Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorVincent CastricUniversité de Lille, Lille, France
- Senior EditorDetlef WeigelMax Planck Institute for Biology Tübingen, Tübingen, Germany
Reviewer #1 (Public Review):
The evolution of dioecy in angiosperms has significant implications for plant reproductive efficiency, adaptation, evolutionary potential, and resilience to environmental changes. Dioecy allows for the specialization and division of labor between male and female plants, where each sex can focus on specific aspects of reproduction and allocate resources accordingly. This division of labor creates an opportunity for sexual selection to act and can drive the evolution of sexual dimorphism.
In the present study, the authors investigate sex-biased gene expression patterns in juvenile and mature dioecious flowers to gain insights into the molecular basis of sexual dimorphism. They find that a large proportion of the plant transcriptome is differentially regulated between males and females with the number of sex-biased genes in floral buds being approximately 15 times higher than in mature flowers. The functional analysis of sex-biased genes reveals that chemical defense pathways against herbivores are up-regulated in the female buds along with genes involved in the acquisition of resources such as carbon for fruit and seed production, whereas male buds are enriched in genes related to signaling, inflorescence development and senescence of male flowers. Furthermore, the authors implement sophisticated maximum likelihood methods to understand the forces driving the evolution of sex-biased genes. They highlight the influence of positive and relaxed purifying selection on the evolution of male-biased genes, which show significantly higher rates of non-synonymous to synonymous substitutions than female or unbiased genes. This is the first report (to my knowledge) highlighting the occurrence of this pattern in plants. Overall, this study provides important insights into the genetic basis of sexual dimorphism and the evolution of reproductive genes in Cucurbitaceae.
Reviewer #2 (Public Review):
Summary:
This study uses transcriptome sequence from a dioecious plant to compare evolutionary rates between genes with male- and female-biased expression and distinguish between relaxed selection and positive selection as causes for more rapid evolution. These questions have been explored in animals and algae, but few studies have investigated this in dioecious angiosperms, and none have so far identified faster rates of evolution in male-biased genes (though see Hough et al. 2014 https://doi.org/10.1073/pnas.1319227111).
Strengths:
The methods are appropriate to the questions asked. Both the sample size and the depth of sequencing are sufficient, and the methods used to estimate evolutionary rates and the strength of selection are appropriate. The data presented are consistent with faster evolution of genes with male-biased expression, due to both positive and relaxed selection.
This is a useful contribution to understanding the effect of sex-biased expression in genetic evolution in plants. It demonstrates the range of variation in evolutionary rates and selective mechanisms, and provides further context to connect these patterns to potential explanatory factors in plant diversity such as the age of sex chromosomes and the developmental trajectories of male and female flowers.
Weaknesses:
The presence of sex chromosomes is a potential confounding factor, since there are different evolutionary expectations for X-linked, Y-linked, and autosomal genes. Attempting to distinguish transcripts on the sex chromosomes from autosomal transcripts could provide additional insight into the relative contributions of positive and relaxed selection.
Reviewer #3 (Public Review):
The potential for sexual selection and the extent of sexual dimorphism in gene expression have been studied in great detail in animals, but hardly examined in plants so far. In this context, the study by Zhao, Zhou et al. al represents a welcome addition to the literature.
Relative to the previous studies in Angiosperms, the dataset is interesting in that it focuses on reproductive rather than somatic tissues (which makes sense to investigate sexual selection), and includes more than a single developmental stage (buds + mature flowers).
Author Response
The following is the authors’ response to the previous reviews.
eLife assessment
This valuable paper examines gene expression differences between male and female individuals over the course of flower development in the dioecious angiosperm Trichosantes pilosa. The authors show that male-biased genes evolve faster than female-biased and unbiased genes. This is frequently observed in animals, but this is the first report of such a pattern in plants. In spite of the limited sample size, the evidence is mostly solid and the methods appropriate for a non-model organism. The resources produced will be used by researchers working in the Cucurbitaceae, and the results obtained advance our understanding of the mechanisms of plant sexual reproduction and its evolutionary implications: as such they will broadly appeal to evolutionary biologists and plant biologists.
