Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorC Brandon OgbunugaforYale University, New Haven, United States of America
- Senior EditorAleksandra WalczakCNRS, Paris, France
Reviewer #2 (Public review):
The manuscript points out that TMB cut-offs are not strong predictors of response to immunotherapy or overall survival. By randomly shuffling TMB values within cohorts to simulate a null distribution of log-rank test p-values, they show that under correction, the statistical significance of previously reported TMB cut-offs for predicting outcomes is questionable. There is a clinical need for a better prediction of treatment response than TMB alone can provide. However, the analysis does not convincingly refute the validity of the well-known pan-cancer correlation between TMB and immunotherapy response. (In a supplemental analysis, the authors attempt to demonstrate a lack of correlation by specifically removing the most supportive cancer types from a pan-cancer correlation test.) The failure to detect significant TMB cut-offs may be due to insufficient power, as the examined cohorts have relatively low sample sizes. A power analysis would be informative of what cohort sizes are needed to detect small to modest effects of TMB on immune response.
The manuscript provides a simple model of immunogenicity that is tailored to be consistent with a claimed lack of relationship between TMB and response to immunotherapy. Under the model, if each mutation that a tumor has acquired has a relatively high probability of being immunogenic (~10%, they suggest), and if 1-2 immunogenic mutations is enough to induce an immune response, then most tumors produce an immune response, and TMB and response should be uncorrelated except in very low-TMB tumors. The question then becomes whether the response is sufficient to wipe out tumor cells in conjunction with immunotherapy, which is essentially the same question of predicting response that motivated the original analysis. While TMB alone is not an excellent predictor of treatment response, the pan-cancer correlation between TMB and response/survival is highly significant, so the model's only independent prediction is wrong. Additionally, experiments to predict and validate neoepitopes suggest that a much smaller fraction of nonsynonymous mutations produce immune responses (1,2).
A key idea that is overlooked in this manuscript is that of survivorship bias: self-evidently, none of the mutations found at the time of sequencing have been immunogenic enough to provoke a response capable of eliminating the tumor. While the authors suggest that immunoediting "is inefficient, allowing tumors to accumulate a high TMB," the alternative explanation fits the neoepitope literature better: most mutations that reach high allele frequency in tumor cells are not immunogenic in typical (or patient-specific) tumor environments. Of course, immunotherapies sometimes succeed in overcoming the evolved immune evasion of tumors. Higher-TMB tumors are likely to continue to have higher mutation rates after sequencing; increased generation of new immunogenic mutations may partially explain their modestly improved responses to therapy.
References:
(1) Wells, D. K. et al. Key Parameters of Tumor Epitope Immunogenicity Revealed Through a Consortium Approach Improve Neoantigen Prediction. Cell 183, 818-834.e13 (2020).
(2) Yadav, M. et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572-576 (2014).
Author response:
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
In their manuscript entitled: "Is tumor mutational burden predictive of response to immunotherapy?", Gurjao and colleagues discuss the use of tumor mutational burden (TMB) as a predictive biomarker for cancer patients to respond to immune checkpoint blockage (ICB). By analyzing a large cohort of 882 patient samples across different tumor types they find either little or no association of TMB to the response of ICB. In addition, they showed that finding the optimal cutoff for patient stratification lead to a severe multiple testing problem. By rigorously addressing this multiple testing problem only non-small cell lung cancer out of 10 cancer types showed a statistically significant association of TMB and response to ICB. Nevertheless, it is clearly shown that in any case the rate of misclassification is too high that TMB alone would qualify as a clinically suitable biomarker for ICB response. Finally, the authors demonstrate with a simple mathematical model that only a few strong immunogenic mutations would be sufficient for an ICB response, thereby showing that also patients with a low TMB score could benefit from immunotherapy. The manuscript is clearly written, the results are well presented and the applied methods are state-of-the-art.
We would like to thank the reviewer for their thoughtful suggestions and efforts towards improving our manuscript. We address below the reviewer’s recommendations.
