Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorMichael CzechUniversity of Massachusetts Chan Medical School, Worcester, United States of America
- Senior EditorBenoit KornmannUniversity of Oxford, Oxford, United Kingdom
Reviewer #1 (Public Review):
Previous reports suggested an association between ceramide accumulation in skeletal muscle and disruption of insulin signaling and metabolic dysregulation. Mechanistically, however, how intracellular ceramide attenuates insulin action and reduces metabolism is not fully understood. It was suggested that insulin receptor (IR) signaling to PI3-K/AKT is inhibited by elevated intracellular ceramide. However, other studies failed to demonstrate an inhibitory effect of ceramide on PI3K/AKT. More recently, a study was published describing that intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle (PMID: 29415895). In the present study, Diaz-Vegas and colleagues used an in vitro system to investigate this topic further and better understand how intracellular ceramide accumulation causes cellular insulin resistance and metabolic dysregulations in cultured myocytes.
The authors applied multiple methods to achieve this goal. Among these procedures are:
1) The overexpression of enzymes involved in mitochondrial ceramide synthesis and degradation;
2) Treatments of myocytes with different pharmacological tools to validate their findings;
3) Mitochondrial proteomics and lipidomics analyses.
The effects of these experimental conditions and treatment on intracellular lipids contents, mitochondrial functions, and insulin signaling in myocytes were then evaluated.
Findings:
The authors' findings indicate that incubation of myocytes with palmitate increases mitochondrial ceramide and reduces the insulin-stimulated GLUT4-HA translocation to the myocyte surface without affecting AKT activation. The elevation in mitochondrial ceramide lowers the coenzyme Q levels e depletes the electron transport chain (ETC) components, impairing mitochondrial respiration. Such mitochondrial dysfunction appears to attenuate the translocation of GLUT4-HA to the plasma membrane of the L6-myotubule. Also, mitochondrial proteomic analysis revealed an association of insulin sensitivity with mitochondrial ceramide and ETC expression levels in human muscle.
Based on these findings, the authors propose a mechanism whereby the building up of ceramide inside mitochondria depletes CoQ and compromise mitochondrial respiratory complexes, raising ROS. The resulting mitochondrial dysfunction causes insulin resistance in cultured myocytes. They postulate that CoQ depletion links ceramides with insulin resistance and define the respirasome as a critical connection between ceramides and mitochondrial dysfunction.
Relevance and critiques:
This original study provides direct evidence that mitochondrial ceramide accumulation depletes CoQ and downregulates multiple ETC components in myocytes. Consequently, elevation in the levels of reactive oxygen species (ROS) and mitochondrial dysfunctions occur. The authors proposed that such mitochondrial dysregulation attenuates insulin-stimulated GLUT4 translocation to the plasma membrane of L6-myotubules. Moreover, mitochondrial ceramide accumulation does not affect insulin action on AKT activation.
Overall, this is a well-done study, showing that in obesity, elevated mitochondrial ceramide suppresses mitochondrial function and attenuates insulin action on glucose transporter GLUT4 translocation into the myocyte surface. The main conclusion is supported by the results presented. The study also applied multiple methods and described several experiments designed to test the author's central hypothesis.
Importantly, these new findings shed light on possible cellular mechanisms whereby ectopic fat deposition in skeletal muscle drives insulin resistance and metabolism dysregulation. The results demonstrating that alterations in mitochondrial ceramide are sufficient to attenuate insulin-stimulated GLUT4 trafficking in cultured myocytes are very interesting. Well-done.
Comments for further discussion and suggestions:
Although the authors' results suggest that higher mitochondrial ceramide levels suppress cellular insulin sensitivity, they rely solely on a partial inhibition (i.e., 30%) of insulin-stimulated GLUT4-HA translocation in L6 myocytes. It would be critical to examine how much the increased mitochondrial ceramide would inhibit insulin-induced glucose uptake in myocytes using radiolabel deoxy-glucose.
Another important question to be addressed is whether glycogen synthesis is affected in myocytes under these experimental conditions. Results demonstrating reductions in insulin-stimulated glucose transport and glycogen synthesis in myocytes with dysfunctional mitochondria due to ceramide accumulation would further support the authors' claim.
In addition, it would be critical to assess whether the increased mitochondrial ceramide and consequent lowering of energy levels affect all exocytic pathways in L6 myoblasts or just the GLUT4 trafficking. Is the secretory pathway also disrupted under these conditions?
