Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorMurali PrakriyaNorthwestern University, Chicago, United States of America
- Senior EditorJohn HuguenardStanford University School of Medicine, Stanford, United States of America
Reviewer #1 (Public Review):
Summary:
The authors study age-related changes in the excitability and firing properties of sympathetic neurons, which they ascribe to age-related changes in the expression of KCNQ (Kv7, "M-type") K+ currents in rodent sympathetic neurons, whose regulation by GPCRs has been most thoroughly studied for over 40 years.
Strengths:
The strengths include the rigor of the current-clamp and voltage-clamp experiments and the lovely, crisp presentation of the data, The separation of neurons into tonic, phasic and adapting classes is also interesting, and informative. The ability to successfully isolate and dissociate peripheral ganglia from such older animals is also quite rare and commendable! There is much useful detail here.
Weaknesses:
Whereas the description of the data are very nice and useful, the manuscript does not provide much in the way of mechanistic insights. As such, the effect is more of an epi-phenomenon of unclear insight, and the authors cannot ascribe changes in signaling mechanisms, such as that of M1 mAChRs to the phenomena that is supported by data.
Reviewer #2 (Public Review):
Summary:
This research shows compelling and detailed evidence showing that aging influences intrinsic membrane properties of peripheral sympathetic motor neurons, which become hyperexcitable. The authors found that sympathetic motor neurons from old mice exhibit increased firing rates (spontaneous and evoked), more depolarized membrane resting potential, and increased rheobase. Furthermore, the study investigates cellular mechanisms underlying age-associated hyperexcitability and shows solid evidence supporting that a decreased activity of KCNQ channels during aging is a major contributor to the increased excitability of sympathetic old neurons. All conclusions of this paper are well supported by the data.
Strengths:
Detailed and rigorous analysis of electrical responses of peripheral sympathetic motor neurons using electrophysiology (perforated patch and whole-cell recordings). The study identifies a decrease in KCNQ current as a cellular mechanism behind age-induced hyperexcitability in sympathetic motor neurons.
Weaknesses:
None, the revised version of the manuscript has addressed all my concerns.
Reviewer #3 (Public Review):
This study described changes in membrane excitability and Na+ and K+ current amplitudes of sympathetic motor neurons in culture. The findings indicate that neurons isolated from aged animals show increased membrane excitability manifested as increased firing rates in response to electrical stimulation and changes in related membrane properties including depolarized resting membrane potential, increased rheobase, and spontaneous firing. By contrast, neuron cultures from young mice show little to no spontaneous firing and relatively low firing rates in response to current injection. These changes in excitability correlate with reductions in the magnitude of KCNQ currents in neurons cultured from aged mice compared to neurons from cultured from young mice. The authors conclude that aging promotes hyperexcitability of sympathetic motor neurons through changes in KCNQ channels.
The electrophysiological cataloging of the neuronal properties is well done, and the experiments are performed using perforated patch recordings which preserves the internal constituents of neurons, providing confidence that the effects seen are not due to washout of regulators from the cells. The main weakness is that this study is a descriptive tabulation of changes in the electrophysiology of neurons in culture, and the effects shown are correlative rather than establishing causality. Pharmacological support is provided indicating that blockade or enhancement of KCNQ reverses the changes in excitability, but the specifics of the effects and relevance to intact preparations are unclear. Additional experiments in slice cultures would provide greater significance on the potential relevance of the findings for intact preparations.
Author response:
The following is the authors’ response to the current reviews.
Reviewer #1:
Summary:
The authors study age-related changes in the excitability and firing properties of sympathetic neurons, which they ascribe to age-related changes in the expression of KCNQ (Kv7, "M-type") K+ currents in rodent sympathetic neurons, whose regulation by GPCRs has been most thoroughly studied for over 40 years.
Strengths:
The strengths include the rigor of the current-clamp and voltage-clamp experiments and the lovely, crisp presentation of the data, The separation of neurons into tonic, phasic and adapting classes is also interesting, and informative. The ability to successfully isolate and dissociate peripheral ganglia from such older animals is also quite rare and commendable! There is much useful detail here.
Thank you for recognizing the effort we put on presenting the data and analyzing the neuronal populations. I also believe the ability to isolate neurons from old animals is worth communicating to the scientific community.
Weaknesses:
Where the manuscript becomes less compelling is in the rapamycin section, which does not provide much in the way of mechanistic insights. As such, the effect is more of an epi-phenomenon of unclear insight, and the authors cannot ascribe a signaling mechanism to it that is supported by data. Thus, this latter part rather undermines the overall impact and central advance of the manuscript. The problem is exacerbated by the controversial and anecdotal nature of the entire mTor/aging field, some of whose findings have very unfortunately had to be recently retracted.
