Peer review process
Revised: This Reviewed Preprint has been revised by the authors in response to the previous round of peer review; the eLife assessment and the public reviews have been updated where necessary by the editors and peer reviewers.
Read more about eLife’s peer review process.Editors
- Reviewing EditorWolf-Dietrich HeyerUniversity of California, Davis, Davis, United States of America
- Senior EditorAdèle MarstonUniversity of Edinburgh, Edinburgh, United Kingdom
Reviewer #1 (Public review):
Overall, the data presented in this manuscript is of good quality. Understanding how cells control RPA loading on ssDNA is crucial to understanding DNA damage responses and genome maintenance mechanisms. The authors used genetic approaches to show that disrupting PCNA binding and SUMOylation of Srs2 can rescue the CPT sensitivity of rfa1 mutants with reduced affinity for ssDNA. In addition, the authors find that SUMOylation of Srs2 depends on binding to PCNA and the presence of Mec1.
Comments on revisions:
I am satisfied with the revisions made by the authors, which helped clarify some points that were confusing in the initial submission.
Reviewer #2 (Public review):
This revised manuscript mostly addresses previous concerns by doubling down on the model without providing additional direct evidence of interactions between Srs2 and PCNA, and that "precise sites of Srs2 actions in the genome remain to be determined." One additional Srs2 allele has been examined, showing some effect in combination with rfa1-zm2.
Many of the conclusions are based on reasonable assumptions about the consequences of various mutations, but direct evidence of changes in Srs2 association with PNCA or other interactors is still missing. There is an assumption that a deletion of a Rad51-interacting domain or a PCNA-interacting domain have no pleiotropic effects, which may not be the case. How SLX4 might interact with Srs2 is unclear to me, again assuming that the SLX4 defect is "surgical" - removing only one of its many interactions.
One point of concern is the use of t-tests without some sort of correction for multiple comparisons - in several figures. I'm quite sceptical about some of the p < 0.05 calls surviving a Bonferroni correction. Also in 4B, which comparison is **? Also, admittedly by eye, the changes in "active" Rad53 seem much greater than 5x. (also in Fig. 3, normalizing to a non-WT sample seems odd).
What is the WT doubling time for this strain? From the FACS it seems as if in 2 h the cells have completed more than 1 complete cell cycle. Also in 5D. Seems fast...
I have one over-arching confusion. Srs2 was shown initially to remove Rad51 from ssDNA and the suppression of some of srs2's defects by deleting rad51 made a nice, compact story, though exactly how srs2's "suppression of rad6" fit in isn't so clear (since Rad6 ties into Rad18 and into PCNA ubiquitylation and into PCNA SUMOylation). Now Srs2 is invoked to remove RPA. It seems to me that any model needs to explain how Srs2 can be doing both. I assume that if RPA and Rad51 are both removed from the same ssDNA, the ssDNA will be "trashed" as suggested by Symington's RPA depletion experiments. So building a model that accounts for selective Srs2 action at only some ssDNA regions might be enhanced by also explaining how Rad51 fits into this scheme.
As a previous reviewer has pointed out, CPT creates multiple forms of damage. Foiani showed that 4NQO would activate the Mec1/Rad53 checkpoint in G1- arrested cells, presumably because there would be single-strand gaps but no DSBs. Whether this would be a way to look specifically at one type of damage is worth considering; but UV might be a simpler way to look.
As also noted, the effects on the checkpoint and on viability are quite modest. Because it isn't clear (at least to me) why rfa1 mutants are so sensitive to CPT, it's hard for me to understand how srs2-zm2 has a modest suppressive effect: is it by changing the checkpoint response or facilitating repair or both? Or how srs2-3KR or srs2-dPIM differ from Rfa1-zm2 in this respect. The authors seem to lump all these small suppressions under the rubric of "proper levels of RPA-ssDNA" but there are no assays that directly get at this. This is the biggest limitation.
Srs2 has also been implicated as a helicase in dissolving "toxic joint molecules" (Elango et al. 2017). Whether this activity is changed by any of the mutants (or by mutations in Rfa1) is unclear. In their paper, Elango writes: "Rare survivors in the absence of Srs2 rely on structure-specific endonucleases, Mus81 and Yen1, that resolve toxic joint-molecules" Given the involvement of SLX4, perhaps the authors should examine the roles of structure-specific nucleases in CPT survival?
Experiments that might clarify some of these ambiguities are proposed to be done in the future. For now, we have a number of very interesting interactions that may be understood in terms of a model that supposes discriminating among gaps and ssDNA extensions by the presence of PCNA, perhaps modified by SUMO. As noted above, it would be useful to think about the relation to Rad6.
Reviewer #3 (Public review):
The superfamily I 3'-5' DNA helicase Srs2 is well known for its role as an anti-recombinase, stripping Rad51 from ssDNA, as well as an anti-crossover factor, dissociating extended D-loops and favoring non-crossover outcome during recombination. In addition, Srs2 plays a key role in in ribonucleotide excision repair. Besides DNA repair defects, srs2 mutants also show a reduced recovery after DNA damage that is related to its role in downregulating the DNA damage signaling or checkpoint response. Recent work from the Zhao laboratory (PMID: 33602817) identified a role of Srs2 in downregulating the DNA damage signaling response by removing RPA from ssDNA. This manuscript reports further mechanistic insights into the signaling downregulation function of Srs2.