Reviewer #2 (Public Review):
The findings reported by Diaz-Vegas et al. extend those described in a previous paper from the same group establishing a role for mitochondrial CoQ depletion in the development of insulin resistance in muscle and adipose tissue (Fazakerley, 2018). In this new report, investigators sought to determine whether CoQ depletion contributes to insulin resistance caused by palmitate exposure and/or intracellular ceramide accumulation. To this end, researchers employed a widely used in vitro model of insulin resistance wherein L6 myocytes develop impaired Glut4 translocation upon exposure to palmitate (in this case, 150 uM for 16 hours). This model was combined with a variety of pharmacologic and genetic manipulations aimed at augmenting or inhibiting CoQ biosynthesis and/or ceramide biosynthesis, specifically in mitochondria. This series of experiments produced a valuable and provocative body of evidence positioning CoQ depletion downstream of mitochondrial ceramide accumulation and necessary for both palmitate- and ceramide-induced insulin resistance in L6 myocytes. Investigators concluded that mitochondrial ceramides, CoQ depletion and respiratory dysfunction are part of a core pathway leading to insulin resistance.
Strengths:
The study provides exciting, first-time evidence linking palmitate-induced insulin resistance to ceramide accumulation within the mitochondria and subsequent depletion of CoQ. Ceramide accumulation specifically in mitochondria was found to be necessary and sufficient to cause insulin resistance in cultured L6 myocytes.
The in vitro experiments featured a set of mitochondrial-targeted genetic manipulations that permitted up/down-regulation of ceramide levels specifically in the mitochondrial compartment. Genetically induced mitochondrial ceramide accumulation led to CoQ depletion, which was accompanied by increased ROS production and diminution of ETC proteins and OXPHOS capacity and impaired insulin action, thereby establishing cause/effect.
Analysis of mitochondria isolated from human muscle biopsies obtained from individuals with disparate metabolic phenotypes revealed a positive correlation between complex I proteins and insulin sensitivity and a negative correlation with mitochondrial ceramide content. While it is likely that many factors contribute to these correlations, the results support the possibility that the ceramide/CoQ mechanism might be relevant to glucose control in humans.
These important findings offer valuable new insights into mechanisms that connect ceramides to insulin resistance and mitochondrial dysfunction, and could inform new therapeutic approaches towards improved glucose control.
Weaknesses:
The mechanistic aspect of the work and conclusions put forth rely heavily on studies performed in cultured myocytes, which are highly glycolytic and generally viewed as a poor model for studying muscle metabolism and insulin action. Nonetheless, the findings provide a strong rationale for moving this line of investigation into mouse gain/loss of function models.
One caveat of the approach taken is that exposure of cells to palmitate alone is not reflective of in vivo physiology. It would be interesting to know if similar effects on CoQ are observed when cells are exposed to a more physiological mixture of fatty acids that includes a high ratio of palmitate, but better mimics in vivo nutrition.
While the utility of targeting SMPD5 to the mitochondria is appreciated, the results in Figure 5 suggest that this manoeuvre caused a rather severe form of mitochondrial dysfunction. This could be more representative of toxicity rather than pathophysiology. It would be helpful to know if these same effects are observed with other manipulations that lower CoQ to a similar degree. If not, the discrepancies should be discussed.
The conclusions could be strengthened by more extensive studies in mice to assess the interplay between mitochondrial ceramides, CoQ depletion and ETC/mitochondrial dysfunction in the context of a standard diet versus HF diet-induced insulin resistance. Does P053 affect mitochondrial ceramide, ETC protein abundance, mitochondrial function, and muscle insulin sensitivity in the predicted directions?
Author Response:
Assessment note: “Whereas the results and interpretations are generally solid, the mechanistic aspect of the work and conclusions put forth rely heavily on in vitro studies performed in cultured L6 myocytes, which are highly glycolytic and generally not viewed as a good model for studying muscle metabolism and insulin action.”
While we acknowledge that in vitro models may not fully recapitulate the complexity of in vivo systems, we believe that our use of L6 myotubes is appropriate for studying the mechanisms underlying muscle metabolism and insulin action. As mentioned below (reviewer 2, point 1), L6 myotubes possess many important characteristics relevant to our research, including high insulin sensitivity and a similar mitochondrial respiration sensitivity to primary muscle fibres. Furthermore, several studies have demonstrated the utility of L6 myotubes as a model for studying insulin sensitivity and metabolism, including our own previous work (PMID: 19805130, 31693893, 19915010).
