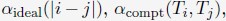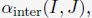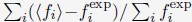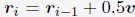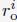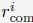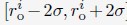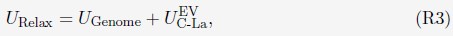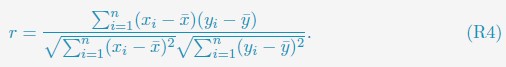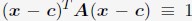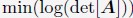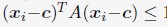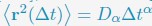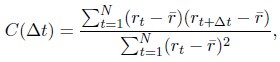Abstract
The intricate structural organization of the human nucleus is fundamental to cellular function and gene regulation. Recent advancements in experimental techniques, including high-throughput sequencing and microscopy, have provided valuable insights into nuclear organization. Computational modeling has played significant roles in interpreting experimental observations by reconstructing high-resolution structural ensembles and uncovering organization principles. However, the absence of standardized modeling tools poses challenges for furthering nuclear investigations. We present OpenNucleome—an open-source software designed for conducting GPU-accelerated molecular dynamics simulations of the human nucleus. OpenNucleome offers particle-based representations of chromosomes at a resolution of 100 KB, encompassing nuclear lamina, nucleoli, and speckles. This software furnishes highly accurate structural models of nuclear architecture, affording the means for dynamic simulations of condensate formation, fusion, and exploration of non-equilibrium effects. We applied OpenNucleome to uncover the mechanisms driving the emergence of “fixed points” within the nucleus—signifying genomic loci robustly anchored in proximity to specific nuclear bodies for functional purposes. This anchoring remains resilient even amidst significant fluctuations in chromosome radial positions and nuclear shapes within individual cells. Our findings lend support to a nuclear zoning model that elucidates genome functionality. We anticipate OpenNucleome to serve as a valuable tool for nuclear investigations, streamlining mechanistic explorations and enhancing the interpretation of experimental observations.
Introduction
The highly complex structural organization of the human nucleus plays a crucial role in the functioning and regulation of our cells.1–10 The complexity arises from the diverse range of nuclear landmarks, such as nucleoli,11 nuclear speckles,8,12 and the nuclear lamina,13 each serving distinct functions. These landmarks provide specialized environments for various nuclear processes, allowing for efficient coordination and regulation of gene expression. Moreover, the spatial arrangement of chromosomes within the nucleus, intertwined with the nuclear landmarks, is critical for proper gene regulation and communication between different genome regions. Disruptions or abnormalities in the nuclear organization can have profound consequences on cellular function and can contribute to the development of diseases, including cancer and genetic disorders.14,15
Recent advancements in experimental techniques have significantly enhanced our understanding of nuclear organization.3,16–20 The advent of high-throughput sequencing-based methods, such as genome-wide chromosome-conformation capture (Hi-C), has unveiled crucial structural elements of the genome,21,22 including chromatin loops,23 topologically associating domains,24,25 and compartments.22 Additionally, sequencing-based techniques such as DamID,26 Chip-Seq,27 and TSA-Seq28 have revealed valuable information regarding interactions between chromosomes and nuclear landmarks. However, it is worth noting that these sequencing methods often offer averaged contacts, which can mask the heterogeneity present across populations, although single-cell techniques are also emerging.29–31 Moreover, translating contact data into spatial positions can be challenging, adding complexity to interpreting experimental findings.
To complement these sequencing approaches, microscopic imaging techniques directly probe the spatial positions within individual nuclei.3,13,32–34 Recent advancements in DNA FISH (Fluorescence In Situ Hybridization) have enabled high-throughput imaging of thousands of loci simultaneously.35,36 These imaging studies have not only confirmed the structural features observed through sequencing techniques but have also provided valuable insights into the heterogeneity present at the single-cell level.
The abundance of available experimental data in the field of nuclear organization provides a fertile ground for structural modeling.37–63 To make sense of this wealth of information, various computational approaches have been introduced, with polymer simulation approaches being extensively utilized. These simulation techniques aid in reconstructing structural ensembles that closely replicate experimental data, offering valuable insights into the mechanisms underlying chromosome folding. In recent studies, these approaches have also been employed to investigate the interplay between the genome and the nuclear lamina, 64–67 as well as nucleoli,68 shedding light on their dynamic relationships.
Despite the progress made in computational modeling, the absence of well-documented software with easy-to-follow tutorials pose a challenge. Many research groups develop their own independent software, which complicates cross-validation and hinders the establishment of best practices for genome modeling. 40,69,70 Moreover, comprehensive models of the entire nucleus, especially at high resolution, remain scarce. Addressing these limitations and fostering collaboration in the scientific community can be achieved through the development of open-source tools. By promoting transparency and accessibility, such tools have the potential to greatly facilitate nuclear modeling and contribute to a more unified and collaborative research environment.
We present OpenNucleome, an open-source software designed for conducting molecular dynamics (MD) simulations of the human nucleus. This software streamlines the process of setting up whole nucleus simulations through just a few lines of Python scripting. Open-Nucleome can unveil intricate, high-resolution structural and dynamic chromosome arrangements at a 100 KB resolution. It empowers researchers to track the kinetics of condensate formation and fusion while also exploring the influence of chemical modifications on condensate stability. Furthermore, it facilitates the examination of nuclear envelope deformation’s impact on genome organization. The software’s modular architecture enhances its adaptability and extensibility. Leveraging the power of OpenMM,71 a GPU-accelerated MD engine, OpenNucleome ensures efficient simulations.
