Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorQiang CuiBoston University, Boston, United States of America
- Senior EditorQiang CuiBoston University, Boston, United States of America
Reviewer #1 (Public review):
Summary:
This paper describes molecular dynamics simulations (MDS) of the dynamics of two T-cell receptors (TCRs) bound to the same major histocompatibility complex molecule loaded with the same peptide (pMHC). The two TCRs (A6 and B7) bind to the pMHC with similar affinity and kinetics, but employ different residue contacts. The main purpose of the study is to quantify via MDS the differences in the inter- and intra-molecular motions of these complexes, with a specific focus on what the authors describe as catch-bond behavior between the TCRs and pMHC, which could explain how T-cells can discriminate between different peptides in the presence of weak separating force.
Strengths:
The authors present extensive simulation data that indicates that, in both complexes, the number of high-occupancy inter-domain contacts initially increases with applied load, which is generally consistent with the authors' conclusion that both complexes exhibit catch-bond behavior, although to different extents. In this way, the paper somewhat expands our understanding of peptide discrimination by T-cells.
Weaknesses:
While generally well supported by data, the conclusions would nevertheless benefit from a more concise presentation of information in the figures, as well as from suggesting experimentally testable predictions.
Reviewer #2 (Public review):
In this work, Chang-Gonzalez and coworkers follow up on an earlier study on the force-dependence of peptide recognition by a T-cell receptor using all-atom molecular dynamics simulations. In this study, they compare the results of pulling on a TCR-pMHC complex between two different TCRs with the same peptide. A goal of the paper is to determine whether the newly studied B7 TCR has the same load-dependent behavior mechanism shown in the earlier study for A6 TCR. The primary result is that while the unloaded interaction strength is similar, A6 exhibits more force stabilization.
This is a detailed study, and establishing the difference between these two systems with and without applied force may establish them as a good reference setup for others who want to study mechanobiological processes if the data were made available, and could give additional molecular details for T-Cell-specialists. As written, the paper contains an overwhelming amount of details and it is difficult (for me) to ascertain which parts to focus on and which results point to the overall take-away messages they wish to convey.
Detailed comments:
(1) In Table 1 - are the values of the extension column the deviation from the average length at zero force (that is what I would term extension) or is it the distance between anchor points (which is what I would assume based on the large values. If the latter, I suggest changing the heading, and then also reporting the average extension with an asterisk indicating no extensional restraints were applied for B7-0, or just listing 0 load in the load column. Standard deviation in this value can also be reported. If it is an extension as I would define it, then I think B7-0 should indicate extension = 0+/- something. The distance between anchor points could also be labeled in Figure 1A.
(2) As in the previous paper, the authors apply "constant force" by scanning to find a particular bond distance at which a desired force is selected, rather than simply applying a constant force. I find this approach less desirable unless there is experimental evidence suggesting the pMHC and TCR were forced to be a particular distance apart when forces are applied. It is relatively trivial to apply constant forces, so in general, I would suggest this would have been a reasonable comparison. Line 243-245 speculates that there is a difference in catch bonding behavior that could be inferred because lower force occurs at larger extensions, but I do not believe this hypothesis can be fully justified and could be due to other differences in the complex.
(3) On a related note, the authors do not refer to or consider other works using MD to study force-stabilized interactions (e.g. for catch bonding systems), e.g. these cases where constant force is applied and enhanced sampling techniques are used to assess the impact of that applied force: https://www.cell.com/biophysj/fulltext/S0006-3495(23)00341-7, https://www.biorxiv.org/content/10.1101/2024.10.10.617580v1. I was also surprised not to see this paper on catch bonding in pMHC-TCR referred to, which also includes some MD simulations: https://www.nature.com/articles/s41467-023-38267-1
(4) The authors should make at least the input files for their system available in a public place (github, zenodo) so that the systems are a more useful reference system as mentioned above. The authors do not have a data availability statement, which I believe is required.