I would strongly recommend to the authors that they end the manuscript with their analysis of the role of M current/KCNQ channels in the numerous age-related changes in sympathetic neuron function that they elegantly report, and save the rapamycin, and possible mTor action, for a separate line of inquiry that the authors could develop in a more thorough and scholarly way.
Whereas the description of the data are very nice and useful, the manuscript does not provide much in the way of mechanistic insights. As such, the effect is more of an epi-phenomenon of unclear insight, and the authors cannot ascribe changes in signaling mechanisms, such as that of M1 mAChRs to the phenomena that is supported by data.
I appreciate the new comment. We had agreed that our rapamycin experiments did not allow to ascribe the mechanism to the signaling pathway of mTOR. The new comment mentions M1 mAChRs signaling as another potential signaling mechanism. Our work centered on determining whether aging altered the function of sympathetic motor neurons and defining the mechanism. We presented evidence showing that the mechanism is a reduction of the M-current. We did not attempt to identify the signaling mechanism linking aging to a reduction in M-current. Therefore, we agree with the reviewer that we do not provide further details on the mechanism and that that remains an open question. However, I find it harsh to say that “the effect is more of an epiphenomenon of unclear insight”. How could we possibly test that the effect of aging on the excitability of these neurons only arises as a secondary effect or that is not causal? How could we test for sufficiency and necessity of aging? How could we modify the state of aging to test for causality? We would have to reverse aging and show that the effect on the excitability is gone. And that is exactly what we tried to do with the rapamycin experiment.
Reviewer #1 (Recommendations For The Authors):
(1) The significance values greater than p < 0.05 do not add anything and distract focus from the results that are meaningful. Fig. 5 is a good example. What does p = 0.7 mean? Or p = 0.6? Does this help the reader with useful information?
I thank Reviewer 1 for raising this question. We have attempted different versions of how we report p values, as we want to make sure to address rigor and transparency in reporting data. As corresponding author, I favor reporting p values for all statistical comparisons. To help the reader identifying what we considered statistically significant, we color coded the p values, with red for p-value<0.05 and black for p-value>0.05. As a reader, seeing a p-value=0.7 allows me to know that the authors performed an analysis comparing these conditions and found the mean not to be different. Not presenting the p-value makes me wonder whether the authors even analyzed those groups. In other words, I value more the ability to analyze the data seeing all p-values than not being distracted by not-significant p-values. This is just my preference.
(2) Fig. 1 is not informative and should be removed.
I thank Reviewer 1 for the suggestion. In previous drafts of the manuscript, this figure was included only as a panel. However, we decided it was better to guide the reader into the scope of our work. This is part of our scientific style and, therefore, we prefer to keep the figure.
(3) The emphasis on a particular muscarinic agonist favored by many ion channel physiologists, oxotremorine, is not meaningful (lines 192, 198). The important point is stimulation of muscarinic AChRs, which physiologically are stimulated by acetylcholine. The particular muscarinic agonist used is unimportant. Unless mandated by eLife, "cholinergic type 1 muscarinic receptors" are usually referred to as M1 mAChRs, or even better is "Gq-coupled M1 mAChRs." I don't think that Kruse and Whitten, 2021 were the first to demonstrate the increase in excitability of sympathetic neurons from stimulation of M1 mAChRs. Please try and cite in a more scholarly fashion.
A) I have modified lines 192 and 198 removing mention to oxotremorine.
B) I have modified the nomenclature used to refer to cholinergic type 1 muscarinic receptors.
C) I cited references on the role of M current on sympathetic motor neuron excitability. I also removed the reference (Kruse and Whitten, 2021) referring only on the temporal correlation between the decrease of KCNQ current with excitability.
(4) The authors may want to use the term "M current" (after defining it) as the current produced by KCNQ2&3-containing channels in sympathetic neurons, and reserve "KCNQ" or "Kv7" currents as those made by cloned KCNQ/Kv7 channels in heterologous systems. A reason for this is to exclude currents KCNQ1-containing channels, which most definitely do not contribute to the "KCNQ" current in these cells. I am not mandating this, but rather suggesting it to conform with the literature.
Thank you for the suggestion. I have modified the text to use the term M current. I maintain the use of KCNQ only when referring to KCNQ channel, such as in the section describing the abundance of KCNQ2.
(5) The section in the text on "Aging reduces KCNQ current" is confusing. Can the authors describe their results and their interpretation more directly?
I am not sure to understand the request. I assumed point 5 and 6 are related and decided to answer point 6.
