Peer review process
Not revised: This Reviewed Preprint includes the authors’ original preprint (without revision), an eLife assessment, public reviews, and a provisional response from the authors.
Read more about eLife’s peer review process.Editors
- Reviewing EditorMelike LakadamyaliUniversity of Pennsylvania, Philadelphia, United States of America
- Senior EditorYamini DalalNational Cancer Institute, Bethesda, United States of America
Reviewer #1 (Public Review):
Summary:
In this study, Gholamalamdari et al. described various aspects of genome organization in relation to nuclear speckles, the nuclear lamina, and nucleoli. Their findings were drawn from the analysis of genomic data sourced from four distinct human cell types. The authors observed significant variation in genome positioning at the lamina and nucleoli across different cell types, whereas contacts with nuclear speckles showed less variability. The data revealed a correlation between gene expression levels and proximity to nuclear speckles, with regions in contact with these speckles coinciding with DNA replication initiation zones. Additionally, the results indicated that the loss of Lamin A and LBR leads to a redistribution of H3K9me3-enriched LADs from the lamina to the nucleolus. Furthermore, a portion of H3K27me3-enriched, partially repressed intergenic LADs (iLADs) was observed to relocate from the nucleolus to the lamina. The study also proposed that these repressed iLADs may compete with LADs for attachment to the nuclear lamina.
Strengths:
The datasets have been thoroughly integrated and exhibit various features of genomic domains interacting with nuclear speckles, the nuclear lamina, and nucleoli, which will be of interest to the field.
Weaknesses:
The weakness of this study lies in the fact that many of the genomic datasets originated from novel methods that were not validated with orthogonal approaches, such as DNA-FISH. Therefore, the detailed correlations described in this work are based on methodologies whose efficacy is not clearly established. Specifically, the authors utilized two modified protocols of TSA-seq for the detection of NADs (MKI67IP TSA-seq) and LADs (LMNB1-TSA-seq). Although these methods have been described in a bioRxiv manuscript by Kumar et al., they have not yet been published. Moreover, and surprisingly, Kumar et al., work is not cited in the current manuscript, despite its use of all TSA-seq data for NADs and LADs across the four cell lines. Moreover, Kumar et al. did not provide any DNA-FISH validation for their methods. Therefore, the interesting correlations described in this work are not based on robust technologies.
An attempt to validate the data was made for SON-TSA-seq of human foreskin fibroblasts (HFF) using multiplexed FISH data from IMR90 fibroblasts (from the lung) by the Zhuang lab (Su et al., 2020). However, the comparability of these datasets is questionable. It might have been more reasonable for the authors to conduct their analyses in IMR90 cells, thereby allowing them to utilize MERFISH data for validating the TSA-seq method and also for mapping NADs and LADs.
Reviewer #2 (Public Review):
Summary:
Golamalamdari, van Schaik, Wang, Kumar Zhang, Zhang, and colleagues study interactions between the speckle, nucleolus, and lamina in multiple cell types (K562, H1, HCT116, and HFF). Their datasets define how interactions between the genome and the different nuclear landmarks relate to each other and change across cell types. They also identify how these relationships change in K562 cells in which LBR and LMNA are knocked out.
Strengths:
Overall, there are a number of datasets that are provided, and several "integrative" analyses are performed. This is a major strength of the paper, and I imagine the datasets will be of use to the community to further probed and the relationships elucidated here further studied. An especially interesting result was that specific genomic regions (relative to their association with the speckle, lamina, and other molecular characteristics) segregate relative to the equatorial plane of the cell.
Weaknesses:
The experiments are largely descriptive, and it is difficult to draw many cause-and-effect relationships. Similarly, the paper would be very much strengthened if the authors provided additional summary statements and interpretation of their results (especially for those not as familiar with 3D genome organization). The study would benefit from a clear and specific hypothesis.
Author response:
Reviewer #1 (Public Review):
Weaknesses:
The weakness of this study lies in the fact that many of the genomic datasets originated from novel methods that were not validated with orthogonal approaches, such as DNA-FISH. Therefore, the detailed correlations described in this work are based on methodologies whose efficacy is not clearly established. Specifically, the authors utilized two modified protocols of TSA-seq for the detection of NADs (MKI67IP TSA-seq) and LADs (LMNB1-TSA-seq). Although these methods have been described in a bioRxiv manuscript by Kumar et al., they have not yet been published. Moreover, and surprisingly, Kumar et al., work is not cited in the current manuscript, despite its use of all TSA-seq data for NADs and LADs across the four cell lines. Moreover, Kumar et al. did not provide any DNA-FISH validation for their methods. Therefore, the interesting correlations described in this work are not based on robust technologies.
