Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorLori SusselUniversity of Colorado Anschutz Medical Campus, Aurora, United States of America
- Senior EditorLori SusselUniversity of Colorado Anschutz Medical Campus, Aurora, United States of America
Reviewer #1 (Public review):
This study by Riegman & George et al. investigates the roles of the chromatin remodeling factor CHD7 and the proneural transcription factor Atoh1 at enhancers in cerebellar granule cells (GCs). Enhancers were categorized based on epigenetic marks and cross-referenced with promoter capture-HiC, ATAC-seq, and expression datasets to identify their long-range target genes, which were found to be enriched for critical neurodevelopmental processes. Differential expression and chromatin accessibility analyses in CHD7 knockout (KO) conditions suggest that this factor regulates a significant number of enhancers. These same enhancers are enriched for proneural transcription factor motifs, with Atoh1 being the most frequently present and likely the most affected. Finally, the direct interaction between CHD7 and Atoh1 was assessed via co-immunoprecipitation in co-transfected cells.
While the paper presents an interesting aspect of enhancer regulation in neurodevelopment, several points warrant attention:
Major Strengths:
The use of chromatin marks increases the resolution of promoter-interacting enhancer regions when integrated with capture-HiC, refining the identification of distal enhancers. Additionally, performing promoter capture-HiC experiments for the first time in this cell type constitutes a valuable resource for the community working on 3D genome organization and neurodevelopment.
Major Weaknesses:
As noted by the authors, limited sequencing depth reduces confidence in the conclusions and may result in missed weaker long-range interactions. Furthermore, the absence of capture-HiC and Atoh1 ChIP-seq experiments in the KO condition prevents direct comparison, thereby limiting the strength of the conclusions.
Additional Consideration:
Caution should be exercised regarding the assumption that every enhancer must physically contact its target promoter. While true for many enhancers, some act in trans through eRNAs or lncRNAs without direct physical contact.
Reviewer #2 (Public review):
Summary:
In this manuscript, the authors aim to identify active, long-range regulatory interactions in cerebellar granule cell progenitors (GCps). As such, the authors perform promoter capture Hi-C to map long-range interactions for all gene promoters, using cells isolated from P7 mouse brain samples. While the resolution of these maps is limited by the relatively large fragment sizes generated from a 6-bp cutter, the authors combine these interactions with other available published datasets, including from their own previous work, (e.g. ATAC-seq and ChIP-seq) to more precisely map putative enhancers within the long-range interacting regions of captured promoters. The paper further focuses on the importance of transcription factor Atoh1 and chromatin remodeller CHD7 in regulation of these putative enhancers in GCps. The authors suggest a direct interaction between CHD7 and Atoh1 by overexpression and co-immunoprecipitation in human embryonic kidney cells.
As stated by the authors, this study represents a valuable resource for researchers interested in the identification of enhancers in GCps cells, and their linked target genes. While broadly descriptive, the study does highlight some gene loci of interest and of biological relevance. For example, through integration of previously published datasets, the study resolves which putative regulatory elements at the Reln locus may regulate its activity.
This manuscript will be of interest to researchers interested in analysing long-distance targets of as well as researchers trying to understand the precise gene regulation in cerebellar development. It may also be of interest to clinical geneticists to interpret novel putative non-coding disease mutations.
Strengths:
The strengths of this manuscript are the integrated approach to identify cell-type specific enhancers utilizing available epigenomic datasets, and leveraging 3D genome topology to directly link them to their target genes. For example for the Reln gene previously implicated in cerebellar phenotypes for CHD7 mutants. The pcHi-C dataset generated in this study provides a valuable reference for the community of enhancer-promoter pairs for a specific cell-type of interest with human disease relevance.
Weaknesses:
The limitations of the study are partially addressed in the text by the authors, including the resolution from the pcHi-C using a 6-bp cutter, the limitation of sequencing depth (more interactions may have been identified with more depth), and the limited of correlation between replicates (likely due to undersampling the library). Page 9 "some additional interactions with the nearest gene promoters might be identified in our pcHi-C dataset with deeper sequencing".
Reviewer #3 (Public review):
Summary:
In this work, Riegman et al. establish the promoter interactome of cerebellar granule cell progenitors (CGPs) and identify thousands of putative enhancers regulating key genes in this cell population. The authors isolate primary CGps cells from the mouse cerebellum and perform promoter capture Hi-C in order to reanalyse previously generated epigenomic datasets (ATAC-seq, H3K4me1/3, H3K27ac) in these cells. They identify 22'797 enhancers interacting with gene promoters. The authors then use CHD7 ChIP-seq experiments to better annotate regulatory regions linked to genes deregulated upon CHD7 loss of function. After observing that CHD7 is frequently co-bound with ATOH1, they compare the binding profiles of ATOH1 and CHD7 together with genes deregulated in loss-of-function datasets, and refine the regulatory elements associated with each of these proteins.
Strengths:
The work is well designed and carefully executed, leading to an enhancer-promoter (E-P) interaction cartography that largely surpasses the current standard in the field. The pc-HiC dataset enables a deeper analysis of previously generated datasets (ChIP-seq and loss-of-function), which clearly improves the understanding of the mechanisms underlying CGps proliferation and differentiation. Moreover, the integration of published loss-of-function datasets for CHD7 and ATOH1 is relatively novel in this type of study and helps reduce the purely descriptive nature of the work. In particular, the analysis sheds light on genes with potential functions in CGps that had not previously been identified, as well as their regulatory connections. Overall, the study is convincing and supports the conclusions presented by the authors.
