Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorMurim ChoiSeoul National University, Seoul, Republic of Korea
- Senior EditorMurim ChoiSeoul National University, Seoul, Republic of Korea
Reviewer #1 (Public Review):
The authors describe a massively parallel reporter assays (MPRA) screen focused on identifying polymorphisms in 5' and 3' UTRs that affect translation efficiency and thus might have a functional impact on cells. The topic is of timely interest, and indeed, several related efforts have recently been published and preprinted (e.g., https://pubmed.ncbi.nlm.nih.gov/37516102/ and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10635273/). This study has several major issues with the results and their presentation.
Major comments:
(1) The main issue is that it appears that the screen has largely failed, yet the reasons for that are unclear, which makes it difficult to interpret. The authors start with a library that includes approximately 6,000 variants, which makes it a medium-sized MPRA. But then, only 483 pairs of WT/mutated UTRs yield high-confidence information, which is already a small number for any downstream statistical analysis, particularly since most don't actually affect translation in the reporter screen setting (which is not unexpected). It is unclear why >90% of the library did not give high-confidence information. The profiles presented as base-case examples in Figure 2B don't look very informative or convincing. All the subsequent analysis is done on a very small set of UTRs that have an effect, and it is unclear to this reviewer how these can yield statistically significant and/or biologically relevant associations.
(2) From the variants that had an effect, the authors go on to carry out some protein-level validations and see some changes, but it is not clear if those changes are in the same direction as observed in the screen.
(3) The authors follow up on specific motifs and specific RBPs predicted to bind them, but it is unclear how many of the hits in the screen actually have these motifs, or how significant motifs can arise from such a small sample size.
(4) It is particularly puzzling how the authors can build a machine learning predictor with >3,000 features when the dataset they use for training the model has just a few dozens of translation-shifting variants.
(5) The lack of meaningful validation experiments altering the SNPs in the endogenous loci by genome editing limits the impact of the results.
Reviewer #2 (Public Review):
Summary:
In their paper "Massively Parallel Polyribosome Profiling Reveals Translation Defects of Human Disease‐Relevant UTR Mutations" the authors use massively parallel polysome profiling to determine the effects of 5' and 3' UTR SNPs (from dbSNP/ClinVar) on translational output. They show that some UTR SNPs cause a change in the polysome profile with respect to the wild-type and that pathogenic SNPs are enriched in the polysome-shifting group. They validate that some changes in polysome profiles are predictive of differences in translational output using transiently expressed luciferase reporters. Additionally, they identify sequence motifs enriched in the polysome-shifting group. They show that 2 enriched 5' UTR motifs increase the translation of a luciferase reporter in a protein-dependent manner, highlighting the use of their method to identify translational control elements.
Strengths:
This is a useful method and approach, as UTR variants have been more difficult to study than coding variants. Additionally, their evidence that pathogenic mutations are more likely to cause changes in polysome association is well supported.
Weaknesses:
The authors acknowledge that they "did not intend to immediately translate the altered polysome profile into an increase or decrease in translation efficiency, as the direction of the shift was not readily evident. Additionally, sedimentation in the sucrose gradient may have been partially affected by heavy particles other than ribosomes." However, shifted polysome distribution is used as a category for many downstream analyses. Without further clarity or subdivision, it is very difficult to interpret the results (for example in Figure 5A, is it surprising that the polysome shifting mutants decrease structure? Are the polysome "shifts" towards the untranslated or heavy fractions?)
Author response:
Public Reviews:
Reviewer #1 (Public Review):
The authors describe a massively parallel reporter assays (MPRA) screen focused on identifying polymorphisms in 5' and 3' UTRs that affect translation efficiency and thus might have a functional impact on cells. The topic is of timely interest, and indeed, several related efforts have recently been published and preprinted (e.g., https://pubmed.ncbi.nlm.nih.gov/37516102/ and https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10635273/). This study has several major issues with the results and their presentation.