(6) Please explain the meaning of the increase in KCNQ2 abundance with age in Fig. 6G. How is this increase in KCNQ2 expression consistent with an increase in excitability? The explanation of "The decrease in KCNQ current and the increase in the abundance of KCNQ2 protein suggest a potential compensatory mechanism that occurs during aging, which we are actively investigating in an independent study." is rather odd, considering that the entire thesis of this paper is that changes in excitability and firing properties are underlied by changes in KCNQ2/3 channel expression/density. Suddenly, is this not the case?? What about KCNQ3? It would be very enlightening if the authors would just quantify the ratio of KCNQ2:KCNQ3 subunits in M-type channels in young and old mice using simple TEA dose/response curves (see Shapiro et al., JNS, 2000; Selyanko et al., J. Physiol., Hadley et al., Br. J. Pharm., 2001 and a great many more). It is also surprising that the authors did not assess or probe for differences in mAChR-induced suppression of M current between SCG neurons of young and old mice. This would seem to be a fundamental experiment in this line of inquiry.
A. Please explain the meaning of the increase in KCNQ2 abundance with age in Fig. 6G. How is this increase in KCNQ2 expression consistent with an increase in excitability? The explanation of "The decrease in KCNQ current and the increase in the abundance of KCNQ2 protein suggest a potential compensatory mechanism that occurs during aging, which we are actively investigating in an independent study." is rather odd, considering that the entire thesis of this paper is that changes in excitability and firing properties are underlied by changes in KCNQ2/3 channel expression/density. Suddenly, is this not the case?? Our interpretation is that the decrease in M current is not caused by a decrease in the abundance of KCNQ (2) channels. We do not claim that changes in excitability are underlied by a reduction in the expression or density of KCNQ2 channels. On the contrary, our working hypothesis is that the reduction in M current is caused by changes in traffic, degradation, posttranslational modifications, or cofactors for KCNQ2 or KCNQ3 channels. We have modified the description in the results section to clarify this concept.
B. What about KCNQ3? Unfortunately, we did not find an antibody to detect KCNQ3 channels. I have added a sentence to state this.
C. KCNQ2:KCNQ3 subunits in M-type channels in young and old mice using simple TEA dose/response curves. This is a great idea. Thank you for the suggestion. Is this a necessary experiment for the acceptance of this manuscript?
D. It is also surprising that the authors did not assess or probe for differences in mAChR-induced suppression of M current between SCG neurons of young and old mice. This would seem to be a fundamental experiment in this line of inquiry. Reviewer 1 is correct. We did not assess for differences in the suppression of M current by mAChR activation. We do not see the connection of this experiment with the scope of the current investigation.
(7) Why do the authors use linopirdine instead of XE-991? Both are dirty drugs hardly specific to KCNQ channels at 25 uM concentrations, but linopirdine less so. The Methods section lists the source of XE991 used in the study, not linopirdine. Is there an error?
A. Why do the authors use linopirdine instead of XE-991? After validation of KCNQ2/3 inhibition by Linopirdine, we found the effect on membrane potential recordings to be reproducible. Linopirdine has also been reported to be reversible. We wanted to assess reversibility on the excitability of young neurons. We did not find the effect to be reversible. We performed experiments applying XE-991 while recording the membrane potential. XE-991 did not show a clear effect. I was not surprised by this. It is very likely that the pharmacological inhibition of one channel leads to the activation of other channel types. This is highlighted in the work by Kimm, Khaliq, and Bean, 2015. “Further experiments revealed that inhibiting either BK or Kv2 alone leads to recruitment of additional current through the other channel type during the action potential as a consequence of changes in spike shape.” In fact, it was quite remarkable that the aged and young phenotypes were mimicked by targeting KCNQ pharmacologically.
B. Both are dirty drugs hardly specific to KCNQ channels at 25 uM concentrations, but linopirdine less so. I have added a sentence to point out that linopirdine is less potent than XE-991. It reads: “We want to point out that linopirdine is less potent than XE-991 and that it has been reported to activate TRPV1 channels (Neacsu and Babes, 2010). Despite this limitation, the application of linopirdine to young sympathetic motor neurons led to depolarization and firing of action potentials.”
C. The Methods section lists the source of XE991 used in the study, not linopirdine. Is there an error? Thank you for pointing out this. I have added information for both retigabine and linopirdine in the Methods section, both were missing.
(8) Can the authors use a more scientific explanation of RTG action than "activating KCNQ channels?" For instance, RTG induces both a negative-shift in the voltage-dependance of activation and a voltage-independent increase in the open probability, both of which differing in detail between KCNQ2 and KCNQ3 subunits. The authors are free to use these exact words. Thus, the degree of "activation" is very dependent upon voltage at any voltages negative to the saturating voltages for channel activation.