Weaknesses:
(1) A substantial part of the manuscript focuses on E-P interactions in CGPs, which gives the impression that this is primarily a genome organisation study. However, in this regard the manuscript does not bring major conceptual novelties. In contrast, the biological insights related to CGPs and the identification of new candidate genes likely represent the most novel aspect of the work. The authors should clarify the central message of the manuscript and reorganise the presentation of the results accordingly.
(2) The numbers presented throughout the manuscript are sometimes confusing. For instance, the authors initially report 106'589 PIF (line 175), but later only 61'928 (line 243) when calling enhancers. The relationship between these numbers is not straightforward. More generally, simplifying the nomenclature used to describe interaction analyses would help emphasise the biological insights rather than the computational framework.
(3) ATAC-seq alone is a relatively poor predictor of enhancers. In this context, H3K27ac would provide a more accurate marker of enhancer activity. This point is particularly important because the authors' data suggest that CHD7 does not function as a pioneer factor capable of opening chromatin. Instead, this role appears to be more closely associated with ATOH1. Therefore, alterations in CHD7 are more likely to affect enhancer activity (reflected by H3K27ac) rather than chromatin accessibility itself. If the authors do not have access to H3K27ac ChIP-seq data, this limitation should be explicitly acknowledged.
(4) The authors do not functionally test most enhancers and instead discuss primarily putative enhancers (with the exception of VISTA-tested elements). Although the term "putative enhancer" appears in some subsections, it is not consistently applied throughout the manuscript. This limitation should be clearly stated early in the manuscript with a sentence such as: "As these regions have not been functionally validated, they should be considered putative enhancers. However, for simplicity, we will refer to them as enhancers throughout the manuscript."
(5) Where feasible, the enhancer identified at the Reln gene should be functionally tested to demonstrate the added value of the approach.
Author response:
General Statements
We thank the reviewers for their careful and supportive reviews of our manuscript. We have addresses all the reviewers comments and extensively revised the manuscript accordingly.
During our revisions, we discovered a bug in the code that calculated the linear genomic distance between the captured promoter regions (bait regions) and the promoter-interacting fragments (PIFs). The error inadvertently halved the distance measurements in the output tables. This has been corrected in the revised manuscript and has resulted in updates to Figure 1B and corrected values in the ‘interaction_distance’ and/or ‘interaction_type’ columns of Supplementary Tables 2, 3, 6 and 8. We thank the reviewers for the opportunity to correct this.
Point-by-point description of the revisions
Reviewer #1 (Evidence, reproducibility and clarity):
In this article, the authors conducted promoter-capture HiC experiments (pcHiC) in Mouse Cerebellar granule cell progenitors (GCps) and obtained a good set of 3D genome interactions map of protein-coding genes' promoters. This dataset was later integrated with ATAC-seq and ChIP-seq experiments to identify putative enhancer regions within promoter-interacting regions, and with higher base-pair resolution than what is obtained by pcHiC experiments. This set of enhancers is then compared to and presented as being more reliable than those present in VISTA enhancer database. In addition, ATAC-seq sites and RNA-seq datasets, both obtained in WT and CHD7 and KO conditions, are integrated to correlate expression of a set of genes to the chromatin accessibility of their distal enhancer(s) which is believed to be promoted by CHD7. The study is completed by focusing on transcription factor motif analysis on CHD7-regulated enhancers which shows an enrichment for proneural transcription factors, with special emphasis on Atoh1 found to be frequently co-recruited with CHD7. Data and methods are well detailed and correctly replicated and will be useful as a resource for the community. The overlap obtained between pcHiC experiments and auto-criticized by the authors is very common and expected in this kind of experiments. In general, the conclusions drawn the article are convincing but some aspects such as comparison to VISTA and the naming of 'enhancers' should be moderated.
We thank the reviewer for their positive and constructive comments. We have amended the manuscript as indicated in detail below.
(1) The comparison of pcHiC-identified enhancers vs. VISTA enhancers should be more balanced, as the two approaches have important conceptual differences. Although VISTA enhancers are based on functional annotation, their target genes might not necessarily be correctly assigned based on the distance. On the other hand, putative enhancer regions identified by pcHiC experiments do not rely on functional testing. So both type of information are useful but can be put in perspective.
We thank the reviewer for making this point. We have amended the text to present a more balanced view e.g. “Using VISTA-designated hindbrain enhancers as an example, we identify the genes most likely regulated directly by these enhancers and update their annotation accordingly.”
(2) To increase the strength of the paper, it would be preferable that authors include simple functional enhancer assays (e.g. CRISPR deletion of contacting enhancer, luciferase assay) to support their perspective since 3D conformation information in KO condition is lacking in the article. Although ideally these experiments should be better performed for a full demonstration, it would be acceptable to at least include a simple functional assay in the WT context to demonstrate that the regulatory regions obtained by crossing genomic data are real enhancers. This point is even more critical knowing that enhancers lacking classical histone marks (H3K27ac+H3K4me1) has been described. The same comment applies to promoter interacting fragments lacking these marks, that could be missing enhancers (i.e enhancers without these marks).