An attempt to validate the data was made for SON-TSA-seq of human foreskin fibroblasts (HFF) using multiplexed FISH data from IMR90 fibroblasts (from the lung) by the Zhuang lab (Su et al., 2020). However, the comparability of these datasets is questionable. It might have been more reasonable for the authors to conduct their analyses in IMR90 cells, thereby allowing them to utilize MERFISH data for validating the TSA-seq method and also for mapping NADs and LADs.
We disagree with the statement that the TSA-seq approach and data has not been validated by orthogonal approaches and with the conclusion that the TSA-seq approach is not robust as summarized here and detailed below in “Specific Comments”. TSA-seq is robust because it is based only on the original immunostaining specificity provided by the primary and secondary antibodies plus the diffusion properties of the tyramide-free radical. TSA-seq has been extensively validated by microscopy and by the orthogonal genomic measurements provided by LMNB1 DamID and NAD-seq. This includes: a) the initial validation by FISH of both nuclear speckle (to an accuracy of ~50 nm) and nuclear lamina TSA-seq and the cross-validation of nuclear lamina TSA-seq with lamin B1 DamID in a first publication (Chen et al, JCB 2018, doi: 10.1083/jcb.201807108); b) the further validation of SON TSA-seq by FISH in a second publication ((Zhang et al, Genome Research 2021, doi:10.1101/gr.266239.120); c) the cross-validation of nucleolar TSA-seq using NAD-seq and the validation by light microscopy of the predictions of differences in the relative distributions of centromeres, nuclear speckles, and nucleoli made from nuclear speckle, nucleolar, and pericentric heterochromatin TSA-seq in the Kumar et al, bioRxiv preprint (which is in a last revision stage involving additional formatting for the journal requirements) doi:https://doi.org/10.1101/2023.10.29.564613; d) the extensive validation of nuclear speckle, LMNB1, and nucleolar TSA-seq generated in HFF human fibroblasts using published light microscopy distance measurements of hundreds of probes generated by multiplexed immuno-FISH MERFISH data (Su et al, Cell 2020, https://doi.org/10.1016/j.cell.2020.07.032), as we described for nucleolar TSA-seq in the Kumar et al, bioRxiv preprint and to some extent for LMNB1 and SON TSA-seq in the current manuscript version (see Specific Comments with attached Author response image 2).
Reviewer 1 raised concerns regarding this FISH validation given that the HFF TSA-seq and DamID data was compared to IMR90 MERFISH measurements. The Su et al, Cell 2020 MERFISH paper came out well after the 4D Nucleome Consortium settled on HFF as one of the two main “Tier 1” cell lines. We reasoned that the nuclear genome organization in a second fibroblast cell line would be sufficiently similar to justify using IMR90 FISH data as a proxy for our analysis of our HFF data. Indeed, there is a high correlation between the HFF TSA-seq and distances measured by MERFISH to nuclear lamina, nucleoli, and nuclear speckles (Author response image 1). Comparing HFF SON-TSA-seq data with published IMR90 SON TSA-seq data (Alexander et al, Mol Cell 2021, doi.org/10.1016/j.molcel.2021.03.006), the HFF SON TSA-seq versus MERFISH scatterplot is very similar to the IMR90 SON TSA-seq versus MERFISH scatterplot. We acknowledge the validation provided by the IMR90 MERFISH is limited by the degree to which genome organization relative to nuclear locales is similar in IMR90 and HFF fibroblasts. However, the correlation between measured microscopic distances from nuclear lamina, nucleoli, and nuclear speckles and TSA-seq scores is already quite high. We anticipate the conclusions drawn from such comparisons are solid and will only become that much stronger with future comparisons within the same cell line.
Author response image 1.