I have modified the text to reflect your suggestion.
(9) Methods: did the authors really use "poly-l-lysine-coated coverslips?" Almost all investigators use poly-D-lysine as a coating for mammalian tissue-culture cells and more substantial coatings such as poly-D-lysine + laminin or rat-tail collagen for peripheral neurons, to allow firm attachment to the coverslip.
That is correct. We used poly-L-lysine-coated coverslips. Sympathetic motor neurons do not adhere to poly-D-Lysine.
(10) As a suggestion, sampling M-type/KCNQ/Kv7 current at 2 kHz is not advised, as this is far faster than the gating kinetics of the channels. Were the signals filtered?
It is correct. Currents were sampled at 2KHz. Data were low-pass filtered at 3 KHz. Our conditions are not far from what is reported by others. Some sample at 10KHz and even 50 KHz. Others do not report the sample frequency.
Reviewer #2:
Weaknesses:
None, the revised version of the manuscript has addressed all my concerns.
I am glad we were able to satisfy previous concerns.
Reviewer #3:
The main weakness is that this study is a descriptive tabulation of changes in the electrophysiology of neurons in culture, and the effects shown are correlative rather than establishing causality.
Allow me to clarify our previous responses and determine how this aligns with your concerns. In the previous revision, Reviewer 3 wrote: “It is difficult to know from the data presented whether the changes in KCNQ channels are in fact directly responsible for the observed changes in membrane excitability.” And suggested to “use of blockers and activators to provide greater relevance.” I assumed these comments were the main concern and that doing such experiments was enough to satisfy the criticism. It is discouraging to see that our experiments did not satisfy the concerns of the reviewer of being correlative.
If Reviewer 3 is referring to stablishing causality between aging and a reduction in M current, I would like to emphasize that such endeavor is complicated as there is not a clear experiment to solve that issue. Our best attempt was to reverse aging with rapamycin, but the recommendation was to remove those experiments.
… but the specifics of the effects and relevance to intact preparations are unclear. Additional experiments in slice cultures would provide greater significance on the potential relevance of the findings for intact preparations.
I apologize for missing this point in the previous revision. The proposed experiments will require an upward microscope coupled to an electrophysiology rig. Unfortunately, I do not have the equipment to do these experiments.
Summary of recommendations from the three reviewers:
Please make corrections as suggested by reviewer 1 to improve the manuscript. Specifically, reviewer 1 suggests making changes to p values in Figure 5,
It is not clear what the suggested changes are. The comment from Reviewer 1 says: The significance values greater than p < 0.05 do not add anything and distract focus from the results that are meaningful. If the suggested change is to remove p values > 0.05, I have explained my rational for keeping those values. If the Journal has a specific format on how to report p-values, I will be happy to make appropriate changes.
and the importance of citing original scholarly works related to effects of increase in excitability of sympathetic neurons by M1 receptors, and the terminology for M currents and KCNQ currents. These changes will improve the manuscript and are strongly recommended.
I cited original papers on that area, and changed the terminology for M current. I kept KCNQ when referring to the channel protein or abundance.
The section dealing with Aging Reduces KCNQ currents seems to contain a lot of extraneous information especially in the last part of the long paragraph and this section should be rewritten for improved clarity… and - the implications or lack thereof - of the correlation of KCNQ with AP firing rates.
A. I removed extraneous information in that section. It now reads: Previous work by our group and others demonstrated that cholinergic stimulation leads to a decrease in M current and increases the excitability of sympathetic motor neurons at young ages \cite{RN67,RN68,RN69,RN71, RN72, RN73, RN74, RN75}. The molecular determinants of the M current are channels formed by KCNQ2 and KCNQ3 in these neurons \cite{RN76, RN77, RN70}. Thus, Figure 6A shows a voltage response (measured in current-clamp mode) and a consecutive M current recording (measured in voltage-clamp mode) in the same neuron upon stimulation of cholinergic type 1 muscarinic receptors. It illustrates the temporal correlation between the decrease of M current with the increase in excitability and firing of APs upon activation with oxotremorine. This strong dependence led us to hypothesize that aging decreases M current, leading to a depolarized RMP and hyperexcitability (Figure 6B). For these experiments, we measured the RMP and evoked activity using perforated patch, followed by the amplitude of M current using a whole-cell voltage clamp in the same cell. We also measured the membrane capacitance as a proxy for cell size. Interestingly, M current density was smaller by 29\% in middle age (7.5 ± 0.7 pA/pF) and by 55\% in old (4.8 ± 0.7 pA/pF) compared to young (10.6 ± 1.5 pA/pF) neurons (Figure 6C-D). The average capacitance was similar in young (30.8 ± 2.2 pF), middle-aged (27.4 ± 1.2 pF), and old (28.8 ± 2.3 pF) neurons (Figure 6E), suggesting that aging is not associated with changes in cell size of sympathetic motor neurons, and supporting the hypothesis that aging alters the levels of M current. Next, we tested the effect on the abundance of the channels mediating M current. Contrary to our expectation, we observed that KCNQ2 protein levels were 1.5 ± 0.1 -fold higher in old compared to young neurons (Figure 6F-G). Unfortunately, we did not find an antibody to detect consistently KCNQ3 channels. We concluded that the decrease in M current is not caused by a decrease in the abundance of KCNQ2 protein.