To address this point, we performed luciferase assays to show that putative enhancers identified with our integrated bioinformatic approach (pcHi-C + ATACseq + H3K4me1 + H3K27ac) do indeed exhibit enhancer activity. For these experiments, we tested these putative fragments in an immortalized cell line SHH-NPD, a GCp-derived cell line generated by Fults laboratory (Jenkins et al. 2014). The results of these experiments are included as Suppl. Fig. 1 in the revised manuscript.
Minor point
- Figure 5B is lacking labels.
We apologise for this oversight – labels have now been added.
Reviewer #1 (Significance):
This article, when completed with possible revision, will be be useful for the community in terms of useful resource of experimentally determined putative enhancers in Cerebellar granule cell progenitors. It also provides some insights into the association of CHD7 and Atoh1 in distal regulation in these cells.
We thank the reviewer for acknowledging the significance of our work.
Reviewer #2 (Evidence, reproducibility and clarity):
In this manuscript, the authors aim to identify active, long-range regulatory interactions in cerebellar granule cell progenitors (GCps). As such, the authors perform promoter capture Hi-C to map long-range interactions for all gene promoters, using cells isolated from P7 mouse brain samples. While the resolution of these maps is limited by the relatively large fragment sizes generated from a 6-bp cutter, the authors combine these interactions with other available published datasets, including from their own previous work, (e.g. ATAC-seq and ChIP-seq) to more precisely map putative enhancers within the long-range interacting regions of captured promoters. The paper further focuses on the importance of transcription factor Atoh1 and chromatin remodeler CHD7 in regulation of these putative enhancers in GCps. The authors suggest a direct interaction between CHD7 and Atoh1 by overexpression and co-immunoprecipitation in human embryonic kidney cells.
As stated by the authors, this study represents a valuable resource for researchers interested in the identification of enhancers in GCps cells, and their linked target genes. While broadly descriptive, the study does highlight some gene loci of interest and of biological relevance. For example, through integration of previously published datasets, the study resolves which putative regulatory elements at the Reln locus may regulate its activity.
We thank the reviewer for their supportive comments.
We provide a summary of our major and minor comments here.
Major comments:
(1) The main take-home messages of the manuscript could be more clearly stated in the introduction to help readers understand the main conclusions of the work.
We have added a sentence to the Introduction to clarify the key take-home messages:
“We report putative distal regulatory elements for >12,000 genes, identify CHD7- and Atoh1-regulated enhancer elements and show that these factors interact and likely co-regulate the expression of key genes in the GCp lineage.”
(2) In the discussion, a previous Hi-C dataset is referred to "Reddy et al. annotated 5,175 promoter-enhancer interactions in GCps using Hi-C without enrichment (Reddy, Majidi et al. 2021)." It would be beneficial to compare the interactions identified previously with the current study (5,175 vs 46,428 interactions).
To address this comment we have performed an additional analysis and include text and Suppl. Figure 3 and Suppl. Table 13 to demonstrate the extent the two datasets compare, overlap and diverge. We have also added additional text to the discussion to highlight the difference and technical considerations between the two approaches and how they complement each other.
The 5,174 enhancer-promoter (E-P) interactions identified by Reddy et al were downloaded and intersected with the 46,428 promoter-accessible PIF regions identified in our study. The new supplementary Figure 3A illustrates that 82% (843/1207) of genes that Reddy et al identifies long-range interacting regions for are represented in our pcHiC dataset. Our pcHiC data contains information on distal interacting regions and potential enhancer regions for an additional 11,511 protein coding genes. Suppl. Figure 3B provides an overview of the Reddy et al E-P interactions that are, and are not identified in the pcHiC. We replicate 38% of Reddy et al’s E-P findings, whilst 53% of the 3229 interactions unique to the Reddy data would not be detected in the pCHiC data due to technical reasons resulting from the capture design and analysis protocol. Of the remaining interactions that are specific to the Reddy data, we identify other distal regions interacting with those same promoters . Suppl. Table 13 details the full comparision of Reddy’s E-P interactions that are found within our dataset.
The differences between the two datasets and the increased number of interactions detected in the pcHiC dataset likely result from the increased enrichment for the captured promoters enabling the detection of interactions that would have been below the detection threshold for the HiC study. In addition there are notable differences in analysis strategies for the two datasets which also contribute to differences in detection of regions. Reddy et al binned the HiC data into 10Kb regions to identify interacting regions and subsequently used chromatin marks to identify possible enhancer and promoter regions within these large regions. In contrast we have used the pCHiC and CHiCAGO algorithm to identify individual HindIII restriction fragments that are proximal to targeted promoter regions (PIFs), and prioritised those that have accessible regions within them which could represent various types of regions that play regulatory roles such as enhancers, CTCF site or facilitator regions, independent of their chromatin mark composition rather than focusing solely on enhancers.
