Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorVolker DötschGoethe University Frankfurt, Frankfurt am Main, Germany
- Senior EditorVolker DötschGoethe University Frankfurt, Frankfurt am Main, Germany
Reviewer #1 (Public Review):
Summary
The authors use an elegant but somewhat artificial heterodimerisation approach to activate the isolated cytoplasmic domains of different receptor kinases (RKs) including the receptor kinase BRI1 and EFR. The developmental RK BRI1 is known to be activated by the co-receptor BAK1. Active BRI1 is then able to phosphorylate downstream substrates. The immune receptor EFR is also an active protein kinase also activated by the co-receptor BAK1. EFR however appears to have little or no kinase activity but seems to use an allosteric mechanism to in turn enable BAK1 to phosphorylate the substrate kinase BIK1. EFR tyrosine phosphorylation by BAK1 appears to trigger a conformational change in EFR, activating the receptor. Likewise, kinase activating mutations can cause similar conformational transitions in EFR and also in BAK1 in vitro and in planta.
Strengths:
I particularly liked The HDX experiments coupled with mutational analysis (Fig. 2) and the design and testing of the kinase activating mutations (Fig. 3), as they provide novel mechanistic insights into the activation mechanisms of EFR and of BAK1. These findings are nicely extended by the large-scale identification of EFR-related RKs from different species with potentially similar activation mechanisms (Fig. 5).
Weaknesses:
In my opinion, there are currently two major issues with the present manuscript. (1) The authors have previously reported that the EFR kinase activity is dispensible for immune signaling (https://pubmed.ncbi.nlm.nih.gov/34531323/) but the wild-type EFR receptor still leads to a much better phosphorylation of the BIK1 substrate when compared to the kinase inactive D849N mutant protein (Fig. 1). (2) How the active-like conformation of EFR is in turn activating BAK1 is poorly characterized, but appears to be the main step in the activation of the receptor complex. Extending the HDX analyses to resting and Rap-activated receptor complexes could be a first step to address this question, but these HDX studies were not carried out due to technical limitations.
Overall this is an interesting study that aims to advance our understanding of the activation mechanisms of different plant receptor kinases with important functions in plant immunity.
Reviewer #2 (Public Review):
Summary:
Transmembrane signaling in plants is crucial for homeostasis. In this study, the authors set out to understand to what extent catalytic activity in the EFR tyrosine kinase is required in order to transmit a signal. This work was driven by mounting data that suggest many eukaryotic kinases do not rely on catalysis for signal transduction, relying instead on conformational switching to relay information. The crucial findings reported here involve the realisation that a kinase-inactive EFR can still activate (ie lead to downstream phosphorylation) of its partner protein BAK1. Using a convincing set of biochemical, mass spectrometric (HD-exchange) and in vivo assays, the team suggest a model in which EFR is likely phosphorylated in the canonical activation segment (where two Ser residues are present), which is sufficient to generate a conformation that can activate BAK1 through dimersation. A model is put forward involving C-helix positioning in BAK1, and the model extended to other 'non-RD' kinases in Arabidopsis kinases that likely do not require kinase activity for signaling.
Strengths:
The work uses logical and well-controlled approaches throughout, and is clear and convincing in most areas, linking data from IPs, kinase assays (including clear 32P-based biochemistry), HD-MX data (from non-phosphorylated EFR) structural biology, oxidative burst data and infectivity assays. Repetitions and statistical analysis all appear appropriate.
Overall, the work builds a convincing story and the discussion does a clear job of explaining the potential impact of these findings (and perhaps an explanation of why so many Arabidopsis kinases are 'pseudokinases', including XPS1 and XIIa6, where this is shown explicitly).
Weaknesses:
No major weaknesses are noted from reviewing the data and the paper follows a logical course built on solid foundations; the use of Tables to explain various experimental data pertinent to the reported studies is appreciated.
(1) The use of a, b,c, d in Figures 2C and 3C etc is confusing to this referee, and is now addressed in the latest version
(2) The debate about kinase v pseudokinases is well over a decade old. For non-experts, the kinase alignments/issues raised are in PMID: 23863165 and might prove useful if cited.