Reviewer #3 (Public review):
Summary:
The paper by Chang-Gonzalez et al. is a molecular dynamics (MD) simulation study of the dynamic recognition (load-induced catch bond) by the T cell receptor (TCR) of the complex of peptide antigen (p) and the major histocompatibility complex (pMHC) protein. The methods and simulation protocols are essentially identical to those employed in a previous study by the same group (Chang-Gonzalez et al., eLife 2024). In the current manuscript, the authors compare the binding of the same pMHC to two different TCRs, B7 and A6 which was investigated in the previous paper. While the binding is more stable for both TCRs under load (of about 10-15 pN) than in the absence of load, the main difference is that, with the current MD sampling, B7 shows a smaller amount of stable contacts with the pMHC than A6.
Strengths:
The topic is interesting because of the (potential) relevance of mechanosensing in biological processes including cellular immunology.
Weaknesses:
The study is incomplete because the claims are based on a single 1000-ns simulation at each value of the load and thus some of the results might be marred by insufficient sampling, i.e., statistical error. After the first 600 ns, the higher load of B7high than B7low is due mainly to the simulation segment from about 900 ns to 1000 ns (Figure 1D). Thus, the difference in the average value of the load is within their standard deviation (9 +/- 4 pN for B7low and 14.5 +/- 7.2 for B7high, Table 1). Even more strikingly, Figure 3E shows a lack of convergence in the time series of the distance between the V-module and pMHC, particularly for B70 (left panel, yellow) and B7low (right panel, orange). More and longer simulations are required to obtain a statistically relevant sampling of the relative position and orientation of the V-module and pMHC.
It is not clear why "a 10 A distance restraint between alphaT218 and betaA259 was applied" (section MD simulation protocol, page 9).
Author response:
Reviewer 1:
Summary:
This paper describes molecular dynamics simulations (MDS) of the dynamics of two T-cell receptors (TCRs) bound to the same major histocompatibility complex molecule loaded with the same peptide (pMHC). The two TCRs (A6 and B7) bind to the pMHC with similar affinity and kinetics, but employ different residue contacts. The main purpose of the study is to quantify via MDS the differences in the inter- and intra-molecular motions of these complexes, with a specific focus on what the authors describe as catch-bond behavior between the TCRs and pMHC, which could explain how T-cells can discriminate between different peptides in the presence of weak separating force.
Strengths:
The authors present extensive simulation data that indicates that, in both complexes, the number of high-occupancy interdomain contacts initially increases with applied load, which is generally consistent with the authors’ conclusion that both complexes exhibit catch-bond behavior, although to different extents. In this way, the paper somewhat expands our understanding of peptide discrimination by T-cells.
The reviewer makes thoughtful assessments of our manuscript. While our manuscript is meant to be a “short” contribution, our significant new finding is that even for TCRs targeting the same pMHC, having similar structures, and leading to similar functional outcomes in conventional assays, their response to applied load can be different. This supports out recent experimental work where TCRs targeting the same pMHC differed in their catch bond characteristics, and importantly, in their response to limiting copy numbers of pMHCs on the antigen-presenting cell (Akitsu et al., Sci. Adv., 2024; cited in our manuscript). Our present manuscript provides the physical basis where two similar TCRs respond to applied load differently. In the revised manuscript, we will make this point clearer.
Weaknesses:
While generally well supported by data, the conclusions would nevertheless benefit from a more concise presentation of information in the figures, as well as from suggesting experimentally testable predictions.
Following the reviewers’ suggestions, we will update figures and use Figure Supplements to make the main figures more concise and to simplify the overall presentation.
Regarding testable predictions, one prediction would be that B7 TCR will exhibit weaker catch bond behavior than A6. This is an important prediction because the two TCRs targeting the same pMHC have similar structures and are functionally similar in conventional assays. This prediction can be tested by single-molecule optical tweezers experiments. We also predict the A6 TCR may perform better when the number of pMHC molecules presented are limited, analogous to our recent experiments on different TCRs, Akitsu et al., Sci. Adv. (2024).