(3) The authors identify an overlap with some of their identified enhancers with those from VISTA. Is this a fair comparison seeing as the enhancer reporters were tested during early embryonic development (e.g. E11.5 and E13.5) and seen to be active in the hindbrain, would these stages be relevant to GCps from P7? Can the authors identify ATAC-seq for example from hindbrain from embryonic stages and determine if the enhancer accessibility profile looks similar to that for the P7 GCps cells?
We thank the reviewer for this important question regarding the developmental relevance of our VISTA comparison and acknowledge that direct comparison between the time point requires careful consideration. Firstly ,to address the question of how similar the chromatin accessibility profiles are between the embryonic and P7 timepoints, we compared the ATAC-seq data from our paper to ENCODE data from the hindbrain. Of the 140 vista enhancers that were intersected with the pCHi-C dataset, 119 were identified from the lacZ studies as active in the hindbrain at E11.5 whilst 21 were identified as active at timepoint E12.5. We compared ENCODE ATAC-seq peaks from the E11.5 (ENCFF743IYX) and E12.5 ( ENCFF198TLF) hindbrain to the GCps from P7 across both the entire genome (global accessibility) as well as specifically +/- 3MB around the VISTA enhancer regions in the PIFs from the pCHiC to assess the conservation of local accessibility profiles.
When looking at the global accessibility profile of embryonic hindbrain versus P7 GCps across the whole genome there was a large degree of overlap with ~85% (E11.5) and ~88% (E12.5) of all ENCODE ATAC peaks overlapping with accessible ATAC summit regions from P7 GCps:
Author response image 1.
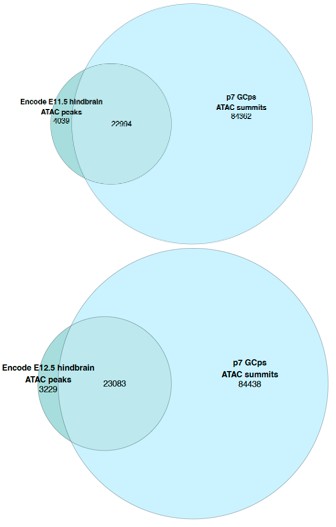
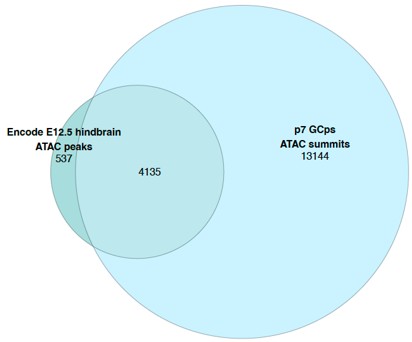
To identify if this was consistent in the immediate chromatin environment of the VISTA enhancers themselves, we compared the accessibility profiles across timepoints in the local environment surrounding the VISTA enhancers. This local environment was defined as a region that added an additional 3MB on either side of all VISTA enhancer positions found in PIFs. 3MB was chosen as the longest interaction found for a single VISTA element was approximately 2.7MB. Consistent with the global analysis a similarly high level of overlap of accessible regions between the timepoints was found for the local chromatin environment in surrounding the VISTA enhancers that were found within PIFs in the pCHiC dataset with ~87% (E11.5) and ~89% (E12.5) of encode detected peaks overlapping with accessible ATAC summit regions from P7 GCps.
Author response image 2.
Regions +/-3MB of VISTA enhancers in PIFs
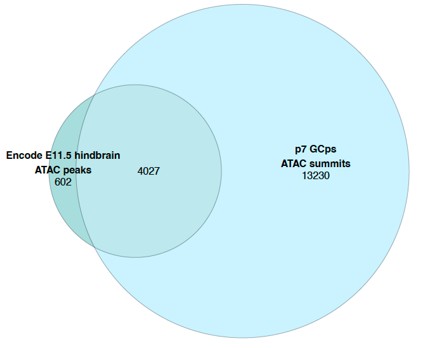
Author response image 3.
Regions +/-3MB of VISTA enhancers in PIFs
Genome browser shots at the three example VISTA loci from Figure 1 further support this approach. In addition to this we also note that a recent study by Chen et al (2024 https://www.nature.com/articles/s41588-024-01681-2) where capture-HiC performed at E11.5 of 935 VISTA enhancers across multiple tissues confirmed that the majority of VISTA enhancer regions (61%) bypass adjacent genes which is consistent with our nearest gene comparison.
(4) The co-IP experiment appears to support the conclusion that Atoh1 and CHD7 can interact, however there are bands in lanes where there should not be (i.e. Input lanes 1 and 4 for FLAG blot). It would be recommended to repeat this result at least once. [Expected time 2-4 weeks].
This experiment has been repeated 3 times with the same result. It is normal for non-specific background bands to appear on Western blot from total cell lysates (inputs) as most antibodies have significant cross-reactivity. The anti-FLAG antibody clearly detects bands above background in lysates where FLAG-tagged CHD7 is expressed. Most critically, despite the presence of non-specific bands in input, FLAG-tagged CHD7 is only detected in immunoprecipitated samples where either FLAG-tagged proteins have been precipitated and FLAG-tagged CHD7 is expressed and HA-tagged Atoh1 has been precipitated when both FLAG-tagged CHD7 and HA-tagged Atoh1 are expressed.
(5) The methods section describes analysis of several datasets, however we could not access the code at the time of review. Do the authors intend to make this code available at the time of publication?