(3) Early on in the paper, the concept of kinases and pseudokinases related to R-spine (and extended R-spine) stability and regulation really needs to be more adequately introduced to explain what comes next; e.g. some of the key work in this area for RAF and Tyr kinases where mutual F-helix Phe amino acid changes are evaluated (conceptually similar to this study of the E-helix Tyr to Phe changes in EFR) should be cited (PMID: 17095602, 24567368 and 26925779).
(4) In my version, some of the experimental text is also currently in the wrong order (and no page numbers, so hard for me to state exactly where in the manuscript); However, I am certain that Figure 2C is mentioned in the text when the data are actually shown in Figure 3C for the EFR-SSAA protein.
(5) Tyr 156 in PKA is not shown in Supplement 1, 2A as suggested in the text; for readers, it will be important to show the alignment of the Tyr residue in other kinases; this has been updated in the second version. Although it is clearly challenging to generate phosphorylated EFR (seemingly through Codon-expansion here?), it appears unlikely that a phosphorylated EFR protein, even semi-pure, couldn't have been assayed to test the idea that the phosphorylation drives/supports downstream signaling. What about a DD or EE mutation, as commonly used (perhaps over-used) in MEK-type studies?
Impact:
The work is an important new step in the huge amount of follow-up work needed to examine how kinases and pseudokinases 'talk' to each other in (especially) the plant kingdom, where significant genetic expansions have occurred. The broader impact is that we might understand better how to manipulate signaling for the benefit of plants and mankind; as the authors suggest, their study is a natural progression both of their own work, and the kingdom-wide study of the Kannan group.
Reviewer #3 (Public Review):
The study presents strong evidence for allosteric activation of plant receptor kinases, which enhances our understanding of the non-catalytic mechanisms employed by this large family of receptors.
Plant receptor kinases (RKs) play a critical role in transducing extracellular signals. The activation of RKs involves homo- or heterodimerization of the RKs, and it is believed that mutual phosphorylation of their intracellular kinase domains initiates downstream signaling. However, this model faces a challenge in cases where the kinase domain exhibits pseudokinase characteristics. In their recent study, Mühlenbeck et al. reveal the non-catalytic activation mechanisms of the EFR-BAK1 complex in plant receptor kinase signaling. Specifically, they aimed to determine that the EFR kinase domain activates BAK1 not through its kinase activity, but rather by utilizing a "conformational toggle" mechanism to enter an active-like state, enabling allosteric trans-activation of BAK1. The study sought to elucidate the structural elements and mutations of EFR that affect this conformational switch, as well as explore the implications for immune signaling in plants. To investigate the activation mechanisms of the EFR-BAK1 complex, the research team employed a combination of mutational analysis, structural studies, and hydrogen-deuterium exchange mass spectrometry (HDX-MS) analysis. For instance, through HDX-MS analysis, Mühlenbeck et al. discovered that the EFR (Y836F) mutation impairs the accessibility of the active-like conformation. On the other hand, they identified the EFR (F761H) mutation as a potent intragenic suppressor capable of stabilizing the active-like conformation, highlighting the pivotal role of allosteric regulation in BAK1 kinase activation. The data obtained from this methodology strengthens their major conclusion. Moreover, the researchers propose that the allosteric activation mechanism may extend beyond the EFR-BAK1 complex, as it may also be partially conserved in the Arabidopsis LRR-RK XIIa kinases. This suggests a broader role for non-catalytic mechanisms in plant RK signaling.
The allosteric activation mechanism was demonstrated for receptor tyrosine kinases (RTKs) many years ago. A similar mechanism has been suggested for the activation of plant RKs, but experimental evidence for this conclusion is lacking. Data in this study represent a significant advancement in our understanding of non-catalytic mechanisms in plant RK signaling. By shedding light on the allosteric regulation of BAK1, the study provides a new paradigm for future research in this area.
Author response:
The following is the authors’ response to the original reviews.
Reviewer #1:
I will summarize my comments and suggestions below.
(1) Abstract:
"Non-catalytic (pseudo)kinase signaling mechanisms have been described in metazoans, but information is scarce for plants." To the best of my understanding EFR is an active protein kinase in vitro and in vivo and cannot be considered a pseudokinase. Consider rephrasing.
We rephrased to: “Non-catalytic signaling mechanisms of protein kinase domains have been described in metazoans, but information is scarce for plants.”