Our work demonstrates the fidelity of the simulated nuclear organizations by faithfully reproducing Hi-C, Lamin B DamID, TSA-Seq, and DNA-MERFISH data. The dynamic insights extracted from this model are pivotal in advancing our understanding of nuclear organization mechanisms. Our findings reveal that inherent heterogeneity in chromosome contacts naturally emerges within single cells. Interestingly, robust contacts between chromosomes and nuclear bodies can also be established due to a coupled self-assembly mechanism. Notably, the resilience of contacts involving nuclear bodies supports a nuclear zoning model for genome function. In the realm of nuclear investigations, we anticipate OpenNucleome to serve as an invaluable tool, seamlessly complementing experimental techniques.
Results
Non-equilibrium nucleus model at 100 KB resolution
We present an open-source implementation of a computational framework that facilitates the structural and dynamical characterization of the human nucleus. This framework builds upon a previous investigation65 but incorporates several significant modifications. Firstly, we enhance the model resolution by a factor of ten, enabling the precise determination of the spatial positioning of each chromatin segment measuring 100 KB in length. Secondly, we present a kinetic scheme for speckles that accounts for the phosphorylation of protein molecules. This inclusion captures the influence of chemical reactions on the stability and dynamics of nuclear bodies. Thirdly, we incorporate explicit nuclear envelope dynamics to explore the impact of large-scale deformations on genome organization. Finally, our implementation into OpenMM offers the advantages of Python Scripting and GPU acceleration, facilitating easy extension and customization. These features will facilitate the broad applicability and adoption of the proposed model.
The nucleus model provides particle-based representations for chromosomes, nucleoli, speckles, and the nuclear envelope. As shown in Fig. 1A and B, each of the 46 chromosomes is represented as a beads-on-a-string polymer, where each bead represents a 100-KB-long genomic segment. Based on Hi-C data, we further assign each bead as compartment A, B, or C to signify euchromatin, heterochromatin, or pericentromeric regions. The lamina was modeled as a spherical enclosure with 10 µm diameter, using discrete particles arranged to represent a mesh grid with covalent bonds linking together nearest neighbors.72 We modeled nucleoli and speckles as liquid droplets that emerge through the spontaneous phase separation of coarse-grained particles, representing protein and RNA molecule aggregates.8,11 These particles exhibited attractive interactions within the same type to promote condensation. More details about the various components of the system can be found in the Supporting Information Section: Components of the whole nucleus model.

Computer model of the human nucleus for structural and dynamical characterizations.
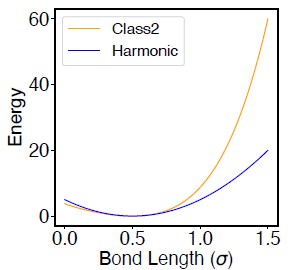
(A) 3D rendering of the nucleus model with particle-based representations for the 46 chromosomes shown as ribbons, the nuclear lamina (grey), nucleoli (cyan), and speckles (yellow). As shown on the right, chromosomes are modeled as beads-on-a-string polymers at a 100 KB resolution, with the beads further categorized into compartment A (red), compartment B (light blue), or centromeric regions (green). (B) Speckle particles undergo chemical modifications concurrent to their spatial dynamics, and the dephosphorylated (dP) particles contribute to droplet formation. (C) Illustration of the ideal and compartment potential that promotes chromosome compaction and microphase separation. Specific interactions between chromosomes and nuclear landmarks are shown on the right.
The energy function of the nucleus model includes three components that account for the self-assembly of chromosomes, the assembly of nuclear bodies, and the coupling between chromosomes and nuclear landmarks. Therefore, the model approximates nuclear organization as a coupled self-assembly process. The chromosome energy function (see Eq. S7 in Supporting Information Section: Hi-C inspired interactions for the diploid human genome) includes terms that account for the polymer connectivity and excluded volume effect, an ideal potential, compartment-specific interactions, and specific interchromosomal interactions. As shown in Fig. 1C, the ideal potential is only applied for beads from the same chromosome to approximate the effect of loop extrusion by Cohesin molecules 73,74 for chromosome compaction and territory formation.75,76 Compartment-specific interactions, on the other hand, promote microphase separation and compartmentalization of euchromatin and heterochromatin. Finally, interchromosomal interactions account for sequence-specific effects that compartment-dependent potentials cannot capture.
Interactions among coarse-grained particles that form nuclear bodies were designed to promote and stabilize the formation of liquid droplets, as has been revealed by many experiments.77–79 We adopted the Lennard-Jones potential for nucleolar particles to mimic the weak, multivalent interactions that arise from protein and RNA molecules that make up the nucleoli. As a first attempt to approximate their complex dynamics, we considered two types of particles that form speckles: phosphorylated (P) and de-phosphorylated (dP). The two types can interconvert via chemical reactions80–82 and dP particles share attractive interactions modeled with the Lennard-Jones potential.
As shown in Fig. 1C, to recognize specific interactions between chromosomes and nuclear landmarks, we introduced contact potentials between them. These potentials are inspired by the experimental techniques that probe the corresponding contacts. The Supporting Information Section: Chromosome-nuclear landmark interactions and Nuclear landmark-nuclear landmark interactions contain more details about all the nuclear landmark related energy functions.
Optimization of model parameters with experimental data
The nucleus model was designed to be interpretable such that energy terms represent physical processes. Furthermore, the expressions of the interaction potentials were also designed such that their parameters can be determined from experimental data via the maximum entropy optimization algorithm.9,83,84 Below, we briefly outline the procedure used for parameter optimization and further details can be found in the Supporting Information Section: Optimization of the whole nucleus model parameters.