Public Reviews:
Reviewer #1 (Public Review):
The evolution of dioecy in angiosperms has significant implications for plant reproductive efficiency, adaptation, evolutionary potential, and resilience to environmental changes. Dioecy allows for the specialization and division of labor between male and female plants, where each sex can focus on specific aspects of reproduction and allocate resources accordingly. This division of labor creates an opportunity for sexual selection to act and can drive the evolution of sexual dimorphism.
In the present study, the authors investigate sex-biased gene expression patterns in juvenile and mature dioecious flowers to gain insights into the molecular basis of sexual dimorphism. They find that a large proportion of the plant transcriptome is differentially regulated between males and females with the number of sex-biased genes in floral buds being approximately 15 times higher than in mature flowers. The functional analysis of sex-biased genes reveals that chemical defense pathways against herbivores are up-regulated in the female buds along with genes involved in the acquisition of resources such as carbon for fruit and seed production, whereas male buds are enriched in genes related to signaling, inflorescence development and senescence of male flowers. Furthermore, the authors implement sophisticated maximum likelihood methods to understand the forces driving the evolution of sex-biased genes. They highlight the influence of positive and relaxed purifying selection on the evolution of male-biased genes, which show significantly higher rates of non-synonymous to synonymous substitutions than female or unbiased genes. This is the first report (to my knowledge) highlighting the occurrence of this pattern in plants. Overall, this study provides important insights into the genetic basis of sexual dimorphism and the evolution of reproductive genes in Cucurbitaceae.
Reviewer #2 (Public Review):
Summary:
This study uses transcriptome sequence from a dioecious plant to compare evolutionary rates between genes with male- and female-biased expression and distinguish between relaxed selection and positive selection as causes for more rapid evolution. These questions have been explored in animals and algae, but few studies have investigated this in dioecious angiosperms, and none have so far identified faster rates of evolution in male-biased genes (though see Hough et al. 2014 https://doi.org/10.1073/pnas.1319227111).
Strengths:
The methods are appropriate to the questions asked. Both the sample size and the depth of sequencing are sufficient, and the methods used to estimate evolutionary rates and the strength of selection are appropriate. The data presented are consistent with faster evolution of genes with male-biased expression, due to both positive and relaxed selection.
This is a useful contribution to understanding the effect of sex-biased expression in genetic evolution in plants. It demonstrates the range of variation in evolutionary rates and selective mechanisms, and provides further context to connect these patterns to potential explanatory factors in plant diversity such as the age of sex chromosomes and the developmental trajectories of male and female flowers.
Weaknesses:
The presence of sex chromosomes is a potential confounding factor, since there are different evolutionary expectations for X-linked, Y-linked, and autosomal genes. Attempting to distinguish transcripts on the sex chromosomes from autosomal transcripts could provide additional insight into the relative contributions of positive and relaxed selection.
Reviewer #3 (Public Review):
The potential for sexual selection and the extent of sexual dimorphism in gene expression have been studied in great detail in animals, but hardly examined in plants so far. In this context, the study by Zhao, Zhou et al. al represents a welcome addition to the literature.
Relative to the previous studies in Angiosperms, the dataset is interesting in that it focuses on reproductive rather than somatic tissues (which makes sense to investigate sexual selection), and includes more than a single developmental stage (buds + mature flowers).
Some aspects of the presentation have been improved in this new version of the manuscript.Specifically:
the link between sex-biased and tissue-biased genes is now slightly clearer,
the limitation related to the de novo assembled transcriptome is now formally acknowledged,
the interpretation of functional categories of the genes identified is more precise,
the legends of supplementary figures have been improved - a large number of typos have been fixed.
in response to this first round of reviews. As I detail below, many of the relevant and constructive suggestions by the previous reviewers were not taken into account in this revision.
For instance:
- Reviewer 2 made precise suggestions for trying to take into account the potential confounding factor of sex-chromosomes. This suggestion was not followed.
For the question of reviewer 2:
The presence of sex chromosomes is a potential confounding factor, since there are different evolutionary expectations for X-linked, Y-linked, and autosomal genes. Attempting to distinguish transcripts on the sex chromosomes from autosomal transcripts could provide additional insight into the relative contributions of positive and relaxed selection.