Using the genetic interaction with mutations in RPA1, mainly rfa1-zm2, the authors test a panel of mutations in Srs2 that affect CDK sites (srs2-7AV), potential Mec1 sites (srs2-2SA), known sumoylation sites (srs2-3KR), Rad51 binding (delta 875-902), PCNA interaction (delta 1159-1163), and SUMO interaction (srs2-SIMmut). All mutants were generated by genomic replacement and the expression level of the mutant proteins was found to be unchanged. This alleviates some concern about the use of deletion mutants compared to point mutations. Double mutant analysis identified that PCNA interaction and SUMO sites were required for the Srs2 checkpoint dampening function, at least in the context of the rfa1-zm2 mutant. There was no effect of this mutants in a RFA1 wild type background. This latter result is likely explained by the activity of the parallel pathway of checkpoint dampening mediated by Slx4, and genetic data with an Slx4 point mutation affecting Rtt107 interaction and checkpoint downregulation support this notion. Further analysis of Srs2 sumoylation showed that Srs2 sumoylation depended on PCNA interaction, suggesting sequential events of Srs2 recruitment by PCNA and subsequent sumoylation. Kinetic analysis showed that sumoylation peaks after maximal Mec1 induction by DNA damage (using the Top1 poison camptothecin (CPT)) and depended on Mec1. This data are consistent with a model that Mec1 hyperactivation is ultimately leading to signaling downregulation by Srs2 through Srs2 sumoylation. Mec1-S1964 phosphorylation, a marker for Mec1 hyperactivation and a site found to be needed for checkpoint downregulation after DSB induction, did not appear to be involved in checkpoint downregulation after CPT damage. The data are in support of the model that Mec1 hyperactivation when targeted to RPA-covered ssDNA by its Ddc2 (human ATRIP) targeting factor, favors Srs2 sumoylation after Srs2 recruitment to PCNA to disrupt the RPA-Ddc2-Mec1 signaling complex. Presumably, this allows gap filling and disappearance of long-lived ssDNA as the initiator of checkpoint signaling, although the study does not extend to this step.
Strengths
(1) The manuscript focuses on the novel function of Srs2 to downregulate the DNA damage signaling response and provide new mechanistic insights.
(2) The conclusions that PCNA interaction and ensuing Srs2-sumoylation are involved in checkpoint downregulation are well supported by the data.
Weaknesses
(1) Additional mutants of interest could have been tested, such as the recently reported Pin mutant, srs2-Y775A (PMID: 38065943), and the Rad51 interaction point mutant, srs2-F891A (PMID: 31142613).
(2) The use of deletion mutants for PCNA and RAD51 interaction is inferior to using specific point mutants, as done for the SUMO interaction and the sites for post-translational modifications.
(3) Figure 4D and Figure 5A report data with standard deviations, which is unusual for n=2. Maybe the individual data points could be plotted with a color for each independent experiment to allow the reader to evaluate the reproducibility of the results.
Comments on revisions:
In this revision, the authors adequately addressed my concerns. The only issue I see remaining is the site of Srs2 action. The authors argue in favor of gaps and against R-loops and ssDNA resulting from excessive supercoiling. The authors do not discuss ssDNA resulting from processing of one-sided DSBs, which are expected to result from replication run-off after CPT damage but are not expected to provide the 3'-junction for preferred PCNA loading. Can the authors exclude PCNA at the 5'-junction at a resected DSB?
Author response:
The following is the authors’ response to the original reviews.
eLife Assessment
This manuscript reports valuable findings on the role of the Srs2 protein in turning off the DNA damage signaling response initiated by Mec1 (human ATR) kinase. The data provide solid evidence that Srs2 interaction with PCNA and ensuing SUMO modification is required for checkpoint downregulation. However, experimental evidence with regard to the model that Srs2 acts at gaps after camptothecin-induced DNA damage is currently lacking. The work will be of interest to cell biologists studying genome integrity but would be strengthened by considering the possible role of Rad51 and its removal.
We thank editors and reviewers for their constructive comments and address their main criticisms below.
(1) Srs2 action sites. Our data provide support to the model that Srs2 removal of RPA is favored at ssDNA regions with proximal PCNA, but not at ssDNA regions lacking proximal PCNA. A prominent example of the former type of ssDNA regions is an ssDNA gap with a 3’ DNA end permissive for PCNA loading. Examples of the latter type of ssDNA sites include those within R-loops and negatively supercoiled regions, both lacking 3’ DNA end required for PCNA loading. The former type of ssDNA regions can recruit other DNA damage checkpoint proteins, such as 9-1-1, which requires a 5’ DNA end for loading; thus, these ssDNA regions are ideal for Srs2’s action in checkpoint dampening. In contrast, ssDNA within supercoiled and Rloop regions, both of which can be induced by CPT treatment (Pommier et al, 2022), lacks the DNA ends required for checkpoint activation. RPA loaded at these sites plays important roles, such as recruiting Rloop removal factors (Feng and Manley, 2021; Li et al, 2024; Nguyen et al, 2017), and they are not ideal sites for Srs2’s checkpoint dampening functions. Based on the above rationale and our data, we suggest that Srs2 removal of RPA is favored only at a subset of ssDNA regions prone to checkpoint activation and can be avoided at other ssDNA regions where RPA mainly helps DNA protection and repair. We have modified the text and model drawing to better articulate the implications of our work, that is, Srs2 can distinguish between two types of ssDNA regions by using PCNA proximity as a guide for RPA removal_._ We noted that the precise sites of Srs2 actions in the genome remain to be determined.
(2) Rad51 in the Srs2-RPA antagonism. In our previous report (Dhingra et al, 2021), we provided several lines of evidence to support the conclusion that Rad51 is not relevant to the Srs2-RPA antagonism, despite it being the best-studied protein that is regulated by Srs2. For example, while rad51∆ rescues the hyperrecombination phenotype of srs2∆ cells as shown by others, we found that rad51∆ did not affect the hypercheckpoint phenotype of srs2∆. In contrast, rfa1-zm1/zm2 have the opposite effects. The differential effects of rad51∆ and rfa1-zm1/zm2 were also seen for the srs2-ATPase dead allele (srs2-K41A). For example, rfa1-zm2 rescued the hyper-checkpoint defect and the CPT sensitivity of srs2-K41A, while rad51∆ had neither effect. These and other data described by Dhingra et al (2021) suggest that Srs2’s effects on checkpoint vs. recombination can be separated and that Rad51 removal by Srs2 is distinct from the Srs2RPA antagonism in checkpoint regulation. Given the functional separation summarized above, in our current work investigating which Srs2 features affect the Srs2-RPA antagonism, we did not focus on the role of Rad51. However, we did examine all known features of Srs2, including its Rad51 binding domain. Consistent with our conclusion summarized above, deleting the Rad51 binding domain in Srs2 (srs2∆Rad51BD) has no effect on rfa1-zm2 phenotype in CPT (Figure 2D). This data provides yet another evidence that Srs2 regulation of Rad51 is separable from the Srs2-RPA antagonism. Our work provides a foundation for future examination of how Srs2 regulates RPA and Rad51 in different manners and if there is a crosstalk between them in specific contexts. We have added this point to the revised text.