Another testable prediction for the conservation of the basic allostery mechanism is to test the Cβ FG-loop deletion mutant located at the hinge region of the β chain, yet its deletion severely impairs the catch bond formation. These predictions will be mentioned and discussed in the updated manuscript.
Reviewer 2:
In this work, Chang-Gonzalez and coworkers follow up on an earlier study on the force-dependence of peptide recognition by a T-cell receptor using all-atom molecular dynamics simulations. In this study, they compare the results of pulling on a TCR-pMHC complex between two different TCRs with the same peptide. A goal of the paper is to determine whether the newly studied B7 TCR has the same load-dependent behavior mechanism shown in the earlier study for A6 TCR. The primary result is that while the unloaded interaction strength is similar, A6 exhibits more force stabilization.
This is a detailed study, and establishing the difference between these two systems with and without applied force may establish them as a good reference setup for others who want to study mechanobiological processes if the data were made available, and could give additional molecular details for T-Cell-specialists. As written, the paper contains an overwhelming amount of details and it is difficult (for me) to ascertain which parts to focus on and which results point to the overall take-away messages they wish to convey.
As mentioned above and as the reviewer correctly pointed out, the condensed appearance of this manuscript arose largely because we intended it to be a Research Advances article as a short follow up study of our previous paper on A6 TCR published in eLife. Most of the analysis scripts for the A6 TCR study are already available on Github. We will additionally deposit sample structures and simulation scripts for the B7 TCR. Trajectory will be provided upon request given their large size.
Regarding the focus issue, it is in part due to the complex nature of the problem, which required simulations under different conditions and multi-faceted analyses. Concisely presenting the complex analyses also has been a challenge in our previous papers on TCR simulations (Hwang et al., PNAS 2020; Chang-Gonzalez et al., eLife, 2024 – both are cited in our manuscript). With updated figures and texts, we expect that the presentation will be a lot clearer. But even in the present form, the reviewer points out the main take-away message well: “The primary result is that while the unloaded interaction strength is similar, A6 exhibits more force stabilization.
Detailed comments:
(1) In Table 1 - are the values of the extension column the deviation from the average length at zero force (that is what I would term extension) or is it the distance between anchor points (which is what I would assume based on the large values. If the latter, I suggest changing the heading, and then also reporting the average extension with an asterisk indicating no extensional restraints were applied for B7-0, or just listing 0 load in the load column. Standard deviation in this value can also be reported. If it is an extension as I would define it, then I think B7-0 should indicate extension = 0+/- something.
The distance between anchor points could also be labeled in Figure 1A.
“Extension” is the distance between anchor points (blue spheres at the ends of the added strands in Fig. 1A). While its meaning should be clear in the section “Laddered extensions” in MD simulation protocol, at first glance it may lead to confusion. In a strict sense, use of “extension” for the distance is a misnomer, but we have used it in our previous two papers (Hwang et al., PNAS 2020; Chang-Gonzalez et al., eLife, 2024), so we prefer to keep it for consistency. Instead, in the caption of Table 1, we will explain its meaning, and also explicitly label it in Fig. 1A, as the reviewer suggested.
Please also note that the no-load case B70 does not have a particular extension that yields zero load on average. It would in fact be very difficult to find such an extension (distance between two anchor points). To simulate the system without load, we separately built a TCR-pMHC complex without added linkers, and held the distal part of pMHC with weak harmonic restraints (explained in sections “Structure preparation” and “Systems without load”). In this way, no external force is applied to TCR as it moves relative to pMHC. We will clarify this when introducing B70 in the Results section.
