Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorPaschalis KratsiosUniversity of Chicago, Chicago, United States of America
- Senior EditorSonia SenTata Institute for Genetics and Society, Bangalore, India
Reviewer #1 (Public review):
Summary:
The researchers aimed to identify which neurotransmitter pathways are required for animals to withstand chronic oxidative stress. This work thus has important implications for disease processes that are caused/linked to oxidative stress. This work identified specific neurotransmitters and receptors that coordinate stress resilience, both prior to and during stress exposure. Further, the authors identified specific transcriptional programs coordinated by neurotransmission that may provide stress resistance.
Strengths:
The manuscript is very clearly written with a well-formulated rationale. Standard C. elegans genetic analysis and rescue experiments were performed to identify key regulators of the chronic oxidative stress response. These findings were enhanced by transcriptional profiling that identified differentially expressed genes that likely affect survival when animals are exposed to stress.
Weaknesses:
Where the gar-3 promoter drives expression was not discussed in the context of the rescue experiments in Fig 7.
Comments on revisions:
This issue has now been appropriately addressed in the revision.
Reviewer #2 (Public review):
In this paper, Biswas et al. describe the role of acetylcholine (ACh) signaling in protection against chronic oxidative stress in C. elegans. They showed that disruption of ACh signaling in either unc-17 mutant or gar-3 mutants led to sensitivity to toxicity caused by chronic paraquat (PQ) treatment. Using RNA seq, they found that approximately 70% of the genes induced by chronic PQ exposure in wild type failed to upregulate in these mutants. The overexpression of gar-3 selectively in cholinergic neurons was sufficient to promote protection against chronic PQ exposure in an ACh-dependent manner. The study points to a previously undescribed role for ACh signaling in providing organism-wide protection from chronic oxidative stress likely through the transcriptional regulation of numerous oxidative stress-response genes. The paper is well-written, and the data are robust, though some conclusions seem preliminary and are not fully support the current data (see below). While the study identifies the muscarinic ACh receptor gar-3 as an important regulator of the response to PQ, the specific neurons in which gar-3 functions were not unambiguously identified, and the sources of ACh that regulate GAR-3 signaling and the identities of the tissues targeted by gar-3 were not addressed.
Comments on revisions:
The authors addressed my comments adequately in their revised submission. Please include representative images to accompany the quantification of the new results presented in Fig S4A.
Author response:
The following is the authors’ response to the original reviews
Reviewer #1 (Public review):
The researchers aimed to identify which neurotransmitter pathways are required for animals to withstand chronic oxidative stress. This work thus has important implications for disease processes that are caused/linked to oxidative stress. This work identified specific neurotransmitters and receptors that coordinate stress resilience, both prior to and during stress exposure. Further, the authors identified specific transcriptional programs coordinated by neurotransmission that may provide stress resistance.
Strengths:
The manuscript is very clearly written with a well-formulated rationale. Standard C. elegans genetic analysis and rescue experiments were performed to identify key regulators of the chronic oxidative stress response. These findings were enhanced by transcriptional profiling that identified differentially expressed genes that likely affect survival when animals are exposed to stress.
We thank the reviewer for their positive assessment.
Weaknesses:
Where the gar-3 promoter drives expression was not discussed in the context of the rescue experiments in Figure 7.
We now provide information about expression using 7.5 kb gar-3 promoter fragment and compare directly with our analysis of endogenous gar-3 expression using the genome-modified gar-3::SL2::GFP strain (Page 16, new Figures 8 and S3).
Reviewer #1 (Recommendations for the authors):
(1) Figure 3B is not mentioned in the text.
Fixed. Figure 3B is now called out on page 10 of the revised manuscript.
(2) The rationale for using the specific PQ concentration was not provided.
We selected this concentration based on its use for chronic assays by other studies in the field to allow for direct comparison with our results. We now clarify this point in the Methods section (Page 26 of the revised text).
(3) Transgenic animals injected with the unc-17βp::gar-3 transgene (25 ng/μL) displayed strikingly increased survival in the presence of 4 mM PQ compared to either gar-3 mutants or wild type (should have a Figure cited here)
Fixed. Figure 9E is now referenced on Page 19 of the revised text.
(4) The text describing Figure 7C details a comparison with the gar-3 single mutant but the graph shows the unc-17 single mutant
Figure 7C is a comparison of the survival of gar-3 single mutants with either wild type or gar-3;ric-3 double mutants as described in the text.