Public Reviews:
Reviewer #1.
Overall, the data presented in this manuscript is of good quality. Understanding how cells control RPA loading on ssDNA is crucial to understanding DNA damage responses and genome maintenance mechanisms. The authors used genetic approaches to show that disrupting PCNA binding and SUMOylation of Srs2 can rescue the CPT sensitivity of rfa1 mutants with reduced affinity for ssDNA. In addition, the authors find that SUMOylation of Srs2 depends on binding to PCNA and the presence of Mec1. Noted weaknesses include the lack of evidence supporting that Srs2 binding to PCNA and its SUMOylation occur at ssDNA gaps, as proposed by the authors. Also, the mutants of Srs2 with impaired binding to PCNA or impaired SUMOylation showed no clear defects in checkpoint dampening, and in some contexts, even resulted in decreased Rad53 activation. Therefore, key parts of the paper would benefit from further experimentation and/or clarification.
We thank the reviewer for the positive comments, and we address her/his remark regarding ssDNA gaps below. In addition, we provide evidence that redundant pathways can mask checkpoint dampening phenotype of the srs2-∆PIM and -3KR alleles.
Major Comments
(1) The central model proposed by the authors relies on the loading of PCNA at the 3' junction of an ssDNA gap, which then mediates Srs2 recruitment and RPA removal. While several aspects of the model are consistent with the data, the evidence that it is occurring at ssDNA gaps is not strong. The experiments mainly used CPT, which generates mostly DSBs. The few experiments using MMS, which mostly generates ssDNA gaps, show that Srs2 mutants lead to weaker rescue in this context (Figure S1). How do the authors explain this discrepancy? In the context of DSBs, are the authors proposing that Srs2 is engaging at later steps of HRdriven DSB repair where PCNA gets loaded to promote fill-in synthesis? If so, is RPA removal at that step important for checkpoint dampening? These issues need to be addressed and the final model adjusted.
Our data provide supports to the model that Srs2 removal of RPA is favored at ssDNA regions with proximal PCNA, but not at ssDNA regions lacking proximal PCNA (Figure 7). A prominent example of the former type is ssDNA gap with 3’ DNA end permissive for PCNA loading. Examples of the latter type of ssDNA sites are present within R-loops and negatively supercoiled regions, and these ssDNA sites lack 3’ DNA ends required for PCNA loading. In principle, the former can recruit other DNA damage checkpoint proteins, such as 9-1-1, which requires 5’ DNA end for loading, thus it is ideal for Srs2’s action in checkpoint dampening. In contrast, ssDNA within supercoiled and R-loop regions, which can be induced by CPT treatment (Pommier et al., 2022), lacks DNA ends required for checkpoint activation. RPA loaded at these sites plays important roles such as recruiting R-loop removal factors (Feng and Manley, 2021; Li et al., 2024; Nguyen et al., 2017), and these are not ideal sites for Srs2 removal of RPA to achieve checkpoint dampening. Our work suggests that Srs2 removal of RPA is favored only at a subset of ssDNA regions prone to checkpoint activation and can be avoided at other ssDNA regions where RPA mainly helps DNA protection and repair. We have modified the text and the model to clarify our conclusions and emphasized that Srs2 can distinguish between two types of ssDNA regions using PCNA proximity as a guide for RPA removal.
We note that in addition to DSBs, CPT also induces both types of ssDNA mentioned above. For example, CPT can lead to ssDNA gap formation upon excision repair or DNA-protein crosslink repair of trapped Top1 (Sun et al, 2020). The resultant ssDNA regions contain 3’ DNA end for PCNA loading, thus favoring Srs2 removal of RPA. CPT treatment also depletes the functional pool of Top1, thus causing topological stress and increased levels of DNA supercoiling and R-loops (Petermann et al, 2022; Pommier et al., 2022). As mentioned above, R-loops and supercoiled regions do not favor Srs2 removal of RPA due to a lack of PCNA loading. We have now adjusted the text to clarify that CPT can lead to the generation of two types of ssDNA regions as stated above. We have also adjusted the model drawing to indicate that while ssDNA gaps can be logical Srs2 action sites, other types of ssDNA regions with proximal PCNA (e.g., resected ssDNA tails) could also be targeted by Srs2. Our work paves the way to determine the precise ssDNA regions for Srs2’s action.
Multiple possibilities should be considered in explaining the less potent suppression of rfa1 mutants by srs2 alleles in MMS compared to CPT conditions. For example, MMS and CPT affect checkpoints differently. While CPT only activates the DNA damage checkpoint, MMS additionally induces DNA replication checkpoint (Menin et al, 2018; Redon et al, 2003; Tercero et al, 2003). It is possible that the Srs2-RPA antagonism is more relevant to the DNA damage checkpoint compared with the DNA replication checkpoint. Further investigation of this possibility among other scenarios will shed light on differential suppression seen here. We have included this discussion in the revised text.