As illustrated in Fig. 2A, starting from a given set of parameters, we first perform MD simulations to produce a collection of 3D structures for the diploid genome and various nuclear bodies. These structures are then transformed into a contact map or contact probabilities between chromatin beads and nuclear landmarks by averaging over homologous chromosomes. Constraints corresponding to different energy terms could be obtained from the simulated results and compared with those estimated from Hi-C, SON TSA-Seq, and Lamin B DamID profiles. Finally, the model parameters were updated based on the difference between simulated and experimental constraints using the adaptive moment estimation (Adam) optimization algorithm.85 The three steps can be repeated with updated parameters to improve the simulation-experiment agreement further.

Overview of the iterative algorithm for parameterizing the nucleus model with experimental data.
Starting from an initial set of parameters, we perform MD simulations to produce an ensemble of nuclear structures. These structures can be transformed into contacts between chromosomes or between chromosomes and nuclear landmarks for direct comparison with experimental data. Differences between simulated and experiment contacts are used to update parameters for additional rounds of optimization if needed.
No quantitative experimental data exists for interactions among nuclear body particles to serve as constraints. We varied the strength of the interaction potential to produce 2-3 nucleoli and ∼ 30 speckle clusters during the simulations (Fig. S1) while ensuring the fluidity of the resulting droplets.
Molecular dynamics simulations with GPU acceleration
We implemented the nucleus model into the MD engine OpenMM.71 OpenMM offers an excellent interface with Python scripting, significantly improving the readability and customizability of the model. The code was designed into functional modules, with different components, such as chromosomes and nuclear landmarks, written as separate classes. This design further facilitates the introduction of additional nuclear components, if desired, with minimal changes to existing code. We provide examples of simulation set up, trajectory analysis, parameter optimization, and introducing new features in the GitHub repository.
Fig. 3A illustrates the workflow for setting up and executing whole nucleus simulations. A configuration file that provides the position of individual particles in the PDB file format is needed to initialize the simulations. This file also contains topological information regarding whether a particle represents chromosomes or nuclear landmarks and the identity of specific chromosomes. The input file can be generated with provided Python scripts by randomly distributing the positions of chromosomes, speckles, and nucleoli, though optimized configurations are also included in the GitHub repository. By default, the lamina particles will be uniformly placed on a sphere of 10 µm in diameter. Upon parsing the configuration file, interactions among various components can be set up with optimized parameters. This step will produce an object that can be used for MD simulations. As shown in Fig. 3B, the workflow only requires a few lines of code. The package also includes analysis scripts to compute contact maps, monitor conformational dynamics, and track nuclear bodies.

OpenNucleome facilitates GPU-accelerated simulations of the human nucleus.
(A) Illustration of workflow for setting up, performing, and analyzing MD simulations. (B) Python scripts setting up whole nucleus simulations. (C) Performance of MD simulations on different number of CPU cores and a single GPU.
A significant benefit of OpenMM71 is its native support of GPU acceleration. As shown in Fig. 3C, the simulation speed with one Nvidia Volta V100 GPU is 150 times faster than that of the 4 Intel Xeon Platinum 8260 CPU cores. Notably, this performance enhancement cannot be achieved by simply increasing the CPU core numbers. For example, the simulation speed with 32 CPU cores is less than twice that of 4 CPU cores, potentially due to the system’s heterogeneous distribution of particles.
Simulations reproduce and predict diverse experimental data
We extensively validated the parameterized nucleus model to examine its biological relevance. MD simulations initialized from 50 different initial configurations were performed to build an ensemble of structures. As mentioned in the following section, a diverse set of initial configurations is essential for reproducing interchromosomal contacts probed in Hi-C. From the simulated structures, we computed various quantities for direct comparison with experimental measurements. Given that the majority of experimental data were analyzed for the haploid genome, we adopted a similar approach by averaging over paternal and maternal chromosomes to facilitate direct comparison. More details on data analysis can be found in the Supporting Information Section: Details of simulation data analysis.
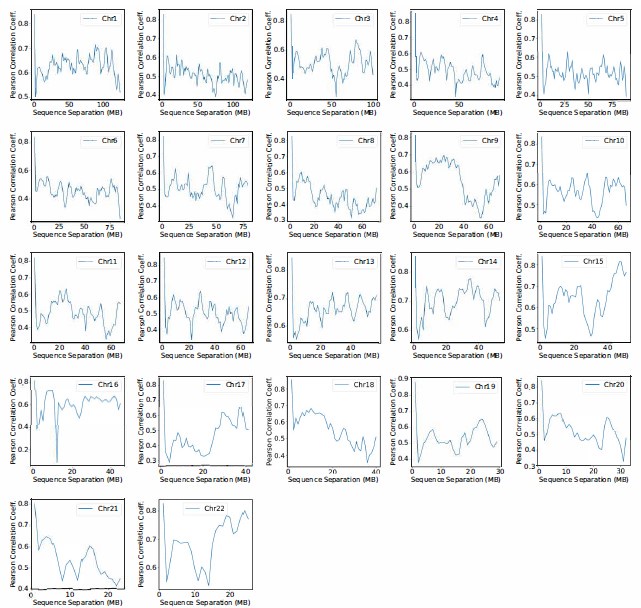
We compared the simulated contact probabilities among chromosomes with Hi-C data. As shown in Fig. 4A and Fig. S2, the simulated and experimental contact maps are highly correlated. The squares along the diagonal support the formation of chromosome territories that promote intrachromosomal contacts, and the apparent checkboard patterns follow the compartmentalization of various chromatin types. We further examined the decay of intra-chromosomal contacts as a function of the sequence separation, which is known to deviate from that of an equilibrium globule.22 As shown in Fig. 4B, the simulated results overlap well with the Hi-C data (orange curve). In addition, the simulated average contact probabilities between various compartment types match values estimated from Hi-C data. Moreover, the simulated and experimental average contact probabilities between pairs of chromosomes agree well, and the Pearson correlation coefficient between the two datasets reaches 0.89.