(2) Page 4: It should be noted, that while membrane associated Rap-RiD systems have been used in planta to activate receptor kinase intracellular domains by promoting interaction with a co-receptor kinase domain, this system does not resemble the actual activation mechanism in the plasma membrane. This would be worth discussing when introducing the system. For example, the first substrates of the RK signaling complex may also be membrane associated and not freely diffuse in solution, which may be important for enzyme-substrate interaction.
We inserted on page 4: “The RiD system was previously applied in planta, maintaining membrane-association by N-terminal myristoylation (Kim et al., 2021). For the in vitro experiments, the myristoylation sites were excluded to facilitate the production of recombinant protein.”
(3) Page 4 and Fig 1: The catalytic Asp in BRI1 is D1027 and not D1009 (https://pubmed.ncbi.nlm.nih.gov/21289069/). Please check and prepare the correct mutant protein if needed.
We clarified this in the text by stating that we mutated the HRD-aspartate to asparagine in all our catalytic-dead mutants: “Kinase-dead variants with the catalytic residue (HRD-aspartate) replaced by asparagine (EFRD849N and BRI1D1009N), had distinct effects […]”. D1027 in BRI1 is the DFG-Asp, which was not mutated in our study.
(4) Page 4 and Fig 1: Is BIK1 a known component of the BR signaling pathway and a direct BRI1 substrate? Or in other words how specific is the trans-phosphorylation assay? In my opinion, a more suitable substrate for BRI1/BAK1 would be BSK1 or BSK3 (for example https://pubmed.ncbi.nlm.nih.gov/30615605/).
Kinase-dead BIK1 is a reported substrate of BRI1. We clarified this in the results section by inserting: “BIK1 was chosen as it is reported substrate of both, EFR/BAK1 and BRI1/BAK1 complexes (Lin et al., 2013).”
(5) Fig. 1B Why is BIK1 D202N partially phosphorylated in the absence of Rap? I would suggest to add control lanes showing BRI1, EFR, FLS2, BAK1 and BIK1 in isolation. Given that a nice in vitro activation system with purified components is available, why not compare the different enzyme kinetics rather than band intensities at only 1 enzyme : substrate ratio?
BIK1 D202N is partially phosphorylated due to the presence of active BAK1 that is capable of transphosphorylating BIK1 D202N as it has been reported in a previous study: (DOI: 10.1038/s41586-018-0471-x).
(6) Page 4 and Fig 1: Is the kinase dead variant of EFR indeed kinase dead? I could still see a decent autorad signal for this mutant when expressed in E. coli (Fig 1 A in Bender et al., 2021; https://pubmed.ncbi.nlm.nih.gov/34531323/)? If this mutant is not completely inactive, could this change the interpretation of the experiments performed with the mutant protein in vitro and in planta in the current manuscript? In my opinion, it could be possible that a partially active EFR mutant can be further activated by BAK1, and in turn can phosphorylate BIK1 D202N. The differences in autorad signal for BRI1D1009?N and EFRD849N is very small, and the entire mechanism hinges on this difference.
We would like to emphasize that the mechanism hinges on the difference between non-dimerized and dimerized kinase domains in the in vitro kinase assay. BRI1 D1009N fails to enhance BIK1 D202N trans-phosphorylation compared to the non-dimerized sample, while EFR D849N is still capable of enhancing BIK1 transphosphorylation upon dimerization as indicated by quantification of autorads (Figure 1B/C). We have also addressed this point in a section on the limitations of our study.
(7) Fig 1B. "Our findings therefore support the hypothesis that EFR increases BIK1 phosphorylation by allosterically activating the BAK1 kinase domain." To the best of my understanding presence of wild-type EFR in the EFR-BAK1 signaling complex leads to much better phosphorylation of BIK1D202N when compared to the EFRD849N mutant. How does that support the allosteric mechanism? By assuming that the D849N mutant is in an inactive conformation and fully catalytically inactive (see above)? Again, I think the data could also be interpreted in such a way that the small difference in autorad signal for BIK1 between BRI1 inactive (but see above) and ERF inactive are due to EFR not being completely kinase dead (see above), rather than EFR being an allosteric regulator. To clarify this point I would suggest to a) perform quantitative auto- and trans-(generic substrate) phosphorylation assays with wt and D849N EFR to derive enzyme kinetic parameters, to (2) include the EFRD849 mutant in the HDX analysis and (3) to generate transgenic lines for EFRD489N/F761H/Y836F // EFRD489N/F761H/SSAA and compare them to the existing lines in Fig. 3.
