. Indeed, they demonstrated that a CELSR1 construct lacking only the cadherin repeats 1–8 retains the ability to activate Gαs, which has been predicted to interact with CELSR1 (Bandekar et al., 2024). Notably, our results showed that a similar Fmi construct lacking the cadherin domains substantially rescues the requirement for Fmi in winner cells during cell competition (Figure 6). These observations suggest that supplying adhesion is not the principal function of Fmi in these events, but are consistent with the possibility that homodimeric adhesion, or interaction with a different ligand, normally activates the receptor, and that the truncated FmiΔCad behaves as a constitutively activated receptor capable of binding to either Gα proteins. While no biochemical characterization of Fmi has been reported, the human orthologs CELSR1-3 have been studied in detail. CELSR2 is autoproteolytically cleaved while CELSRs 1 and 3 are not, yet all three couple to GαS (Huong Bui et al., 2023). Additional efforts will be required to determine the functional ligand(s) for Fmi and whether it signals similarly. Furthermore, AGPCRs participate in a wide variety of developmental and physiologic events through diverse effectors (Einspahr and Tilley, 2022; Sreepada et al., 2022). The pathway by which Fmi participates in cell competition remains to be explored.
Fmi, cell competition, and cancer
When the first examples of supercompetition were observed in Myc clones and the Hippo pathway (Moreno and Basler, 2004; de la Cova et al., 2004; Ziosi et al., 2010; Neto-Silva et al., 2010), they hinted at the possibility that tumors, which behave like supercompetitors, could use similar mechanisms to outcompete wildtype cells. Understanding tumor competition may open new avenues for early detection and therapy (Baker and Li, 2008; Moreno, 2008).
Research in Drosophila and mammals has shown that cell competition plays a dual role during tumorigenesis. Cells harboring mutations in proto-oncogenes or tumor-suppressor genes often behave as losers (Maruyama and Fujita, 2017; Morata and Calleja, 2020; Kanda and Igaki, 2020). Through the process of EDAC, epithelial tissues use cell competition to eliminate transformed pre-neoplastic cells by removing them from the tissue via directed cell death or extrusion (Kon et al., 2017; Watanabe et al., 2018). Pre-neoplastic cells that escape EDAC may accumulate additional mutations to become malignant tumors (Watanabe et al., 2018). Malignant tumors not only escape EDAC, but acquire properties that allow them to outcompete wildtype cells, facilitating invasion and metastasis (Suijkerbuijk et al., 2016; Kohashi et al., 2021). Furthermore, competition between clones within tumors further selects far more aggressive tumor behavior (Parker et al., 2021).
Another commonality between developmental and oncogenic cell competition is the involvement of the transmembrane protein Flower (Fwe). In both Drosophila and mammals, multiple isoforms of Fwe signal fitness; expression of FweLose isoforms mark losers for elimination (Rhiner et al., 2010; Merino et al., 2013; Levayer et al., 2015; Madan et al., 2019). Forced expression of FweLose induces cell competition and elimination of the loser, suggesting that Fwe comparison is involved in the sensing and/or initiation of differential fitness. This contrasts with Fmi, whose differential expression does not act as a trigger for competition, but which is needed in winners to allow them to win, suggesting that Fmi is involved after sensing in the execution of functions necessary to manifest winner behavior.