Simulated structures reproduce contact frequencies between chromosomes and between chromosomes and nuclear landmarks.
(A) Comparison between simulated (top right) and experimental (bottom left) whole-genome contact probability maps with Pearson correlation coefficient r = 0.89. Zoom-ins of various regions are provided in Fig. S2. (B) Comparison between simulated and experimental average contact frequencies, including average contacts between genomic loci from the same chromosomes at a given separation (top), average contacts between genomic loci classified into different compartment types (middle), and average contacts between various chromosome pairs (bottom). (C) Comparison between simulated and experimental Lamin-B DamID (top) and SON TSA-Seq signals (bottom), with Pearson correlation coefficients of haploid chromosomes shown on the right.
We further examined the contacts between chromosomes and nuclear landmarks. As illustrated in Fig. 4C, the simulated Lamin-B DamID signals for chromosome 7 match well with the experimental results, capturing the complex contact pattern that weaves chromatin towards and away from the nuclear envelope. Similarly, SON TSA-Seq data that quantify the contact between chromosomes and speckles are well captured by simulated structures. The anti-correlation between DamID and TSA-Seq is clearly visible. The observed agreement between simulation and experimental results is not limited to any particular chromosome. Good agreements are achieved for all chromosomes.
The simulations also provide 3D representations of the nucleus that can be compared with DNA-MERFISH data.35 We found that the simulated radius of gyration of individual chromosomes matches well with experimental values (Fig. 5A). The simulated and experimental average normalized chromosome radial positions also correlate strongly, as shown in Fig. 5B. We note that while the sequencing results presented in Fig. 4 were used for model parameterization, the MERFISH data were not. Therefore, the simulation results here are de novo predictions, and their agreement with experimental data strongly supports the coupled assembly mechanism used for designing the energy function.

Structural and dynamical predictions of the nucleus model match results from microscopy imaging.
(A) Comparison between the simulated and experimental radius of gyration, Rg, for haploid chromosomes. The Pearson correlation coefficient between the two, r, is shown in the legend. (B) Comparison between the simulated and experimental normalized radial positions for haploid chromosomes, with their Pearson correlation coefficient shown in the legend. Detailed definition of the normalized radial positions is provided in the Supporting Information Section: Computing simulated normalized chromosome radial positions. (C) Mean-squared displacements, MSDs, as a function of time are shown for selected telomeres. (D) The probability distribution of the anomalous exponent, α, obtained from fitting the MSDs curves for all telomeres with the expression, ⟨r2(Δt)⟩ = DαΔtα.
A significant advantage of MD simulation-based models is the dynamical information they naturally produce. We measured the dynamics of telomeres by tracking the mean square displacements (MSDs), ⟨r2(Δt)⟩, as a function of time. In Fig. 5C, we plot representative MSD trajectories over a one-hour timescale. In line with previous research, 86–88 telomeres display anomalous subdiffusive motion. When fitted with the equation ⟨r2(Δt)⟩ = DαΔtα, these trajectories yield a spectrum of α values, with a peak around 0.59. The exponent and the diffusion coefficient Dα = (27±11)×10−4µm2·s−α both match well with the experimental values,89,90 upon setting the nucleoplasmic viscosity as 1Pa · s (see Supporting Information Section: Mapping the reduced time unit to real time for more details).
The good agreement in the dynamics of individual loci further inspired us to examine the diffusion of whole chromosomes. In particular, we plotted the normalized chromosome radial positions as a function of time in Fig. 6A. Remarkably, we found that chromosomes appear arrested and no significant changes in their positions are observed over timescales comparable to the cell cycle (see also Fig. S12). Therefore, our simulations predict that large-scale movements of chromosomes are unlikely during the G1 phase.

Heterogeneity and conserved features of nuclear organizations.
(A) Normalized chromosome radial positions as a function of simulation time. (B) Contacts between chromosome 1 and 2 from two independent simulation trajectories show significant variations. (C) Genome-wide in silico Lamin B DamID (top) and SON TSA-Seq (bottom) profiles computed from two independent trajectories. Pearson correlation coefficients, r, are provided on each plot. (D) Pairwise Person correlation coefficients between interchromosomal contact matrices (left), genome-wide Lamin B DamID profiles (middle), and genome-wide SON TSA-Seq profiles (right) determined from independent trajectories. The averages excluding the diagonals of the three datasets are 0.06, 0.53, and 0.72.
Heterogeneity and robustness of the simulated conformational ensemble
The lack of relaxation of chromosome radial positions suggests the importance of starting configurations used to initialize the simulations. Statistical averages of the resulting ensemble of nuclear structures depend crucially on these starting configurations. Using an optimization procedure, we selected them from 1000 configurations to maximize the agreement with experimental lamin-B DamID and interchromosomal contact probabilities. The Supporting Information Section: Initial configurations for simulations provides more details on preparing the 1000 initial configurations.
We selected a total of 50 starting configurations to initiate independent simulations. Smaller sets of starting configurations are not sufficient to reproduce the interchromosomal contact probabilities, as shown in Fig. S3B. Notably, different sets of 50 configurations selected from independent trials show significant overlap (Fig. S3D), supporting the robustness of the selection protocol in detecting conserved features of genome organization.