Yes once the publication is approved all code will be made available along with conda environment yaml files to replicate the software environment in which the analysis was performed.
(6) Page 7 "replicate one and two, respectively". Can the authors clarify the number of biological replicates performed for pcHi-C?
Two biological replicates were performed for pcHiC which were then bioinformatically combined into a ‘superset’ for CHiCAGO interaction calling as is standard practice for pcHiC data (see e.g. Cairns et al, 2016. We have revised the text to make this clearer.
Minor comments:
(1) Page 3 "controlling the expression of 577 genes in GCps" - the authors do not provide evidence that these enhancers control gene expression directly, this should be reworded.
Thank you. We have reworded to: “contacting the promoters of 577 genes” to indicate that these were identified using pcHi-C and not functional assays.
(2) Page 5 "where transient amplifying divisions exponentially expand GCps" - at what stages of embryonic/postnatal development are GCps first detected, and when do they amplify and then differentiate?
GCps that form the EGL are specified in the rhombic lip from E13.5 (Machold, 2005 and Wang, 2005) and a clear EGL can be observed in the cerebellar anlage from E14 (Ben-Arie, 1997) of development. They amplify from this stage and differentiation, induced by neurogenic factors like NeuroD1 is visible from P0 onwards (Miyata, 1999). We have amended the text to include this additional information: “GCps that form the EGL are specified in the rhombic lip from E13.5 (Ben-Arie et al, 1997; Machold & Fishell, 2005) and a clear EGL can be observed in the cerebellar anlage from E14 (Ben-Arie et al., 1997) of development. They amplify from this stage and differentiation, induced by neurogenic factors like NeuroD1 is visible from P0 onwards (Miyata et al, 1999).”
(3) Page 7 "identified 164,387 unique and significant interactions" - how is an interaction defined, a single read, or evidenced by a certain number of reads. "promoter interacting fragments or PIFs" - is PIF referring to a single read evidencing an interaction?
An interaction is defined by the CHiCAGO algorithm. The number of reads needed to score an interaction depends on the both the distance away that PIF is from the promoter (this is modelled using a distance-dependent component that accounts for decay of contact frequence with genomic distance) and also includes a component that models how the sequence or other technical artifacts might influence the capture bias of some sequences compared to others. For each promoter a background model is generated of the expected number of reads that would be captured based on the above considerations and if the number of reads for those regions exceeds this background model by a certain threshold the interaction is deemed significant using a p-value like score. In practice this means that regions further from the promoter will often require less reads to signify a significant interaction compared to regions that are much closer to the promoter. The significant PIFs in the dataset are all evidenced by a minimum of 3 reads in at least one biological replicate. We have included a short explanation of this in the methods of the revised manuscript for clarity.
The maximum reads in a single replicate library for a specific PIF was 1557, and the median number of reads per PIF was 17.
(4) Page 8. What is the distinct between PIFs and "promoter interacting regions (PIRs)"? These could be better defined in the text.
Thank you for picking up this discrepancy, we were using PIR and PIF interchany. We have amended the manuscript to refer to PIFs consistently throughout.
(5) Figure 1C-F. Labels "Random" and "PIFs" don't line up well with the two bars.
Thank you, this has been corrected.
(6) Page 9. Could the authors show some representative images for the "VISTA hindbrain enhancers" (e.g. for Figure 1I-K).
We have inserted representative images showing in vivo activity of these enhancers in mouse embryos from the VISTA enhancer site.
(7) Fig 2G, Page 11 "The 12,354 genes that were linked to a PIF containing an ATAC-seq peak were found to have a higher median expression level than the 2,049 genes that had PIFs that did not coincide with ATAC-seq peaks" - is this significant?
Apologies for this oversight. We have performed a two-sided t-test on the log transformed TPMs between the two groups and have included the significance in the revised figure (p=1.8 e-40).
(8) "Gene Ontology analysis of genes with accessible PIFs revealed a significant enrichment for 119 biological processes" - can you include the GO terms in a supplementary table? Is there a way to prioritise down the 12,354 genes to a shorter more significant list of genes, this seems a long list to include in GO analysis.
We have included a supplementary table with this data in the revised manuscript (Suppl. Table 6). We included all 12,354 genes in this analysis as the point of this analysis was to demonstrate that developmental processes are enriched in the PIFs with accessible chromatin, compared to the genes where only PIFs without ATAC were identified.
(9) Page 11 - "The chromatin remodelling factor CHD7 is essential for normal expansion of GCps in the postnatal mouse cerebellum (Whittaker et al., 2017b) and deletion of Chd7 from GCps results in striking cerebellar hypoplasia and polymicrogyria (Feng et al., 2017; Reddy et al., 2021; Whittaker et al., 2017b). CHD7 haploinsufficiency is also sufficient to cause cerebellar hypoplasia and foliation defects both in mouse models and in the context of CHARGE syndrome in humans (Whittaker et al, 2017a; Yu et al, 2013)." - this appears more suitable for the introduction.
Thank you, we have moved this text to the Introduction.
(10) Page 12 "the majority of which (4,663/5,369) displayed decreased accessibility when Chd7 is depleted". This was difficult to understand initially - which are expected to be the direct effects? Increased or decreased accessibility? Perhaps it would be better to focus only on the decreased accessibility sites?