Reviewer #2 (public comments)
In this paper, Biswas et al. describe the role of acetylcholine (ACh) signaling in protection against chronic oxidative stress in C. elegans. They showed that disruption of ACh signaling in either unc-17 mutants or gar-3 mutants led to sensitivity to toxicity caused by chronic paraquat (PQ) treatment. Using RNA seq, they found that approximately 70% of the genes induced by chronic PQ exposure in wild type failed to upregulate in these mutants. The overexpression of gar-3 selectively in cholinergic neurons was sufficient to promote protection against chronic PQ exposure in an ACh-dependent manner. The study points to a previously undescribed role for ACh signaling in providing organism-wide protection from chronic oxidative stress, likely through the transcriptional regulation of numerous oxidative stressresponse genes. The paper is well-written, and the data are robust, though some conclusions seem preliminary and do not fully support the current data. While the study identifies the muscarinic ACh receptor gar-3 as an important regulator of the response to PQ, the specific neurons in which gar-3 functions were not unambiguously identified, and the sources of ACh that regulate GAR-3 signaling and the identities of the tissues targeted by gar-3 were not addressed, limiting the scope of the study.
We thank the reviewer for their positive assessment. We provide additional data and discussion of the points raised by the reviewer in the revised manuscript. In particular, as suggested by the reviewer, we conducted additional tissue-specific rescue experiments to try to better define GAR-3 site of action. We found that specific rescue of gar-3 expression in either cholinergic motor neurons or muscles each provide partial rescue. In addition, we quantified the expression of the nhr-185 and fbxa-73 genes, identified as upregulated by PQ in our RNA-seq studies, following oxidative stress (new Fig. S4). We observed increased expression of both genes following PQ exposure, providing independent confirmation for transcriptional upregulation of these genes as part of the stress response. See the responses to points #1 and #3 below for additional details.
Major Comments:
(1) The site of action of cholinergic signaling for protection from PQ was not adequately explored. The authors' conclusion that cholinergic motor neurons are protective is based on studies using overexpression of gar-3 and an unc-17 allele that may selectively disrupt ACh in cholinergic motor neurons (Figure 9F), but these approaches are indirect. To more directly address the site of action, the authors should conduct rescue experiments using well-defined heterologous promoters. Figure 7G shows that gar-3 expressed under a 7.5 kb promoter fragment fully rescues the defect of gar-3 mutants, but the authors did not report where this promoter fragment is expressed, nor did they conduct rescue experiments of the specific tissues where gar-3 is known to be expressed (cholinergic neurons, GABAergic neurons, pharynx, or muscles). UNC-17 rescue experiments could also be useful to address the site of action. Does expression of unc-17 selectively in cholinergic motor neurons rescue the stress sensitivity of unc-17 mutants (or restore resistance to gar-3(OE); unc-17 mutants)? These experiments may also address whether ACh acts in an autocrine or paracrine manner to activate gar-3, which would be an important mechanistic insight to this study that is currently lacking.
We performed additional rescue experiments using heterologous promoters to drive gar-3 expression in cholinergic neurons or muscle and found that each provided a small, but significant degree of rescue as assessed from Kaplan-Meier survival curves. These results are presented in Figure 8 of the revised manuscript. We have not conducted similar unc-17 rescue experiments; however, we point out that cellspecific unc-17 knockdown by RNAi using the unc-17b promoter (expression largely restricted to ventral cord ACh motor neurons) increases sensitivity to PQ in our long-term survival assays (Figure 3A). Combined with our analysis of unc-17(e113) mutants, we believe these results support a requirement for unc-17 expression in cholinergic motor neurons.
(2) The genetic pan-neuronal silencing experiments presented in Figure 1 motivated the subsequent experiments, but the authors did not relate these observations to ACh/gar-3 signaling. For example, the authors did not address whether silencing just the cholinergic motor neurons at the different times tested has the same effects on survival as pan-neuronal silencing.