While the ensemble as a whole is relatively robust, individual configurations with the ensemble exhibit significant differences. For example, the Lamin B DamID profiles produced from different trajectories are only weakly correlated (Fig. 6C), with an average correlation coefficient of 0.53. These weak correlations result from significant differences in the normalized radial positions of chromosomes, as can be seen in representative configurations from two simulation trajectories (Fig. 6B). The fluctuations of normalized radial positions cause changes in contacts between chromosomes as well, resulting in little correlation between interchromosomal contact matrices (Fig. 6D).
We examined genome organizations reported by Su et al. 35 and found a similar variation of interchromosomal contact probabilities across individual cells (Fig. S3A, D). Notably, the simulated configurations capture the fluctuations of interchromosomal contacts observed in DNA-MERFISH data, further supporting the biological relevance of the reported in silico structures.
Despite the differences in interchromosomal contacts across trajectories, high conservation of connections between chromosomes and speckles can be observed in individual simulations. For example, the average correlation coefficient between in silico SON TSA-Seq profiles produced from different trajectories is 0.72, much higher than the corresponding value for Lamin B DamID profiles. Conservation of contacts between chromosomes and nuclear bodies (zones) across individual cells has indeed been reported in a previous study that simultaneously images chromatin and various subnuclear structures.36
Nuclear deformation preserves chromosome-nuclear body contacts
Numerous studies have highlighted the remarkable influence of nuclear shape on the positioning of chromosomes and the regulation of gene expression.53,91 The nucleus, once regarded as a mere compartment for DNA storage, is increasingly recognized as a dynamic and intricately structured organelle. To better understand the interplay between nuclear shape and genome organization as a fundamental mechanism that shapes the transcriptional landscape, we performed additional simulations in which the nuclear lamina was altered from a sphere into more ellipsoidal shapes by applying a force along the z-axis (Fig. 7A). More details about these simulations can be found in the Section: Nuclear envelope deformation simulations of the Supporting Information.

Nuclear deformations influence genome organization while preserving chromatin-speckle contacts.
(A) Illustration of force induced nuclear envelope deformation. The nuclear lamina is modeled as a particle mesh where neighboring lamina particles are covalently bonded together. (B) Example nucleus conformations at different strengths of applied force. (C) Pearson correlation coefficients between results from simulations of deformed nuclei and those from a spherical nucleus for interchromosomal contacts (left), DamID profiles (middle), and TSA-Seq (right). The values at zero force were computed from two independent simulations starting from the same initial configurations.
As illustrated in Fig. 7B, the presence of external forces resulted in significant alterations in nuclear shape. We conducted two independent simulations with different force strengths, leading to varying degrees of deformation in the nuclear lamina. This deformation, in turn, caused a reorganization of chromosomes, affecting their normalized radial positions and pairwise contacts (see Fig. S4 and Fig. 7C). We observed that more deformed nuclei exhibited lower correlation coefficients for interchromosomal contacts compared to results obtained from simulations in a spherical nucleus. Similarly, the DamID profiles exhibited significant variations upon nucleus deformation, whereas TSA-Seq signals were much less affected and remained highly correlated with the results from the spherical nucleus simulations.
Therefore, it appears that speckles, and potentially other nuclear condensates, can dynamically reorganize in response to changes in chromosome conformations to maintain contacts with genomic loci. This robustness in nuclear body contacts may be essential for ensuring the robust functioning of the genome in a population of cells with significant variability in nuclear shape.
Conclusions and Discussion
We introduced a computational model, OpenNucleome, to facilitate simulations for the human nucleus at high structural and temporal resolution. We conducted extensive crossvalidation with experimental data to support the biological relevance of simulated 3D structures. Implementing the model into the MD package, OpenMM enables GPU acceleration for long-timescale simulations. Tutorials in the format of Python Scripts with extensive documentation are provided to facilitate the adoption of the model by the community.
Our software enhances the capabilities of existing genome simulation tools. 40,69,70 Specifically, OpenNucleome aligns with the design principles of Open-MiChroM, 70 prioritizing open-source accessibility while expanding simulation capabilities to the entire nucleus. Similar to software from the Alber lab,69 OpenNucleome offers high-resolution genome organization that faithfully reproduces a diverse range of experimental data. Furthermore, beyond static structures, OpenNucleome facilitates dynamic simulations with explicit representations of various nuclear condensates, akin to the model developed by Fujishiro and Sasai 40.
A significant advantage of OpenNucleome lies in its predictive power for dynamical information. For example, the model succeeded in reproducing the subdiffusive behavior of telomeres. We further showed that the dynamics of individual chromosomes are slow and their radial positions do not relax over the time course of a cell cycle. This is consistent with previous theoretical estimations on chromosome dynamics 92 and recent observations of solid behavior of chromatin in vivo.93 Live cell experiments that directly track the positions of multiple chromosomes could further validate/falsify this prediction. We anticipate the model will greatly facilitate the investigation of the dynamics of genomic loci and nuclear bodies and the interpreting of live cell imaging results.
Slow chromosome dynamics and a lack of conformational relaxation naturally result in the heterogeneity of chromosome radial positions across individual cells. This heterogeneity raises doubts about the notion that chromosome radial positions provide robust and reliable mechanisms for gene regulation.2,94–96 Instead, our results support the nuclear zoning model for gene regulation,36 where specific loci function as “fixed points” anchored to certain nuclear bodies in all cells. This anchoring mechanism robustly creates the desired molecular environment surrounding these genomic segments. Unlike chromosome radial positions, contacts between genomic loci and speckles can be robustly established in individual cells, as shown in our simulations. It was achieved through a nucleation process that attracts speckle particles towards specific loci due to specific interactions. Nucleation occurs much more rapidly than chromosome rearrangement due to the smaller size of speckle particles. The coupled self-assembly mechanism for chromosomes and nuclear bodies can similarly facilitate the formation of other nuclear zones for different kinds of fixed points.