(2) As in the previous paper, the authors apply ”constant force” by scanning to find a particular bond distance at which a desired force is selected, rather than simply applying a constant force. I find this approach less desirable unless there is experimental evidence suggesting the pMHC and TCR were forced to be a particular distance apart when forces are applied. It is relatively trivial to apply constant forces, so in general, I would suggest this would have been a reasonable comparison. Line 243-245 speculates that there is a difference in catch bonding behavior that could be inferred because lower force occurs at larger extensions, but I do not believe this hypothesis can be fully justified and could be due to other differences in the complex.
There is indeed experimental evidence that the TCR-pMHC complex operates under constant separation. The spacing between a T-cell and an antigen-presenting cell is maintained by adhesion molecules such as the CD2CD58 pair, as explained in our paper on the A6 TCR, (Chang-Gonzalez et al., eLife, 2024; please see the bottom paragraph on page 4 of the paper). In in vitro single-molecule experiments, pulling to a fixed separation and holding is also commonly done. Detailed comparison between constant extension vs. constant force simulations is definitely a subject of our future study. We will clarify these points when explaining about the constant extension (or separation).
Regarding line 243–245, we agree with the reviewer that without further tests, lower forces at larger extensions per se cannot be an indicator that B7 forms a weaker catch bond. But with additional insight, it does have an indirect relevance. In addition to fewer TCR-pMHC contacts (Fig. 1C of our manuscript), the intra-TCR contacts are also reduced compared to those of A6 (Fig. 1D vs. Chang-Gonzalez et al., eLife, 2024, Fig. 8A,B, first column; reproduced in the figure in our response to reviewer 3 below). This shows that the B7 TCR forms a looser complex with pMHC compared to A6. With its higher compliance, the B7 TCR-pMHC complex needs to be under a greater extension than A6 to apply comparable levels of force, and it would be more difficult to achieve load-induced stabilization of the TCR-pMHC interface, hence a weaker catch bond. We will add this point when explaining the weaker catch bond behavior of B7.
(3) On a related note, the authors do not refer to or consider other works using MD to study force-stabilized interactions (e.g. for catch bonding systems), e.g. these cases where constant force is applied and enhanced sampling techniques are used to assess the impact of that applied force: https://www.cell.com/biophysj/fulltext/S0006-3495(23)00341-7, https://www.biorxiv.org/content/10.1101/2024.10.10.617580v1. I was also surprised not to see this paper on catch bonding in pMHC-TCR referred to, which also includes some MD simulations: https://www.nature.com/articles/s41467-023-38267-1
We thank the reviewer for bringing the three papers to our attention, which are:
(1) Languin-Cattoën, Sterpone, and Stirnemann, Biophys. J. 122:2744 (2023): About bacterial adhesion protein FimH.
(2) Peña Ccoa, et al., bioRxiv (2024): About actin binding protein vinculin.
(3) Choi et al., Nat. Comm. 14:2616 (2023): About a mathematical model of the TCR catch bond.
Catch bond mechanisms of FimH and vinculin are different from that of TCR in that FimH and vinculin have relatively well-defined weak- and strong-binding states where there are corresponding crystal structures. Availability of the end-state structures enable using simulation approaches such as enhanced sampling of individual states and studying the transition between the two states. In contrast, TCR does not have any structurally well-defined weakor strong-binding states, which requires a different approach. As demonstrated in our current manuscript as well as in our previous two papers (Hwang et al., PNAS 2020; Chang-Gonzalez et al., eLife, 2024), our microsecond-long simulations of the complex under realistic pN-level loads and a combination of analysis methods are effective for elucidating the catch bond mechanism of TCR. In the revised manuscript, we will cite the two papers, to compare the TCR catch bond mechanism with those of FimH and vinculin, which will offer a broader perspective.