Empirically, the analyses could be expanded by an attempt to distinguish between genes on the autosomes and the sex chromosomes. Genotypic patterns can be used to provisionally assign transcripts to XY or XX-like behavior when all males are heterozygous and all females are homozygous (fixed X-Y SNPs) and when all females are heterozygous and males are homozygous (lost or silenced Y genes). Comparing such genes to autosomal genes with sex-biased expression would sharpen the results because there are different expectations for the efficacy of selection on sex chromosomes. See this paper (Hough et al. 2014; https://www.pnas.org/doi/abs/10.1073/pnas.1319227111), which should be cited and does in fact identify faster substitution rates in Y-linked genes.
Authors’ response: We have cited Hough et al. (2014) and Sandler et al. (2018) in the revised manuscript. We agree that the presence of sex chromosomes is potentially a confounding factor. By adopting methods in Hough et al. (2014) and Sandler et al. (2018), we tried to distinguish transcripts on sex chromosomes from autosomal chromosomes. For a total of 2,378 unbiased genes, we found that 36 genes were putatively sex chromosomal genes, 20 of which were exclusively heterozygous and homozygous for males and females, respectively; while the other 16 genes showing an opposite genotyping patterns between males and females. For 343 male-biased genes, only three ones exhibit a pattern of potentially sex-linked. For the 1,145 female-biased genes, we identified 19 genes which might located on the sex chromosomes. Among the 19 genes, five genes were exclusively heterozygous for males and exclusively homozygous for females, while reversed genotyping patterns presented in the other 14 genes. So, sex-linked genes may contribute relatively little to rapid evolution of male-biased genes. An alternative explanation is that the results could be unreliable due to small sample sizes. Thus, we did not describe them in the Results section. We will investigate the issue when whole genome sequences and population datasets become available in the near future.
- Reviewer 1 & 3 indicated that results were mentioned in the discussion section without having been described before. This was not fixed in this new version.
For the question of reviewer 1:
2) Paragraph (407-416) describes the analysis of duplicated genes under relaxed selection but there is no mention of this in the results.
Authors’ response: Following this suggestion, in the Results section, we have added a sentence, “We also found that most of them were members of different gene families generated by gene duplication (Table S13)” on line 310-311 in the revised manuscript (Rapid_evolution_of_malebiased_genes_Trichosanthes_pilosa_Tracked_change_2023_11_06.docx).
For the question of reviewer 1:
38- line 417-424. The discussion should not contain new results.
Authors’ response: Thank you for pointing out this. In the Results section, we have added a few sentences as following: “Similarly, given that dN/dS values of sex-biased genes were higher due to codon usage bias, lower dS rates would be expected in sex-biased genes relative to unbiased genes (Ellegren & Parsch, 2007; Parvathy et al., 2022). However, in our results, the median of dS values in male-biased genes were much higher than those in female-biased and unbiased genes in the results of ‘free-ratio’ (Fig. S4A, female-biased versus male-biased genes, P = 6.444e-12 and malebiased versus unbiased genes, P = 4.564e-13) and ‘two-ratio’ branch model (Fig. S4B, femalebiased versus male-biased genes, P = 2.2e-16 and male-biased versus unbiased genes, P = 9.421e08, respectively). ” on line 323-331, and consequently, removed the following sentence, “femalebiased vs male-biased genes, P = 6.444e-12 and male-biased vs unbiased genes, P = 4.564e-13” and “female-biased versus male-biased genes, P = 2.2e-16 and male-biased versus unbiased genes, P = 9.421e-08, respectively” in the Discussion section.
- Reviewer 1 asked for a comparison between the number of de novo assembled unigenes in this transcriptome and the number of genes in other Cucurbitaceae species. I could not see this comparison reported.
Authors’ response: In the first revision, we described only percentages. We have now added the number of genes. We modify this part as follows: “The majority of unigenes were annotated by homologs in species of Cucurbitaceae (61.6%, 36,375), including Momordica charantia (16.3%, 9,625), Cucumis melo (11.9%, 7,027), Cucurbita pepo (11.9%, 7,027), Cucurbita moschata (11.5%, 6,791), Cucurbita maxima (10.1%, 5,964) and other species (38.4%, 22,676) (Fig. S1C).”.
- Reviewer 1 pointed out that permutation tests were more appropriate, but no change was made to the manuscript.
