Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorStephan PlessUniversity of Copenhagen, Copenhagen, Denmark
- Senior EditorKenton SwartzNational Institute of Neurological Disorders and Stroke, Bethesda, United States of America
Reviewer #1 (Public Review):
Despite numerous studies on quinidine therapies for epilepsies associated with GOF mutant variants of Slack, there is no consensus on its utility due to contradictory results. In this study Yuan et al. investigated the role of different sodium selective ion channels on the sensitization of Slack to quinidine block. The study employed electrophysiological approaches, FRET studies, genetically modified proteins and biochemistry to demonstrate that Nav1.6 N- and C-tail interacts with Slack's C-terminus and significantly increases Slack sensitivity to quinidine blockade in vitro and in vivo. This finding inspired the authors to investigate whether they could rescue Slack GOF mutant variants by simply disrupting the interaction between Slack and Nav1.6. They find that the isolated C-terminus of Slack can reduce the current amplitude of Slack GOF mutant variants co-expressed with Nav1.6 in HEK cells and prevent Slack induced seizures in mouse models of epilepsy. This study adds to the growing list of channels that are modulated by protein-protein interactions, and is of great value for future therapeutic strategies.
I have a few comments with regard to how Nav1.6 sensitize Slack to block by quinidine.
It is not clear to me if the Slack induced current amplitude varies depending on the specific Nav subtype. To this end, it would be valuable to test if Slack open probability is affected by the presence of specific Nav subtypes. Nav induced differences in Slack current amplitude and open probability could explain why individual Nav subtypes show varied ability to sensitize Slack to quinidine blockade.
It has previously been shown that INaP (persistent sodium current) is important for inducing Slack currents. Here the authors show that INaT (transient sodium current) of Nav1.6 is necessary for the sensitization of Slack to quinidine block whereas INaP surprisingly has no effect. The authors then show that the N-tail together with C-tail of Nav1.6 can induce same effect on Slack as full-length Nav1.6 in presence of high intracellular concentrations of sodium. However, it is not clear to me how the isolated N- and C-tail of Nav1.6 can induce sensitization of Slack to quinidine by interacting with C-terminus of Slack, while sensitization also is dependent on INaT. The authors speculate on different slack open conformation, but one could speculate if there is a missing link, such as an un-identified additional interacting protein that causes the coupling.
Reviewer #2 (Public Review):
This is a very interesting paper about the coupling of Slack and Nav1.6 and the insight this brings to the effects of quinidine to treat some epilepsy syndromes.
Slack is a sodium-activated potassium channel that is important to hyperpolarization of neurons after an action potential. Slack is encoded by KNCT1 which has mutations in some epilepsy syndromes. These types of epilepsy are treated with quinidine but this is an atypical antiseizure drug, not used for other types of epilepsy. For sufficient sodium to activate Slack, Slack needs to be close to a channel that allows robust sodium entry, like Nav channels or AMPA receptors. but more mechanistic information is not available. Of particular interest to the authors is what allows quinidine to be effective in reducing Slack.
In the manuscript, the authors show that Nav, not AMPA receptors, are responsible for Slack's sensitization to quinidine blockade, at least in cultured neurons (HeK293, primary cortical neurons). Most of the paper focuses on the evidence that Nav1.6 promotes Slack sensitivity to quinidine.
The paper is very well written although there are reservations about the use of non-neuronal cells or cultured primary neurons rather than a more intact system. I also have questions about the figures. Finally, riluzole is not a selective drug, so the limitations of this drug should be discussed. On a minor point, the authors use the term in vivo but there are no in vivo experiments.
Reviewer #3 (Public Review):
Yuan et al., set out to examine the role of functional and structural interaction between Slac and NaVs on the Slack sensitivity to quinidine. Through pharmacological and genetic means they identify NaV1.6 as the privileged NaV isoform in sensitizing Slac to quinidine. Through biochemical assays, they then determine that the C-terminus of Slack physically interacts with the N- and C-termini of NaV1.6. Using the information gleaned from the in vitro experiments the authors then show that virally-mediated transduction of Slack's C-terminus lessens the extent of SlackG269S-induced seizures. These data uncover a previously unrecognized interaction between a sodium and a potassium channel, which contributes to the latter's sensitivity to quinidine.