Mutations of proteins, especially those that require conformational plasticity for their function can have pleiotropic effects as the mutation may affect the conformational plasticity and consequently catalytic and non-catalytic functions that depend on the conformational plasticity. In such cases, it is difficult to fully untangle catalytic and non-catalytic functions. Coming back to EFR D849N, the D849N mutation may also impact the non-catalytic function by altering the conformational plasticity, explaining the difference observed in EFR vs EFR D849N. As you rightly suggested, HDX would be a way to address this but would still not clarify whether catalytic activity contributes to activation. We instead attempted to produce analog sensitive EFR variants for in vivo characterization of EFR-targeted catalytic inhibition. Unfortunately, we failed in producing an analog-sensitive variant for which we could show ATP-analog binding. To address your concern, we inserted a section on limitations of the study.
(8) Fig. 2B,C, supplement 3 C,D. Has it been assessed if the different EFR versions were expressed to similar protein levels and still localized to the PM?
Localization of the mutant receptors has not been explicitly evaluated by confocal microscopy. However, the selected mutation EFRF761H is shown to accumulate in stable Arabidopsis lines (Figure 3 – Supplement 1C) and BAK1 could be coIPed by all EFR variants upon elf18-treatment (Figure 3 B), indicating plasma membrane localization.
(9) How the active-like conformation of EFR is in turn activating BAK1 is poorly characterized, but appears to be the main step in the activation of the receptor complex. Extending the HDX analyses to resting and Rap-activated receptor complexes could be a first step to address this question. I tried to come up with an experimental plan to test if indeed the kinase activity of BAK1 and not of EFR is essential for signal propagation, but this is a complex issue. You would need to be able to mimic an activated form of EFR (which you can), to make sure its inactive (possibly, see above) and likewise to engineer a catalytically inactive form of BAK1 in an active-like state (difficult). As such a decisive experiment is difficult to implement, I would suggest to discuss different possible interpretations of the existing data and alternative scenarios in the discussion section of the manuscript.
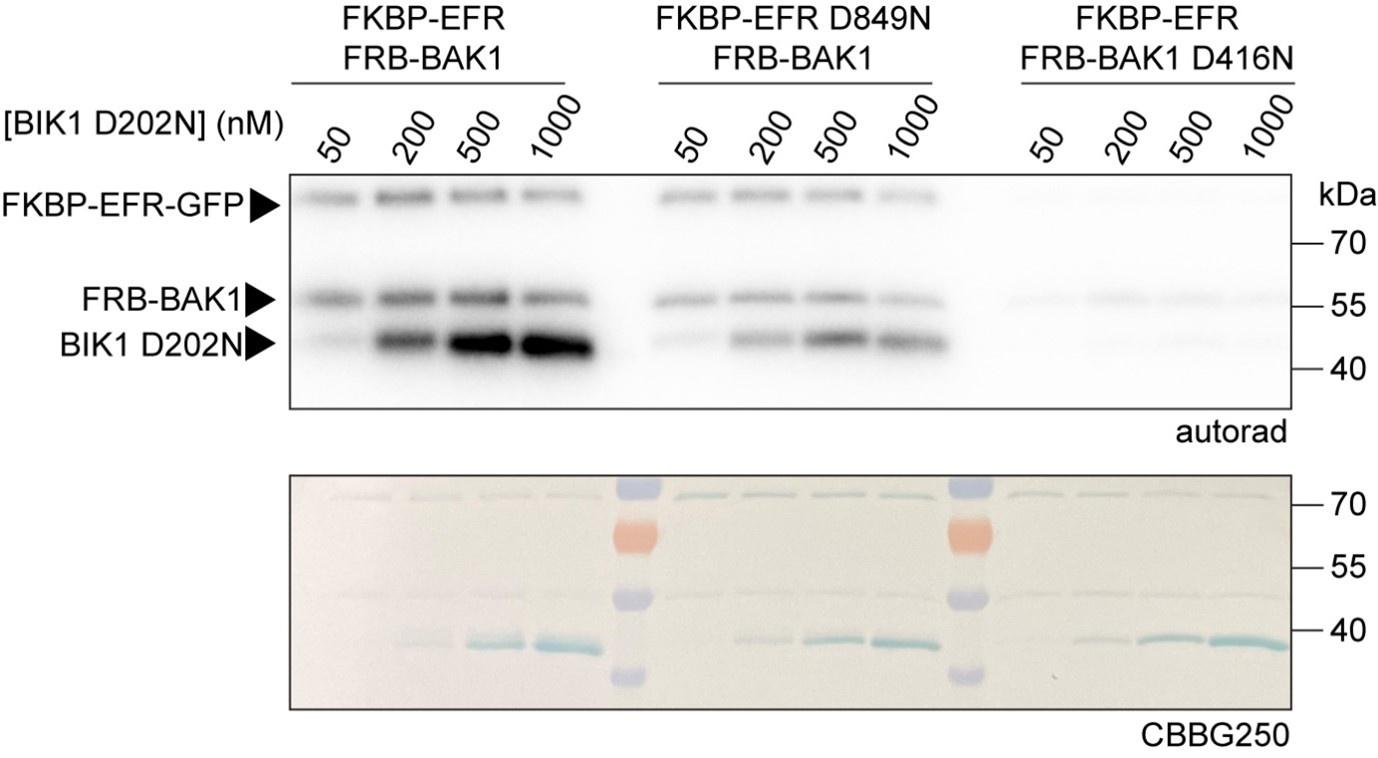
