Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorMurim ChoiSeoul National University, Seoul, Republic of Korea
- Senior EditorMurim ChoiSeoul National University, Seoul, Republic of Korea
Reviewer #1 (Public review):
Summary:
Howard et al. performed deep mutational scanning on the MC4R gene, using a reporter assay to investigate two distinct downstream pathways across multiple experimental conditions. They validated their findings with ClinVar data and previous studies. Additionally, they provided insights into the application of DMS results for personalized drug therapy and differential ligand responses across variant types.
Strengths:
They captured over 99% of variants with robust signals and investigated subtle functionalities, such as pathway-specific activities and interactions with different ligands, by refining both the experimental design and analytical methods.
Weaknesses:
While the study generated informative results, it lacks a detailed explanation regarding the input library, replicate correlation, and sequencing depth for a given number of cells. Additionally, there are several questions that it would be helpful for authors to clarify.
(1) It would be helpful to clarify the information regarding the quality of the input library and experimental replicates. Are variants evenly represented in the library? Additionally, have the authors considered using long-read sequencing to confirm the presence of a single intended variant per construct? Finally, could the authors provide details on the correlation between experimental replicates under each condition?
(2) Since the functional readout of variants is conducted through RNA sequencing, it seems crucial to sequence a sufficient number of cells with adequate sequencing saturation. Could the authors clarify the coverage depth used for each RNA-seq experiment and how this depth was determined? Additionally, how many cells were sequenced in each experiment?
(3) It appears that the frequencies of individual RNA-seq barcode variants were used as a proxy for MR4C activity. Would it be important to also normalize for heterogeneity in RNA-seq coverage across different cells in the experiment? Variability in cell representation (i.e., the distribution of variants across cells) could lead to misinterpretation of variant effects. For example, suppose barcode_a1 represents variant A and barcode_b1 represents variant B. If the RNA-seq results show 6 reads for barcode_a1 and 7 reads for barcode_b1, it might initially appear that both variants have similar effect sizes. However, if these reads correspond to 6 separate cells each containing 1 copy of barcode_a1, and only 1 cell containing 7 copies of barcode_b1, the interpretation changes significantly. Additionally, if certain variants occupy a larger proportion of the cell population, they are more likely to be overrepresented in RNA sequencing.
(4) Although the assay system appears to effectively represent MC4R functionality at the molecular level, we are curious about the potential disparity between the DMS score system and physiological relevance. How do variants reported in gnomAD distribute within the DMS scoring system?
(5) To measure Gq signaling, the authors used the GAL4-VPR relay system. Is there additional experimental data to support that this relay system accurately represents Gq signaling?
(6) Identifying the variants responsive to the corrector was impressive. However, we are curious about how the authors confirmed that the restoration of MC4R activity was due to the correction of the MC4R protein itself. Is there a possibility that the observed effect could be influenced by other factors affected by the corrector? When the corrector was applied to the cells, were any expected or unexpected differential gene expression changes observed?
(7) As mentioned in the introduction, gain-of-function (GoF) variants are known to be protective against obesity. It would be interesting to see further studies on the observed GoF variants. Do the authors have any plans for additional research on these variants?
Reviewer #2 (Public review):
Overview
In this manuscript, the authors use deep mutational scanning to assess the effect of ~6,600 protein-coding variants in MC4R, a G protein-coupled receptor associated with obesity. Reasoning that current deep mutational scanning approaches are insufficiently precise for some drug development applications, they focus on articulating new, more precise approaches. These approaches, which include a new statistical model and innovative reporter assay, enable them to probe molecular phenotypes directly relevant to the development of drugs that target this receptor with high precision and statistical rigor.
They use the resulting data for a variety of purposes, including probing the relationship between MC4R's sequence and structure, analyzing the effect of clinically important variants, identifying variants that disrupt downstream MC4R signaling via one but not both pathways, identifying loss of function variants are amenable to a corrector drug and exploring how deep mutational scanning data could guide small molecule drug optimization.
Strengths
The analysis and statistical framework developed by the authors represent a significant advance. In particular, the study makes use of barcode-level internally replicated measurements to more accurately estimate measurement noise.
The framework allows variant effects to be compared across experimental conditions, a task that is currently hard to do with rigor. Thus, this framework will be applicable to a large number of existing and future deep mutational scanning experiments.