We have previously shown that the majority of differentially accessible regions in Chd7-deficient GCps show decreased accessibility. Chromatin remodelling by CHD7 could conceptually reduce or increase accessibility of a particular locus and the only way to infer direct effects are by identifying regions to which CHD7 is recruited.
Approximately ~9% of the sites that decreased in accessibility overlapped with regions bound by CHD7 (464/4663), whilst ~2% of sites that increased in accessibility overlapped with regions of CHD7 binding (14/706). Whilst it is likely that the majority of directly regulated sites decrease in chromatin accessibility when CHD7 is removed, the number of sites that increases in accessibility is small but observed and should be included for completeness.
(11) The analysis in Fig 3A reveals that only a small number of CHD7-bound enhancers show differential accessibility and altered linked gene expression upon CHD7-knock down. This requires a little more discussion - why do so many sites change in accessibility compared to the number of sites which change accessibility or are associated with gene expression change?
Identifying CHD7-regulated enhancers is challenging, mostly due to the inefficiency of CHD7 ChIP-seq. The low quality of available CHD7 ChIP-seq data has made it particularly difficult to identify CHD7 peaks. However, the integration of this data with ATAC-seq accessibility, chromatin modification and pcHi-C data has allowed us to identify a subset of enhancers that are most likely directly regulated by CHD7. However, given these technical limitations, we would be hesitant to conclude from the present data that the majority of chromatin accessibility changes in enhancers in Chd7-deficient GCps are indirect. We have added the following text to the discussion to indicate this: “Identifying CHD7-regulated enhancers is challenging, mostly due to the inefficiency of CHD7 ChIP-seq. The low quality of available CHD7 ChIP-seq data has made it particularly difficult to identify CHD7 peaks. However, integrating CHD7 ChIP-seq data with ATAC-seq accessibility, histone modification ChIP-seq and pcHi-C data has allowed us to identify a subset of enhancers that are most likely directly regulated by CHD7. However, given these technical limitations, we would be hesitant to conclude from the present data that the majority of chromatin accessibility changes in enhancers in Chd7-deficient GCps are indirect, as suggested by the data in Fig. 3A.”
(12) Page 12 - "Over-representation analysis confirmed an enrichment of genes linked to nervous system development" - could this and the GO term analysis be included in a supplementary figure?
We have included these results as Suppl. Table 7 in the revised manuscript.
(13) Fig 3D - what does the arrow represent in the chromatin schematic?
The arrow in the schematic indicates chromatin remodelling – we have clarified this in the figure legend and added headings to these panels to indicate the 3 different types of elements: Direct CHD7 targets, Indirect targets and CHD7-bound elements.
(14) Fig 3G does not appear to be referenced in the text. The value of the Upset plots in the main figure 3 wasn't very clear, perhaps these could be moved to the supplement? Is there a clearer plot to support the conclusion "CHD7 primarily regulates enhancers".
We apologise, the panels were mis-labeled in the text. This has now been corrected. We hope that the amendments in response to point 13 above now clarifies these findings showing that direct CHD7 targets are characterised by active enhancer marks.
(15) Page 14 "putative consensus sites for proneural bHLH TAL-family of proteins Neurog2, Neurod2, Neurod1, and, Atoh1 in elements" - HOCOMOCO motifs are only shown for Atoh1 and Nhlh1. It may be valuable to show the sites for all the listed TFs. What does white represent in the heatmap in Fig 3H? This plot is difficult to interpret, and also relatively small in the figure but appears important to conclusions. Perhaps Fig 3H could be made more prominent?
Thank you for highlighting that the white boxes might be confusing. The white blocks indicate that these motifs do not pass threshold for significantly enriched in the dataset based on the p and q values.This has now been clarified in the figure legend.
We have enlarged panel H to make more prominent.
(16) Page 15 - "Myb was the only motif specific to CHD7 bound regions that changed in accessibility compared to those that exhibited accessibility changes without CHD7 binding or CHD7 binding without accessibility changes (Suppl. Fig. 1)." I couldn't interpret this sentence, requires clarifying.
We agree that this description is confusing and since it is difficult to draw clear conclusions about the significance of enhancers with Myb motifs in this context, we have removed this sentence from the revised manuscript.
(17) Page 16 and Fig 4B - a discussion of why both up and down regulated genes are detected for Atoh1 depletion? Which class of genes are expected to be directly regulated (the down-regulated genes)?
Like most transcription factors, ATOH1 may be able to function as both a repressor and activator depending on the context. Although the majority of genes are downregulated in Atoh1-defivcient cells, suggesting that Atoh1 functions as an activator in most cases, our analysis have identified several up-regulated genes that contain Atoh1 ChIP-seq peaks in their cognate enhancers (See Suppl. Table 7), consistent with these also being direct Atoh1 targets.
(18) Fig 5B - the genomic traces are not labelled in this figure.
Thank you, labels have been added.
(19) Page 17 - "Pathway enrichment analysis of the 22 genes compared to all genes that were expressed in GCps shows a significant enrichment of terms: Hypoplasia of the pons (HP:0012110 P=0.006) and Abnormal pons morphology (HP:0007361 P=0.016) from human phenotype ontology, due to the presence of Reln, Dcc, Mab21l1 and Gli2." - this analysis should be included in the supplementary tables.