Reviewer #1 (Recommendations For The Authors):
(1) The method used for mutation call can also influence the TMB score. Mutation data was downloaded from public databases and not re-called for this study, a potential caller bias could be present. What was the calling strategy of the used data sets? For the present study, I don't think that this is crucial because different callers or post-call processing would be used at different sites to determine TMB. I think it should the mutation calling bias should also be discussed in the manuscript as another shortcoming for TMB as a biomarker for ICB response.
We thank the reviewer for this comment. Mutational data was not aggregated across studies and caller bias would thus not have any impact on the results of this manuscript. In addition, we further clarified the role of mutation calling bias in the Discussions section.
“Although attractive and scalable, TMB does not consider the effect of specific mutations (missense, frameshift etc), their presentation and clonality (19), nor the state of the tumour, its microenvironment, and interactions with the immune system that can be integrated into potentially better predictors of response to ICB (43, 44). In addition, another major limitation of TMB is the lack of standardized measures. This includes the lack of standard sequencing methods to assess TMB: TMB can be measured from Whole-Exome sequencing, Whole-Genome sequencing, targeted panel and even RNA sequencing. This also includes biases introduced by using different mutation calling pipelines resulting in different TMB, sequencing depth and different characteristics of the samples (e.g. low purity samples typically yield lower TMB).”
(2) In their mathematical model of neoantigens and immunogenicity it is assumed that the probability of a mutation to be immunogenic is constant for all mutations. In reality this is certainly not satisfied. However, the central conclusion from the model still holds. I think that this is important to discuss in the manuscript.
We thank the reviewer for this suggestion and now consider the case where each mutation has its own probability p(i) of being immunogenic.
“Our model shows that achieving about constant 𝑃{𝑖𝑚𝑚𝑢𝑛𝑒 𝑟𝑒𝑠𝑝𝑜𝑛𝑠𝑒} for 𝑁 > 10 − 20 mutations, requires  and
and  . The same argument holds when each mutation has its own probability to be immunogenic 𝑝(𝑖), then
. The same argument holds when each mutation has its own probability to be immunogenic 𝑝(𝑖), then 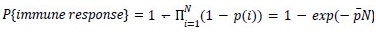 , where is the mean probability of a mutation to be immunogenic. Thus only the average probability of a mutation to be immunogenic matters. In summary, we find that the model agrees with clinical data if individual non-synonymous mutations have, on average, 𝑝~10 − 20% chance for triggering an immune response.”
, where is the mean probability of a mutation to be immunogenic. Thus only the average probability of a mutation to be immunogenic matters. In summary, we find that the model agrees with clinical data if individual non-synonymous mutations have, on average, 𝑝~10 − 20% chance for triggering an immune response.”
(3) In the mathematical formula on page 8, C_N^k is the binomial coefficient. This should be stated or written out.
Thank you for pointing this out. Corrected.
“Due to immunodominance, only a few 𝑘crit immunogenic mutations are sufficient to elicit a full k𝑐𝑟𝑖𝑡 immune response. Hence, the probability for a cancer with 𝑁 (=TMB) mutations to elicit an immune response is then the probability of having 𝑘 or more immunogenic mutations among :
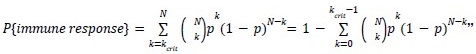
which is the CDF of a binomial distribution.”
(4) The mathematical model provides an explanation that tumors with a low TMB can also respond on ICB. It cannot explain tumors with high TMB lacking ICB response. An explanation of this phenomenon is discussed in the paper but I think also the impact of the tumor immune microenvironment should be mentioned here.
As we explained in the presentation of the model, even immunogenic tumors elicit response to ICB with some probability. In the revision we write:
“𝑃{𝑐𝑙𝑖𝑛𝑖𝑐𝑎𝑙 𝑟𝑒𝑠𝑝𝑜𝑛𝑠𝑒} = 𝑃{𝑐𝑙𝑖𝑛𝑖𝑐𝑎𝑙 𝑟𝑒𝑠𝑝𝑜𝑛𝑠𝑒|𝑖𝑚𝑚𝑢𝑛𝑒 𝑟𝑒𝑠𝑝𝑜𝑛𝑠𝑒} · 𝑃{𝑖𝑚𝑚𝑢𝑛𝑒 𝑟𝑒𝑠𝑝𝑜𝑛𝑠𝑒}, where 𝑃{𝑐𝑙𝑖𝑛𝑖𝑐𝑎𝑙 𝑟𝑒𝑠𝑝𝑜𝑛𝑠𝑒|𝑖𝑚𝑚𝑢𝑛𝑒 𝑟𝑒𝑠𝑝𝑜𝑛𝑠𝑒} is the probability of clinical response, given that cancer elicits an immune response which is complex and depends on many factors including tumor immune microenvironment. Yet the prerequisite for the clinical response is the immune response 𝑃{𝑖𝑚𝑚𝑢𝑛𝑒 𝑟𝑒𝑠𝑝𝑜𝑛𝑠𝑒} that we focus on.”