The authors refine their existing barcode transcription-based assay for GPCR signaling, and develop a clever "relay" new reporter system to boost signaling in a particular pathway. They show that these reporters can be used to measure both gain of function and loss of function effects, which many deep mutational scanning approaches cannot do.
The use of systematic approaches to integrate and then interrogate high-dimensional deep mutational scanning data is a big strength. For example, the authors applied PCA to the variant effect results from reporters for two different MC4R signaling pathways and were able to discover variants that biased signaling through one or the other pathway. This approach paves the way for analyses of higher dimensional deep mutational scans.
The authors use the deep mutational scanning data they collect to map how different variants impact small molecule agonists activate MC4R signaling. This is an exciting idea, because developing small-molecule protein-targeting therapeutics is difficult, and this manuscript suggests a new way to map small-molecule-protein interactions.
Weaknesses
The authors derive insights into the relationship between MC4R signaling through different pathways and its structure. While these make sense based on what is already known, the manuscript would be stronger if some of these insights were validated using methods other than deep mutational scanning.
Likewise, the authors use their data to identify positions where variants disrupt MC4R activation by one small molecule agonist but not another. They hypothesize these effects point to positions that are more or less important for the binding of different small molecule agonists. The manuscript would be stronger if some of these insights were explored further.
Impact
In this manuscript, the authors present new methods, including a statistical framework for analyzing deep mutational scanning data that will have a broad impact. They also generate MC4R variant effect data that is of interest to the GPCR community.
Author response:
We thank the reviewers for their support of this work and insightful recommendations for how to improve it. We have provided specific responses to each reviewer comment below. To summarize how we intend to address the requested revisions:
Many of the reviewers’ comments requested additional technical or quality details about the DMS libraries or assays (e.g., number of cells tested, number of sequencing reads, assay replication, assay sensitivity, library balance), and we provide additional information and analyses that we can incorporate into the relevant portions of the text, supplementary tables, and supplementary figures to address these questions.
Some comments asked to clarify nomenclature/wording or provide additional labels to images, and we will make these changes as requested.
A few questions would require additional experimental data to address. Where experiments have already been performed, we will incorporate those results or cite relevant work previously reported in the literature.
Reviewer 1:
Summary
Howard et al. performed deep mutational scanning on the MC4R gene, using a reporter assay to investigate two distinct downstream pathways across multiple experimental conditions. They validated their findings with ClinVar data and previous studies. Additionally, they provided insights into the application of DMS results for personalized drug therapy and differential ligand responses across variant types.
Strengths
They captured over 99% of variants with robust signals and investigated subtle functionalities, such as pathway-specific activities and interactions with different ligands, by refining both the experimental design and analytical methods.
Weaknesses
While the study generated informative results, it lacks a detailed explanation regarding the input library, replicate correlation, and sequencing depth for a given number of cells.
Additionally, there are several questions that it would be helpful for authors to clarify.
(1) It would be helpful to clarify the information regarding the quality of the input library and experimental replicates. Are variants evenly represented in the library? Additionally, have the authors considered using long-read sequencing to confirm the presence of a single intended variant per construct? Finally, could the authors provide details on the correlation between experimental replicates under each condition?
Are variants evenly represented in the library?
We strive to achieve as evenly balanced library as possible at every stage of the DMS process (e.g., initial cloning in E. coli through integration into human cells). Below is a representative plot showing the number of barcodes per amino acid variant at each position in a given ~60 amino acid subregion of MC4R, which highlights how evenly variants are represented at the E. coli cloning stage.
Author response image 1.
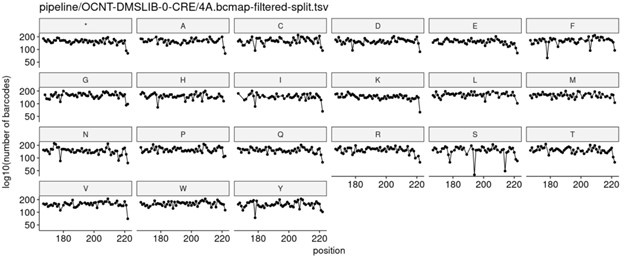
We also make similar measurements after the library is integrated into HEK293T cell lines, and see similarly even coverage across all variants, as shown in the plot below.
Author response image 2.