These results have been included as Suppl. Table 12 in the revised manuscript.
(20) Do the authors have a suggestion for which domains of Atoh1 and CHD7 could be interacting? Could the authors design truncated constructs for overexpression in HEK cells to test this hypothesis? [Expected time 4-6 weeks, interesting but not essential to do experimental work here].
We agree this is an interesting question. Our collaborator, Professor Peter Scambler (UCL) has performed a yeast two hybrid screen for CHD7 interacting proteins in a mouse E11.5 library using the CHD7 BRK domain (aa 2521-2708) as bait. The screen had a single hit, which encompassed the N-term 127aa of ATOH1 (personal communication). This observation supports our co-IP data and suggests that the N-terminus of ATOH1 interacts with the BRK domain of CHD7 but further validation will be needed to confirm this.
(21) Page 28 "Differential accessibility analysis was performed using DESeq2 (v 1.22.1)" and Page 19 "Whereas chromatin accessibility at some of these enhancers were affected by Chd7-deficiency" - what were the cutoffs used for looking at differentially accessible regions? Complete loss of accessibility or a quantitative change?
Quantitative change rather than complete loss was used. Thresholds based on adjusted p-values (padj<0.05) were used as indicated in the methods.
Requested comments on referencing:
- "Long-range" - how do the authors define long-range? Can this be referenced. CO? good reference here.- look to CHiCAGO paper
- "When chromatin conformation or 3D organisation data is not available, studies typically assign regulatory elements to the nearest gene promoter" - needs referencing.
- "Many of these 22 genes regulated by CHD7 and Atoh1 have established critical roles in cerebellar development, including Neurod2, Pax6 and Gli2 (Fig. 5B)" - needs referencing. "from human phenotype ontology, due to the presence of Reln, Dcc, Mab21l1 and Gli2" - needs referencing.
Thank you, references have been added.
- "active enhancers (H3K27ac+, H3K4me1+), promoters (H3K27ac+, H3K4me3+), regulatory elements (H3K27ac+, H3K4me1+, H3K4me3+), or poised enhancers (H3K4me1+)" - needs referencing.
Thank you, references have been added.
- Reference required in main text for VISTA (e.g. Visel et al., 2007)
Thank you, reference added.
Reviewer #2 (Significance):
The strengths of this manuscript are the integrated approach to identify cell-type specific enhancers utilizing available epigenomic datasets, and leveraging 3D genome topology to directly link them to their target genes. For example for the Reln gene previously implicated in cerebellar phenotypes for CHD7 mutants. The pcHi-C dataset generated in this study provides a valuable reference for the community of enhancer-promoter pairs for a specific cell-type of interest with human disease relevance.
We thank the reviewer for recognising the potential value of our work to the community.
The limitations of the study are partially addressed in the text by the authors, including the resolution from the pcHi-C using a 6-bp cutter, the limitation of sequencing depth (more interactions may have been identified with more depth), and the limitated of correlation between replicates (likely due to undersampling the library). Page 9 "some additional interactions with the nearest gene promoters might be identified in our pcHi-C dataset with deeper sequencing".
We thank the reviewer for highlighting our acknowledgements of the potential limitations of our work.
Additional limitations include the use of the VISTA browser mouse LacZ embryos to validate some of their enhancers, the limitation here being that the VISTA browser tests enhancers at embryonic stages (focused at E11.5 and E13.5) while the GCps cells were collected at P7. The LacZ images from VISTA are also not shown. The HEK cells used for the co-IP could be seen as a limitation as these are not relevant cells for the cell state studied, the authors could clarify their use of these cells.
We thank the reviewer for their careful assessment of the limitations of our study. We have now included images of the VISTA enhancers in Fig. 1I,J,K. Rather than a limitation, using irrelevant cells for co-IP might be seen as a better approach, as conceivably the chances of an indirect interaction between the two proteins being tested by a bridging complex is less in an irrelevant cell types that might not contain such complexes. Either way, HEK293T cells is the standard laboratory model for co-IP studies as they can be transfected with ease.
The study reported here is largely based on previous work from the authors (Whittaker et al 2017b). This study reported that the chromatin remodelling factor CHD7 is essential for normal expansion of GCps in the postnatal mouse cerebellum and deletion of CHD7 from GCps resulted in the phenotype of cerebellar hypoplasia. This study also largely leverages previously published datasets from the Whittaker et al 2017b (e.g. CHD7 deletion data) and reanalyses it in the light of the new pcHi-C datasets.
This manuscript will be of interest to researchers interested in analysing long-distance targets of as well as researchers trying to understand the precise gene regulation in cerebellar development. It may also be of interest to clinical geneticists to interpret novel putative non-coding disease mutations.
We thank the reviewer for highlighting the wide interest of our manuscript.
In assessing this manuscript, my expertise lies in models of human development and gene regulation, with a focus on enhancer function.