Reviewer #2 (Public Review):
The manuscript points out that TMB cut-offs are not strong predictors of response to immunotherapy or overall survival. By randomly shuffling TMB values within cohorts to simulate a null distribution of log-rank test p-values, they show that under correction, the statistical significance of previously reported TMB cut-offs for predicting outcomes is questionable.
We would like to thank the reviewer for their thoughtful suggestions and efforts towards improving our manuscript.
There is a clinical need for a better prediction of treatment response than TMB alone can provide. However, no part of the analysis challenges the validity of the well-known pan-cancer correlation between TMB and immunotherapy response.
We address the pan-cancer correlation in the supplemental text and Figure S3. We realized the supplemental text was missing in eLife submission and included in the bioRxiv only. We apologize for this oversight. In particular, we show that the “well-known pan-cancer correlation” is largely based on a few outlier cancer subtypes - MSI colorectal cancers and uveal/ ocular melanomas. We show that when we remove these cancer types from the pan-cancer dataset, the correlation becomes non-significant for the remaining 15 cancer types.
The failure to detect significant TMB cut-offs may be due to insufficient power, as the examined cohorts have relatively low sample sizes. A power analysis would be informative of what cohort sizes are needed to detect small to modest effects of TMB on immune response.
Since we see no effect, we cannot perform a power analysis. Moreover, increasing cohort sizes cannot increase the effect -- dramatic misclassification of responders (the fraction of responders below the treatment cutoff) would remain the same, making TMB unsuitable for clinical decision-making.
The manuscript provides a simple model of immunogenicity that is tailored to be consistent with a claimed lack of relationship between TMB and response to immunotherapy. Under the model, if each mutation that a tumor has acquired has a relatively high probability of being immunogenic (~10%, they suggest), and if 1-2 immunogenic mutations is enough to induce an immune response, then most tumors produce an immune response, and TMB and response should be uncorrelated except in very low-TMB tumors.
Contrary to reviewer’s suggestion, our modeling is not tailored to be consistent with the lack of association between TMB and response. On the contrary, we found the model has two regimes: the first regime (where p<<1) in which higher TMB leads to a higher probability of response, which doesn’t agree with the data , and the second regime (p~0.1) in which cancers with TMB>10-20 are immunogenic, consistent with the clinical data.
We further expanded on these key points in the Results:
“The model shows two different behaviors. If individual mutations are unlikely to be immunogenic (𝑝 ≪ 1) , e.g. due to a low probability of being presented, the probability of response increases gradually with TMB (Figure 5B). The neoantigen theory generally expects such gradual increase in immunogenicity of cancer with TMB. Yet, available data (Figure 2) don’t show such a trend.
On the contrary, if mutations are more likely to be immunogenic 𝑝~0. 1, the probability of response quickly saturates (Figure 5C), making such tumors respond to ICB irrespective of TMB, as we observed in clinical data.”
We also expanded on these key points in the Introduction:
“We develop a simple model that is based on the neoantigen theory and find that it has two regimes. In one regime, the probability of response increases gradually with TMB, as commonly believed. Yet in the other, the probability of response saturates after a few mutations, making a chance to respond independent of TMB. Our analysis of the clinical data is consistent with the latter regime. Thus our model shows that the neoantigen theory is fully consistent with the lack of association between TMB and response.”
The question then becomes whether the response is sufficient to wipe out tumor cells in conjunction with immunotherapy, which is essentially the same question of predicting response that motivated the original analysis. While TMB alone is not an excellent predictor of treatment response, the pan-cancer correlation between TMB and response/survival is highly significant, so the model's only independent prediction is wrong.