We addressed your concern whether BAK1 kinase activity is essential for signaling propagation by pairing EFRF761H and BAK1D416N (Figure 4 Supplement 2 C) which fails to induce signaling. In this case, EFRF761H is in its activated conformation but cannot activate downstream signaling. We also attempted to address your concern by an in vitro kinase assay by pairing EFR and BAK1D416N and using a range of concentrations of the substrate BIK1D202N. We observed that catalytic activity of BAK1 but not EFR was essential for BIK1 phosphorylation. However, this experiment does not address whether activated EFR can efficiently propagate signaling in the absence of BAK1 catalytic activity. In the limitations of the study section, we now discuss the catalytic importance of EFR for signaling activation.
Author response image 1.
BIK1 trans-phosphorylation depends on BAK1 catalytic activity. Increasing concentrations of BIK1 D202N were used as substrate for Rap-induced dimers of EFR-BAK1, EFR D849N-BAK1, and EFR-BAK1 D416N respectively. BIK1 trans-phosphorylation depended on the catalytic activity of BAK1. Proteins were purified from E. coli λPP cells. Three experiments yielded similar results of which a representative is shown here.
Reviewer #2:
All of my suggestions are minor.
Figure 1B, I think it would be more useful to readers to explain the amino acid in the D-N change, rather than just call it D-to-N? Also, please label the bands on the stained gel; the shift on FKBP-BRI1 and FKBP-EFR are noticeable on the Coomassie stain.
We implemented your suggestions.
Figure 1-Supplement 1. There is still a signal in pS612 BAK1 (it states 'also failed to induce BAK1 S612 phosphorylation' in the text, which is not quite correct). Also, could mention the gel shift seen in BAK1, which appears absent in Y836F.
We corrected the text which now states: “To test whether the requirement for Y836 phosphorylation is similar, we immunoprecipitated EFR-GFP and EFRY836F-GFP from mock- or elf18-treated seedlings and probed co-immunoprecipitated BAK1 for S612 phosphorylation. EFRY836F also obstructed the induction of BAK1 S612 phosphorylation (Figure 1 – Supplement 1), indicating that EFRY836F and EFRSSAA impair receptor complex activation.” The gel shift of BAK1 you pointed out was not observed in replications and thus we prefer not to comment on it.
Figure 2 and 3 are full of a, b, c,d's, which I don't understand. Sorry
We used uppercase letters to indicate subpanels and lowercase letters to indicate the results of the statistical testing. In the figure caption, we have clarified that the lowercase letters refer to statistical comparisons.