Evidence is accumulating that a human ortholog of Fmi, CELSR3, is expressed at high levels in a range of solid tumors, including lung, prostate, pancreatic, hepatic, ovarian, and colorectal cancers, and in some cases has been shown to be associated with poor prognosis (Katoh and Katoh, 2007; Erkan et al., 2010; Asad et al., 2014; Goryca et al., 2018; Li et al., 2021; Chen et al., 2021c). Recently, CELSR1 upregulation has also been linked to poor ovarian cancer prognosis, likely by promoting proliferation, migration, and invasion (Zuo et al., 2023). If CELSR1/3 are promoting winner cell behavior in these tumors, as might be predicted from its function in Drosophila, this could provide the rationale for future efforts to understand the mechanism by which Fmi/CELSR3 facilitates cell competition, with the goal of identifying an intervention that could blunt or perhaps even eliminate the aggressiveness of an array of highly morbid cancers.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Drosophila melanogaster) | fmi | FlyBase | FBgn0024836 | Also known as stan |
| Gene (D. melanogaster) | fz | FlyBase | FBgn0001085 | |
| Gene (D. melanogaster) | dsh | FlyBase | FBgn0000499 | |
| Gene (D. melanogaster) | vang | FlyBase | FBgn0015838 | Also known as stbm |
| Gene (D. melanogaster) | pk | FlyBase | FBgn0003090 | |
| Gene (D. melanogaster) | dgo | FlyBase | FBgn0086898 | |
| Gene (D. melanogaster) | scrib | FlyBase | FBgn0263289 | |
| Gene (D. melanogaster) | myc | FlyBase | FBgn0262656 | |
| Gene (D. melanogaster) | Ras85D | FlyBase | FBgn0003205 | |
| Strain, strain background (Escherichia coli) | 5-alpha High Efficiency | NEB | C2987H | |
| Genetic reagent (D. melanogaster) | W RNAi | BDSC | 33623 | |
| Genetic reagent (D. melanogaster) | Fmi RNAi | BDSC | 26022 | |
| Genetic reagent (D. melanogaster) | Fz RNAi | BDSC | 31311 | |
| Genetic reagent (D. melanogaster) | Dsh RNAi | BDSC | 31306 | |
| Genetic reagent (D. melanogaster) | Vang RNAi | BDSC | 34354 | |
| Genetic reagent (D. melanogaster) | Pk RNAi | BDSC | 32413 | |
| Genetic reagent (D. melanogaster) | Scrib RNAi | BDSC | 39073 | |
| Transfected construct (D. melanogaster) | Arm-fmiΔCad | This paper | Located in chromosome 2R | |
| Antibody | Rabbit polyclonal α-pHis3 | Millipore | 1:100 | |
| Antibody | Rabbit monoclonal α-Dcp1 | Cell Signaling | RRID:AB_2721060 | 1:100 |
| Antibody | Mouse monoclonal α-LacZ | Promega | 1:500 | |
| Antibody | Rabbit polyclonal α-Cas3 | AbCam | 1:200 | |
| Antibody | 488-Goat polyclonal α-rabbit | Thermo Scientific | RRID:AB_3251385 | 1:500 |
| Antibody | 546-Goat polyclonal α-mouse | Thermo Scientific | RRID:AB_2535765 | 1:500 |
| Antibody | 546-Goat polyclonal α-rabbit | Thermo Scientific | RRID:AB_2534077 | 1:500 |
| Antibody | 647-Donkey polyclonal α-mouse | Thermo Scientific | RRID:AB_162542 | 1:500 |
| Commercial assay or kit | HiFi DNA assembly kit | New England Biolabs | ||
| Chemical compound, drug | Alexa 350 phalloidin | Thermo Scientific | 1:500 | |
| Chemical compound, drug | Alexa 635 phalloidin | Thermo Scientific | 1:500 | |
| Chemical compound, drug | DAPI | Invitrogen | 1 μg/mL | |
| Chemical compound, drug | Vectashield | Vector Labs | ||
| Software, algorithm | Fiji | https://fiji.sc | RRID:SCR_002285 | |
| Software, algorithm | Counting macros | https://github.com/iPabloSB/Nuclear-counts; Shcherbina and Sanchez Bosch, 2023 | ||
| Software, algorithm | Prism 10 | GraphPad | RRID:SCR_002798 |
Resource availability
Lead contact
Request a detailed protocolFurther information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact (jaxelrod@stanford.edu).
Materials availability
Request a detailed protocolPlasmid and fly lines generated in this study are available from the lead contact upon request.
Experimental model and study participant details
Fly stocks and husbandry
Request a detailed protocolFlies were maintained in standard fly food in a temperature-controlled incubator at either 25°C or 18°C. Egg collections were performed over a timespan of 24 or 48 hr. Vials with eggs were kept at 25°C until heat-shocked for clone generation or dissected.
Heat-shock was performed for 15 min on late first instar and early second instar larvae, 48 hr after egg laying (AEL). Vials were kept at 25°C after heat-shock until larvae were dissected.