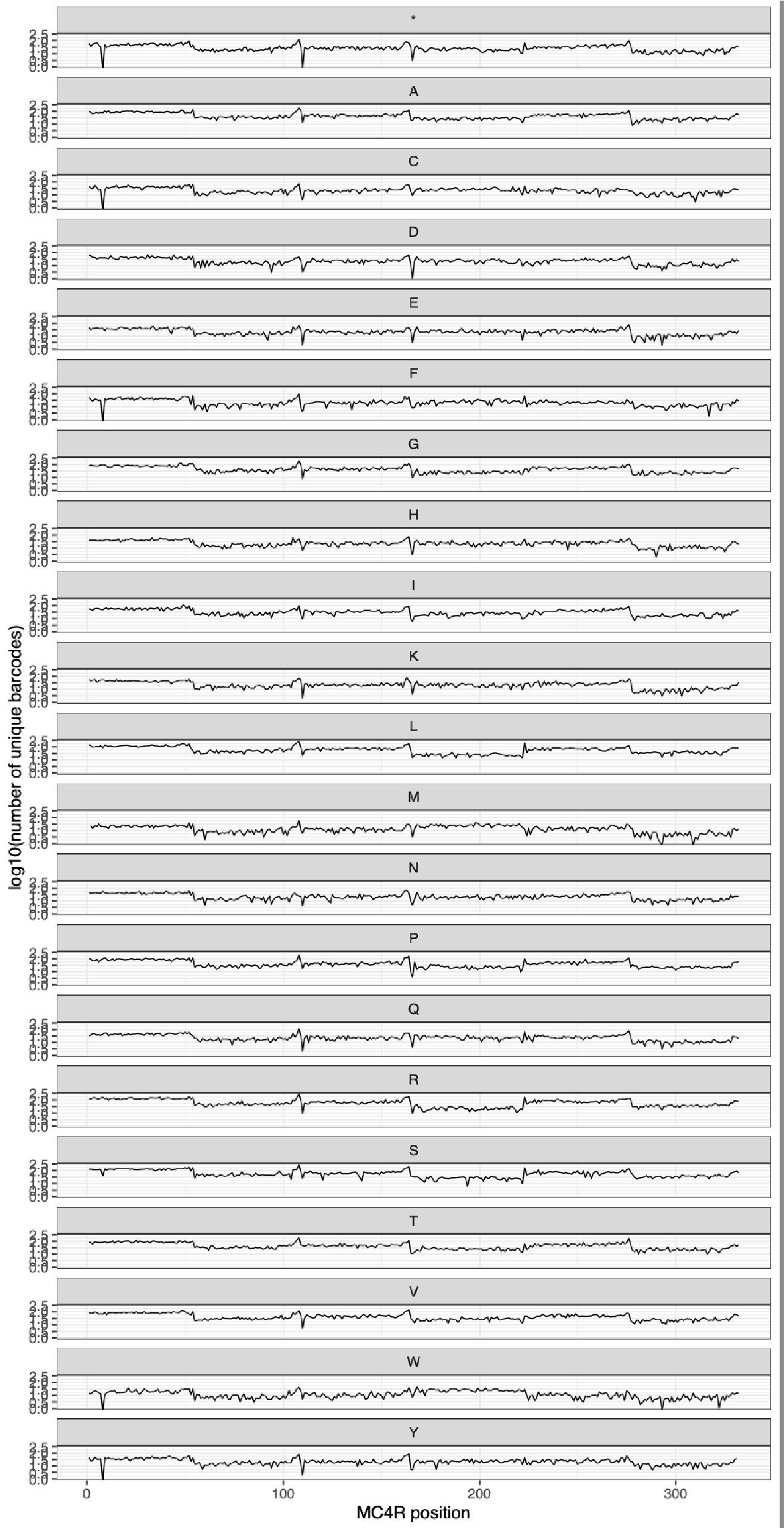
Additionally, have the authors considered using long-read sequencing to confirm the presence of a single intended variant per construct?