Figure 2 A. If each point on the x-axis is one amino acid, I think it would again be useful to name the amino acids that the gold or purple or blue colored lines extend through.
Each point stands for a peptide which are sorted by position of their starting amino acid from N-terminus to C-terminus. We now added plots of HDX for individual peptides that correspond to the highlighted region in subpanel A.
Figure Supplement 1 is very small for what it is trying to show, even on the printed page. If this residue were to be phosphorylated, what would happen to the H-bond?
We suppose that VIa-Tyr phosphorylation would break the H-bond and causes displacement of the aC-b4 loop. Recent studies, published after our submission, highlight the importance of this loop for substrate coordination and ATP binding. Thus, phosphorylation of VIa-Tyr and displacing this loop may render the kinase rather unproductive. We have expanded the discussion to include this point.
Figure 2B: Tyr 836 is not present in any of the alignments in Figure 2A. This should be rectified, because the text talks about the similarity to Tyr 156 in PKA.
We have adjusted the alignments such that they now contain the VIa-Tyr residues of EFR and PKA.
Figure 4D. Is there any particular reason that these Blots are so hard to compare or FKBP and BAK1?
We assume it is referred to Figure 4 – Supplement 2 D. FKBP-EFR and FRB-BAK1 both are approximately the size of RubisCo, the most abundant protein in plant protein samples and which overlay the FKBP- and FRB-tagged kinase. Thus, it is difficult to detect these proteins.
Reviewer #3:
(1) The paper reporting the allosteric activation mechanism of EGFR should be cited.
Will be included.
(2)The authors showed that "Rap addition increased BIK1 D202N phosphorylation when the BRI1 or EFR kinase domains were dimerized with BAK1, but no such effect was observed with FLS2". Please explain why FLS2 failed to enhance BIK1 transphosphorylation by Rap treatment?
Even though BIK1 is a reported downstream signaling component of FLS2/BAK1, it might be not the most relevant downstream signaling component and rather related RLCKs, like PBL1, might be better substrates for dimerized FLS2/BAK1. We haven’t tested this, however. Alternatively, the purified FLS2 kinase domain might be labile and quickly unfolds even though it was kept on ice until the start of the assay, or the N-terminal FKBP-tag may disrupt function. As the reason for our observation is not clear, we have removed FLS2 in vitro dimerization experiments from the manuscript.
(3) Based solely on the data presented in Figure 1, it can be concluded that EFR's kinase activity is not required to facilitate BIK1 transphosphorylation. Therefore, the title of Figure 1, "EFR Allosterically Activates BAK1," may be inappropriate.
We have changed the figure title to: “EFR facilitates BIK1 trans-phosphorylation by BAK1 non-catalytically.”
(4) In Figure 1- Supplement 1, I could not find any bands in anti-GFP and anti-BAK1 pS612 of input. Please redo it.
Indeed, we could not detect protein in the input samples of this experiment. BAK1 S612 phosphorylation is an activation mark and not necessarily expected to be abundant enough for detection in input samples. EFR-GFP, however, is usually detected in input samples and is reported in Macho et al. 2014 from which manuscript these lines come. Why EFR-GFP is not detected in this set of experiments is unclear but, in our opinion, does not detract from the conclusions drawn since similar amounts of EFR-GFP are pulled-down across all samples.
(5) For Figure 2A, please mark the structure represented by each color directly in the figure.
We have made the suggested change.
(6) Please modify "EFRF761/Y836F and EFRF761H/SSAA restore BIK1 trans-phosphorylation" to "EFRF761H/Y836F and EFRF761H/SSAA restore BIK1 trans-phosphorylation".
Thank you for spotting this. We changed it.
(7) The HDX-MS analysis demonstrated that the EFR (Y836F) mutation inhibits the formation of the active-like conformation. Conversely, the EFR (F761H) mutation serves as a potent intragenic suppressor, significantly stabilizing the active-like conformation. Confirming through HDX-MS conformational testing that the EFR (Y836F F761H) double mutation does not hinder the formation of the active-like EFR kinase conformation would greatly strengthen the conclusions of the article.
Response: We agree that this is beneficial, and we attempted to do it but failed to produce enough protein for HDX-MS analysis. We stated this now in an extra section of the paper (“Limitations of the study”).