In addition, we have provided evidence of the similarities between L6 cells overexpressing SMPD5 and human muscle biopsies at protein levels and the reproducibility of the negative correlation between ceramide and Coenzyme Q observed in L6 cells in vivo, specifically in the skeletal muscle of mice in chow diet. These findings support the relevance of our in vitro results to in vivo muscle metabolism.
Finally, we will supplement our findings by demonstrating a comparable relationship between ceramide and Coenzyme Q in mice exposed to a high-fat diet, to be shown in Supplementary Figure 4 H-I. Further animal experiments will be performed to validate our cell-line based conclusions. We hope that these additional results address the concerns raised by the reviewer and further support the relevance of our in vitro findings to in vivo muscle metabolism and insulin action.
Points from reviewer 1:
- Although the authors' results suggest that higher mitochondrial ceramide levels suppress cellular insulin sensitivity, they rely solely on a partial inhibition (i.e., 30%) of insulin-stimulated GLUT4-HA translocation in L6 myocytes. It would be critical to examine how much the increased mitochondrial ceramide would inhibit insulin-induced glucose uptake in myocytes using radiolabel deoxy-glucose.
Response: The primary impact of insulin is to facilitate the translocation of glucose transporter type 4 (GLUT4) to the cell surface, which effectively enhances the maximum rate of glucose uptake into cells. Therefore, assessing the quantity of GLUT4 present at the cell surface in non-permeabilized cells is widely regarded as the most reliable measure of insulin sensitivity (PMID: 36283703, 35594055, 34285405). Additionally, plasma membrane GLUT4 and glucose uptake are highly correlated. Whilst we have routinely measured glucose uptake with radiolabelled glucose in the past, we do not believe that evaluating glucose uptake provides a better assessment of insulin sensitivity than GLUT4.
We will clarify the use of GLUT4 translocation in the Results section:
“...For this reason, several in vitro models have been employed involving incubation of insulin sensitive cell types with lipids such as palmitate to mimic lipotoxicity in vivo. In this study we will use cell surface GLUT4-HA abundance as the main readout of insulin response...”
- Another important question to be addressed is whether glycogen synthesis is affected in myocytes under these experimental conditions. Results demonstrating reductions in insulin-stimulated glucose transport and glycogen synthesis in myocytes with dysfunctional mitochondria due to ceramide accumulation would further support the authors' claim.
Response: We have carried out supplementary experiments to investigate glycogen synthesis in our insulin-resistant models. Our approach involved L6-myotubes overexpressing the mitochondrial-targeted construct ASAH1 (as described in Fig. 3). We then challenged them with palmitate and measured glycogen synthesis using 14C radiolabeled glucose. Our observations indicated that palmitate suppressed insulin-induced glycogen synthesis, which was effectively prevented by the overexpression of ASAH1 (N = 5, * p<0.05). These results provide additional evidence highlighting the role of dysfunctional mitochondria in muscle cell glucose metabolism.
These data will be added to Supplementary Figure 4K and the results modified as follows:
“Notably, mtASAH1 overexpression protected cells from palmitate-induced insulin resistance without affecting basal insulin sensitivity (Fig. 3E). Similar results were observed using insulin-induced glycogen synthesis as an ortholog technique for Glut4 translocation. These results provide additional evidence highlighting the role of dysfunctional mitochondria in muscle cell glucose metabolism (Sup. Fig. 5K). Importantly, mtASAH1 overexpression did not rescue insulin sensitivity in cells depleted…”
We will add to the method section:
“L6 myotubes overexpressing ASAH were grown and differentiated in 12-well plates, as described in the Cell lines section, and stimulated for 16 h with palmitate-BSA or EtOH-BSA, as detailed in the Induction of insulin resistance section.
On day seven of differentiation, myotubes were serum starved in plain DMEM for 3 and a half hours. After incubation for 1 hour at 37C with 2 µCi/ml D-[U-14C]-glucose in the presence or absence of 100 nM insulin, glycogen synthesis assay was performed, as previously described (Zarini S. et al., J Lipid Res, 63(10): 100270, 2022).”
- In addition, it would be critical to assess whether the increased mitochondrial ceramide and consequent lowering of energy levels affect all exocytic pathways in L6 myoblasts or just the GLUT4 trafficking. Is the secretory pathway also disrupted under these conditions?