Major comments:
(1) The main issue is that it appears that the screen has largely failed, yet the reasons for that are unclear, which makes it difficult to interpret. The authors start with a library that includes approximately 6,000 variants, which makes it a medium-sized MPRA. But then, only 483 pairs of WT/mutated UTRs yield high-confidence information, which is already a small number for any downstream statistical analysis, particularly since most don't actually affect translation in the reporter screen setting (which is not unexpected). It is unclear why >90% of the library did not give highconfidence information. The profiles presented as base-case examples in Figure 2B don't look very informative or convincing. All the subsequent analysis is done on a very small set of UTRs that have an effect, and it is unclear to this reviewer how these can yield statistically significant and/or biologically relevant associations.
To make sure our final results are technically and statistically sound, we applied stringent selection criteria and cutoffs in our analytics workflow. First, from our RNA-seq dataset, we filtered the UTRs with at least 20 reads in a polysome profile across all three repeated experiments. Secondly, in the following main analysis using a negative binomial generalized linear model (GLM), we further excluded the UTRs that displayed batch effect, i.e. their batch-related main effect and interaction are significant. We believe our measure has safeguarded the filtered observations (UTRs) from the (potential) high variation of our massively parallel translation assays and thus gives high confidence to our results.
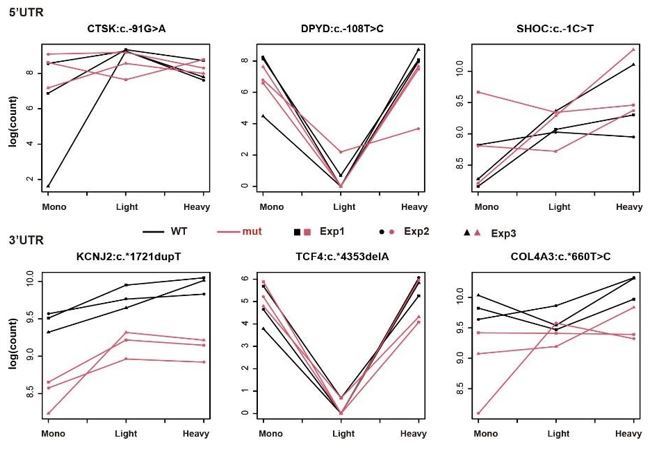
Regarding the interpretation of Figure 2B, since we aimed to identify the UTRs whose interaction term of genotype and fractions is significant in our generalized linear model, it is statistically conventional to double-check the interaction of the two variables using such a graph. For instance, in the top left panel of Figure 2B (5'UTR of ANK2:c.-39G>T), we can see that read counts of WT samples congruously decreased from Mono to Light, whereas the read counts of mutant samples were roughly the same in the two fractions – the trend is different between WT and mutant. Ergo, the distinct distribution patterns of two genotypes across three fractions in Figure 2B offer the readers a convincing visual supplement to our statistics from GLM.
In contrast to Figure 2B, the graphs of nonsignificant UTRs (shown below) reveal that the trends between the two genotypes are similar across the 'Mono and Light' and 'Light and Heavy' polysome fractions. Importantly, our analysis remains unaffected by differential expression levels between WT and mutant, as it specifically distinguishes polysome profiles with different distributions. This consistent trend further supports the lack of interaction between genotype and polysome fractions for these UTRs.
Author response image 1.
Figure: Examples of non-significant UTR pairs in massively parallel polysome profiling assays.
(2) From the variants that had an effect, the authors go on to carry out some protein-level validations and see some changes, but it is not clear if those changes are in the same direction as observed in the screen.
To infer the directionality of translation efficiency from polysome profiles, a common approach involves pooling polysome fractions and comparing them with free or monosome fractions to identify 'translating' fractions. However, this method has two major potential pitfalls: (i) it sacrifices resolution and does not account for potential bias toward light or heavy polysomes, and (ii) it fails to account for discrepancies between polysome load and actual protein output (as discussed in https://doi.org/10.1016/j.celrep.2024.114098 and https://doi.org/10.1038/s41598-019-47424-w). Therefore, our analysis focused on the changes within polysome profiles themselves. 'Significant' candidates were identified based on a significant interaction between genotype and polysome distribution using a negative binomial generalized linear model, without presupposing the direction of change on protein output.