(2) The data in Figure 3 showing that Srs2 mutants reduce Rad53 activation in the rfa1-zm2 mutant are confusing, especially given the claim of an anti-checkpoint function for Srs2 (in which case Srs2 mutants should result in increased Rad53 activation). The authors propose that Rad53 is hyperactivated in rfa1-zm2 mutant because of compromised ssDNA protection and consequential DNA lesions, however, the effects sharply contrast with the central model. Are the authors proposing that in the rfa1-zm2 mutant, the compromised protection of ssDNA supersedes the checkpoint-dampening effect? Perhaps a schematic should be included in Figure 3 to depict these complexities and help the reader. The schematic could also include the compensatory dampening mechanisms like Slx4 (on that note, why not move Figure S2 to a main figure?... and even expand experiments to better characterize the compensatory mechanisms, which seem important to help understand the lack of checkpoint dampening effect in the Srs2 mutants)
Partially defective alleles often do not manifest null phenotype. In this case, while srs2∆ increases Rad53 activation (Dhingra et al., 2021), srs2-∆PIM and -3KR did not (Figure 3A-3B). However, srs2-∆PIM did increase Rad53 activation when combined with another checkpoint dampening mutant slx4RIM (now Figure 4B-4C). This result suggests that defects of partially defective srs2 alleles can be masked by Slx4. Further, srs2-∆PIM and 3KR rescued rfa1-zm2’s checkpoint abnormality (now Figure 3B-3C), suggesting that Srs2 binding to PCNA and its sumoylation contribute to the Srs2-RPA antagonism in the DNA damage checkpoint response.
Partially defective alleles that impair specific features of a protein without producing null phenotype have been used widely to reveal biological mechanisms. For example, a partially defective allele of the checkpoint protein Rad9 perturbing binding to gamma-H2A (rad9-K1088M) does not cause DNA damage sensitivity on its own, due to the compensation from other checkpoint factors (Hammet et al, 2007). However_, rad9-K1088M_ rescues the DNA damage sensitivity and persistent G2/M checkpoint of slx4 mutants, providing strong evidence for the notion that Slx4 dampens checkpoint via regulating Rad9 (Ohouo et al, 2013).
We have now indicated that our model highlights the checkpoint recovery process and does not depict another consequence of the Srs2-RPA antagonism, that is, rfa1 DNA binding mutants can lead to increased levels of DNA lesions and consequently stronger checkpoint activation, which are rescued by lessening Srs2’s ability to strip RPA from DNA (Dhingra et al., 2021). We have stated these points more clearly in the text and added a schematic (Figure 3A) to outline the genetic relationship and interpretations. We also moved Figure S2 to the main figures (Figure 4), as suggested by the reviewer. Better characterizing the compensatory mechanisms among the multiple checkpoint dampening pathways requires substantial amounts of work that will be pursued in the future.
(3) The authors should demarcate the region used for quantifying the G1 population in Figure 3B and explain the following discrepancy: By inspection of the cell cycle graph, all mutants have lower G1 peak height compared to WT (CPT 2h). However, in the quantification bar graph at the bottom, ΔPIM has higher G1 population than the WT.
We now describe how the G1 region of the FACS histogram was selected to derive the percentage of G1 cells in Figure 3B (now Figure 3C). Briefly, the G1 region from the “G1 sample” was used to demarcate the G1 region of the “CPT 2h” sample. We noticed that a mutant panel was mistakenly put in the place of wild-type, and this error is now corrected. The conclusion remains that srs2-∆PIM and srs2-3KR improved rfa1-zm2 cells’ ability to exit G2/M, while they themselves do not show difference from the wild-type control for the percentage of G1 cells after 2hr CPT treatment. We have added statistics in Figure 3C that support this conclusion.
Reviewer #2:
This is an interesting paper that delves into the post-translational modifications of the yeast Srs2 helicase and proteins with which it interacts in coping with DNA damage. The authors use mutants in some interaction domains with RPA and Srs2 to argue for a model in which there is a balance between RPA binding to ssDNA and Srs2's removal of RPA. The idea that a checkpoint is being regulated is based on observing Rad53 and Rad9 phosphorylation (so there are the attributes of a checkpoint), but evidence of cell cycle arrest is lacking. The only apparent delay in the cell cycle is the re-entry into the second S phase (but it could be an exit from G2/M); but in any case, the wild-type cells enter the next cell cycle most rapidly. No direct measurement of RPA residence is presented.
We thank the reviewer for the helpful comments. Previous studies have shown that CPT does not induce the DNA replication checkpoint, and thus does not slow down or arrest S phase progression; however, CPT does induce the DNA damage checkpoint, which causes a delay (not arrest) in G2/M phase and re-entering into the second G1 (Menin et al., 2018; Redon et al., 2003). Our result is consistent with these findings, showing that CPT induces G2/M delay but not arrest. We have now made this point clearer in the text.
We have previously reported chromatin-bound RPA levels in rfa1-zm2, srs2, and their double mutants, as well as in vitro ssDNA binding by wild-type and mutant RPA complexes (Dhingra et al., 2021). These data showed that Srs2 loss or its ATPase dead mutant led to 4-6-fold increase of RPA levels on chromatin, which was rescued by rfa1-zm2 (Dhingra et al., 2021). On its own, rfa1-zm2 did not cause defective chromatin association, despite modestly reducing ssDNA binding in vitro (Dhingra et al., 2021). This discrepancy could be due to a lack of sensitivity of the chromatin fractionation assay in revealing moderate changes of RPA residence on DNA in vivo. Our functional assays (Figure 2-3) were more effective in identifying the Srs2 features pertaining to RPA regulation.
Strengths:
Data concern viability assays in the presence of camptothecin and in the post-translational modifications of Srs2 and other proteins.
Weaknesses:
There are a couple of overriding questions about the results, which appear technically excellent. Clearly, there is an Srs2-dependent repair process here, in the presence of camptothecin, but is it a consequence of replication fork stalling or chromosome breakage? Is repair Rad51-dependent, and if so, is Srs2 displacing RPA or removing Rad51 or both? If RPA is removed quickly what takes its place, and will the removal of RPA result in lower DDC1-MEC1 signaling?
Srs2 can affect both the checkpoint response and DNA repair processes in CPT conditions. However, rfa1zm2 mainly affects the former role of Srs2; this allows us to gain a deeper understanding of this role, which is critical for cell survival in CPT (Dhingra et al., 2021). Building on this understanding, our current study identified two Srs2 features that could afford spatial and temporal regulation of RPA removal from DNA, providing a rationale for how cells can properly utilize an activity that can be beneficial yet also dangerous if it were to lack regulation. Study of Srs2-mediated DNA repair in CPT conditions, either in Rad51-dependent or -independent manner, to deal with replication fork stalling or DNA breaks will require studies in the future.