The following fly lines were used for the experiments:
y, w, hsp-Flp act5C-Gal4, UAS-nGFP to generate wing disc clones.
ey-Flp; if /SM5^; TM2/^TM6b to generate eye disc clones and whole tumors.
Bloomington TRiP UAS-RNAi lines RRID:BDSC_33623 (w control), RRID:BDSC_26022 (fmi), RRID:BDSC_31311 (fz), RRID:BDSC_31306 (dsh), RRID:BDSC_34354 (vang), RRID:BDSC_32413 (pk), RRID:BDSC_39073 (scrib).
PCP mutant alleles FRT42D, fmiE45/CyO, FRT42D, fmiE59/CyO, FRT2A, fzR52/TM6b, FRT42D, vangA3, FRT42D dgo380, FRT42D pkpk-sple13.
UAS-scrib RNAi; act5C>CD2>Gal4, UAS-RFP, UAS-RasV12/TM6b to make whole eye tumors.
UAS-scrib RNAi FRT42D, tub-Gal80; act5C>CD2>Gal4, UAS-RFP, UAS-RasV12/TM6b to generate tumor clones.
UAS-scrib RNAi, FRT42D, tub-nRFP/FRT42D tub-Gal80 to generate scrib RNAi eye disc clones.
FRT42D fmiE59/FRT42D, tub-nRFP, tub-Gal80 to generate wing disc twin spots.
UAS-dMyc/TM6b for eye and wing disc supercompetitor clones.
FRT42D fmiE59, arm-fmiΔCad to rescue wing disc clones.
Genotypes of experimental models
Request a detailed protocolFigure 1:
(B) ey-Flp; act5C>CD2>Gal4, UAS-nRFP / TM6b.
(C–H) ey-Flp; UAS-scrib RNAi / UAS-DCR2; act5C>CD2>Gal4, UAS-nRFP, UAS-RasV12/TM6b crossed to the homozygous UAS-RNAi line noted above.
(J) ey-Flp; FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4, UAS-nRFP/TM6b.
(K) ey-Flp; FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/UAS-scrib RNAi.
(L) ey-Flp; FRT42D tub-Gal80/FRT42D fmiE45; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/UAS-scrib RNAi.
(M) ey-Flp; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/UAS-scrib RNAi; fzR52 FRT2A/tub-Gal80 FRT2A.
(N) ey-Flp; FRT42D tub-Gal80/FRT42D dgo380; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/UAS-scrib RNAi.
(O) ey-Flp; FRT42D tub-Gal80/FRT42D pkpk-sple13; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/UAS-scrib RNAi.
(P) ey-Flp; FRT42D tub-Gal80/FRT42D vangA3; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/UAS-scrib RNAi.
Figure 2:
(A) ey-Flp; FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4 UAS-nRFP/UAS-dMyc.
(B) ey-Flp; FRT42D tub-Gal80/FRT42D fmiE59; act5C>CD2>Gal4 UAS-nRFP/UAS-dMyc.
(C) ey-Flp; FRT42D fmiE59 tub-Gal80/FRT42D tub-nRFP; act5C>CD2>Gal4 UAS-nGFP/UAS-dMyc.
(E) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D; UAS-dMyc / +.
(F) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D fmiE59; UAS-dMyc / +.
(G) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D fmiE59 tub-nRFP tub-Gal80/FRT42D; UAS-dMyc / +.
Figure 3:
(A–C) ey-Flp; FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4 UAS-nRFP / +.
(D–F) ey-Flp; FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4 UAS-nRFP/UAS-scrib RNAi pucE69.
(G–I) ey-Flp; FRT42D tub-Gal80/FRT42D fmiE59; act5C>CD2>Gal4 UAS-nRFP/UAS-scrib RNAi pucE69.
(J–K) ey-Flp; FRT42D fmiE59 tub-Gal80/FRT42D; act5C>CD2>Gal4 UAS-nRFP/UAS-scrib RNAi pucE69.
Figure 4:
(A–C) ey-Flp; UAS-scrib RNAi FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/pucEE69.
(E–G) ey-Flp; UAS-scrib RNAi FRT42D tub-Gal80/FRT42D fmiE59; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/pucEE69.