The third paper (Choi, 2023) proposes a mathematical model to analyze extensive sets of data, and also perform new experiments and additional simulations. Of note, their model assumptions are based mainly on the steered MD (SMD) simulation in their previous paper (Wu, et al., Mol. Cell. 73:1015, 2019). In their model, formation of a catch bond (called catch-slip bond in Choi’s paper) requires partial unfolding of MHC and tilting of the TCR-pMHC interface. While further studies are needed to find whether those changes are indeed required, even so, the question remains regarding how the complex in the fully folded state can bear load and enter such a state in the first place. Our current and previous simulation studies suggest a mechanism by which ligand- and load-dependent responses occur as the first obligatory step of catch bond formation, after which partial unfolding and/or extensive conformational transitions may occur, as described in our recent paper (Akitsu et al., Sci. Adv., 2024). In the revised manuscript, we will cite Wu’s paper and briefly explain the above.
(4) The authors should make at least the input files for their system available in a public place (github, zenodo) so that the systems are a more useful reference system as mentioned above. The authors do not have a data availability statement, which I believe is required.
As mentioned above, we will make sample input files and coordinates available on Github. Data availability statement will be added.
Reviewer 3:
Summary:
The paper by Chang-Gonzalez et al. is a molecular dynamics (MD) simulation study of the dynamic recognition (load-induced catch bond) by the T cell receptor (TCR) of the complex of peptide antigen (p) and the major histocompatibility complex (pMHC) protein. The methods and simulation protocols are essentially identical to those employed in a previous study by the same group (Chang-Gonzalez et al., eLife 2024). In the current manuscript, the authors compare the binding of the same pMHC to two different TCRs, B7 and A6 which was investigated in the previous paper. While the binding is more stable for both TCRs under load (of about 10-15 pN) than in the absence of load, the main difference is that, with the current MD sampling, B7 shows a smaller amount of stable contacts with the pMHC than A6.
Strengths:
The topic is interesting because of the (potential) relevance of mechanosensing in biological processes including cellular immunology.
Weaknesses:
The study is incomplete because the claims are based on a single 1000-ns simulation at each value of the load and thus some of the results might be marred by insufficient sampling, i.e., statistical error. After the first 600 ns, the higher load of B7high than B7low is due mainly to the simulation segment from about 900 ns to 1000 ns (Figure 1D). Thus, the difference in the average value of the load is within their standard deviation (9 +/- 4 pN for B7low and 14.5 +/- 7.2 for B7high, Table 1). Even more strikingly, Figure 3E shows a lack of convergence in the time series of the distance between the V-module and pMHC, particularly for B70 (left panel, yellow) and B7low (right panel, orange). More and longer simulations are required to obtain a statistically relevant sampling of the relative position and orientation of the V-module and pMHC.
The reviewer uses data points during the last 100 ns to raise an issue with sampling. But since we are using realistic pN range forces, force fluctuates more slowly. In fact, in our simulation of B7high, while the force peaks near 35 pN at 500 ns (Fig. 1D of our manuscript; reproduced as panels C and D below), the contact heat map shows no noticeable changes around 500 ns (Fig. 2C of our manuscript). Thus, a wider time window must be considered rather than focusing on instantaneous force.
We believe the reviewer’s concern about sampling arose also due to a lack of clear explanation. Author response image 1 below contains panels from our earlier eLife paper on the A6 TCR. Panels A and B are from Fig. 8 of the A6 paper, and panels C and D are from Fig. 1D of our present manuscript. The high-load simulations in both cases (outlined circles) fluctuate widely in force so that one might argue that sampling was insufficient. However, unless one is interested in finding the precise value of force for a given extension, sampling in our simulations was reasonable enough to distinguish between high- and low-force behaviors. To support this, we show panel E below, which is from Appendix 3–Fig. 1 of our A6 paper. Added to this panel are the average forces and standard deviations of B7low and B7high from Table 1 of our manuscript (red squares). Please note that all of the data were measured after 500 ns. Except for Y8Alow and dFGlow of A6 (explained below), all of the data points lie on nearly a straight line.
Author response image 1.