Moreover, it is worth noting that in single-strand annealing, which is ostensibly Rad51 independent, a defect in completing repair and assuring viability is Srs2-dependent, but this defect is suppressed by deleting Rad51. Does deleting Rad51 have an effect here?
We have previously shown that rad51∆ did not rescue the hyper-checkpoint phenotype of srs2∆ cells in CPT conditions, while rfa1-zm1 and -zm2 did (Dhingra et al., 2021). This differential effect was also seen for the srs2 ATPase-dead allele (Dhingra et al., 2021). These and other data described by Dhingra et al (2021) suggest that Srs2’s effects on checkpoint vs. recombination are separable at least in CPT condition, and that the Srs2-RPA antagonism in checkpoint regulation is not affected by Rad51 removal (unlike in SSA).
Neither this paper nor the preceding one makes clear what really is the consequence of having a weakerbinding Rfa1 mutant. Is DSB repair altered? Neither CPT nor MMS are necessarily good substitutes for some true DSB assay.
We have previously showed that rfa1-zm1/zm2 did not affect the frequencies of rDNA recombination, gene conversation, or direct repeat repair (Dhingra et al., 2021). Further, rfa1-zm1/zm2 did not suppress the hyperrecombination phenotype of srs2∆, while rad51∆ did (Dhingra et al., 2021). In a DSB system, wherein the DNA repeats flanking the break were placed 30 kb away from each other, srs2∆ led to hyper-checkpoint and lethality, both of which were rescued by rfa1-zm mutants (Dhingra et al., 2021). In this assay, rfa1-zm1/zm2 did not show sensitivity, suggesting largely proficient DNA repair. Collectively, these data suggest that moderately weakening DNA binding of Rfa1 does not lead to detectable effect on the recombinational repair examined thus far, rather it affects Srs2-mediated checkpoint downregulation. In-depth studies of rfa1-zm mutations in the context of various DSB repair steps will be interesting to pursue in the future.
With camptothecin, in the absence of site-specific damage, it is difficult to test these questions directly. (Perhaps there is a way to assess the total amount of RPA bound, but ongoing replication may obscure such a measurement). It should be possible to assess how CPT treatment in various genetic backgrounds affects the duration of Mec1/Rad53-dependent checkpoint arrest, but more than a FACS profile would be required.
Quantitative measurement of RPA residence time on DNA in cellular context and the duration of the
Mec1/Rad53-mediated cell cycle delay/arrest will be informative but requires further technology development. Our current work provides a foundation for such quantitative assessment.
It is also notable that MMS treatment does not seem to yield similar results (Fig. S1).
Figure S1 showed that srs2-∆PIM and srs2-3KR had weaker suppression of rfa1-zm2 growth on MMS plates than on CPT plates. Multiple possibilities should be considered in explaining the less potent suppression of rfa1 mutants by srs2 in MMS compared with CPT conditions. For example, MMS and CPT affect checkpoints differently. While CPT only activates the DNA damage checkpoint, MMS additionally induces DNA replication checkpoint (Menin et al., 2018; Redon et al., 2003; Tercero et al., 2003). It is therefore possible that the Srs2RPA antagonism is more relevant for the DNA damage checkpoint control compared with the DNA replication checkpoint. Further investigation of this possibility will shed light on differential suppression seen here. We have included this discussion in the revised text.
Reviewer #3:
The superfamily I 3'-5' DNA helicase Srs2 is well known for its role as an anti-recombinase, stripping Rad51 from ssDNA, as well as an anti-crossover factor, dissociating extended D-loops and favoring non-crossover outcome during recombination. In addition, Srs2 plays a key role in ribonucleotide excision repair. Besides DNA repair defects, srs2 mutants also show a reduced recovery after DNA damage that is related to its role in downregulating the DNA damage signaling or checkpoint response. Recent work from the Zhao laboratory (PMID: 33602817) identified a role of Srs2 in downregulating the DNA damage signaling response by removing RPA from ssDNA. This manuscript reports further mechanistic insights into the signaling downregulation function of Srs2.
Using the genetic interaction with mutations in RPA1, mainly rfa1-zm2, the authors test a panel of mutations in Srs2 that affect CDK sites (srs2-7AV), potential Mec1 sites (srs2-2SA), known sumoylation sites (srs2-3KR), Rad51 binding (delta 875-902), PCNA interaction (delta 1159-1163), and SUMO interaction (srs2SIMmut). All mutants were generated by genomic replacement and the expression level of the mutant proteins was found to be unchanged. This alleviates some concern about the use of deletion mutants compared to point mutations. The double mutant analysis identified that PCNA interaction and SUMO sites were required for the Srs2 checkpoint dampening function, at least in the context of the rfa1-zm2 mutant. There was no effect of these mutants in a RFA1 wild-type background. This latter result is likely explained by the activity of the parallel pathway of checkpoint dampening mediated by Slx4, and genetic data with an Slx4 point mutation affecting Rtt107 interaction and checkpoint downregulation support this notion. Further analysis of Srs2 sumoylation showed that Srs2 sumoylation depended on PCNA interaction, suggesting sequential events of Srs2 recruitment by PCNA and subsequent sumoylation. Kinetic analysis showed that sumoylation peaks after maximal Mec1 induction by DNA damage (using the Top1 poison camptothecin (CPT)) and depended on Mec1. These data are consistent with a model that Mec1 hyperactivation is ultimately leading to signaling downregulation by Srs2 through Srs2 sumoylation. Mec1-S1964 phosphorylation, a marker for Mec1 hyperactivation and a site found to be needed for checkpoint downregulation after DSB induction did not appear to be involved in checkpoint downregulation after CPT damage. The data are in support of the model that Mec1 hyperactivation when targeted to RPA-covered ssDNA by its Ddc2 (human ATRIP) targeting factor, favors Srs2 sumoylation after Srs2 recruitment to PCNA to disrupt the RPA-Ddc2-Mec1 signaling complex. Presumably, this allows gap filling and disappearance of long-lived ssDNA as the initiator of checkpoint signaling, although the study does not extend to this step.