(I–K) ey-Flp; FRT42D tub-Gal80/FRT42D fmiE59; act5C>CD2>Gal4 UAS-nRFP/UAS-dMyc.
(L) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D; UAS-dMyc / +.
(M) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D fmiE59; UAS-dMyc / +.
Figure 5:
(A, B) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D vangA3; UAS-dMyc / +.
Figure 6:
(A) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D; UAS-dMyc / +.
(B) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D fmiE59; UAS-dMyc / +.
(C) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D fmiE59 arm-fmiΔCad; UAS-dMyc / +.
Figure 1—figure supplement 1:
(A, B) ey-Flp; FRT42D tub-Gal80/FRT42D tub-nRFP; act5C>CD2>Gal4/UAS-nGFP.
Figure 1—figure supplement 2:
(A) ey-Flp; scrib RNAi FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4, UAS-nRFP, UAS-RasV12/w RNAi.
(B) ey-Flp; scrib RNAi FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4, UAS-nRFP, UAS-RasV12/fmi RNAi.
(C) ey-Flp; FRT42D / FRT42D GMR-Hid, l(2)CL-R; act5C>CD2>Gal4, UAS-nRFP, UAS-RasV12/scrib RNAi.
(D) ey-Flp; FRT42D fmiE59/FRT42D GMR-Hid, l(2)CL-R; act5C>CD2>Gal4, UAS-nRFP, UAS-RasV12/scrib RNAi.
Figure 2—figure supplement 1:
(A) ey-Flp; FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4/UAS-nRFP.
(B) ey-Flp; FRT42D tub-Gal80/FRT42D fmiE59; act5C>CD2>Gal4/UAS-nRFP.
(D, E) hsp-Flp tub-Gal4, UAS-nGFP; FRT42D tub-nRFP tub-Gal80/FRT42D fmiE59; MKRS / TM6b.
Figure 4—figure supplement 1:
(A–C) ey-Flp; FRT42D tub-Gal80/FRT42D fmiE45; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/UAS-scrib RNAi.
Figure 4—figure supplement 2:
(A–C) ey-Flp; UAS-scrib RNAi FRT42D tub-Gal80/FRT42D; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/pucEE69.
(D–F) ey-Flp; UAS-scrib RNAi FRT42D tub-Gal80/FRT42D fmiE59; act5C>CD2>Gal4, UAS-nRFP UAS-RasV12/pucEE69.
Figure 5—figure supplement 1:
(A–F) hsp-Flp, tub-Gal4, UAS-nGFP; FRT42D tub-Gal80/FRT42D; UAS-dMyc / +.
Method details
Generation of arm-fmiΔCad
Request a detailed protocolTo make the arm-fmiΔCad construct, we used cDNA from the fmi isoform A (stan-RA) terminally tagged with an HA tag, fused using an SGGGGS linker (fmi::HA). We subcloned by Gibson assembly a PCR fragment containing the coding sequence for fmi::HA lacking the first 1328 aa, which contain the 9 cadherin domains (fmiΔ1-1328) into a pCaSpeR4 vector backbone with the armadillo promoter and an Fz 5’UTR. The pCaSpeR4-armP-fmiΔCad construct was introduced into flies by P-element integration, and we used a fly line carrying the construct on chromosome arm 2R.
Wing imaginal disc dissection and immunohistochemistry
Request a detailed protocolThird instar wandering larvae (120 hr AEL) were dissected by transversally cutting the larva in two halves. The posterior half was discarded, and the anterior part was inverted, after which the fat body and digestive tissue (mouth hooks, salivary glands, and gut) were removed. Inverted larvae were fixed in 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS)+0.02% Triton X-100 (PBS-T) for 30 min at room temperature (RT). Fixed larvae were then washed three times with PBS-T and then stained.
For antibody staining, larvae were stained with primary antibodies diluted in PBS-T+3% normal donkey serum (NDS) overnight at 4°C, and then stained with secondary antibodies and DAPI for 1 hr at room temperature. Stained larvae were then washed three times with PBS. Wing discs were carefully removed from the inverted larva and mounted in Vectashield mounting medium (Vector Labs).