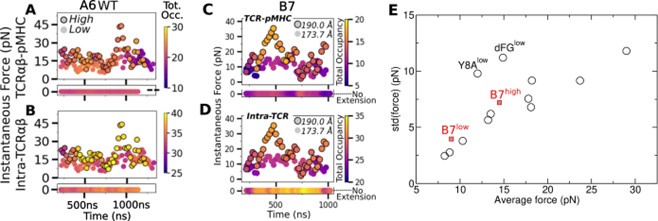
Thermodynamically, the force and position of the restraint (blue spheres in Fig. 1A of our manuscript) form a pair of generalized force and the corresponding spatial variable in equilibrium at temperature 300 K, which is akin to the pressure P and volume V of an ideal gas. If V is fixed, P fluctuates. Denoting the average and std of pressure as ⟨P⟩ and ∆P, respectively, Burgess showed that ∆P/⟨P⟩ is a constant (Eq. 5 of Burgess, Phys. Lett. A, 44:37; 1973). In the case of the TCRαβ-pMHC system, although individual atoms are not ideal gases, since their motion leads to the fluctuation in force on the restraints, the situation is analogous to the case where pressure arises from individual ideal gas molecules hitting the confining wall as the restraint. Thus, the near-linear behavior in panel E above is a consequence of the system being many-bodied and at constant temperature. The linearity is also an indirect indicator that sampling of force was reasonable. The fact that A6 and B7 data show a common linear profile further demonstrates the consistency in our force measurement. That said, the B7 data points (red in panel E) are elevated slightly above nearby A6 data points. This is consistent with B7 forming an overall weaker complex, both at the TCR-pMHC interface (panels A vs. C) and within intra-TCR interfaces (panels B vs. D), which can be seen by the wider ranges of color bars in panels A and B for A6 compared to panels C and D for B7.
About the two outliers of A6, Y8Alow is for an antagonist peptide and dFGlow is the Cβ FG-loop deletion mutant. Interestingly, both cases had reduced numbers of contacts with pMHC, which likely caused a wider conformational motion, hence greater fluctuation in force.
A similar argument applies to Fig. 3E of our manuscript. If precise values of the V-module to pMHC distance were needed, longer or duplicate simulations would be necessary, however, Fig. 3E as it currently stands clearly shows that B7high maintains more stable interface compared to B7low, which is consistent with all other measures we used, such as Fig. 3B (Hamming distance), Fig. 3C (buried surface area), and Fig. 4A–E (Vα-Vβ motion and CDR3 distance). They are also consistent with our simulations of A6.
Thus, rather than relying on peculiarities of individual trajectories, we analyze data in multiple ways and draw conclusions based on features that are consistent across different simulations. Please also note that reviewer 1 mentioned that our conclusions are “generally well supported by data.”
We will update our manuscript to concisely explain the above and also will add Panel E above as a supplement of Fig. 1.
It is not clear why ”a 10 A distance restraint between alphaT218 and betaA259 was applied” (section MD simulation protocol, page 9).
αT218 and βA259 are the residues attached to a leucine-zipper handle in in vitro optical trap experiments (Das, et al., PNAS 2015). In T cells, those residues also connect to transmembrane helices. Author response image 2 is a model of N15 TCR used in experiments in Das’ paper, constructed based on PDB 1NFD. Blue spheres represent Cα atoms corresponding to αT218 and βA259 of B7 TCR. Their distance is 6.7 ˚A. The 10-˚A distance restraint in simulation was applied to mimic the presence of the leucine zipper that prevents excessive separation of the added strands. The distance restraint is a flat-bottom harmonic potential which is activated only when the distance between the two atoms exceeds 10 ˚A, which we did not clarify in our original manuscript. The same restraint was used in our previous studies on JM22 and A6 TCRs.
We will add the figure as a supplement of Fig. 1, cite Das’ paper, and also update description of the distance restraint in the MD simulation protocol section.
Author response image 2.