Strengths
(1) The manuscript focuses on the novel function of Srs2 to downregulate the DNA damage signaling response and provide new mechanistic insights.
(2) The conclusions that PCNA interaction and ensuing Srs2-sumoylation are involved in checkpoint downregulation are well supported by the data.
We thank the reviewer for carefully reading our work and for his/her positive comments.
Weaknesses
(1) Additional mutants of interest could have been tested, such as the recently reported Pin mutant, srs2Y775A (PMID: 38065943), and the Rad51 interaction point mutant, srs2-F891A (PMID: 31142613).
Residue Y775 of Srs2 was shown to serve as a separation pin in unwinding D-loops and dsDNA with 3’ overhang in vitro; however, srs2-Y775A lacks cellular phenotype in assays for gene conversion, crossover, and genetic interactions. As such, the biological role of this residue has not been clear. In addressing reviewer’s comment, we obtained srs2-Y775A, and the control strains as described in the recent publication (Meir et al, 2023). While srs2-Y775A on its own did not affect CPT sensitivity, it improved rfa1-zm_2 mutant growth on media containing CPT. This result suggests that Y775 can influence RPA regulation during in checkpoint dampening. Given that truncated Srs2 (∆Cter 276 a.a.) containing Y775A showed normal RPA stripping activity _in vitro, it is possible that cellular assay using rfa1-zm2 is more sensitive for revealing defect of this activity or full-length protein is required for manifest Y775A effect. Future experiments distinguishing these possibilities can provide more clarity. Nevertheless, our result reveals the first phenotype of Srs2 separation pin mutant. We have added this new result (Figure S4) and our interpretation.
We have already included data showing that a srs2 mutant lacking the Rad51 binding domain (srs2∆Rad51BD, ∆875-902) did not affect rfa1-zm2 growth in CPT nor caused defects in CPT on its own (Figure 2D). This data suggest that Rad51 binding is not relevant to the Srs2-RPA antagonism in CPT, a conclusion fully supported by data in our previous study (Dhingra et al., 2021). Collectively, these findings do not provide a strong rationale to test a point mutation within the Rad51BD region.
(2) The use of deletion mutants for PCNA and RAD51 interaction is inferior to using specific point mutants, as done for the SUMO interaction and the sites for post-translational modifications.
We generally agree with this view. However, it is less of a concern in the context of the Rad51 binding site mutant (srs2-∆Rad51BD) since it behaved as the wild-type allele in our assays. The srs2-∆PIM mutant (lacking 4 amino acids) has been examined for PCNA binding in vitro and in vivo (Kolesar et al, 2016; Kolesar et al, 2012); to our knowledge no detectable defect was reported. Thus, we believe that this allele is suitable for testing whether Srs2’s ability to bind PCNA is relevant to RPA regulation.
(3) Figure 4D and Figure 5A report data with standard deviations, which is unusual for n=2. Maybe the individual data points could be plotted with a color for each independent experiment to allow the reader to evaluate the reproducibility of the results.
We have included individual data points as suggested and corrected figure legend to indicate that three independent biological samples per genotype were examined in both panels.
References:
Dhingra N, Kuppa S, Wei L, Pokhrel N, Baburyan S, Meng X, Antony E, Zhao X (2021) The Srs2 helicase dampens DNA damage checkpoint by recycling RPA from chromatin. Proc Natl Acad Sci U S A 118: e2020185118.
Feng S, Manley JL (2021) Replication Protein A associates with nucleolar R loops and regulates rRNA transcription and nucleolar morphology. Genes Dev 35: 1579-1594.
Fiorani S, Mimun G, Caleca L, Piccini D, Pellicioli A (2008) Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle 7: 493-499.
Hammet A, Magill C, Heierhorst J, Jackson SP (2007) Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep 8: 851-857.
Kolesar P, Altmannova V, Silva S, Lisby M, Krejci L (2016) Pro-recombination role of Srs2 protein requires SUMO (Small Ubiquitin-like Modifier) but is independent of PCNA (Proliferating Cell Nuclear Antigen) interaction. J Biol Chem 291: 7594-7607.
Kolesar P, Sarangi P, Altmannova V, Zhao X, Krejci L (2012) Dual roles of the SUMO-interacting motif in the regulation of Srs2 sumoylation. Nucleic Acids Res 40: 7831-7843.
Li Y, Liu C, Jia X, Bi L, Ren Z, Zhao Y, Zhang X, Guo L, Bao Y, Liu C et al (2024) RPA transforms RNase H1 to a bidirectional exoribonuclease for processive RNA-DNA hybrid cleavage. Nat Commun 15: 7464.
Meir A, Raina VB, Rivera CE, Marie L, Symington LS, Greene EC (2023) The separation pin distinguishes the pro- and anti-recombinogenic functions of Saccharomyces cerevisiae Srs2. Nat Commun 14: 8144.
Memisoglu G, Lanz MC, Eapen VV, Jordan JM, Lee K, Smolka MB, Haber JE (2019) Mec1(ATR) autophosphorylation and Ddc2(ATRIP) phosphorylation regulates dna damage checkpoint signaling. Cell Rep 28: 1090-1102 e1093.
Menin L, Ursich S, Trovesi C, Zellweger R, Lopes M, Longhese MP, Clerici M (2018) Tel1/ATM prevents degradation of replication forks that reverse after Topoisomerase poisoning. EMBO Rep 19: e45535.
Nguyen HD, Yadav T, Giri S, Saez B, Graubert TA, Zou L (2017) Functions of Replication Protein A as a sensor of R loops and a regulator of RNaseH1. Mol Cell 65: 832-847 e834.
Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB (2013) DNA-repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493: 120-124.
Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, Ulrich HD (2005) Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 19: 123-133.
Petermann E, Lan L, Zou L (2022) Sources, resolution and physiological relevance of R-loops and RNA-DNA hybrids. Nat Rev Mol Cell Biol 23: 521-540.