Authors’ response: Thank you for your suggestion. In the first revision, we have indirectly responded to the issues. Wilcoxon rank sum test is more commonly used for all comparisons between sex-biased and unbiased genes in many papers. Additionally, we tested datasets using permutation t-tests, which is consistent with the results of Wilcoxon rank sum test. For example, we found that only in floral buds, there are significant differences in ω values in the results of ‘free-ratio’ (female-biased versus male-biased genes, P = 0.04282 and male-biased versus unbiased genes, P = 0.01114) and ‘two-ratio’ model (female-biased versus male-biased genes, P = 0.01992 and male-biased versus unbiased genes, P = 0.02127, respectively). We also described these results in the Results section accordingly (line 278-284).
- Reviewer 3 pointed out the small sample size (both for the RNA-seq and the phylogenetic analysis), but again this limitation is not acknowledged very clearly.
Authors’ response: Sorry, we acknowledged that our sample size was relatively small. In the revised version, we have added a sentence as follows, “Additionally, our sample size is relatively small, and may provide low power to detect differential expression.” in the Discussion section.
- Reviewer 1 & 3 pointed out that Fig 3 was hard to understand and asked for clarifications that I did not see in the text and the figure in unchanged.
Authors’ response: Thank you for your suggestions. We have revised the manuscript to clarify the meaning of the acronym (F1TGs, F2TGs, M1TGs, M2TGs, F1BGs, F2BGs, M1BGs and M2BGs) and presented the number of genes. We have added two labels, indicating that panels A and B correspond to males and C and D to females in Fig. 3.
- Reviewer 3 suggested to combine all genes with sex-bias expression when evaluating the evolutionary rate, in addition to the analyses already done. This suggestion was not followed.
For the question of reviewer 3:line 196 and following: In these analyses, I could not understand the rationale for keeping buds vs mature flowers as separate analyses throughout. Why not combine both and use the full set of genes showing sex-bias in any tissue? This would increase the power and make the presentation of the results a lot more straightforward.
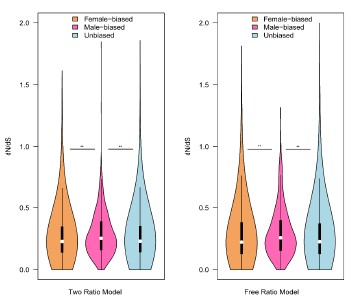
Authors’ response: Thank you for your suggestions. In the first revision, we tried to respond to the issues. First, we observed strong sexual dimorphism in floral buds, such as racemose versus solitary, early-flowering versus late-flowering. Second, as you pointed out earlier, “the dataset is interesting in that it focuses on reproductive rather than somatic tissues (which makes sense to investigate sexual selection), and includes more than a single developmental stage (buds + mature flowers)”, we totally agree with you on this point. Third, according to your suggestions, we combined all genes with sex-bias expression to evaluate the evolutionary rates. We found significant differences (please see a Figure below) in ω values in the results of ‘free-ratio’ (female-biased versus male-biased genes, P =0.005622 and male-biased versus unbiased genes, P = 0.001961) and ‘two-ratio’ model (female-biased versus male-biased genes, P = 0.008546 and male-biased versus unbiased genes, P = 0.009831, respectively) using Wilcoxon rank sum test. However, the significance is lower than previous results in floral buds due to sex-biased genes of mature flower joined, especially compared to the results of “free-ratio model”. Additionally, we also test all combined genes with sex-bias expression using permutation t-test. Unfortunately, there are no significant differences in ω values expect for male-biased versus unbiased genes in the results of ‘free-ratio’ model (P = 0.03034) and ‘two-ratio’ model (P = 0.0376), respectively. To a certain extent, the combination of all genes with sex-bias expression may cover the signals of rapid evolution of sex-biased genes in floral buds. Therefore, these results are not described in our manuscript. In the near future, we would like to make further investigations through more development stages of flowers and new technologies (e.g. Single-Cell method, See Murat et al., 2023) in each sex to consolidate the conclusion, and it is hoped that we could find more meaningful results.
Author response image 1.
- Reviewer 3 pointed out that hand-picking specific categories of genes was not statistically valid, and in fact not necessary in the present context. This was not changed.
For the question of reviewer3: removing genes on a post-hoc basis seems statistically suspicious to me. I don't think your analysis has enough power to hand-pick specific categories of genes, and it is not clear what this brings here. I suggest simply removing these analyses and paragraphs.