Despite the heterogeneity in chromosome positions and interchromosomal contacts, the ensemble of nuclear structures as a whole is not random and exhibits conserved features. For example, on average, certain chromosomes remain closer to the nuclear envelope than others (see Fig. 5B). Similarly, the average contact frequency between certain chromosome pairs is higher than others, though this trend can be frequently violated in individual cells. How such conserved features arise as cells exit from the mitotic phase remains unclear and would be interesting for further explorations.
Methods
Molecular dynamics simulation details
We used the software package OpenMM71 to perform MD simulations in reduced units at constant temperature (T = 1.0). Unless otherwise specified, we froze the lamina particles and only propagated the dynamics of chromatin, nucleoli, and speckles.
Two integration schemes were used with a time step of dt = 0.005 to efficiently generate structural ensembles and produce realistic dynamical information, respectively. For simulations used in parameter optimization and building structural ensembles, we employed the Langevin integrator with a damping coefficient of γ−1 = 10.0. In the case of MSD calculations shown in Fig. 6, we utilized Brownian dynamics with a damping coefficient of γ−1 = 0.01. The higher damping coefficient provides a better approximation to the viscous nucleus environment, while the smaller value in the Langevin integrator facilitates conformational sampling with faster diffusion rates.
We employed the semi-grand Monte Carlo technique97 to simulate chemical transitions between two types of speckle particles. At every 4000 simulation steps, we attempt a total of NSp chemical reactions that converts one type of speckle particles to the other type with a probability of 0.2. NSp corresponds to the total number of speckle particles, and the switching probability was chosen to be comparable to the experimental phosphorylation rate. More details on the speckle dynamics are provided in the Supporting Information Section: Speckles as phase-separated droplets undergoing chemical modifications.
When deforming the nuclear envelope, we unfroze the lamina particles and evolved them dynamically as the rest of the nucleus. Bonded interactions among lamina particles held the nuclear envelope together as a particle mesh. A harmonic force along the z-axis was introduced to compress the particle mesh. More details are provided in the Supporting Information Section: Nuclear envelope deformation simulations.
For simulations used to optimize parameters, a total of 50 independent 3-million-step-long trajectories were performed. Configurations were recorded at every 2000 simulation steps for analysis. The first 500,000 steps of each trajectory were discarded as equilibration. For production simulations, we performed 50 independent 10-million-step long trajectories starting from different initial configurations. Nuclear structures were again recorded at every 2000 steps to determine statistical averages presented in the paper. An additional 8 simulations of 30 million steps in length were performed to compute telomere MSDs.
We mapped the reduced units to real units with the conversion of length scale σ = 385 nm and the time scale in Brownian dynamics simulations τ = 0.65 s. These conversions were determined by as detailed in the Supporting Information Section: Unit Conversion.
Experimental data processing and analysis
We obtained the in situ Hi-C data, SON TSA-seq data, and Lamin-B DamID data of HFF cell lines from the 4DN data portal. The intra and interchromosomal interactions were calculated at 100 KB resolution with VC SQRT normalization applied to the interaction matrices. Hi-C data extraction and normalization were performed using Juicer tools.98 We followed the same processing and normalization method described in Ref. 99 to analyze TSA-seq data. Two biological replicates of Lamin-B DamID data were merged and the normalized counts over Dam-only control were used for analysis. The SON TSA-Seq and Lamin-B DamID data were processed at the 25 KB resolution and the average values at the 100 KB resolution were used in Fig. 4 for model validation.
Supporting information
Acknowledgements
This work was supported by the National Institutes of Health (Grant R35GM133580).
Competing interests
Authors declare that they have no competing interests.
Data and materials availability
Hi-C data (https://data.4dnucleome.org, accession number: 4DNFIB59T7NN). SON TSA-seq data (https://data.4dnucleome.org, accession number: pulldown data 4DNEX6U8TS3Y, control data 4DNEXI7XUWFK). Lam-inB DamID data (https://data.4dnucleome.org, accession number 4DNESXZ4FW4T). The software is available at: available at https://github.com/ZhangGroup-MITChemistry/OpenNucleome.