Response: As the secretory pathway primarily involves the synthesis and transportation of soluble proteins that are secreted into the extracellular space, and given that the majority of cellular transmembrane proteins (excluding those of the mitochondria) use this pathway to arrive at their ultimate destination, we believe that the question posed by the reviewer is highly challenging and beyond the scope of our research. We will add this to the discussion:
“...the abundance of mPTP associated proteins suggesting a role of this pore in ceramide induced insulin resistance (Sup. Fig. 6E). In addition, it is yet to be determined whether the trafficking defect is specific to Glut4 or if it affects the exocytic-secretory pathway more broadly…”
Points from reviewer 2:
- The mechanistic aspect of the work and conclusions put forth rely heavily on studies performed in cultured myocytes, which are highly glycolytic and generally viewed as a poor model for studying muscle metabolism and insulin action. Nonetheless, the findings provide a strong rationale for moving this line of investigation into mouse gain/loss of function models.
Response: The relative contribution of the anaerobic (glycolysis) and aerobic (mitochondria) contribution to the muscle metabolism can change in L6 depending on differentiation stage. For instance, Serrage et al (PMID30701682) demonstrated that L6-myotubes have a higher mitochondrial abundance and aerobic metabolism than L6-myoblasts. Others have used elegant transcriptomic analysis and metabolic characterisation comparing different skeletal muscle models for studying insulin sensitivity. For instance, Abdelmoez et al in 2020 (PMID31825657) reported that L6 myotubes exhibit greater insulin-stimulated glucose uptake and oxidative capacity compared with C2C12 and Human Mesenchymal Stem Cells (HMSC). Overall, L6 cells exhibit higher metabolic rates and primarily rely on aerobic metabolism, while C2C12 and HSMC cells rely on anaerobic glycolysis. It is worth noting that L6 myotubes are the cell line most closely related to adult human muscle when compared with other muscle cell lines (PMID31825657). Our presented results in Figure 6 H and I provide evidence for the similarities between L6 cells overexpressing SMPD5 and human muscle biopsies. Additionally, in Figure 3J-K, we demonstrate the reproducibility of the negative correlation between ceramide and Coenzyme Q observed in L6 cells in vivo, specifically in the skeletal muscle of mice in chow diet. Furthermore, we have supplemented these findings by demonstrating a comparable relationship in mice exposed to a high-fat diet, as shown in Supplementary Figure 4 H-I (refer to point 4). We will clarify these points in the Discussion:
“In this study, we mainly utilised L6-myotubes, which share many important characteristics with primary muscle fibres relevant to our research. Both types of cells exhibit high sensitivity to insulin and respond similarly to maximal doses of insulin, with Glut4 translocation stimulated between 2 to 4 times over basal levels in response to 100 nM insulin (as shown in Fig. 1-4 and (46,47)). Additionally, mitochondrial respiration in L6-myotubes have a similar sensitivity to mitochondrial poisons, as observed in primary muscle fibres (as shown in Fig. 5 (48)). Finally, inhibiting ceramide production increases CoQ levels in both L6-myotubes and adult muscle tissue (as shown in Fig. 2-3). Therefore, L6-myotubes possess the necessary metabolic features to investigate the role of mitochondria in insulin resistance, and this relationship is likely applicable to primary muscle fibres”.
We will also add additional data - in point 2 - from differentiated human myocytes that are consistent with our observations from the L6 models. Additional experiments are in progress to further extend these findings.
- One caveat of the approach taken is that exposure of cells to palmitate alone is not reflective of in vivo physiology. It would be interesting to know if similar effects on CoQ are observed when cells are exposed to a more physiological mixture of fatty acids that includes a high ratio of palmitate, but better mimics in vivo nutrition.
Response: Palmitate is widely recognized as a trigger for insulin resistance and ceramide accumulation, which mimics the insulin resistance induced by a diet in rodents and humans. Previous studies have compared the effects of a lipid mixture versus palmitate on inducing insulin resistance in skeletal muscle, and have found that the strong disruption in insulin sensitivity caused by palmitate exposure was lessened with physiologic mixtures of fatty acids, even with a high proportion of saturated fatty acids. This was associated, in part, to the selective partitioning of fatty acids into neutral lipids (such as TAG) when muscle cells are exposed to physiologic lipid mixtures (Newsom et al PMID25793412). Hence, we think that using palmitate is a better strategy to study lipid-induced insulin resistance in vitro. We will add to results:
“In vitro, palmitate conjugated with BSA is the preferred strategy for inducing insulin resistance, as lipid mixtures tend to partition into triacylglycerides (33)”.
We are also performing additional in vivo experiments to add to the physiological relevance of the findings.
- While the utility of targeting SMPD5 to the mitochondria is appreciated, the results in Figure 5 suggest that this manoeuvre caused a rather severe form of mitochondrial dysfunction. This could be more representative of toxicity rather than pathophysiology. It would be helpful to know if these same effects are observed with other manipulations that lower CoQ to a similar degree. If not, the discrepancies should be discussed.