We agree long-read sequencing would be an excellent way to confirm that our constructs contain a single intended variant. However, we elected for an alternate method (outlined in more detail in Jones et al. 2020) that leverages multiple layers of validation. First, the oligo chip-synthesized portions of the protein containing the variants are cloned into a sequence-verified plasmid backbone, which greatly decreases the chances of spuriously generating a mutation in a different portion of the protein. We then sequence both the oligo portion and random barcode using overlapping paired end reads during barcode mapping to avoid sequencing errors and to help detect DNA synthesis errors. At this stage, we computationally reject any constructs that have more than one variant. Given this, the vast majority of remaining unintended variants would come from somatic mutations introduced by the E. coli cloning or replication process, which should be low frequency. We have used our in-house full plasmid sequencing method, OCTOPUS, to sample and spot check this for several other DMS libraries we have generated using the same cloning methods. We have found variants in the plasmid backbone in only ~1% of plasmids in these libraries. Our statistical model also helps correct for this by accounting for barcode-specific variation. Finally we believe this provides further motivation for having multiple barcodes per variant, which dilutes the effect of any unintended additional variants.
Finally, could the authors provide details on the correlation between experimental replicates under each condition?
Certainly! In general, the Gs reporter had higher correlation between replicates than the Gq system (r ~ 0.5 vs r ~ 0.4). The plots below show two representative correlations at the RNA-seq stage of read counts for barcodes between the low a-MSH conditions. One important advantage of our statistical model is that it’s able to leverage information from barcodes regardless of the number of replicates they appear in.
Author response image 3.
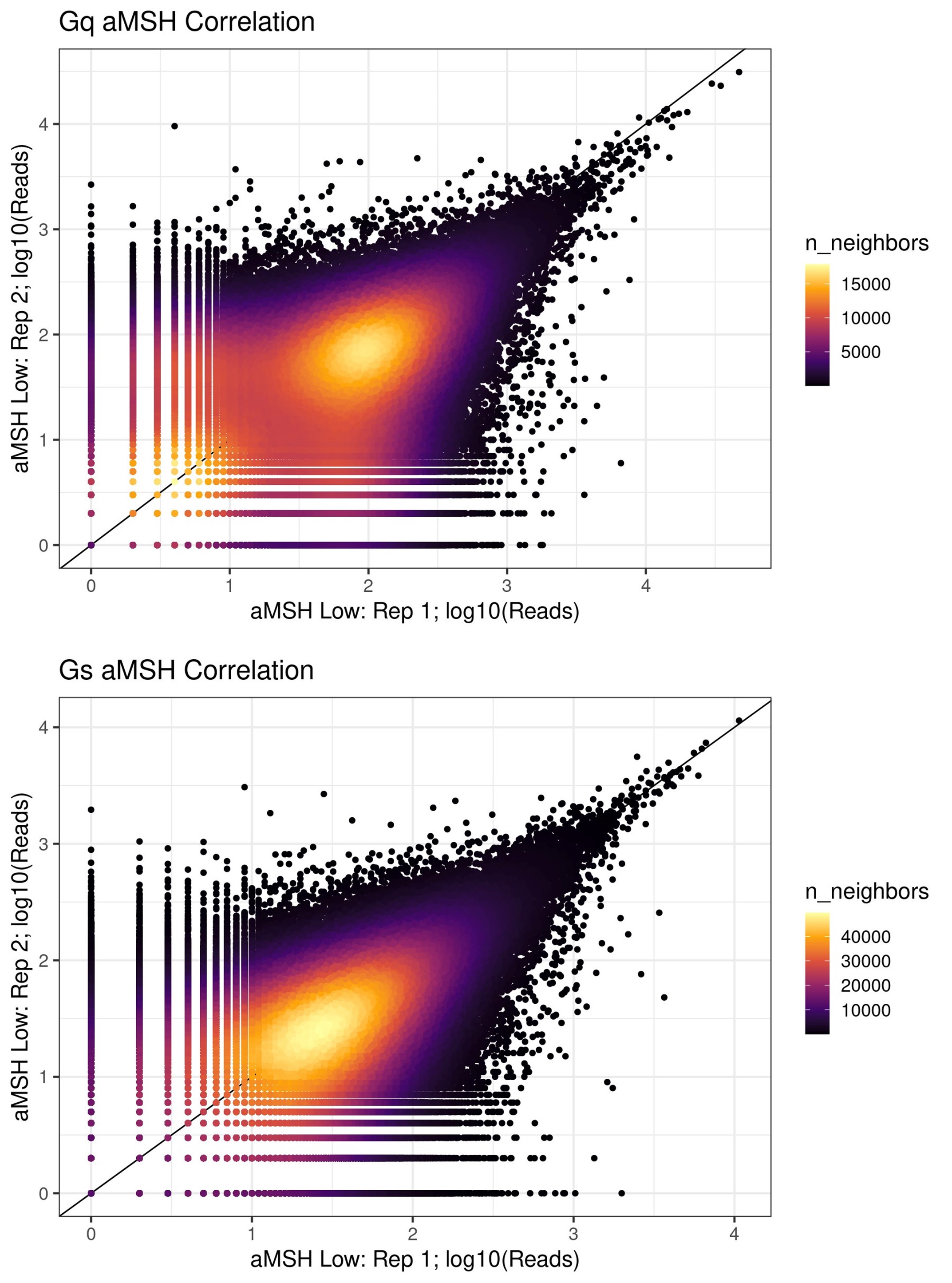
Since the functional readout of variants is conducted through RNA sequencing, it seems crucial to sequence a sufficient number of cells with adequate sequencing saturation. Could the authors clarify the coverage depth used for each RNA-seq experiment and how this depth was determined? Additionally, how many cells were sequenced in each experiment?