References
- (1)The 4D nucleome projectNature 549:219–226Google Scholar
- (2)Chromatin organization and transcriptional regulationCurrent opinion in genetics & development 23:89–95Google Scholar
- (3)The spatial organization of the human genomeAnnual review of genomics and human genetics 14:67–84Google Scholar
- (4)The 3D genome in transcriptional regulation and pluripotencyCell stem cell 14:762–775Google Scholar
- (5)The 3D genome as moderator of chromosomal communicationCell 164:1110–1121Google Scholar
- (6)Developmental enhancers and chromosome topologyScience 361:1341–1345Google Scholar
- (7)Molecular basis and biological function of variability in spatial genome organizationScience 365:eaaw9498Google Scholar
- (8)Genome organization around nuclear specklesCurrent opinion in genetics & development 55:91–99Google Scholar
- (9)Multiscale modeling of genome organization with maximum entropy optimizationJ. Chem. Phys 155:010901Google Scholar
- (10)From Nucleosomes to Compartments: Physicochemical Interactions Underlying Chromatin OrganizationAnnual Review of Biophysics 53Google Scholar
- (11)The nucleolus as a multiphase liquid condensateNature reviews Molecular cell biology 22:165–182Google Scholar
- (12)Nuclear speckles: a model for nuclear organellesNature reviews Molecular cell biology 4:605–612Google Scholar
- (13)Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repressionCell 169:780–791Google Scholar
- (14)Cytokines and their relationship to the symptoms and outcome of cancerNature Reviews Cancer 8:887–899Google Scholar
- (15)Chromatin organization is a major influence on regional mutation rates in human cancer cellsnature 488:504–507Google Scholar
- (16)Genome-wide mapping and analysis of chromosome architectureNature reviews Molecular cell biology 17:743–755Google Scholar
- (17)Chromosome conformation capture and beyond: toward an integrative view of chromosome structure and functionMolecular cell 77:688–708Google Scholar
- (18)How the genome folds: the biophysics of four-dimensional chromatin organizationAnnual review of biophysics 48:231–253Google Scholar
- (19)Understanding 3D genome organization by multidisciplinary methodsNature Reviews Molecular Cell Biology 22:511–528Google Scholar
- (20)Imaging specific genomic DNA in living cellsAnnu Rev Biophys Google Scholar
- (21)Capturing chromosome conformationscience 295:1306–1311Google Scholar
- (22)Comprehensive mapping of long-range interactions reveals folding principles of the human genomescience 326:289–293Google Scholar
- (23)A 3D map of the human genome at kilobase resolution reveals principles of chromatin loopingCell 159:1665–1680Google Scholar
- (24)Chromatin domains: the unit of chromosome organizationMolecular cell 62:668–680Google Scholar
- (25)Structural and functional diversity of topologically associating domainsFEBS letters 589:2877–2884Google Scholar
- (26)[16] DamID: mapping of in vivo protein– genome interactions using tethered DNA adenine methyltransferaseMethods in enzymology 410:342–359Google Scholar
- (27)ChIP–seq: advantages and challenges of a maturing technologyNature reviews genetics 10:669–680Google Scholar
- (28)Mapping 3D genome organization relative to nuclear compartments using TSA-Seq as a cytological rulerJournal of Cell Biology 217:4025–4048Google Scholar
- (29)Spatiotemporal single-cell analysis of gene expression in the mouse suprachiasmatic nucleusNature neuroscience 23:456–467Google Scholar
- (30)Massively multiplex single-cell Hi-CNature methods 14:263–266Google Scholar
- (31)Single-cell Hi-C reveals cell-to-cell variability in chromosome structureNature 502:59–64Google Scholar
- (32)Spatially resolved, highly multiplexed RNA profiling in single cellsScience 348:aaa6090Google Scholar
- (33)Super-resolution imaging reveals distinct chromatin folding for different epigenetic statesNature 529:418–422Google Scholar
- (34)Identification of gene positioning factors using high-throughput imaging mappingCell 162:911–923Google Scholar
- (35)Genome-scale imaging of the 3D organization and transcriptional activity of chromatinCell 182:1641–1659Google Scholar
- (36)Integrated spatial genomics reveals global architecture of single nucleiNature 590:344–350Google Scholar
- (37)Data-driven polymer model for mechanistic exploration of diploid genome organizationBiophysical Journal 119:1905–1916Google Scholar
- (38)Predicting three-dimensional genome organization with chromatin statesPLoS computational biology 15:e1007024Google Scholar
- (39)Integrative genome modeling platform reveals essentiality of rare contact events in 3D genome organizationsNature Methods 19:938–949Google Scholar
- (40)Generation of dynamic three-dimensional genome structure through phase separation of chromatinProceedings of the National Academy of Sciences 119:e2109838119Google Scholar
- (41)From Hi-C contact map to three-dimensional organization of interphase human chromosomesPhysical Review X 11Google Scholar
- (42)Exploring the three-dimensional organization of genomes: interpreting chromatin interaction dataNature Reviews Genetics 14:390–403Google Scholar
- (43)Modeling epigenome folding: formation and dynamics of topologically associated chromatin domainsNucleic acids research 42:9553–9561Google Scholar
- (44)Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcriptionCell 157:950–963Google Scholar
- (45)De novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architectureProceedings of the National Academy of Sciences 114:12126–12131Google Scholar
- (46)Polymer simulations of heteromorphic chromatin predict the 3D folding of complex genomic lociMolecular cell 72:786–797Google Scholar
- (47)Chromatin organization by an interplay of loop extrusion and compartmental segregationBiophysical Journal 114:30aGoogle Scholar
- (48)Polymer physics predicts the effects of structural variants on chromatin architectureNature genetics 50:662–667Google Scholar
- (49)Interphase human chromosome exhibits out of equilibrium glassy dynamicsNature communications 9:3161Google Scholar
- (50)Bottom–up modeling of chromatin segregation due to epigenetic modificationsProceedings of the National Academy of Sciences 115:12739–12744Google Scholar
- (51)From Effective Interactions Extracted Using Hi-C Data to Chromosome Structures in Conventional and Inverted NucleiPRX Life 1:013010Google Scholar