Response: We conducted a staining procedure using the mitochondrial marker mitoDsRED to observe the effect of SMPD5 overexpression on cell toxicity. The resulting images, displayed in the figure below (Author response image 1), demonstrate that the overexpression of SMPD5 did not result in any significant changes in cell morphology or impact the differentiation potential of our myoblasts into myotubes.
Author response image 1.
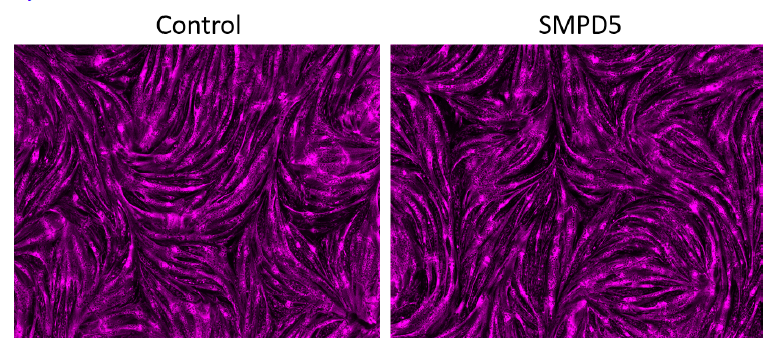
In addition, we evaluated cell viability in HeLa cells following exposure to SACLAC (2 uM) to induce CoQ depletion (left panel). Specifically, we measured cell death by monitoring the uptake of Propidium iodide (PI) as shown in the right panel. Our results demonstrated that Saclac-induced CoQ depletion did not lead to cell death at the doses used for CoQ depletion (Author response image 2).
Author response image 2.
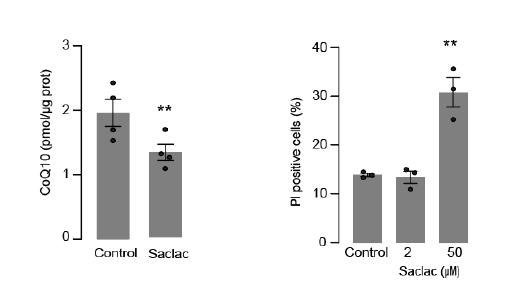
Therefore, we deemed it improbable that the observed effect is caused by cellular toxicity, but rather represents a pathological condition induced by elevated levels of ceramides. We will add to discussion:
“...downregulation of the respirasome induced by ceramides may lead to CoQ depletion. Despite the significant impact of ceramide on mitochondrial respiration, we did not observe any indications of cell damage in any of the treatments, suggesting that our models are not explained by toxic/cell death events.”
- The conclusions could be strengthened by more extensive studies in mice to assess the interplay between mitochondrial ceramides, CoQ depletion and ETC/mitochondrial dysfunction in the context of a standard diet versus HF diet-induced insulin resistance. Does P053 affect mitochondrial ceramide, ETC protein abundance, mitochondrial function, and muscle insulin sensitivity in the predicted directions?
Response: We would like to note that the metabolic characterization and assessment of ETC/mitochondrial function in these mice (both fed a high-fat (HF) and chow diet, with or without P053) were previously published (Turner N, PMID30131496). In addition to this, we have conducted targeted metabolomic and lipidomic analyses to investigate the impact of P053 on ceramide and CoQ levels in HF-fed mice. As illustrated in the figures below (Author response image 3), the administration of P053 led to a reduction in ceramide levels (left panel) and an increase in CoQ levels (right panel) in HF-fed mice, which is consistent with our in vitro findings.
Author response image 3.
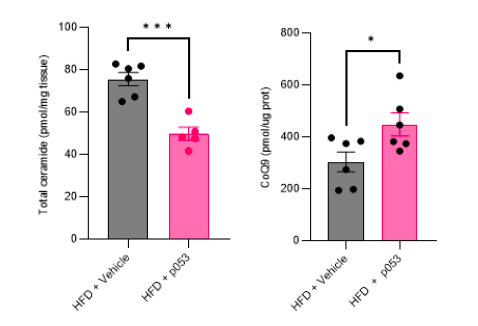
We will add to results:
“…similar effect was observed in mice exposed to a high fat diet for 5 wks (Supp. Fig. 4H-I further phenotypic and metabolic characterization of these animals can be found in (41))”
We will further perform more in-vivo studies to corroborate these findings.