Scatterplots showing the correlation between TSA-seq and MERFISH microscopic distances. Top: IMR90 SON TSA-seq (from Alexander et al, Mol Cell 2021) (left) and HFF SON TSA-seq (right) (x-axis) versus distance to nuclear speckles (y-axis). Bottom: HFF Lamin B1 TSA-seq (x-axis) versus distance to nuclear lamina (y-axis) (left) and HFF MKI67IP (nucleolar) TSA-seq (x-axis) versus distance to nucleolus (y-axis) (right).
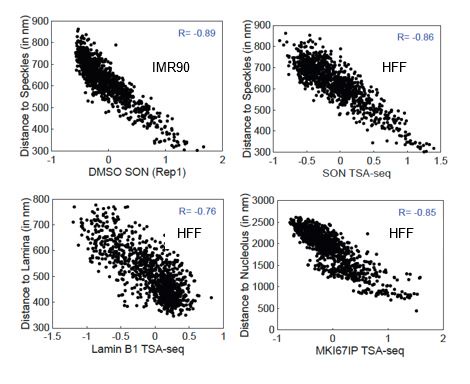
In our revision, we will add justification of the use of IMR90 fibroblasts as a proxy for HFF fibroblasts through comparison of available data sets.
Reviewer #2 (Public Review):
Weaknesses:
The experiments are largely descriptive, and it is difficult to draw many cause-and-effect relationships. Similarly, the paper would be very much strengthened if the authors provided additional summary statements and interpretation of their results (especially for those not as familiar with 3D genome organization). The study would benefit from a clear and specific hypothesis.
We acknowledge that this study was hypothesis-generating rather than hypothesis-testing in its goal. This research was funded through the NIH 4D-Nucleome Consortium, which had as its initial goal the development, benchmarking, and validation of new genomic technologies. Our Center focused on the mapping of the genome relative to different nuclear locales and the correlation of this intranuclear positioning of the genome with functions- specifically gene expression and DNA replication timing. By its very nature, this project has taken a discovery-driven versus hypothesis-driven scientific approach. Our question fundamentally was whether we could gain new insights into nuclear genome organization through the integration of genomic and microscopic measurements of chromosome positioning relative to multiple different nuclear compartments/bodies and their correlation with functional assays such as RNA-seq and Repli-seq.
Indeed, as described in this manuscript, this study resulted in multiple new insights into nuclear genome organization as summarized in our last main figure. We believe our work and conclusions will be of general interest to scientists working in the fields of 3D genome organization and nuclear cell biology. We anticipate that each of these new insights will prompt future hypothesis-driven science focused on specific questions and the testing of cause-and-effect relationships.
Given the extensive scope of this manuscript, we were limited in the extent that we could describe and summarize the background, data, analysis, and significance for every new insight. In our editing to reach the eLife recommended word count, we removed some of the explanations and summaries that we had originally included.
As suggested by Reviewer 2, in our revision we will add back additional summary and interpretation statements to help readers unfamiliar with 3D genome organization.
Specific Comments in response to Reviewer 1:
(1) We disagree with the comment that TSA-seq has not been cross-validated by other orthogonal genomic methods. In the first TSA-seq paper (Chen et al, JCB 2018, doi: 10.1083/jcb.201807108), we showed a good correlation between the identification of iLADs and LADs by nuclear lamin and nuclear speckle TSA-seq and the orthogonal genomic method of lamin B1 DamID, which is reproduced using our new TSA-seq 2.0 protocol in this manuscript. Similarly, in the Kumar et al, bioRxiv preprint (doi:https://doi.org/10.1101/2023.10.29.564613), we showed a general agreement between the identification of NADs by nucleolar TSA-seq and the orthogonal genomic method of NAD-seq. (We expect this preprint to be in press soon; it is now undergoing a last revision involving only reformatting for journal requirements.) Additionally, we also showed a high correlation between Hi-C compartments and subcompartments and TSA-seq in the Chen et al, JCB 2018 paper. Specifically, there is an excellent correlation between the A1 Hi-C subcompartment and Speckle Associated Domains as detected by nuclear speckle TSA-seq. Additionally, the A2 Hi-C subcompartment correlated well with iLAD regions with intermediate nuclear speckle TSA-seq scores, and the B2 and B3 Hi-C subcompartments with LADs detected by both LMNB TSA-seq and LMNB1 DamID. More generally, Hi-C A and B compartment identity correlated well with predictions of iLADs versus LADs from nuclear speckle and nuclear lamina TSA-seq.