Pommier Y, Nussenzweig A, Takeda S, Austin C (2022) Human topoisomerases and their roles in genome stability and organization. Nat Rev Mol Cell Biol 23: 407-427.
Redon C, Pilch DR, Rogakou EP, Orr AH, Lowndes NF, Bonner WM (2003) Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO Rep 4: 678-684.
Sun Y, Saha S, Wang W, Saha LK, Huang SN, Pommier Y (2020) Excision repair of topoisomerase DNAprotein crosslinks (TOP-DPC). DNA Repair (Amst) 89: 102837.
Tercero JA, Longhese MP, Diffley JFX (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323-1336.
Reviewer #1 (Recommendations For The Authors):
(1) "the srs2-ΔPIM (Δ1159-1163 amino acids)". "11" should not be italic.
Corrected.
(2) "the srs2-SIMmut (1170 IIVID 1173 to 1170 AAAAD 1173)". "1173" should be 1174.
Corrected.
(3) Can Slx4-RIM mutant rescue rfa1-zm2 CPT sensitivity?
We found that unlike srs2∆, slx4∆ failed to rescue rfa1-zm2 CPT sensitivity (picture on the right). On the other hand, slx4∆ counteracts Rad9-dependent Rad53 activation as shown by Ohouo et al (2013).
Author response image 1.
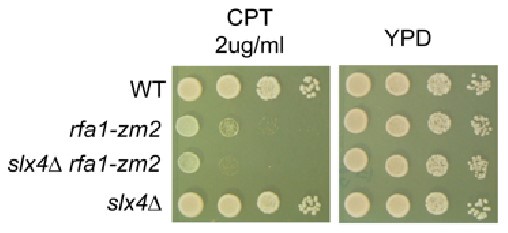
(4) One genotype (rfa1-zm2 srs2-3KR) is missing in Figure 5B.
Corrected.
(5) In Fig. S2C, FACS plots do not match the bar graph (see major concern 3).
Corrected and is described in more detail in Major Concern #3.
Reviewer #2 (Recommendations For The Authors):
Figure 1. The colors in A are not well-conserved in B.
Colors for srs2-7AV and -2SA in panel B are now matched with those in panel A.
Figure 2. Is srs2-SIMmut the same as srs2-sim?
This mutant allele is now referred to as srs2-SIMmut throughout the text and figures.
The suppression of rfa1-zm2 and (less strongly) rfa-t33 by the Srs2 mutants is interesting. Based on previous data, the suppression is apparently mutual, though it isn't shown here, unless we misunderstand.
We have previously shown that rfa1-zm2 and srs2∆ showed mutual suppression (Dhingra et al 2021 PNAS) and have included an example in Figure S1A. Unlike srs2∆, srs2-∆PIM and -3KR showed little damage sensitivity and DDC defects, likely due to the compensation by the Slx4-mediated checkpoint dampening (detailed in the Public Review section). Suppression is not applicable toward mutants lacking a phenotype, though the mutants could confer suppression when there is a functional relationship with another mutant, as we see here toward rfa1-zm2.
Is Srs2 interaction with PCNA dependent on its ubiquitylation or SUMO? Does PCNA mutant K164R mimic this mutation? (this may well be known; our ignorance).
It was known that Srs2 can bind unmodified PCNA, though SUMO enhances this interaction; however, a very small percentage of PCNA is sumoylated in cells and PCNA sumoylation affects both Srs2-dependent and independent processes (e.g., (Papouli et al, 2005). As such, the genetic interaction of K164R with rfa1-zm2 can be difficult to interpret.
Why srs2-7AV or srs2-sim make rfa1-zm2 even more sensitive is also not obvious. The authors take refuge in the statement that Srs2 "has multiple roles in cellular survival of genotoxic stress" but don't attempt to be more precise.
Our understanding of srs2-7AV and -sim is limited; thus, more specific speculation cannot be made at this time.
Figure 3. It is striking (Figure 3A) that all the cells have reached G2 an hour after releasing from alpha-factor arrest, even though presumably CPT treatment must impair replication. It is even more striking that there is apparently no G2/M arrest in the presumably damaged cells as the WT (Figure 3B) has the most rapid progression through the cell cycle. How does this compare with cells in the absence of CPT? The idea that CPT is triggering Rad53-mediated response is hard to understand if there is in fact no delay in the cell cycle. Instead, the several mutants appear to delay re-entry into S... Or maybe it is actually an exit from G2/M?
This phenomenon needs a better explanation.
CPT does not induce the DNA replication checkpoint nor S phase delay, explaining apparent G2 content by the one hour time point; however, CPT does induce the DNA damage checkpoint, and a delay (not arrest) in G2/M (Menin et al., 2018; Redon et al., 2003; Tercero et al., 2003). We confirmed these findings. In our hand, wildtype G1 cells released into the cell cycle in the absence of CPT complete the first cell cycle within 80 minutes, such that most cells are in the second G1 phase by 90 min. In contrast, when wild-type cells were treated with CPT, G2/M exit was only partial at 120min (e.g., Figure 3B). These features differentiate CPT treatment from MMS treatment, which induces both types of checkpoints and lengthening the time that cells reach G2. We have highlighted this unique feature of CPT in checkpoint induction.
What is "active Rad53"? If the authors mean they are using a phospho-specific Ab versus Rad53, they should explain this. It's impossible to know if total Rad53 is altered from Figure 3A. A blot with an antibody that detects both phosphorylated and nonphosphorylated Rad53 would help.
The F9 antibody used here detects phosphorylated Rad53 forms induced by Mec1 activation and does not detect unphosphorylated Rad53 (Fiorani et al, 2008). We changed “active Rad53” to “phosphorylated Rad53”. We used Pgk1 as a loading control to ensure equal loading, which help to quantify the relative amount of “active Rad53” in cells. This method has been used widely in the field.
Also is there a doublet of Rad53 in the right two lanes and in WT? Rad53 often shows more than one slowmigrating species, so this isn't necessarily a surprise. Were both forms used in quantitation?