Authors’ response: Thank you for your suggestions. We have changed them accordingly. We removed a part of the following paragraph, “To confirm the contributions of positive selection and relaxed selection to rapid rates of male-biased genes in floral buds, we generated three datasets of OGs by excluding different sets of genes. Specifically, we excluded 18 relaxed selective male-biased genes (5.23%), 98 positively selected male-biased genes (28.57%), and 112 male-biased genes (32.65%) under positive and relaxed selection from 343 OGs (Fig. S4). We observed that after excluding male-biased genes under relaxed purifying selection, the median (0.264) decreased by 0.34% compared to the median (0.265) of all OGs (Fig. S4A-B). However, after excluding positively selected male-biased genes, the median (0.236) was reduced by 11% (Fig. S4A, C) in the results of ‘free-ratio’ branch model. This pattern was consistent with the results of ‘two-ratio’ branch model as well (Fig. S4E-G).” on line 290 to 300.
However, we kept the following paragraph, “We also analyzed female-biased and unbiased genes that underwent positive and relaxed selection in floral buds (Tables S6-S10). We identified 216 (18.86%) positively selected, and 69 (6.03%) relaxed selective female-biased genes from 1,145 OGs, respectively. Similarly, we found 436 (18.33%) positively selected, and 43 (1.81%) unbiased genes under relaxed selection from 2,378 OGs, respectively. Notably, male-biased genes have a higher proportion (10%) of positively selected genes compared to female-biased and unbiased genes. However, relaxed selective male-biased genes have a higher proportion (3.24%) than unbiased genes, but about 0.8% lower than that of female-biased genes.”. In this way, we can compare the proportion of sex-biased genes that have undergone positive selection and release selection among female-biased genes, unbiased genes and male-biased genes in floral buds in the Discussion section.
- Reviewer 1 asked for all data to be public, but I could not find in the manuscript where the link to the data on ResearchGate was provided.
Authors’ response: We have added a link in the Data Availability section.
- Reviewers 1 & 3 pointed out that since only two tissues were compared, the claims on pleiotropy should have been toned down, but no change was made to the text.
Authors’ response: Thank you for your suggestions. We revised “due to low pleiotropic constraints” to “due to low evolutionary constraints” and revised “low pleiotropy” to “low constraints”.
- Reviewer 1 asked for a clarification on which genes are plotted on the heatmap of Fig3C and an explanation of the color scale. No change was made.
Authors’ response: Sorry for the confusion. Actually, Reviewer 1 asked that “Fig. 2C, which genes are plotted on the heatmap and what is the color scale corresponding to?” In the previous revision, we have revised them (See Fig. 2 Sex-biased gene expression for floral buds and flowers at anthesis in males and females of Trichosanthes pilosa). Sex-biased genes (the union of sex-biased genes in F1, M1, F2 and M2) are plotted on the heatmap. The color gradient represents from high to low (from red to green) gene expression.
- Reviewer 1 asked for panel B in Fig S5 and S6 to be removed. They are still there. They asked for abbreviations to be explained in the legend of Fig S8. This was not done. They asked for details about columns headers. Such detailed were not added. They asked for more recent references on line 53-56: this was not done.
Authors’ response: We have removed panel B in Fig. S5 and S6. We explained abbreviations in text and Fig. S8. We added more details about the column headers in Supplementary Table S4, S5, S6, S7, S8, S9 and S10. We also added more recent references on line 53-56.
Recommendations for the authors:
Reviewer #3 (Recommendations For The Authors):
Authors’ response: Thank you for your suggestions. We have revised/fixed these issues following your concerns and suggestions.
Line 46-48 would be clearer as « Sexual dimorphism is the condition where sexes of the same species exhibit different morphological, ecological and physiological traits in gonochoristic animals and dioecious plants, despite male and female individuals sharing the same genome except for sex chromosomes or sex-determining loci »
Authors’ response: Thanks. We have revised it accordingly.
Line 50: replace «in both » by «between the two »
Authors’ response: We have revised it.
Line 51: « genes exclusively » -> « genes expressed exclusively »
Authors’ response: We have revised it.
Line 58: « in many animals » -> « in several animal species »
Authors’ response: We have revised it to “in some animal species”.
Line 58: « to which » -> « of this bias »
Authors’ response: We have revised it.
Line 64: « Most dioecious plants possess homomorphic sex-chromosomes that are roughly similar in size when viewed by light microscopy. » : a reference is missing
Authors’ response: We have added the reference.