B. and - the implications or lack thereof - of the correlation of KCNQ with AP firing rates. I am not sure to understand the request on the section of the correlation of KCNQ with AP firing rate. I divided the long paragraph.
The apparent lack of correlation between KCNQ current and KCNQ2 protein needs to be better explained. This is a central part of the study and this result undercuts the premise of the paper.
Indeed, total KCNQ2 protein abundance increases while M current decreases. We do not claim in our work that changes in excitability are caused by a reduction in the expression or density of KCNQ2 channels. On the contrary, our current working hypothesis is that the reduction in M current is caused by changes in traffic, degradation, posttranslational modifications, or cofactors for KCNQ2 or KCNQ3 channels. I have modified the description in the results section and discussion to clarify this concept.
Additionally, the poor specificity of Linordipine for KCNQ should be pointed out in the limitations.
I pointed this limitation. It reads: We want to point out that linopirdine is less potent than XE-991 and that it has been reported to activate TRPV1 channels (Neacsu and Babes, 2010). Despite this limitation, the application of linopirdine to young sympathetic motor neurons led to depolarization and firing of action potentials.
Finally, the editor notes that the author response should not contain ambiguities in what was addressed in the revision. In the original summary of consolidated revisions that were requested, one clearly and separately stated point (point 4) was that experiments in slice cultures should be strongly considered to extend the significance of the work to an intact brain preparation. The author response letter seems to imply that this was done, but this is not the case. The author response seems to have combined this point with another separate point (point 3) about using KCNQ drugs, and imply that all concerns were addressed. Authors should be clear about what revisions were in fact addressed.
As corresponding author, and direct responsible of the document provided for the reply to the reviewers, I apologize for my mistake. After reviewing this comment, I realized I did not respond to the Major points in the section of the Recommendations for the authors from Reviewer 3. I missed that entire section. My previous responses addressed the Public review of reviewer 3. When doing so, I did not separate the sentences, omitting the request on performing the experiment in slices.
The following is the authors’ response to the original reviews.
Reviewer #1
Summary:
The authors study age-related changes in the excitability and firing properties of sympathetic neurons, which they ascribe to age-related changes in the expression of KCNQ (Kv7, "M-type") K+ currents in rodent sympathetic neurons, whose regulation by GPCRs has been most thoroughly studied for over 40 years. The authors suggest the ingestion of rapamycin may partially reverse the age-related decrease in M-channel expression. With the rapamycin part included, it is unclear how this work will impact the field of age-related neuronal dysfunction, as the mechanistic information is not strong.
Strengths:
The strengths include the rigor of the current-clamp and voltage-clamp experiments, the lovely, crisp presentation of the data, and the expert statistics. The separation of neurons into tonic, phasic, and adapting classes is also interesting, and informative. The writing is also elegant, and crisp. The above is especially true of the manuscript up until the part dealing with the effects of rapamycin, which becomes less compelling.
We appreciate the thoughtful comments and constructive feedback to improve the impact of the manuscript.
Weaknesses:
Where the manuscript becomes less compelling is in the rapamycin section, which does not provide much in the way of mechanistic insights. As such, the effect is more of an epi-phenomenon of unclear insight, and the authors cannot ascribe a signaling mechanism to it that is supported by data. Thus, this latter part rather undermines the overall impact and central advance of the manuscript. The problem is exacerbated by the controversial and anecdotal nature of the entire mTor/aging field, some of whose findings have very unfortunately had to be recently retracted.
I would strongly recommend to the authors that they end the manuscript with their analysis of the role of M current/KCNQ channels in the numerous age-related changes in sympathetic neuron function that they elegantly report, and save the rapamycin, and possible mTor action, for a separate line of inquiry that the authors could develop in a more thorough and scholarly way.