Reviewer #3 (Evidence, reproducibility and clarity):
Riegman et al have explored the gene regulatory landscapes of cerebellar granule cell progenitors (GCps). They have generated promoter capture Hi-C data to identify regions that interact with promoters in these cells. In addition they generate ATACseq data in wild-type and CDH7 knock-out cells. They integrate these data to identify enhancers that potentially regulate genes in GCps. In addition, the authors identify an interaction between CHD7 and ATOH1, whose binding sites also overlap in the genome.
The dataset can be potentially interesting for people studying cerebellar development.
I have a few concerns regarding the paper. The most pressing one is that the authors seem to equate interactions in pcHi-C with regulation. This is problematic for two reasons. First whether interaction equates regulation is still debated and whether this can be detected with a low-resolution C-method (i.e. using HindIII) is a further point of contention.
We thank the reviewer for pointing this out. We agree and apologise for not being clear in our manuscript. We have made the necessary amendments to indicate that pcHi-C by itself only assess proximity in the nucleus, not function.
We acknowledge the limitations of the pcHi-C method, including that resolution is limited by the use of a restriction enzyme. However, we (see e..g. Suppl. Fig. 1) and others (see e.g. Freire-Pritchett et al (2017) and Mifsud et al (2015)) have used this approach successfully to identify functional enhancer elements.
The second issue has to do with the way the pcHi-C data is interpreted. What is detected as a significant interaction by Chicago are regions that have a contact frequence above background. This means that local regions with a (much) higher contact frequency may not be called as significant. When we follow the logic that contact frequency is related to gene activation (which may not necessarily be true) whether a fragment is more frequently contacted than the background should not matter (relative contact frequency), rather it should be interpreted based on the absolute contact frequency.
The reviewer is right that local regions will have a higher contact frequency and that local contacts aren’t always captured by the CHiCAGO model. However, the purpose of this study was to prioritise the identification of distal elements that are not captured by existing methods including nearest gene annotation.
There are a number of reasons why absolute contact frequency might not be an appropriate measure to infer gene regulation: 1) Many factors can affect the absolute contact frequency including the proportion of cells that are exhibiting active transcription at that time across a population, especially if expression is limited to a small number of this population at that time. 2) Absolute contact frequency assumes that more contact results in more regulation which is not necessarily true and would depend on the combination of factors that are associated with that regulatory element. Figure 1 from https://www.nature.com/articles/s41596-023-00817-8 - Figure 1 – Micro capture C show that regions with low absolute contact frequency compared to adjacent regions have potential to regulate gene expression, as have other studies that have used CHiCAGO to identify regulatory elements. 3) The sequence of some fragments makes them more likely to captured or enriched in the HiC protocol, which the relative contact frequency above background controls for.
This becomes relevant because the authors claim that 80% of enhancers are wrongly annotated based on their metrics. The only way to correctly annotate an enhancer is to knock it out and checking the effect on genes in the vicinity. Therefore, to claim that their method can correctly annotate enhancer is grossly overstated, particularly when considering the issues with contact frequency stated above. Therefore, claims like 80% of enhancers are wrongly annotated should be removed from the paper. The authors should discuss how to annotate enhancers, in the Discussion and what the proper method is for annotations.
We have amended the text to indicate that we do not suggest that VISTA enhancers are wrongly annotated but incompletely assigned. We apologise for making this suggestion in the first draft. There is however complementary evidence from Cheng et al (2024), now referenced in the revised manuscript, that also find 60% of the VISTA enhancers skip their adjacent gene. It is also well established in the literature that nearest genes are not always regulated.
Other points:
- The authors claims that PIFs have 2.14 and 2.69 fold enrichment of H3K4me1 and H3K27ac sites. Did the authors use the whole genome as background. If so, they should take into account that promoter are more likely in regions of high gene density, which are more dense in active marks. It would be better to perform local, circular permuation of the the PIFs around the promoter.
The reviewer is correct that a whole genome background is not an appropriate background for testing enrichment of active marks within PIFs. Fortunately, this is taken into account in the CHiCAGO enrichment test which selects the background from fragments that are matched to the same distance of the PIFs to account for the observation that promoters are more likely in regions of high gene density and are therefore more enriched for active chromatin modifications.
- The authors talk about "lead PIF", which is the fragment with the "most significant CHICAGO score". What does this mean? Something is significant or not, despite common misuse of the term there is no gradient of significance.
The reviewer makes a good point here and we apologise for the oversight in wording and have corrected the text to be more specific that the lead PIF is the one with the highest ChiCAGO score.
- In the GO analysis the categories with the lowest p-value are presented, but this biases for large categories. It would be more relevant to also select for and show the enrichment scores.
We agree with the reviewer that a drawback of GO analysis is that it biases for large categories and that if by ‘enrichment score’ the reviewer means the –log10(p-value) we have included that in the supplementary tables which also includes the size of the category and number of genes detected in it.
Reviewer #3 (Significance):
The study provides a dataset that may be interesting for people studying cerebellar development. In that sense the data is mostly interesting from a fundamental viewpoint. The data seem of good quality.
The authors claim that they a very sizeable fraction of enhancers are misannotated, but I do not believe that this is correct.
We thank the reviewer for pointing this out. We apologise for creating the impression that VISTA enhancers are incorrectly annotated. We have amended the text to reflect that these are incompletely annotated.
My expertise is 3D genome, bioinformatics.