- (52)Live imaging of chromatin distribution reveals novel principles of nuclear architecture and chromatin compartmentalizationSci. Adv 7Google Scholar
- (53)Shaping the genome via lengthwise compaction, phase separation, and lamina adhesionNucleic Acids Res 50:1–14Google Scholar
- (54)Phase Separation and Correlated Motions in Motorized GenomeJ. Phys. Chem. B 126:5619–5628Google Scholar
- (55)Chromosome positioning from activity-based segregationNucleic Acids Res 42:4145–4159Google Scholar
- (56)From 1D sequence to 3D chromatin dynamics and cellular functions: A phase separation perspectiveNucleic Acids Res 46:9367–9383Google Scholar
- (57)Mesoscale Liquid Model of Chromatin Recapitulates Nuclear Order of EukaryotesBiophys. J 118:2130–2140Google Scholar
- (58)Deciphering the molecular mechanism of the cancer formation by chromosome structural dynamicsPLOS Comput. Biol 17:e1009596Google Scholar
- (59)Four-dimensional chromosome reconstruction elucidates the spatiotemporal reorganization of the mammalian X chromosomeProc. Natl. Acad. Sci 118Google Scholar
- (60)Quantifying Chromosome Structural Reorganizations during Differentiation, Reprogramming, and TransdifferentiationPhys. Rev. Lett 129:068102Google Scholar
- (61)Polymer folding through active processes recreates features of genome organizationProceedings of the National Academy of Sciences 120:e2221726120Google Scholar
- (62)High-resolution single-cell 3D-models of chromatin ensembles during Drosophila embryogenesisNat. Commun 12:1–12Google Scholar
- (63)Predicting scale-dependent chromatin polymer properties from systematic coarse-grainingNat. Commun 14:4108Google Scholar
- (64)Mesoscale phase separation of chromatin in the nucleusElife 10Google Scholar
- (65)Compartmentalization with nuclear landmarks yields random, yet precise, genome organizationBiophysical Journal 122:1376–1389Google Scholar
- (66)The interplay of chromatin phase separation and lamina interactions in nuclear organizationBiophysical Journal 120:5005–5017Google Scholar
- (67)Separate roles for chromatin and lamins in nuclear mechanicsNucleus 9:119–124Google Scholar
- (68)Chromatin network retards nucleoli coalescenceNature Communications 12:6824Google Scholar
- (69)Evaluating the role of the nuclear microenvironment in gene function by population-based modelingNature Structural & Molecular Biology :1–14Google Scholar
- (70)A scalable computational approach for simulating complexes of multiple chromosomesJournal of molecular biology 433:166700Google Scholar
- (71)OpenMM 7: Rapid development of high performance algorithms for molecular dynamicsPLoS computational biology 13:e1005659Google Scholar
- (72)HP1α is a chromatin crosslinker that controls nuclear and mitotic chromosome mechanicsElife 10:e63972Google Scholar
- (73)Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomesProceedings of the National Academy of Sciences 112:E6456–E6465Google Scholar
- (74)Formation of chromosomal domains by loop extrusionCell reports 15:2038–2049Google Scholar
- (75)Transferable model for chromosome architectureProceedings of the National Academy of Sciences 113:12168–12173Google Scholar
- (76)Genomic energy landscapesBiophysical journal 112:427–433Google Scholar
- (77)Cajal bodies, nucleoli, and speckles in the Xenopus oocyte nucleus have a low-density, sponge-like structureMolecular biology of the cell 16:202–211Google Scholar
- (78)Surface fluctuations and coalescence of nucleolar droplets in the human cell nucleusPhysical review letters 121Google Scholar
- (79)Nucleolar dynamics and interactions with nucleoplasm in living cellsElife 8:e47533Google Scholar
- (80)Ephemeral protein binding to DNA shapes stable nuclear bodies and chromatin domainsBiophysical journal 112:1085–1093Google Scholar
- (81)Mechanisms for active regulation of biomolecular condensatesTrends in Cell Biology 30:4–14Google Scholar
- (82)Modelling the compartmentalization of splicing factorsJournal of theoretical biology 239:298–312Google Scholar
- (83)Learning the Formation Mechanism of Domain-Level Chromatin States with Epigenomics DataBiophys. J 116:2047–2056Google Scholar
- (84)Efficient Hi-C inversion facilitates chromatin folding mechanism discovery and structure predictionBiophys. J 122:3425–3438Google Scholar
- (85)Adam: A method for stochastic optimizationarXiv Google Scholar
- (86)Anomalous diffusion, spatial coherence, and viscoelasticity from the energy landscape of human chromosomesProceedings of the National Academy of Sciences 115:7753–7758Google Scholar
- (87)Transient anomalous diffusion of telomeres in the nucleus of mammalian cellsPhysical review letters 103:018102Google Scholar
- (88)Chromatin mechanics dictates subdiffusion and coarsening dynamics of embedded condensatesBiophysical Journal 120:318aGoogle Scholar
- (89)Loss of lamin A function increases chromatin dynamics in the nuclear interiorNature communications 6:8044Google Scholar
- (90)Compartmentalization of telomeres through DNA-scaffolded phase separationDevelopmental cell 57:277–290Google Scholar
- (91)Interphase chromosomes of the Aedes aegypti mosquito are liquid crystalline and can sense mechanical cuesNature Communications 14:326Google Scholar
- (92)Structure and dynamics of interphase chromosomesPLoS Comput. Biol 4:e1000153Google Scholar
- (93)Condensed chromatin behaves like a solid on the mesoscale in vitro and in living cellsCell 183:1772–1784Google Scholar
- (94)Chromatin structure: does the 30-nm fibre exist in vivo?Current opinion in cell biology 22:291–297Google Scholar
- (95)Nuclear organization of the genome and the potential for gene regulationNature 447:413–417Google Scholar
- (96)The meaning of gene positioningCell 135:9–13Google Scholar
- (97)Scalable parallel Monte Carlo algorithm for atomistic simulations of precipitation in alloysPhysical Review B 85:184203Google Scholar
- (98)Juicer provides a one-click system for analyzing loop-resolution Hi-C experimentsCell systems 3:95–98Google Scholar
- (99)TSA-seq reveals a largely conserved genome organization relative to nuclear speckles with small position changes tightly correlated with gene expression changesGenome research 31:251–264Google Scholar
Article and author information
Author information
Version history
- Sent for peer review:
- Preprint posted:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.93223. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2024, Lao et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.