Our study indicates that TMB is a very poor predictor (writing that it’s “not an excellent predictor of treatment response” is understatement). Moreover we show that a widely believed “pan-cancer correlation” is shaky as well (Supplemental text and Figure S3). So we don’t see any contradictions between the model and the data.
Additionally, experiments to predict and validate neoepitopes suggest that a much smaller fraction of nonsynonymous mutations produce immune responses1,2.
We agree with the reviewer. That’s exactly what the model suggests.
A key idea that is overlooked in this manuscript is that of survivorship bias: self-evidently, none of the mutations found at the time of sequencing have been immunogenic enough to provoke a response capable of eliminating the tumor. While the authors suggest that immunoediting "is inefficient, allowing tumors to accumulate a high TMB," the alternative explanation fits the neoepitope literature better: most mutations that reach high allele frequency in tumor cells are not immunogenic in typical (or patient-specific) tumor environments. Of course, immunotherapies sometimes succeed in overcoming the evolved immune evasion of tumors. Higher-TMB tumors are likely to continue to have higher mutation rates after sequencing; increased generation of new immunogenic mutations may partially explain their modestly improved responses to therapy.
We disagree with reviewers' assertion that survivorship bias could explain observed phenomena. If immunogenic mutations that arise during cancer development were eliminated (by purifying selection, i.e. reduced fitness or cellular death) then observed mutations would carry noticeable signatures of purifying selection. On the contrary, cancer genomic data shows incredibly weak signals of purifying selection on non-synonymous mutations (Weghorn and Sunyaev, Nature Genetics 2017), and observed passenger mutations are practically indistinguishable from random in their effect on proteins (McFarland et al PNAS 2013).
We do agree with the statement that “most mutations … in tumor cells are not immunogenic”. In fact that’s exactly what our model predicts: (1-p)~90% of mutations in the model are non-immunogenic, while remaining p~10% being sufficient to trigger an immune response. We clarify this in the text of the paper: “On the contrary, if mutations are more likely to be immunogenic 𝑝~0. 1, the probability of response quickly saturates (Figure 5C), making such tumors respond to ICB irrespective of TMB, as we observed in clinical data. ”
Reviewer #2 (Recommendations For The Authors):
Abstract
Defining TMB as "number of non-synonymous mutations": while TMB is not consistently defined throughout the literature, it is usually given as a rate rather than a total count, and sometimes synonymous mutations are included. Consider adopting the definition used by the TMB Harmonization Project: "number of somatic mutations per megabase of interrogated genomic sequence.3"
We thank the reviewer for their comment,
Be more specific about your findings, so that abstract readers can get some understanding of your proposed explanation for the "immunogenicity of neoantigens and the lack of association between TMB and response."
We thank the reviewer for their comment. We modified the abstract to explain that the theory we developed expands the neoantigen theory yet can be consistent with the observed lack of association between TMB and response:
"Second, we develop a model that expands the neoantigen theory and can be consistent with both immunogenicity of neoantigens and the lack of association between TMB and response. Our analysis shows that the use of TMB in clinical practice is not supported by available data and can deprive patients of treatment to which they are likely to respond.”
Introduction
Again, consider using a more standard definition of TMB.
We thank the reviewer for their comment. Our study did not seek to harmonize TMB across the datasets and we thus used the total number of mutations rather than the mutational rate often used for comparison across different datasets.
Expand the introduction to provide a preview of the purpose and direction of your analysis. The current draft reveals only that the analysis will relate to TMB.
We expanded the introduction providing the motivation, the approach, and the summary of main findings.
“Using a biomarker to stratify and prioritize patients for treatment runs a risk of depriving patients who have a chance to respond to a life-saving treatment. High variability of response makes relying on a predictor particularly risky. Hence, we revisit original data that were used to establish correlation between TMB and response. We tested TMB as a predictor of both binary responder/non-responder labels from original clinical studies, as well as continuous survival data. We also investigated whether a TMB threshold could distinguish patients with high and low survival after multiple hypothesis testing. We find that no TMB threshold performs better on the clinical data than on randomized ones.
