Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorYu ZhaoInstitute of Radiation Medicine, Chinese Academy of Medical Sciences and Peking Union Medical College, Tianjin, China
- Senior EditorCaigang LiuShengjing Hospital of China Medical University, Shenyang, China
Reviewer #1 (Public review):
Summary:
Identifying drugs that target specific disease phenotypes remains a persistent challenge. Many current methods are only applicable to well-characterized small molecules, such as those with known structures. In contrast, methods based on transcriptional responses offer broader applicability because they do not require prior information about small molecules. Additionally, they can be rapidly applied to new small molecules. One of the most promising strategies involves the use of "drug response signatures"-specific sets of genes whose differential expression can serve as markers for the response to a small molecule. By comparing drug response signatures with expression profiles characteristic of a disease, it is possible to identify drugs that modulate the disease profile, indicating a potential therapeutic connection.
This study aims to prioritize potential drug candidates and to forecast novel drug combinations that may be effective in treating triple-negative breast cancer (TNBC). Large consortia, such as the LINCS-L1000 project, offer transcriptional signatures across various time points after exposing numerous cell lines to hundreds of compounds at different concentrations. While this data is highly valuable, its direct applicability to pathophysiological contexts is constrained by the challenges in extracting consistent drug response profiles from these extensive datasets. The authors use their method to create drug response profiles for three different TNBC cell lines from LINCS.
To create a more precise, cancer-specific disease profile, the authors highlight the use of single-cell RNA sequencing (scRNA-seq) data. They focus on TNBC epithelial cells collected from 26 diseased individuals compared to epithelial cells collected from 10 healthy volunteers. The authors are further leveraging drug response data to develop inhibitor combinations.
Strengths:
The authors of this study contribute to an ongoing effort to develop automated, robust approaches that leverage gene expression similarities across various cell lines and different treatment regimens, aiming to predict drug response signatures more accurately. The authors are trying to address the gap that remains in computational methods for inferring drug responses at the cell subpopulation level.
Weaknesses:
One weakness is that the authors do not compare their method to previous studies. The authors develop a drug response profile by summarizing the time points, concentrations, and cell lines. The computational challenge of creating a single gene list that represents the transcriptional response to a drug across different cell lines and treatment protocols has been previously addressed. The Prototype Ranked List (PRL) procedure, developed by Iorio and co-authors (PNAS, 2010, doi:10.1073/pnas.1000138107), uses a hierarchical majority-voting scheme to rank genes. This method generates a list of genes that are consistently overexpressed or downregulated across individual conditions, which then hold top positions in the PRL. The PRL methodology was used by Aissa and co-authors (Nature Comm 2021, doi:10.1038/s41467-021-21884-z) to analyze drug effects on selective cell populations using scRNA-seq datasets. They combined PRL with Gene Set Enrichment Analysis (GSEA), a method that compares a ranked list of genes like PRL against a specific set of genes of interest. GSEA calculates a Normalized Enrichment Score (NES), which indicates how well the genes of interest are represented among the top genes in the PRL. Compared to the method described in the current manuscript, the PRL method allows for the identification of both upregulated and downregulated transcriptional signatures relevant to the drug's effects. It also gives equal weight to each cell line's contribution to the drug's overall response signature.
The authors performed experimental validation of the top two identified drugs; however, the effect was modest. In addition, the effect on TNBC cell lines was cell-line specific as the identified drugs were effective against BT20, whose transcriptional signatures from LINCS were used for drug identification, but not against the other two cell lines analyzed. An incorrect choice of genes for the signature may result in capturing similarities tied to experimental conditions (e.g., the same cell line) rather than the drug's actual effects. This reflects the challenges faced by drug response signature methods in both selecting the appropriate subset of genes that make up the signature and in managing the multiple expression profiles generated by treating different cell lines with the same drug.
Reviewer #2 (Public review):
Summary:
In their study, Osorio and colleagues present 'retriever,' an innovative computational tool designed to extract disease-specific transcriptional drug response profiles from the LINCS-L1000 project. This tool has been effectively applied to TNBC, leveraging single-cell RNA sequencing data to predict drug combinations that may effectively target the disease. The public review highlights the significant integration of extensive pharmacological data with high-resolution transcriptomic information, which enhances the potential for personalized therapeutic applications.
Strengths:
A key finding of the study is the prediction and validation of the drug combination QL-XII-47 and GSK-690693 for the treatment of TNBC. The methodology employed is robust, with a clear pathway from data analysis to experimental confirmation.