The conclusions of this paper are mostly well supported by data, but some aspects of functional and structural studies in vivo as well as physically interaction need to be clarified and extended.
1) Immunolabeling of the hippocampus CA1 suggests sodium channels as well as Slac colocalization with AnkG (Fig 3A). Proximity ligation assay for NaV1.6 and Slac or a super-resolution microscopy approach would be needed to increase confidence in the presented colocalization results. Furthermore, coimmunoprecipitation studies on the membrane fraction would bolster the functional relevance of NaV1.6-Slac interaction on the cell surface.
2) Although hippocampal slices from Scn8a+/- were used for studies in Fig. S8, it is not clear whether Scn8a-/- or Scn8a+/- tissue was used in other studies (Fig 1J & 1K). It will be important to clarify whether genetic manipulation of NaV1.6 expression (Fig. 1K) has an impact on sodium-activated potassium current, level of surface Slac expression, or that of NaV1.6 near Slac.
3) Did the epilepsy-related Slac mutations have an impact on NaV1.6-mediated sodium current?
4) Showing the impact of quinidine on persistent sodium current in neurons and on NaV1.6-expressing cells would further increase confidence in the role of persistent sodium current on sensitivity of Slac to quinidine.
Author Response:
Reviewer #1 (Public Review):
Despite numerous studies on quinidine therapies for epilepsies associated with GOF mutant variants of Slack, there is no consensus on its utility due to contradictory results. In this study Yuan et al. investigated the role of different sodium selective ion channels on the sensitization of Slack to quinidine block. The study employed electrophysiological approaches, FRET studies, genetically modified proteins and biochemistry to demonstrate that Nav1.6 N- and C-tail interacts with Slack's C-terminus and significantly increases Slack sensitivity to quinidine blockade in vitro and in vivo. This finding inspired the authors to investigate whether they could rescue Slack GOF mutant variants by simply disrupting the interaction between Slack and Nav1.6. They find that the isolated C-terminus of Slack can reduce the current amplitude of Slack GOF mutant variants co-expressed with Nav1.6 in HEK cells and prevent Slack induced seizures in mouse models of epilepsy. This study adds to the growing list of channels that are modulated by protein-protein interactions, and is of great value for future therapeutic strategies.
I have a few comments with regard to how Nav1.6 sensitize Slack to block by quinidine.
(1) It is not clear to me if the Slack induced current amplitude varies depending on the specific Nav subtype. To this end, it would be valuable to test if Slack open probability is affected by the presence of specific Nav subtypes. Nav induced differences in Slack current amplitude and open probability could explain why individual Nav subtypes show varied ability to sensitize Slack to quinidine blockade.
We appreciate the reviewer for raising this point. In order to address whether the whole-cell current amplitudes of Slack varies depending on the specific NaV subtype, we examined Slack current amplitudes upon co-expression of Slack with specific NaV subtypes in HEK293 cells. The results have shown that there are no significant differences in Slack current amplitudes upon co-expression of Slack with different NaV channel subtypes (Author response image 1), suggesting whole-cell Slack current amplitudes cannot explain the varied ability of NaV subtypes to sensitize Slack to quinidine blockade. To investigate the effect of different NaV channel subtypes on Slack open probability, we will perform the single-channel recordings in the future studies.
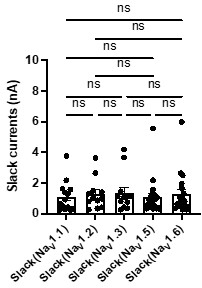
Author response image 1.
The amplitudes of Slack currents upon co-expression of Slack with specific NaV subtypes in HEK293 cells. ns, p > 0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
(2) It has previously been shown that INaP (persistent sodium current) is important for inducing Slack currents. Here the authors show that INaT (transient sodium current) of Nav1.6 is necessary for the sensitization of Slack to quinidine block whereas INaP surprisingly has no effect. The authors then show that the N-tail together with C-tail of Nav1.6 can induce same effect on Slack as full-length Nav1.6 in presence of high intracellular concentrations of sodium. However, it is not clear to me how the isolated N- and C-tail of Nav1.6 can induce sensitization of Slack to quinidine by interacting with C-terminus of Slack, while sensitization also is dependant on INaT. The authors speculate on different slack open conformation, but one could speculate if there is a missing link, such as an un-identified additional interacting protein that causes the coupling.