(3) The authors follow up on specific motifs and specific RBPs predicted to bind them, but it is unclear how many of the hits in the screen actually have these motifs, or how significant motifs can arise from such a small sample size.
We calculated the Δmotif enrichment in significant UTRs versus nonsignificant UTRs using Fisher’s exact test. For example, the enrichment of the Δ‘AGGG’ motif in 3’ UTRs is shown below:
Author response table 1.

This test yields a P-value of 0.004167 by Fisher’s exact test. The P-values and Odds ratios of Δmotifs in relation to polysome shifting are included in Supplementary Table S4, and we will update the detailed motif information in the revised Supplementary Table S4.
(4) It is particularly puzzling how the authors can build a machine learning predictor with >3,000 features when the dataset they use for training the model has just a few dozens of translation-shifting variants.
We understand the concern regarding the relatively small number of translation-shifting variants compared to the large number of features. To address this, we employed LASSO regression, which, according to The Elements of Statistical Learning by Hastie, Tibshirani, and Friedman, is particularly suitable for datasets where the number of features 𝑝𝑝 is much larger than the number of samples 𝑁𝑁. LASSO effectively performs feature selection by shrinking less important coefficients to zero, allowing us to build a robust and generalizable model despite the limited number of variants.
(5) The lack of meaningful validation experiments altering the SNPs in the endogenous loci by genome editing limits the impact of the results.
We plan to assess the endogenous effect by generating CRISPR knock-in clones carrying the UTR variant.
Reviewer #2 (Public Review):
Summary:
In their paper "Massively Parallel Polyribosome Profiling Reveals Translation Defects of Human Disease‐Relevant UTR Mutations" the authors use massively parallel polysome profiling to determine the effects of 5' and 3' UTR SNPs (from dbSNP/ClinVar) on translational output. They show that some UTR SNPs cause a change in the polysome profile with respect to the wild-type and that pathogenic SNPs are enriched in the polysome-shifting group. They validate that some changes in polysome profiles are predictive of differences in translational output using transiently expressed luciferase reporters. Additionally, they identify sequence motifs enriched in the polysome-shifting group. They show that 2 enriched 5' UTR motifs increase the translation of a luciferase reporter in a proteindependent manner, highlighting the use of their method to identify translational control elements.
Strengths:
This is a useful method and approach, as UTR variants have been more difficult to study than coding variants. Additionally, their evidence that pathogenic mutations are more likely to cause changes in polysome association is well supported.
Weaknesses:
The authors acknowledge that they "did not intend to immediately translate the altered polysome profile into an increase or decrease in translation efficiency, as the direction of the shift was not readily evident. Additionally, sedimentation in the sucrose gradient may have been partially affected by heavy particles other than ribosomes." However, shifted polysome distribution is used as a category for many downstream analyses. Without further clarity or subdivision, it is very difficult to interpret the results (for example in Figure 5A, is it surprising that the polysome shifting mutants decrease structure? Are the polysome "shifts" towards the untranslated or heavy fractions?)
Our approach, combining polysome fractionation of the UTR library with negative binomial generalized linear model (GLM) analysis of RNA-seq data, systematically identifies variants that affect translational efficiency. The GLM model is specifically designed to detect UTR pairs with significant interactions between genotype and polysome fractions, relying solely on changes in polysome profiles to identify variants that disrupt translation. Consequently, our analytical method does not determine the direction of translation alteration.
Following the massively parallel polysome profiling, we sought to understand how these polysomeshifting variants influence the translation process. To do this, we examined their effects on RNA characteristics related to translation, such as RBP binding and RNA structure. In Figure 5A, we observed a notable trend in significant hits within 5’ UTRs—they tend to increase ΔG (weaker folding energy) in response to changes in polysome profiles, regardless of whether protein production increases or decreases (Fig. 3).