This will be addressed by incorporating the following details into the manuscript:
We seeded 17 million cells per replicate at the start of each assay and, with a doubling of ~1.5x over the course of the assay, harvested ~25.5 million cells per replicate for RNA extraction and sequencing. We found this sufficient to get at least ~30-60x cellular coverage per amino acid variant.
Total mapped reads per replicate at RNA-seq stage
- Gs/CRE: 9.1-18.2 million mapped reads, median=12.3
- Gq/UAS: 8.6-24.1 million mapped reads, median=14.5
- Gs/CRE+Chaperone: 6.4-9.5 million mapped reads, median=7.5
Reads per barcode distribution
- Median read counts of 8, 10, and 6 reads per sample per barcode for Gs/CRE, Gq/UAS, and Gs/CRE+Chaperone assays, respectively.
Barcodes per variant distribution
- As reported, the median number of barcodes per variant across samples (the “median of medians”) is 56 for Gs/CRE and 28 for Gq/UAS
- Additionally, it is 44 for Gs/CRE+Chaperone
It appears that the frequencies of individual RNA-seq barcode variants were used as a proxy for MR4C activity. Would it be important to also normalize for heterogeneity in RNA-seq coverage across different cells in the experiment? Variability in cell representation (i.e., the distribution of variants across cells) could lead to misinterpretation of variant effects. For example, suppose barcode_a1 represents variant A and barcode_b1 represents variant B. If the RNA-seq results show 6 reads for barcode_a1 and 7 reads for barcode_b1, it might initially appear that both variants have similar effect sizes. However, if these reads correspond to 6 separate cells each containing 1 copy of barcode_a1, and only 1 cell containing 7 copies of barcode_b1, the interpretation changes significantly. Additionally, if certain variants occupy a larger proportion of the cell population, they are more likely to be overrepresented in RNA sequencing.
We account for this heterogeneity in several ways. First, as shown above (Response to Reviewer 1, Question 1), we aim to have even representation of variants within our libraries. Second, we utilize compositional control conditions like forskolin or unstimulated conditions to obtain treatment-independent measurements of barcode abundance and, consequently, of mutant-vs-WT effects that are due to compositional rather than biological variability. We expect that variability observed under these controls is due to subtle effects of molecular cloning, gene expression, and stochasticity. Using these controls, we observe that mutant-vs-WT effects are generally close to zero in these normalization conditions (e.g., in untreated Gq, see Supplementary Figure 3) as compared to drug-treated conditions. For example, pre-mature stops behave similar to WT in normalization conditions. This indicates that mutant abundance is relatively homogenous. Where there are barcode-dependent effects on abundance, we can use information from these conditions to normalize that effect. Finally, our mixed-effect model accounts for barcode-specific deviations from the expected mutant effect (e.g. a “high count” barcode consistently being high relative to the mean).
Although the assay system appears to effectively represent MC4R functionality at the molecular level, we are curious about the potential disparity between the DMS score system and physiological relevance. How do variants reported in gnomAD distribute within the DMS scoring system?
Figure 2D shows DMS scores (variant effect on Gs signaling) relative to human population frequency for all MC4R variants reported in gnomAD as of January 8, 2024.
To measure Gq signaling, the authors used the GAL4-VPR relay system. Is there additional experimental data to support that this relay system accurately represents Gq signaling?