Weaknesses:
However, several issues need to be addressed. The predictive accuracy of 'retriever' is contingent upon the quality and comprehensiveness of the LINCS-L1000 and single-cell datasets utilized, which is an important caveat as these datasets may not fully capture the heterogeneity of patient responses to treatment. While the in vitro validation of the drug combinations is promising, further in vivo studies and clinical trials are necessary to establish their efficacy and safety. The applicability of these findings to other cancer types also warrants additional investigation. Expanding the application of 'retriever' to a broader range of cancer types and integrating it with clinical data will be crucial for realizing its potential in personalized medicine. Furthermore, as the study primarily focuses on kinase inhibitors, it remains to be seen how well these findings translate to other drug classes.
Author response:
Reviewer 1:
Summary:
Identifying drugs that target specific disease phenotypes remains a persistent challenge. Many current methods are only applicable to well-characterized small molecules, such as those with known structures. In contrast, methods based on transcriptional responses offer broader applicability because they do not require prior information about small molecules. Additionally, they can be rapidly applied to new small molecules. One of the most promising strategies involves the use of “drug response signatures”-specific sets of genes whose differential expression can serve as markers for the response to a small molecule. By comparing drug response signatures with expression profiles characteristic of a disease, it is possible to identify drugs that modulate the disease profile, indicating a potential therapeutic connection.
This study aims to prioritize potential drug candidates and to forecast novel drug combinations that may be effective in treating triple-negative breast cancer (TNBC). Large consortia, such as the LINCS-L1000 project, offer transcriptional signatures across various time points after exposing numerous cell lines to hundreds of compounds at different concentrations. While this data is highly valuable, its direct applicability to pathophysiological contexts is constrained by the challenges in extracting consistent drug response profiles from these extensive datasets. The authors use their method to create drug response profiles for three different TNBC cell lines from LINCS.
To create a more precise, cancer-specific disease profile, the authors highlight the use of single-cell RNA sequencing (scRNA-seq) data. They focus on TNBC epithelial cells collected from 26 diseased individuals compared to epithelial cells collected from 10 healthy volunteers. The authors are further leveraging drug response data to develop inhibitor combinations.
Strengths:
The authors of this study contribute to an ongoing effort to develop automated, robust approaches that leverage gene expression similarities across various cell lines and different treatment regimens, aiming to predict drug response signatures more accurately. The authors are trying to address the gap that remains in computational methods for inferring drug responses at the cell subpopulation level.
Weaknesses:
One weakness is that the authors do not compare their method to previous studies. The authors develop a drug response profile by summarizing the time points, concentrations, and cell lines. The computational challenge of creating a single gene list that represents the transcriptional response to a drug across different cell lines and treatment protocols has been previously addressed. The Prototype Ranked List (PRL) procedure, developed by Iorio and co-authors (PNAS, 2010, doi:10.1073/pnas.1000138107), uses a hierarchical majority-voting scheme to rank genes. This method generates a list of genes that are consistently overexpressed or downregulated across individual conditions, which then hold top positions in the PRL. The PRL methodology was used by Aissa and co-authors (Nature Comm 2021, doi:10.1038/s41467-021-21884-z) to analyze drug effects on selective cell populations using scRNA-seq datasets. They combined PRL with Gene Set Enrichment Analysis (GSEA), a method that compares a ranked list of genes like PRL against a specific set of genes of interest. GSEA calculates a Normalized Enrichment Score (NES), which indicates how well the genes of interest are represented among the top genes in the PRL. Compared to the method described in the current manuscript, the PRL method allows for the identification of both upregulated and downregulated transcriptional signatures relevant to the drug’s effects. It also gives equal weight to each cell line’s contribution to the drug’s overall response signature.
The authors performed experimental validation of the top two identified drugs; however, the effect was modest. In addition, the effect on TNBC cell lines was cell-line specific as the identified drugs were effective against BT20, whose transcriptional signatures from LINCS were used for drug identification, but not against the other two cell lines analyzed. An incorrect choice of genes for the signature may result in capturing similarities tied to experimental conditions (e.g., the same cell line) rather than the drug’s actual effects. This reflects the challenges faced by drug response signature methods in both selecting the appropriate subset of genes that make up the signature and managing the multiple expression profiles generated by treating different cell lines with the same drug.
We appreciate the reviewer’s thoughtful feedback and their suggestion to refer to the Prototype Ranked List (PRL) manuscript. Unfortunately, since this methodology for the PRL isn’t implemented in an open-source package, direct comparison with our approach is challenging. Nonetheless, we investigated whether using ranks would yield similar results for the most likely active drug pairs identified by retriever. To do this, we calculated and compared the rankings of the average effect sizes provided by retriever. Although the Spearman (ρ \= 0.98) correlation coefficient was high, we observed that key genes are disadvantaged when using ranks compared to effect sizes. This difference is particularly evident in the gene set enrichment analysis, where using average ranks identified only one pathway as statistically significantly enriched. The code to replicate these analyses is available at https://github.com/dosorio/L1000-TNBC/blob/main/Code/.