We used the following primary antibodies: rabbit α-pHis3 (Millipore), 1:100; rabbit α-Dcp1 (Cell Signaling), 1:100. Secondary staining was performed with Thermo Scientific 546-goat α-rabbit, 1:500.
Immunohistochemistry of third instar larval eye discs
Request a detailed protocolDiscs dissected from late third instar larvae were fixed for 5–15 min in 4% PFA in PBS at 4°C. Fixed eye discs were washed two times in PBS-T. After blocking for 1 hr in 5% bovine serum albumin in PBS-T at 4°C, discs were incubated with primary antibodies in the blocking solution overnight at 4°C. Incubations with secondary antibodies were done for 90 min at room temperature in PBS-T. Secondary antibody was washed three times with PBS-T. Incubations in phalloidin (1:200) and DAPI (1 µg/mL), if required, were done in PBS-T for 15 min followed by washing at room temperature before mounting. Stained samples were mounted in 15 μL Vectashield mounting medium (Vector Labs). We used the following primary antibodies: mouse anti-LacZ (1:500 dilution, Promega) rabbit α-Dcp1 (1:500 dilution, Cell Signaling), mouse α-Fmi (1:200 dilution, DSHB). We used the following secondary antibodies from Thermo Scientific: 488-goat α-rabbit, 546-goat α-mouse, 594-donkey α-mouse, 647-donkey α-mouse, Alexa 635- and Alexa 350-conjugated phalloidin.
Image acquisition
Request a detailed protocolImages of whole discs were taken with a Leica SP8 system equipped with a White Light Laser and HyD detectors. A ×40, NA 1.5 Leica objective and 1.51 refractive index immersion oil were used. Image stacks were taken in 8-bit at 1024×1024 px resolution, and a pinhole of 1 airy unit (AU), using a z-step of 0.3 μM. For automated image quantification pipelines, residual tissue from the leg or haltere discs was removed in Fiji (fiji.sc, Schindelin et al., 2012), as well as the peripodial cells at the apical section of the Z-stack.
Adult eyes and RFP/GFP signal from pupae and eye discs isolated from late third instar larvae were imaged on a Leica MZ16F Stereomicroscope.
Quantification and statistical analysis
Eye disc clone analysis
Request a detailed protocolEye imaginal discs representative slices were selected to measure the size of RFP+ clones. To do so, the GFP+ clone area was divided by the non-fluorescent eye disc area to obtain the ratio of GFP+ clone vs non-GFP twin and plotted as the log10(ratio) on violin plots including all data points, median, and quartiles. Statistical analysis was performed in GraphPad Prism 10. Data was analyzed using unpaired, two-tailed t-tests.
Wing disc clone analysis
Request a detailed protocolWing imaginal disc GFP clone and RFP twin spot cell counts were obtained using a set of automated macros written for ImageJ (Sanchez Bosch and Axelrod, 2024). Cell ratios were obtained as the fraction of GFP+ cells vs RFP+ twin spot cells. Cell ratios were transformed as the log10(ratio) and graphed as violin plots including all data points, median, and quartiles. Statistical analysis was performed in GraphPad Prism 10. Data was analyzed using either unpaired, two-tailed t-test (Figure 5), or an ordinary one-way ANOVA with a Tukey’s multiple comparisons test (Figures 2—4 and 6).
Apoptosis quantification
Request a detailed protocolApoptotic cells marked with positive Dcp1 staining were scored when located one to three cells away from a clone boundary, as those were arbitrarily deemed as caused by cell competition. Cell counts were plotted individually, with each disc’s WT and GFP cells plotted side-by-side, linked by a straight line. The difference in apoptosis between WT and >>Myc (or >>Myc, fmiE59), GFP+ cells in each disc was then calculated (GFP minus WT cells) and plotted on the right. The mean difference of the analysis was also plotted in the same graph as a dashed line. The statistical differences were obtained using a paired, two-tailed t-test.