We agree with the reviewer in that we cannot ascribe a signaling mechanism to the reversibility observed with rapamycin. Therefore, we are following the recommendation of the reviewer and have removed the rapamycin section.
We want to emphasize that, in the aging field, any advancement in the knowledge of how drugs such as rapamycin reverse age-associated phenotypes is of crucial importance. These drugs, commonly referred to as aging interventions, include rapamycin, calorie restriction, elamipretide, and metformin. We could have used any of these interventions. And yet, the cellular and molecular mechanisms for each one of these anti-aging drugs are unknown.
We want to note that, although the nature of the mTOR field is controversial, the effect of rapamycin in extending lifespan and improving health is not. At least these authors have not been able to find retracted papers on that subject or notices from the NIA alerting on this issue. We kindly request the reviewer to provide the references related to rapamycin that were retracted so we can evaluate how that affects the rigor of the premise for our future work.
As authors, we also find it important to note that we are confident of our observations regarding the effect of rapamycin, and that we are not removing this section because we are retracting our claims. We will use these data to continue our research of the mechanism behind the effect of aging on sympathetic motor neurons.
Reviewer #2:
Summary:
This research shows compelling and detailed evidence showing that aging influences intrinsic membrane properties of peripheral sympathetic motor neurons such that they become more excitable. Furthermore, the authors present convincing evidence that the oral administration of the anti-aging drug Rapamycin partially reversed hyperexcitability in aged neurons. This study also investigates the molecular mechanisms underlying age-associated hyperexcitability in mouse sympathetic motor neurons. In that regard, the authors found an age-associated reduction of an outward current having properties similar to KCNQ2/Q3 potassium current. They suggested a reduction of KCNQ2/Q3 current density in aged neurons as a potential mechanism behind their overactivity.
Strengths:
Detailed and rigorous analysis of electrical responses of peripheral sympathetic motor neurons using electrophysiology (perforated patch and whole-cell recordings). Most of the conclusions of this paper are well supported by the data.
We thank the reviewer for valuing our effort to present a detailed and rigorous analysis.
Weaknesses:
(1) The identity of the age-associated reduced current as KCNQ2/Q3 is not corroborated by pharmacology (blocking the current with the specific blocker XE-991).
We have performed experiments using blockers of KCNQ channels. See responses below.
(2) The manuscript does not include a direct test of the reduction of KCNQ current as the mechanism behind age-induced hyperexcitability.
Thank you for raising this point. We have performed experiments blocking KCNQ channels with Linopiridine in young neurons and found that the pharmacological reduction of KCNQ current was enough to depolarize the cell and, in some cases, elicit the firing of action potentials. We present the results in a new figure. We also added the description in the Results section.
Reviewer #3:
This is a descriptive study of membrane excitability and Na+ and K+ current amplitudes of sympathetic motor neurons in culture. The main findings of the study are that neurons isolated from aged animals show increased membrane excitability manifested as increased firing rates in response to electrical stimulation and changes in related membrane properties including depolarized resting membrane potential, increased rheobase, and spontaneous firing. By contrast, neuron cultures from young mice show little to no spontaneous firing and relatively low firing rates in response to current injection. These changes in excitability correlate with significant reductions in the magnitude of KCNQ currents in aged neurons compared to young neurons. Treating cultures with the immunosuppressive drug, rapamycin, which has known antiaging effects in model animals appears to reverse the firing rates in aged neurons and enhance KCNQ current. The authors conclude that aging promotes hyperexcitability of sympathetic motor neurons.
The electrophysiological cataloging of the neuronal properties is generally well done, and the experiments are performed using perforated patch recordings which preserve the internal constituents of neurons, providing confidence that the effects seen are not due to washout of regulators from the cells.
The main weakness is that this study is a descriptive tabulation of changes in the electrophysiology of neurons in culture, and the effects shown are correlative rather than establishing causality. It is difficult to know from the data presented whether the changes in KCNQ channels are in fact directly responsible for the observed changes in membrane excitability.
We appreciate the constructive criticism. In an attempt to assess whether changes in KCNQ are in fact directly responsible for the changes in membrane excitability, we have performed experiments blocking KCNQ channels with Linopirdine in young neurons and found that the pharmacological reduction of KCNQ current was enough to depolarize the cell and, in some cases, elicit the firing of action potentials. Conversely, we activated KCNQ channels in old neurons with retigabine and found that the pharmacological activation was enough to hyperpolarize the membrane potential and stop the firing of action potentials. This effect was reversible. These two experiments provide solid evidence to our statement that age-associated reduction of KCNQ activity is responsible for the hyperexcited state in sympathetic motor neurons. We present the results in a new figure (Figure 8). We also added the description in the Results section.