We used the pan-neuronal silencing to motivate further analysis of various neurotransmitter systems. Our genetic studies implicate both glutamatergic and cholinergic systems in protective responses to oxidative stress. The effects of pan-neuronal silencing on survival during long-term PQ exposure may therefore be derived solely from cholinergic neurons, glutamatergic neurons, or a combination of both neuronal populations. Distinguishing between these possibilities may be quite complicated and is not central to the main message of our paper. We therefore suggest this additional analysis lies outside the scope of this revision. Nonetheless, to address the reviewer’s point, in the revised text we expand our discussion relating the pan-neuronal silencing results to our analysis of ACh signaling (pages 21-22).
(3) It is assumed that protection occurs through inter-tissue signaling of ACh to target tissues, where it impacts gene expression. While this is a reasonable assumption, it has not been directly shown here. It is recommended that the authors examine GFP reporter expression of a sampling of the genes identified in this study (including proteasomal genes that the authors highlight) that are regulated by unc-17 and gar-3. This would serve to independently confirm the RNAseq data and to identify target tissues that are subject to gene expression regulation by ACh, which would significantly strengthen the study.
Agreed. To address this question, we investigated expression of the nhr-185 and fbxa-73 genes implicated as upregulated by oxidative stress in our RNA-seq studies. Consistent with our RNA-seq findings, we observed significantly increased expression of a nhr-185pr::GFP transcriptional reporter, primarily in the pharynx and anterior intestine, following 48 hrs of PQ exposure. These results support transcriptional upregulation of expression in these tissues as part of the stress response. fbxa-73 was among the proteasomal genes implicated as oxidative stress-responsive by RNA-seq. Consistent with this finding, by quantitative RT-PCR we observed a significant increase in fbxa-73 expression in wild type animals following 48 hrs of PQ treatment. These new results provide independent confirmation of the gene expression changes we observed by RNA-seq and are now included in new Figure S4 and discussed on Pages 17-18 of the revised manuscript.
Reviewer #2 (Recommendations for the authors):
(1) As an independent way of addressing whether enhanced ACh signaling is sufficient for protection, the authors could examine stress resistance in ace mutants, as was reported in PMID: 39097618, or in mutants with increased ACh secretion.
We thank the reviewer for this suggestion. We are pursuing the impacts of increased cholinergic activation in a separate study. We are pursuing experiments along the lines the reviewer suggests as one facet of this independent study. Our findings here provide evidence that increasing GAR-3 signaling in ACh motor neurons by cell-specific overexpression enhances protection.
(2) To address the specificity of ACh signaling by gar-3 for this response, the authors could report survival data for mutants lacking each of the other two mACh receptors, gar-1 and gar-2.
We thank the reviewer for this suggestion. We now include new data showing that gar-3;gar-2 double mutants have similar survival to gar-3 single mutants in the presence of PQ new Figure 7F). We agree that further studies of additional GPCRs (e.g. gar-1 and metabotropic glutamate receptors) will be required to definitively establish specificity for GAR-3 and we now acknowledge this point on page 15 of the revised text.
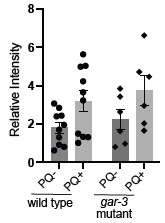
(3) Do carbonylation levels correlate with toxicity? For example, do gar-3 mutants have more carbonylation and gar-3 OE have less?
This is an interesting question. To try to address this, we performed additional protein carbonylation experiments for unc-17 and gar-3 mutants. We found a similar increase in protein carbonylation following PQ exposure for gar-3 mutants as observed for wild type; however, we also noted a higher level a batch-to-batch variability for gar-3 compared with wild type and are therefore hesitant to draw firm conclusions. We have not included these data in the revised manuscript but provide them for the reviewer’s information here (Author response image 1 shows our prior N2 data for comparison). We were not able to conduct similar experiments for unc-17 mutants because we noted local starvation when the animals were grown at the high density required to obtain the protein quantities needed for these experiments.
Author response image 1.
(4) Citations in text for Figures 4A and 8A are missing.
Fixed. Figures 4A and 8A (now 9A) are cited on pages 10 and 17 of the revised text, respectively.
(5) Figures 4-6 and 8 have limited information content. Condense or move to supplementary.
While we acknowledge the reviewer’s viewpoint here, we believe that the analyses of the transcriptional responses described in Figures 4-6 and 8 are central to the study. To address reviewers’ comments, we have included a new Figure 8 and merged previous Figures 8 and 9 (new Figure 9) in the revised manuscript.
(6) "expression of" is repeated in "Finally, transgenic expression of expression of a wild-type GAR-3::YFP"
Fixed.