Cell proliferation quantification
Request a detailed protocolClone proliferation ratios were measured in ImageJ. First, we obtained the number of GFP+ cells vs the cells outside the GFP+ clones by using the same macros used to quantify GFP clones and then counting the total cells in the disc by counting the DAPI nuclei (Sanchez Bosch and Axelrod, 2024), and then subtracting the GFP+ cells from the total DAPI cell counts. Then, pHis3-positive cells were located by using the 3D Find maxima function from the ImageJ 3D Suite (https://mcib3d.frama.io/3d-suite-imagej/). To do so, the pHis3 channel was processed as follows: (1) specks were removed by using the Remove Outliers (radius of 5, threshold of 50), then the background was removed with a 2Px 3D Gaussian Blur and the Subtract background function (rolling ball radius = 10 px, with sliding parabolic and disabled smoothing). Last, the pHis3+ peaks were found using the 3D Maxima finder (minimum threshold of 5 px, with a XY and Z radius of 3 px and discarding all peaks below the noise level of 20).
GFP+, pHis3 cells were obtained by first creating a binary mask of the GFP channel by smoothing the image with a 5 px 3D Gaussian blur and then using a Li threshold to create the mask. To ensure proper quantification, correct thresholding of the clones was visually assessed and adjusted when needed. Then, all pHis3 peaks that were inside the GFP+ clone were counted as proliferative GFP+ cells. Last the proliferative ratio of GFP+ cells was obtained by dividing the fraction of pHis3 cells inside GFP clones by the fraction of pHis3 cells outside of GFP+ clones. The log10 of the proliferative ratio was plotted as a violin plot representing each disc as a data point and indicating the median and quartiles. Statistical analysis was performed in GraphPad Prism 10. Data was analyzed using an ordinary one-way ANOVA and the groups were compared to each other using a Tukey’s multiple comparisons test.
Data availability
All images acquired for the study are available in the BioStudies database under accession number S-BIAD1513. The code for the ImageJ/Fiji macros used in this study can be accessed and downloaded from GitHub (copy archived at Shcherbina and Sanchez Bosch, 2023). Numerical data for all graphs in this study is provided in Supplementary file 1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
ArrayExpressID S-BIAD1513. Flamingo participates in multiple models of cell competition.
References
-
Planar signaling and morphogenesis in DrosophilaDevelopmental Cell 2:525–535.https://doi.org/10.1016/s1534-5807(02)00176-4
-
Cell competition: how to eliminate your neighboursDevelopment 141:988–1000.https://doi.org/10.1242/dev.079129
-
Cell competition and its possible relation to cancerCancer Research 68:5505–5507.https://doi.org/10.1158/0008-5472.CAN-07-6348
-
Emerging mechanisms of cell competitionNature Reviews. Genetics 21:683–697.https://doi.org/10.1038/s41576-020-0262-8
-
Planar cell polarity in development and diseaseNature Reviews. Molecular Cell Biology 18:375–388.https://doi.org/10.1038/nrm.2017.11
-
The involvement of noncanonical Wnt signaling in cancersBiomedicine & Pharmacotherapy 133:110946.https://doi.org/10.1016/j.biopha.2020.110946
-
Increased expression of CELSR3 indicates a poor prognostic factor for Prostate CancerJournal of Cancer 12:1115–1124.https://doi.org/10.7150/jca.49567
-
Pathophysiological impact of the adhesion G protein-coupled receptor familyAmerican Journal of Physiology. Cell Physiology 323:C640–C647.https://doi.org/10.1152/ajpcell.00445.2021
-
Regulation of imaginal disc growth by tumor-suppressor genes in DrosophilaAnnual Review of Genetics 40:335–361.https://doi.org/10.1146/annurev.genet.39.073003.100738
-
Characterization of the interface between normal and transformed epithelial cellsNature Cell Biology 11:460–467.https://doi.org/10.1038/ncb1853
-
Mechanism of tumor-suppressive cell competition in fliesCancer Science 111:3409–3415.https://doi.org/10.1111/cas.14575
-
WNT/PCP signaling pathway and human cancer (review)Oncology Reports 14:1583–1588.