Furthermore, a notable omission seems to be the analysis of Ca2+ currents which have been widely linked to alterations in membrane properties in aging.
We thank the reviewer for the comment. We did omit to include data on our studies of calcium currents. We agree that the study of the effect of calcium currents is relevant as it can influence the afterhyperpolarization. Furthermore, we believe that potential effects on calcium currents need to be studied in relation to other physiological processes that depend on calcium, including excitation-transcription coupling, calcium handling, and neurotransmitter release. Adding this information to this manuscript would only contribute to the tabulation of effects that we observe in sympathetic motor neurons with aging. As our main goal was to determine the ion channels responsible for the hyperexcited state, voltage-gated calcium channels or other calcium sources could have reflected a more indirect mechanism as compared to changes in sodium or potassium currents. We will continue our investigation on calcium currents and report our observations in the future, but for now, we have decided to leave it out of this work.
As well, additional experiments in slice cultures would provide greater significance on the potential relevance of the findings for intact preparations. Finally, experiments using KCNQ blockers and activators could provide greater relevance that the observed changes in KCNQ are indeed connected to changes in membrane excitability.
We are happy to report that we have performed these experiments and that the results strengthen the conclusion that changes in KCNQ are connected to changes in membrane excitability.
Recommendations for the authors:
We recommend the following essential revisions summarized from the reviews:
(1) Is the change in KCNQ current responsible for the altered membrane excitability? What happens to membrane excitability when KCNQ is partially blocked (see reviewer 2 comment below)? Conversely, what happens to the excitability of aged neurons if KCNQ is activated (e.g., with retigabine)? (see reviewer 3 comment below). Results of these important experiments are needed to support the argument that KCNQ underlies the alterations in firing and membrane excitability.
We have responded to this point. Thank you for the suggested experiments. In summary, the new experiments show that blocking KCNQ channels in young neurons lead to depolarization, and in some cases, the firing of action potentials. Conversely, the activation of KCNQ channels in aged neurons leads to hyperpolarization and a cease of firing. We have added a new figure and reported the results in the Results section.
(2) Rapamycin experiments are underdeveloped and weak. These should be further developed by examining the effects of KCNQ blockers to see if their effects on membrane excitability are reversed. Also, see comment 2 from reviewer 1.
We have followed the recommendation by reviewer 1 and removed the section on rapamycin.
(3) The study should examine voltage-gated calcium currents to determine potential changes in these currents with aging. See reviewer 3 comments.
We thank the reviewer for the comment. We performed preliminary experiments and found that aging impacts calcium currents. However, we omitted to include the data. In our opinion, the changes in calcium currents are outside the scope of this work, as the changes could be related to physiological processes that go beyond the control of firing. Effects on calcium currents need to be studied in relation to other physiological processes that depend on calcium, including excitation-transcription coupling, calcium handling, and neurotransmitter release. The study of the relationship between changes in calcium currents and those physiological processes would require multiple experiments and detailed analysis. We will continue our investigation on calcium currents and report our observations in the future, but for now, we have decided to leave it out of this work.
We have also edited suggestions in the Figures and Legends.
(2) In Fig.4 panel H, Y-axis must be # AP at 100 pA.
We corrected the axis in Figure 4H.
(3) In Legend Fig. 5, the number of cells for each subpopulation (n) needs to be corrected. In plots F-I, n= 9, 7, and 3 seem to be the number of adapting cells for 12-, 64- and 115w-old, respectively, instead of the number of single, phasic, and old cells for 12-week-old mice. A similar correction seems to be needed for 64-week-old and 115-week-old.
We corrected the n number in Figure 5.
(4) In Figure 6 panel C, it would be helpful for a reader to align the voltage protocol depicted with the current shown.
We have aligned the voltage protocol to the current traces.
(5) In the legend of Figure 7, the description of panel A ends with "Magnitude of voltage step to elicit each trace is shown in black", however in panel A there is no voltage depiction. In the description of panel D, "N = X animals, n=x cells" must be corrected.
We have modified the legend to clarify. It now reads: “Text at the right of each current trace corresponds to the voltage used to elicit that current.”