Line 67: remove « that »
Authors’ response: We have revised it.
line 96: change to: « only the five above-mentioned studies »
Authors’ response: We have revised it.
Line 97: remove « the »
Authors’ response: We have revised it.
Line 111: « Drosophia » -> Drosophila
Authors’ response: We have revised it.
Line 114: exhibiting -> « exhibited »
Authors’ response: We have revised it.
Line 115: suggest -> « suggesting »
Authors’ response: We have revised it.
Line 117: « studies in plants have rarely reported elevated rates of sex-biased genes » : is it « rarely » or « never » ?
Authors’ response: We have revised to “never”.
Line 143: « It’s » -> « Its »
Authors’ response: We have revised it.
Line 143-146: say whether the male parts (e.g. anthers) are still present in females flowers, and the female parts (pistil+ ovaries) in the male flowers, or whether these respective organs are fully aborted.
Authors’ response: We have added the following sentence, “The male parts (e. g., anthers) of female flowers, and the female parts (e. g., pistil and ovaries) of male flowers are fully aborted” in line 148150 of the Introduction section.
Line 158: this is now clearer, but please specify whether you are talking about 12 floral buds in total, or 12 per individual (i.e. 72 buds in total).
Authors’ response: We have revised it to “Using whole transcriptome shotgun sequencing, we sequenced floral buds and flowers at anthesis from female and male of dioecious T. pilosa. We set up three biological replicates from three female and three male plants, including 12 samples in total (six floral buds and six flowers at anthesis)”.
Line 194-198: These sentences are unclear and hard to link to the figure. Consider changing for « In male plants, the number of tissue-biased genes in flowers at anthesis (M2TGs: n = 2795) was higher than that in floral buds (M1TGs: n = 1755, Fig. 3A and 3B). Figure 3 is also very hard to read. Adding a label on the side to indicate that panels A and B correspond to male-biased genes and C and D to female-biased genes could be useful.
Authors’ response: Thank you for your suggestions. We have revised the text to clarify the meaning of the acronym (F1TGs, F2TGs, M1TGs, M2TGs, F1BGs, F2BGs, M1BGs and M2BGs) and presented the number of genes. We have added two labels, indicating that panels A and B correspond to males and C and D to females in Figure 3.
Line 208: explain the approach: e.g. « We then compared rates of protein evolution among malebiased, female-biased and unbiased genes. To do this, we sequenced floral bud transcriptomes from the closely related T. anguina, as well as two more distant outgroups, T. kirilowii and Luffa cylindrica. T. kirilowii is a dioecious species like T. pilosa, and the other two are monoecious. We identified one-to-one orthologous groups (OGs) for 1,145 female-biased, 343 male-biased, and 2,378 unbiased genes. »
Authors’ response: We have revised this paragraph to the following, “We compared rates of protein evolution among male-biased, female-biased and unbiased genes in four species with phylogenetic relationships (((T. anguina, T. pilosa), T. kirilowii), Luffa cylindrica), including dioecious T. pilosa, dioecious T. kirilowii, monoecious T. anguina in Trichosanthes, together with monoecious Luffa cylindrica. To do this, we sequenced transcriptomes of T. pilosa. We also collected transcriptomes of T. kirilowii, as well as genomes of T. anguina and Luffa cylindrica.”
Line 220: « the same ω value was in all branches » -> « all branches are constrained to have the same ω value ».
Authors’ response: We have revised it.
Line 221: « results of the 'two-ratio' branch model ... »
Authors’ response: We have revised it.
Line 235: add a few words to explain why the effect size is bigger than for buds, but still is not significant: e.g. «possibly because of limited statistical power due to the low number of sex-biased genes in flowers at anthesis »
Authors’ response: We have revised this to “However, there is no statistically significant difference in the distribution of ω values using Wilcoxon rank sum tests for female-biased versus male-biased genes (P = 0.0556), female-biased versus unbiased genes (P = 0.0796), and male-biased versus unbiased genes (P = 0.3296) possibly because of limited statistical power due to the low number of sex-biased genes in flowers at anthesis.” in line 260-261.
Line 255: explain in plain English what the « A model » is. This was already requested in the previous version.
Authors’ response: We have revised “A model” to “classical branch-site model A”.