-
Sequential oncogenic mutations influence cell competitionCurrent Biology 31:3984–3995.https://doi.org/10.1016/j.cub.2021.06.064
-
Tissue crowding induces caspase-dependent competition for spaceCurrent Biology 26:670–677.https://doi.org/10.1016/j.cub.2015.12.072
-
Systematic expression analysis of the CELSR family reveals the importance of CELSR3 in human lung adenocarcinomaJournal of Cellular and Molecular Medicine 25:4349–4362.https://doi.org/10.1111/jcmm.16497
-
Cell Competition in CarcinogenesisCancer Research 82:4487–4496.https://doi.org/10.1158/0008-5472.CAN-22-2217
-
Cell competition in mammals - novel homeostatic machinery for embryonic development and cancer preventionCurrent Opinion in Cell Biology 48:106–112.https://doi.org/10.1016/j.ceb.2017.06.007
-
Minutes: mutants of Drosophila autonomously affecting cell division rateDevelopmental Biology 42:211–221.https://doi.org/10.1016/0012-1606(75)90330-9
-
Cell competition and tumorigenesis in the imaginal discs of DrosophilaSeminars in Cancer Biology 63:19–26.https://doi.org/10.1016/j.semcancer.2019.06.010
-
Cell competition: A historical perspectiveDevelopmental Biology 476:33–40.https://doi.org/10.1016/j.ydbio.2021.02.012
-
Is cell competition relevant to cancer?Nature Reviews. Cancer 8:141–147.https://doi.org/10.1038/nrc2252
-
Loss of Scribble causes cell competition in mammalian cellsJournal of Cell Science 125:59–66.https://doi.org/10.1242/jcs.085803
-
Emerging roles of adhesion G protein-coupled receptorsBiochemical Society Transactions 49:1695–1709.https://doi.org/10.1042/BST20201144
-
Automated counting of Drosophila imaginal disc cell nucleiBiology Open 13:bio060254.https://doi.org/10.1242/bio.060254
-
Fiji: an open-source platform for biological-image analysisNature Methods 9:676–682.https://doi.org/10.1038/nmeth.2019
-
SoftwareNuclear-counts, version swh:1:rev:395e310bab4766707bf3a66a6892c4511f3f2e79Software Heritage.
-
Planar cell polarity signaling: from fly development to human diseaseAnnual Review of Genetics 42:517–540.https://doi.org/10.1146/annurev.genet.42.110807.091432
-
Adhesion G protein-coupled receptor gluing action guides tissue development and diseaseJournal of Molecular Medicine 100:1355–1372.https://doi.org/10.1007/s00109-022-02240-0
-
Genetic and developmental mechanisms underlying the formation of the Drosophila compound eyeDevelopmental Dynamics 241:40–56.https://doi.org/10.1002/dvdy.22738
-
Cellular and molecular mechanisms underlying planar cell polarity pathway contributions to cancer malignancySeminars in Cell & Developmental Biology 81:78–87.https://doi.org/10.1016/j.semcdb.2017.09.026
-
Mechanisms and mechanics of cell competition in epitheliaNature Reviews. Molecular Cell Biology 14:581–591.https://doi.org/10.1038/nrm3639
-
Planar cell polarity signaling: the developing cell’s compassCold Spring Harbor Perspectives in Biology 1:a002964.https://doi.org/10.1101/cshperspect.a002964
-
Epithelial microRNA-9a regulates dendrite growth through Fmi-Gq signaling in Drosophila sensory neuronsDevelopmental Neurobiology 76:225–237.https://doi.org/10.1002/dneu.22309
Article and author information
Author details
Pablo Sanchez Bosch

Funding
National Institutes of Health (R35GM131914)
- Jeffrey D Axelrod
Swiss National Science Foundation (P400PB_199258)
- Pablo Sanchez Bosch
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We would like to thank members of the Axelrod lab for fruitful discussions. Stocks obtained from the Bloomington Drosophila Stock Center (NIH P40OD018537) were used in this study. This work was funded by NIH R35GM131914 (JDA) and the Swiss National Science Foundation P400PB_199258 (PSB). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Reviewed Preprint version 3:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.98535. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2024, Sanchez Bosch, Cho et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,402
- views
-
- 60
- downloads
-
- 3
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 3
- citations for umbrella DOI https://doi.org/10.7554/eLife.98535
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Flamingo participates in multiple models of cell competition
eLife 13:RP98535.
https://doi.org/10.7554/eLife.98535.4




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}