Both forms are used for quantification.
Figure 4A. Is there a di-SUMO form above the band marked Srs2-Su? Is this known? Is it counted?
Mono-sumoylated form of Srs2 is the most abundant form of sumoylated Srs2, though we detected a sumoylated Srs2 band that can represent its di-sumo form. We did quantify both forms in the plot.
B. The dip at 1.5 h in Rad9-P is curious. It would be useful to know what % of Rad9 is phosphorylated in a repair-defective (rad52?) background with CPT treatment. And would such rad52 cells show a long arrest?
This dip is reproducible and may reflect that a population of cells escape G2/M delay at this timepoint.
Figure 5. It seems clear that the autophosphorylation site of Mec1, which was implicated in turning off a longdelayed G2/M arrest has no effect here, but presumably, a kinase-dead Mec1 (or deletion) does? The idea that a checkpoint is being regulated seems to come more from an assumption than from any direct data; as noted above, the only apparent delay in the cell cycle is the re-entry into S. There clearly is Rad53 and Rad9 phosphorylation so there are the attributes of a checkpoint. If PI3KK phosphorylation is important, can this be accomplished by Tel1 as well as Mec1?
A mec1 helicase dead or null would not activate the checkpoint at the first place, therefore will not be useful to address whether Mec1 autophosphorylation is implicated in turning off checkpoint. A recent study from the Haber lab provided evidence that Mec1 autophosphorylation at S1964 helps to turn off the checkpoint in a DSB situation (Memisoglu et al, 2019). The role of Tel1 in checkpoint dampening will be interesting to examine in the future.
Figure 6. Two Rfa1 phospho-sites don't appear to be important, but do the known multiple phosphorylations of Rfa2 play a role?
Figure 6D examined three Rfa2 phosphorylation sites and found no genetic interaction with srs2∆.
Summary: There are a lot of interesting data here, but they don't strongly support the author's model in the absence of a more direct way to monitor RPA binding and removal. This could be done using some sitespecific damage, but hard to do with CPT or MMS (which themselves don't appear to have the same effect). The abstract suggests Srs2 is "temporally and spatially regulated to both allow timely checkpoint termination and to prevent superfluous RPA removal." But where is the checkpoint termination if there's no evident checkpoint? And "superfluous" is probably not the right word (= unnecessary); probably the authors intend "excessive"? As noted above, it also isn't clear if the displacement is of RPA or of Rad51, which normally replaces RPA and which is well-known to be itself displaced by Srs2. Again, if CPT is causing enough damage to kill orders of magnitudes of cells (are the plate and liquid concentrations comparable, we suddenly wonder) then why isn't there some stronger evidence for a cell cycle response to the DDC?
As described in the Public Review section, we have previously shown that a lack of Srs2-mediated checkpoint downregulation leads to a 4-6 fold increase of RPA on chromatin, which was rescued by rfa1-zm2 (Dhingra et al., 2021). On its own, rfa1-zm2 did not cause defective chromatin association in our assays, despite modestly reducing ssDNA binding in vitro (Dhingra et al., 2021). This discrepancy could be due to a lack of sensitivity of chromatin fractionation assay in revealing moderate changes of RPA residence on DNA. Considering this, we decided to employ functional assays (Figure 2-3) that are more effective in identifying the specific Srs2 features pertaining to RPA regulation.
We respectfully disagree with the reviewer’s point that there is “no evident checkpoint” in CPT. Previous studies have shown that CPT induces the DNA damage checkpoint as evidenced by Mec1 activation and phosphorylation of Rad53 and Rad9, and delaying exit from G2/M (Dhingra et al., 2021; Menin et al., 2018; Redon et al., 2003). Our data are fully consistent with these reports. It is important to note that DNA damage checkpoint can manifest at a range of strengths depending on the genotoxic conditions and treatment, but the fundamental principles are the same. For example, we found that the Srs2-RPA antagonism not only affects the checkpoint downregulation in CPT, but also does so in MMS treatment and in a DSB system. We focused on CPT condition in this work, since CPT only induces the DNA damage checkpoint but not DNA replication checkpoint while MMS induces both. Further investigating the Srs2-RPA antagonism in a DSB system can be interesting to pursue in the future.
We believe that “superfluous removal” is appropriately used when discussing RPA regulation at genomic sites wherein it supports ssDNA protection and DNA repair, rather than DDC. Examples of these sites include R-loops and negatively supercoiled regions. These sites lack 3’ and 5’ DNA ends at the ss-dsDNA junctions for loading PCNA and the 9-1-1 checkpoint factors, and thus are not designated for checkpoint regulation.
We addressed the reviewer’s point regarding Rad51 in the Public Review section. We disagree with reviewer’s view that “Rad51 normally replaces RPA”. RPA is involved in many more processes than Rad51 wherein it is not replaced by Rad51.
Regarding toxicity of CPT, our view is that it stems from a combination of checkpoint regulation and other processes that also involve the Srs2-RPA antagonism. While this work focused on the checkpoint aspect of this antagonism, future studies will be conducted to address the latter.
One reference is entered as Lee Zhou and Stephen J. Elledge as opposed to "Zhou and Elledge."
Corrected.
Reviewer #3 (Recommendations For The Authors):
(1) It would be nice to see the additional point mutants (srs2-Y775A, srs2-F891A) be tested, as they showed little to no phenotypes in the previously reported analyses, which did not specifically test the function surveyed here.
This point is addressed in the Public Reviews section.
(2) Maybe the caveat of using deletion versus point mutations could be discussed.
This point is addressed in the Public Reviews section.
(3) Please plot individual data points of the two independent experiments in Figures 4D and 5A so that the reader can evaluate reproducibility. N=2 does not really allow deriving SD.
This point is addressed in the Public Reviews section and three individual data points are now included in both panels.
(4) It will help the reader to have the exact strains used in each experiment listed in each figure legend. Minor point.
The strain table is now updated to address this point.
(5) Page 7 middle paragraph: The reference to Figure 4A in line 11 should probably be Figure S3A.
Corrected.