The full Gq reporter uses an NFAT response element from the IL-2 promoter to regulate the expression of the GAL4-VPR relay. In this system, the activation of Gq signaling results in the activation of the NFAT response element, and this signal is then amplified by the GAL4-VPR relay. The NFAT response element has been previously well-validated to respond to the activation of Gq signaling (e.g., PMID: 8631834). We will add this reference to the text to further support the use of the Gq assay.
Identifying the variants responsive to the corrector was impressive. However, we are curious about how the authors confirmed that the restoration of MC4R activity was due to the correction of the MC4R protein itself. Is there a possibility that the observed effect could be influenced by other factors affected by the corrector? When the corrector was applied to the cells, were any expected or unexpected differential gene expression changes observed?
While we do not directly measure whether Ipsen-17 has effects on other signaling processes, previous work has shown that Ipsen-17 treatment does not indirectly alter signaling kinetics such as receptor internalization (Wang et al., 2014). Furthermore, our analysis methods inherently account for this by normalizing variant effects to WT signaling levels. Any observed rescue of a given variant inherently means that the variant is specifically more responsive to Ipsen-17 than WT, and the fact that different variants exhibit different levels of rescue is reassuring that the mechanism is on target to MC4R. Lastly, Ipsen-17 is known to be an antagonist of alpha-MSH activity and is thought to bind directly to the same site on MC4R (Wang et al., 2014).
As mentioned in the introduction, gain-of-function (GoF) variants are known to be protective against obesity. It would be interesting to see further studies on the observed GoF variants. Do the authors have any plans for additional research on these variants?
We agree this would be an excellent line of inquiry, but due to changes in company priorities we unfortunately do not have any plans for additional research on these variants.
Reviewer 2:
Overview
In this manuscript, the authors use deep mutational scanning to assess the effect of ~6,600 protein-coding variants in MC4R, a G protein-coupled receptor associated with obesity. Reasoning that current deep mutational scanning approaches are insufficiently precise for some drug development applications, they focus on articulating new, more precise approaches. These approaches, which include a new statistical model and innovative reporter assay, enable them to probe molecular phenotypes directly relevant to the development of drugs that target this receptor with high precision and statistical rigor.
They use the resulting data for a variety of purposes, including probing the relationship between MC4R's sequence and structure, analyzing the effect of clinically important variants, identifying variants that disrupt downstream MC4R signaling via one but not both pathways, identifying loss of function variants are amenable to a corrector drug and exploring how deep mutational scanning data could guide small molecule drug optimization.
Strengths
The analysis and statistical framework developed by the authors represent a significant advance. In particular, the study makes use of barcode-level internally replicated measurements to more accurately estimate measurement noise.
The framework allows variant effects to be compared across experimental conditions, a task that is currently hard to do with rigor. Thus, this framework will be applicable to a large number of existing and future deep mutational scanning experiments.
The authors refine their existing barcode transcription-based assay for GPCR signaling, and develop a clever "relay" new reporter system to boost signaling in a particular pathway. They show that these reporters can be used to measure both gain of function and loss of function effects, which many deep mutational scanning approaches cannot do.
The use of systematic approaches to integrate and then interrogate high-dimensional deep mutational scanning data is a big strength. For example, the authors applied PCA to the variant effect results from reporters for two different MC4R signaling pathways and were able to discover variants that biased signaling through one or the other pathway. This approach paves the way for analyses of higher dimensional deep mutational scans.
The authors use the deep mutational scanning data they collect to map how different variants impact small molecule agonists activate MC4R signaling. This is an exciting idea, because developing small-molecule protein-targeting therapeutics is difficult, and this manuscript suggests a new way to map small-molecule-protein interactions.
Weaknesses
The authors derive insights into the relationship between MC4R signaling through different pathways and its structure. While these make sense based on what is already known, the manuscript would be stronger if some of these insights were validated using methods other than deep mutational scanning.
Likewise, the authors use their data to identify positions where variants disrupt MC4R activation by one small molecule agonist but not another. They hypothesize these effects point to positions that are more or less important for the binding of different small molecule agonists. The manuscript would be stronger if some of these insights were explored further.
Impact
In this manuscript, the authors present new methods, including a statistical framework for analyzing deep mutational scanning data that will have a broad impact. They also generate MC4R variant effect data that is of interest to the GPCR community.