Line 258: explain in plain English what the « foreground 2b ω value » corresponds to
Authors’ response: We have revised to as follows, “foreground 2b ω value” to “foreground ω >1”. Additionally, we also added the sentence “The classical branch-site model assumes four site classes (0, 1, 2a, 2b), with different ω values for the foreground and background branches. In site classes 2a and 2b, the foreground branch undergoes positive selection when there is ω > 1.” in line 624-627.
Line 259: explain how these different approaches complement each other rather than being redundant. This was also already requested in the previous version.
Authors’ response: Sorry. We have now revised it as follows, “As a complementary approach, we utilized the aBSREL and BUSTED methods that are implemented in HyPhy v.2.5 software, which avoids false positive results by classical branch-site models due to the presence of rate variation in background branches, and detected significant evidence of positive selection.” in line 292-295.
Line 270: remove « dramatically », and also remove « or eliminated at both gene-wide and genomewide levels », as well as « relative to positive selection »
Authors’ response: Thank you for your suggestions. We have revised it.
Line 290-309: remove this section - this was already pointed out in the previous reviews as a « ad hoc » procedure, and this point has already been made clear with the RELAX analysis.
Authors’ response: Thank you for your suggestions. We revised this section accordingly. We remove the following paragraph, “To confirm the contributions of positive selection and relaxed selection to rapid rates of male-biased genes in floral buds, we generated three datasets of OGs by excluding different sets of genes. Specifically, we excluded 18 relaxed selective male-biased genes (5.23%), 98 positively selected male-biased genes (28.57%), and 112 male-biased genes (32.65%) under positive and relaxed selection from 343 OGs (Fig. S4). We observed that after excluding malebiased genes under relaxed purifying selection, the median (0.264) decreased by 0.34% compared to the median (0.265) of all OGs (Fig. S4A-B). However, after excluding positively selected malebiased genes, the median (0.236) was reduced by 11% (Fig. S4A, C) in the results of ‘free-ratio’ branch model. This pattern was consistent with the results of ‘two-ratio’ branch model as well (Fig. S4E-G).” on line 334-344.
However, we kept the other parts “We also analyzed female-biased and unbiased genes that underwent positive and relaxed selection in floral buds (Tables S6-S10). We identified 216 (18.86%) positively selected, and 69 (6.03%) relaxed selective female-biased genes from 1,145 OGs, respectively. Similarly, we found 436 (18.33%) positively selected, and 43 (1.81%) unbiased genes under relaxed selection from 2,378 OGs, respectively. Notably, male-biased genes have a higher proportion (10%) of positively selected genes compared to female-biased and unbiased genes. However, relaxed selective male-biased genes have a higher proportion (3.24%) than unbiased genes, but about 0.8% lower than that of female-biased genes.”. In this way, we can compare the proportion of sex-biased genes that have undergone positive selection and release selection among female-biased genes, unbiased genes and male-biased genes in floral buds in the Discussion sections.
Line 348: Here you talk about « Numerous studies », but then only report three studies. Please clarify.
Authors’ response: Thank you for your suggestions. We have revised it to “Several studies”.
Line 352: Cut the sentence: « In contrast, the wind-pollinated dioecious plant Populus balsamifera ... »
Authors’ response: Thank you for your suggestions. We have revised it.
Line 357: « In contrast to the above studies... »: If I understand correctly, this is not in contrast to the observation in Populus balsamifera. Please clarify.
Authors’ response: Thank you for your suggestions. We have revised to “Similar to the above study of Populus balsamifera.”.
Line 420: « our results » -> « we »; « that underwent » -> « undergoing »
Authors’ response: Thank you for your suggestions. We have revised it.
Figure 3 is very hard to read and poorly labeled (see my comments on line 194 above). It is also hard to link to the text, since the numbers reported in the text are actually not present in the figure unless the readers makes some calculations themselves. This should be improved. Also, the use of acronyms (e.g. M1BG, F2TG etc.) contributes to making the text very difficult to read. The acronyms should at least be explained very clearly in the text when they are used.
Authors’ response: Thank you for your suggestions. We have revised the text to clarify the meaning of the acronym (F1TGs, F2TGs, M1TGs, M2TGs, F1BGs, F2BGs, M1BGs and M2BGs) and give the number of genes. We have added two labels, indicating that panels A and B correspond to males and C and D to females in Figure 3.