We further show that irrespective of the strategy to choose the threshold, even if we were to employ the optimal TMB cutoff, it would still lead to about 25% of responders falling below the treatment prioritization threshold. In addition, we re-examine the pan-cancer association of TMB with response rate to ICB.
“Finally we revisit the neoantigen theory that was the rationale for using TMB as a predictor of response to immunotherapy. The theory stipulates that non-synonymous mutations can lead to the production of unique antigens (_neo_antigens) that are recognized by the immune system as foreign, triggering the immune response to cancer. The theory further assumes that the more mutations a cancer has, the more likely it triggers the immune system, and the more likely it will benefit from immunotherapy. We develop a simple model that is based on the neoantigen theory and find that it has two regimes. In one regime, the probability of response increases gradually with TMB, as commonly believed. Yet in the other, the probability of response saturates after a few mutations, making a chance to respond independent of TMB. Our analysis of the clinical data is consistent with the latter regime. Thus our model shows that the neoantigen theory is fully consistent with the lack of association between TMB and response.”
Section: Is TMB associated with response after treatment?
The claim that after excluding melanoma and some colorectal cancers, there is no relationship between TMB and response rates in pan-cancer studies cites references 12 and 14. In reference 12 (Yarchoan et al.), it is clear from glancing at their Figure 1 that a pan-cancer correlation between TMB and response would remain with these cancer types excluded. This discrepancy requires explanation. "Supplementary text" is cited for this claim, but it was not included in the file that I received.
We address the pan-cancer correlation in the supplemental text and Figure S3. While the figure was available, we realized the supplemental text was missing in eLife submission. We apologize for this oversight.
Plots of survival and TMB do not show "visible correlation": Please strengthen this claim with an appropriate statistical test.
We expand the figure caption to explain the following:
“Plots of progression-free survival and TMB for melanoma and lung cancer ICB cohorts show the lack of correlation or of an obvious TMB cutoff. Computing a simple correlation for survival and censored data cannot correctly represent the dependence since patients who are alive live longer than the reported survival, and limiting correlation to patients who are dead would bias the analysis. Thus other survival statistics are used through the paper.”
Section: Model reconciles neoantigen theory and data
Page 8: In the probability formula, the C term is not defined. My guess is that it means choose(N, k).
Please clarify.
Thank you for pointing this out. Corrected using more conventional notation.
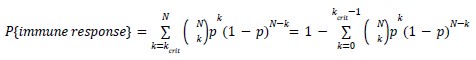
which is the CDF of a binomial distribution.
Page 8: Assuming the above, P(immune response) = P(X >= k_crit); where X~Bin(N, p). The formula should be explicitly introduced in terms of the CDF of the binomial distribution to prevent readers from thinking the wheel is being re-invented.
We thank the reviewer for pointing this out, we modified the equation in the text to make it easier to see this point (see above). We refrain from going further since the CDF of a binomial distribution doesn’t have a closed form and can only be written as the regularized incomplete beta function.
Page 9: Missing word in "allowing cancers with as little as mutations to be"
We thank the reviewer for pointing this out, we modified the text accordingly.
See comments in public review. In brief, I think a convincing case is made regarding the significance of TMB cut-offs as predictors of survival within cancer types, but frankly this elementary model is not compelling.
Section: Materials and Methods
In the manuscript, it is stated that TMB is accepted as reported by data sources. Since most of the comparisons in the manuscript are within-data-source, that is acceptable. However, it should be ensured that TMB measurements are comparable between samples within each source. For example, when TMB is reported as a total mutation count, it can be verified that all samples have the same coverage, or measurement can be converted to mutations per megabase of coverage. In the same vein, if this manuscript's definition of TMB only includes nonsynomous mutations, it should be confirmed that the TMB reported by data sources excludes synonymous mutations.
We thank the reviewer for their comment. We leverage total TMB as reported in the original studies claiming an association between TMB and response/ survival.
Figure S2: Instead of writing "the Youden index associated cutoffs is also plotted," it can be stated that the asterisk represents the Youden index cutoff, or a legend can be added that provides this information.
We thank the reviewer for pointing this out, we modified the text accordingly.