We fully agree the importance of investigating the detailed mechanism underlying the sensitization of Slack to quinidine blockade mediated by the N- and C-termini of NaV1.6. Regarding the possibility of additional interacting proteins (“missing link”) that mediate the coupling between Slack and NaV1.6, our GST-pull down assays involving Slack and the N- and C-termini of NaV1.6 (Fig. S7) suggest a direct interaction between Slack and NaV1.6 channels. This finding leads us to consider the possibility of additional interacting proteins might be excluded. In order to further address these questions, we plan to employ structural biological methods, such as cryo-electron microscopy (cryo-EM).
Reviewer #2 (Public Review):
This is a very interesting paper about the coupling of Slack and Nav1.6 and the insight this brings to the effects of quinidine to treat some epilepsy syndromes.
Slack is a sodium-activated potassium channel that is important to hyperpolarization of neurons after an action potential. Slack is encoded by KNCT1 which has mutations in some epilepsy syndromes. These types of epilepsy are treated with quinidine but this is an atypical antiseizure drug, not used for other types of epilepsy. For sufficient sodium to activate Slack, Slack needs to be close to a channel that allows robust sodium entry, like Na channels or AMPA receptors. but more mechanistic information is not available. Of particular interest to the authors is what allows quinidine to be effective in reducing Slack.
In the manuscript, the authors show that Nav, not AMPA receptors are responsible for Slack activation, at least in cultured neurons (HeK293, primary cortical neurons). Most of the paper focuses on the evidence that Nav1.6 promotes Slack sensitivity to quinidine.
(1) The paper is very well written although there are reservations about the use of non-neuronal cells or cultured primary neurons rather than a more intact system.
We appreciate the reviewer's positive evaluation of our work. We acknowledge that utilizing a more intact system would provide valuable insights into the inhibitory effect of quinidine on Slack-NaV1.6. However, there are certain challenges associated with studying Slack currents in their entirety.
First, in our experiments, isolating Slack currents from Na+-activated K+ currents in an intact system is challenging as selective inhibitors for Slick are currently unavailable. To address this, we propose using Slick gene knockout mice to specifically measure Slack currents under physiological conditions in the future investigations. Second, we have observed that the interaction between Slack and NaV1.6 primarily occurs at the axon initial segment of neurons. This poses a difficulty when using brain slices for measurements, as employing the whole-cell voltage-clamp technique to assess Slack at the axon initial segment may introduce systemic errors.
We believe that testing the pharmacological effects of quinidine on Slack-NaV1.6 in primary neurons remains the optimal approach. Although non-neuronal cells or cultured primary neurons may not fully replicate the complexity of an intact system, they still provide valuable insights into the interactions between Slack and NaV1.6, and the effects of quinidine.
(2) I also have questions about the figures.
We will make the necessary modifications and clarifications based on the reviewer's comments:
(3) Finally, riluzole is not a selective drug, so the limitations of this drug should be discussed.
We thank the reviewer for raising this point. We will discuss the limitations of riluzole in our revised version of the manuscript.
(4) On a minor point, the authors use the term in vivo but there are no in vivo experiments.
We thanks the reviewer for raising this point. In our experiments, although we did not conduct experiments directly in living organisms, our results demonstrated the co-immunoprecipitation of NaV1.6 with Slack in homogenates from mouse cortical and hippocampal tissues (Fig. 3C). This result may support that the interaction between Slack and NaV1.6 occurs in vivo.
Reviewer #3 (Public Review):
Yuan et al., set out to examine the role of functional and structural interaction between Slack and NaVs on the Slack sensitivity to quinidine. Through pharmacological and genetic means they identify NaV1.6 as the privileged NaV isoform in sensitizing Slack to quinidine. Through biochemical assays, they then determine that the C-terminus of Slack physically interacts with the N- and C-termini of NaV1.6. Using the information gleaned from the in vitro experiments the authors then show that virally-mediated transduction of Slack's C-terminus lessens the extent of SlackG269S-induced seizures. These data uncover a previously unrecognized interaction between a sodium and a potassium channel, which contributes to the latter's sensitivity to quinidine.