Author response image 1.
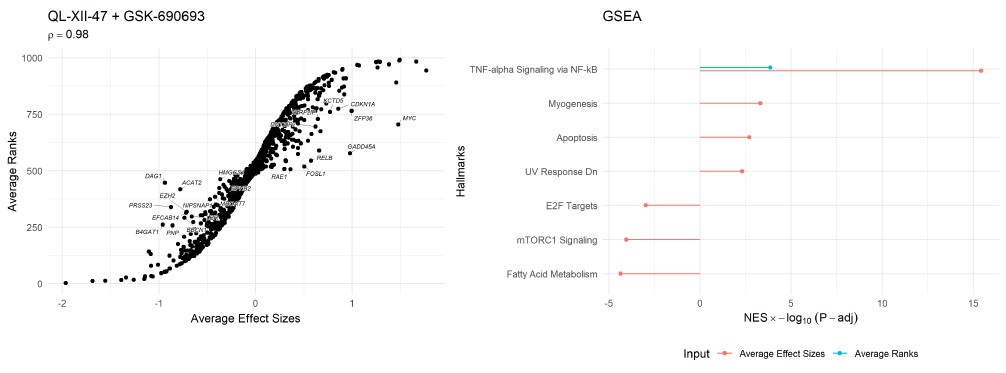
Given the similarity in purpose between retriever and the PRL approach, we have added the following statement to the introduction: “Previously, this goal was approached using a majority-voting scheme to rank genes across various cell types, concentrations, and time points. This approach generates a prototype ranked list (PRL) that represents the consistent ranks of genes across several cell lines in response to a specific drug.”
Regarding the experimental validation, we believe there is a misunderstanding about the evidence we provided. We would like to claridy that we used three different TNBC cell lines: CAL120, BT20, and DU4475. It’s important to note that CAL120 and DU4475 were not included in the signature generation process. Despite this, we observed effects that exceeded the additive effects expectations, particularly in the CAL120 cell line (Figure 5, Panel F).
Reviewer 2:
Summary:
In their study, Osorio and colleagues present ‘retriever,’ an innovative computational tool designed to extract disease-specific transcriptional drug response profiles from the LINCS-L1000 project. This tool has been effectively applied to TNBC, leveraging single-cell RNA sequencing data to predict drug combinations that may effectively target the disease. The public review highlights the significant integration of extensive pharmacological data with high-resolution transcriptomic information, which enhances the potential for personalized therapeutic applications.
Strengths:
A key finding of the study is the prediction and validation of the drug combination QL-XII-47 and GSK-690693 for the treatment of TNBC. The methodology employed is robust, with a clear pathway from data analysis to experimental confirmation.
Weaknesses:
However, several issues need to be addressed. The predictive accuracy of ’retriever’ is contingent upon the quality and comprehensiveness of the LINCS-L1000 and single-cell datasets utilized, which is an important caveat as these datasets may not fully capture the heterogeneity of patient responses to treatment. While the in vitro validation of the drug combinations is promising, further in vivo studies and clinical trials are necessary to establish their efficacy and safety. The applicability of these findings to other cancer types also warrants additional investigation. Expanding the application of ’retriever’ to a broader range of cancer types and integrating it with clinical data will be crucial for realizing its potential in personalized medicine. Furthermore, as the study primarily focuses on kinase inhibitors, it remains to be seen how well these findings translate to other drug classes.
We thank the reviewer for their thoughtful and constructive feedback. We appreciate your insights and agree that several important considerations need to be addressed.
We recognize that the predictive accuracy of retriever depends on the LINCS-L1000 and single-cell datasets. These resources may not fully represent the complete range of transcriptional responses to disease and treatment across different patients. As you mentioned, this is an important limitation. However, we believe that by extrapolating the evaluation of the most likely active compound to each individual patient, we can help address this issue. This approach will provide valuable insights into which patients in the study are most likely to respond positively to treatment.
On the in-vitro validation of drug combinations, we agree that while promising, these results are not sufficient on their own to establish clinical efficacy. Additional in-vivo studies will be essential in assessing the therapeutic potential and safety of these combinations, and clinical trials will be an important next step to validate the translational impact of our findings.
Lastly, we appreciate the reviewer’s comment about the focus of our study on kinase inhibitors. This result was unexpected, as we tested the full set of compounds from the LINCS-L1000 project. We agree that exploring other top candidates, including different drug classes, will be important for assessing how broadly retriever approach can be applied.