New Figure 8
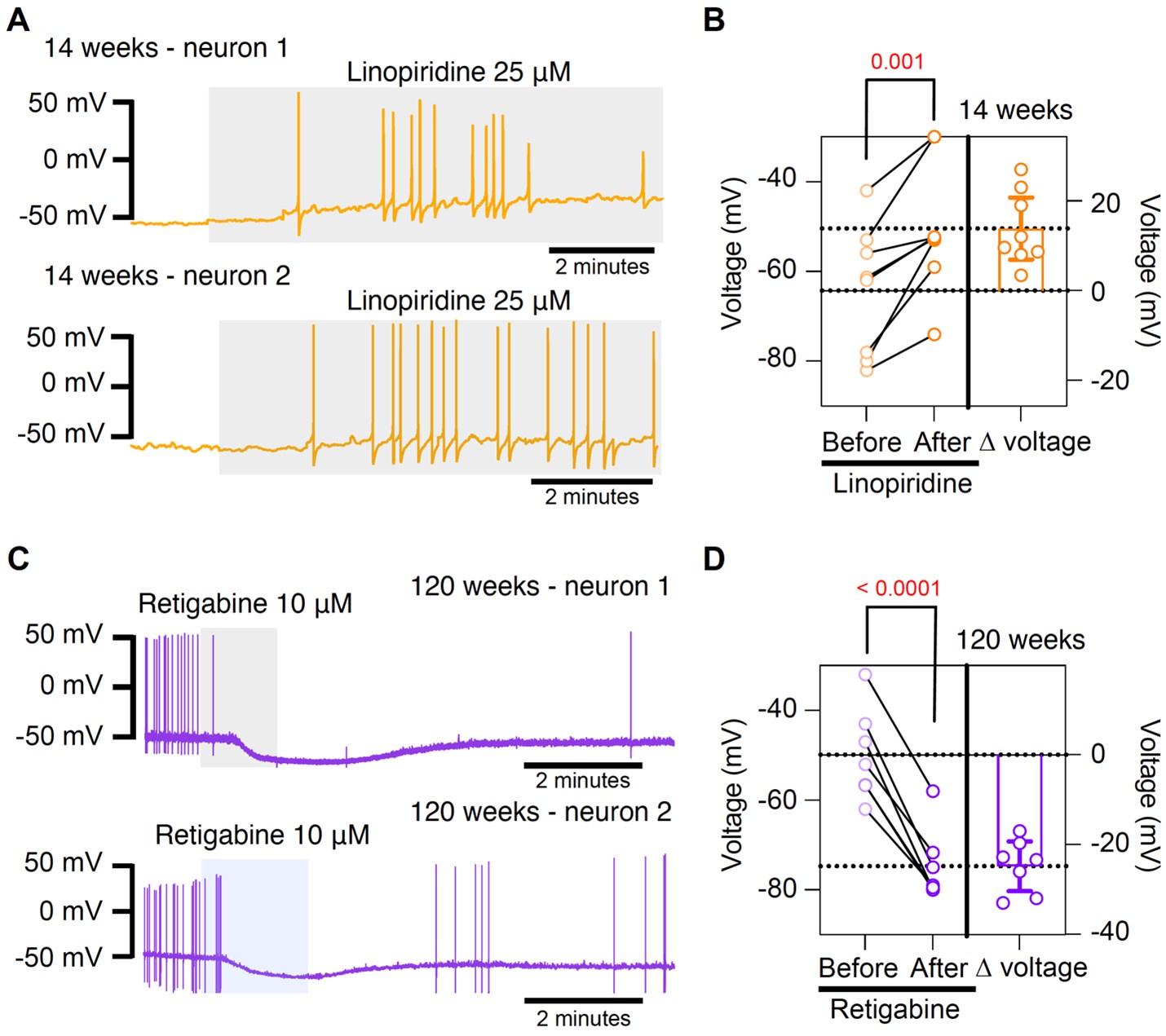
Author response image 1.
Pharmacological inhibition and activation of KCNQ channels mimic the age-dependent phenotype. A. Membrane potential recordings from two young neurons treated with 25 μM linopirdine during the time illustrated by the light gray box. No holding current was applied. B. Left: Summary of the resting membrane potential measured before (light orange) and after (dark orange) the application of linopirdine. Right: Summary of the depolarization produced by linopirdine calculated by subtracting the post-drug voltage from the pre-drug voltage (V). Data points are from N = 2 animals, n = 8 cells, 14-week-old mice. C. Membrane potential recordings from two aged neurons treated with 10 μM retigabine during the time illustrated by the light gray box. No holding current was applied. D. Left: Summary of the resting membrane potential measured before (light purple) and after (dark purple) the application of retigabine. Right: Summary of the hyperpolarization produced by retigabine calculated by subtracting the post-drug voltage from the pre-drug voltage (V). Data points are from N = 2 animals, n = 7 cells, 120-week-old mice. P-values are shown at the top of the graphs.