The conclusions of this paper are mostly well supported by data, but some aspects of functional and structural studies in vivo as well as physically interaction need to be clarified and extended.
(1) Immunolabeling of the hippocampus CA1 suggests sodium channels as well as Slack colocalization with AnkG (Fig 3A). Proximity ligation assay for NaV1.6 and Slack or a super-resolution microscopy approach would be needed to increase confidence in the presented colocalization results. Furthermore, coimmunoprecipitation studies on the membrane fraction would bolster the functional relevance of NaV1.6-Slac interaction on the cell surface.
We thank the reviewer for good suggestions. We acknowledge that employing proximity ligation assay and high-resolution techniques would significantly enhance our understanding of the localization of the Slack-NaV1.6 coupling.
At present, the technical capabilities available in our laboratory and institution do not support high-resolution testing. However, we are enthusiastic about exploring potential collaborations to address these questions in the future. Furthermore, we fully recognize the importance of conducting co-immunoprecipitation (Co-IP) assays from membrane fractions. While we have already completed Co-IP assays for total protein and quantified the FRET efficiency values between Slack and NaV1.6 in the membrane region, the Co-IP assays on membrane fractions will be conducted in our future investigations.
(2) Although hippocampal slices from Scn8a+/- were used for studies in Fig. S8, it is not clear whether Scn8a-/- or Scn8a+/- tissue was used in other studies (Fig 1J & 1K). It will be important to clarify whether genetic manipulation of NaV1.6 expression (Fig. 1K) has an impact on sodium-activated potassium current, level of surface Slack expression, or that of NaV1.6 near Slack.
We thank the reviewer for pointing this out. In Fig. 1G,J,K, primary cortical neurons from homozygous NaV1.6 knockout (Scn8a-/-) mice were used. We will clarify this information in the revised manuscript. In terms of the effects of genetic manipulation of NaV1.6 expression on IKNa and surface Slack expression, we compared the amplitudes of IKNa measured from homozygous NaV1.6 knockout (NaV1.6-KO) neurons and wild-type (WT) neurons. The results showed that homozygous knockout of NaV1.6 does not alter the amplitudes of IKNa (Author response image 2). The level of surface Slack expression will be tested further.
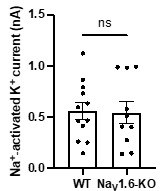
Author response image 2.
The amplitudes of IKNa in WT and NaV1.6-KO neurons (data from manuscript Fig. 1K). ns, p > 0.05, unpaired two-tailed Student’s t test.
(3) Did the epilepsy-related Slack mutations have an impact on NaV1.6-mediated sodium current?
We thank the reviewer’s question. We examined the amplitudes of NaV1.6 sodium current upon expression alone or co-expression of NaV1.6 with epilepsy-related Slack mutations (K629N, R950Q, K985N). The results showed that the tested epilepsy-related Slack mutations do not alter the amplitudes of NaV1.6 sodium current (Author response image 3).
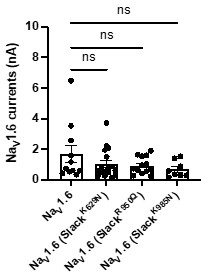
Author response image 3.
The amplitudes of NaV1.6 sodium currents upon co-expression of NaV1.6 with epilepsy-related Slack mutant variants (SlackK629N, SlackR950Q, and SlackK985N). ns, p>0.05, one-way ANOVA followed by Bonferroni’s post hoc test.
4) Showing the impact of quinidine on persistent sodium current in neurons and on NaV1.6-expressing cells would further increase confidence in the role of persistent sodium current on sensitivity of Slack to quinidine.
We appreciate the reviewer’s question. Previous studies have shown that quinidine can inhibit persistent sodium currents at low concentrations1. In our experiments, blocking persistent sodium currents by application of riluzole in the bath solution showed no significant effects on the sensitivity of Slack to quinidine blockade upon co-expression of Slack with NaV1.6 (Fig. 2F,H). This result suggested that persistent sodium currents were not involved in the sensitization of Slack to quinidine blockade.
- Ju YK, Saint DA, Gage PW. Effects of lignocaine and quinidine on the persistent sodium current in rat ventricular myocytes. Br J Pharmacol. Oct 1992; 107(2):311-6. doi:10.1111/j.1476-5381.1992.tb12743.x