(2) In the Chen et al, JCB 2018 paper we also qualitatively and quantitatively validated TSA-seq using FISH. Qualitatively, we showed that both nuclear speckle and nuclear lamin TSA-seq correlated well with distances to nuclear speckles versus the nuclear lamina, respectively, measured by immuno-FISH.
Quantitatively, we showed that SON TSA-seq could be used to estimate the microscopic mean distance to nuclear speckles with mean and median residuals of ~50 nm. First, we used light microscopy to show that the spreading of tyramide-biotin signal from a point-source of TSA staining fits well with the exponential decay predicted theoretically by reaction-diffusion equations assuming a steady rate of tyramide-biotin free radical generation by the HRP enzyme and a constant probability throughout the nucleus of free-radical quenching (through reaction with protein tyrosine residues and nucleic acids). Second, we used the exponential decay constant measured by light microscopy together with FISH measurements of mean speckle distance for several genomic regions to fit an exponential function and to predict distance to nuclear speckles genome-wide directly from SON TSA-seq sequencing reads. Third, we used this approach to test the predictions against a new set of FISH measurements, demonstrating an accuracy of these predictions of ~50 nm.
(3) The importance of the quantitative validation by immuno-FISH of using TSA-seq to estimate mean distance to nuclear speckles is that it demonstrates the robustness of the TSA-seq approach. Specifically, it shows how the TSA-seq signal is predicted to depend only on the specificity of the primary and secondary antibody staining and the diffusion properties of the tyramide-biotin free radicals produced by the HRP peroxidase. This is fundamentally different from the significant dependence on antibodies and choice of marker proteins for molecular proximity assays such as DamID, ChIP-seq, and Cut and Run/Tag which depend on molecular proximity for labeling and/or pulldown of DNA.
This robustness leads to specific predictions. First, it predicts similar TSA-seq signals will be produced using antibodies against different marker proteins against the same nuclear compartment. This is because the exponential decay constant (distance at which the signal drops by one half) for the spreading of the TSA is in the range of several hundred nm, as measured by light microscopy for several TSA staining conditions. Indeed, we showed in the Chen et al, JCB 2018 paper that antibodies against two different nuclear speckle proteins produced very similar TSA-seq signals while antibodies against LMNB versus LMNA also produced very similar TSA-seq signals. Similarly, we showed in the Kumar et al preprint that antibodies against four different nucleolar proteins showed similar TSA-seq signals, with the highest correlation coefficients for the TSA-seq signals produced by the antibodies against two GC nucleolar marker proteins and the TSA-seq signals produced by the antibodies against two FC/DFC nucleolar marker proteins.
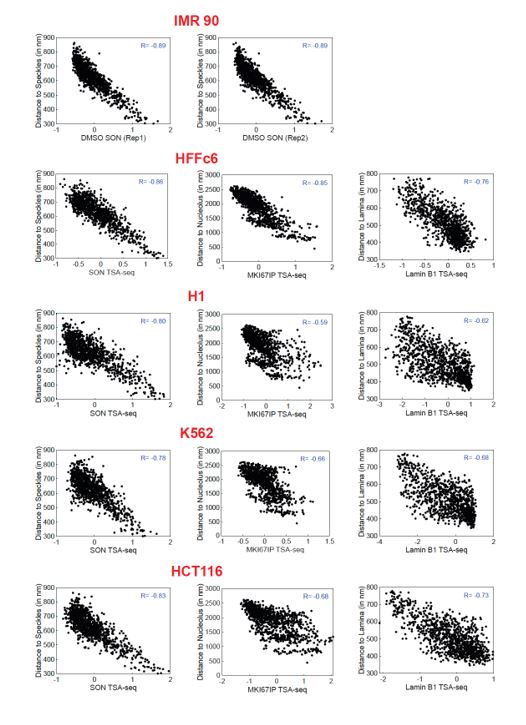
Author response image 2.
Comparison of TSA-seq data from different cell lines versus IMR90 MERFISH. The observed correlation between SON (nuclear speckle) TSA-seq versus MERFISH is nearly as high for TSA-seq data from HFF as it is for TSA-seq data from the IMR90 cell line (Alexander et al, Mol Cell 2021) in which the MERFISH was performed. The correlations for SON, LMNB1 (nuclear lamina) and MKI67IP (nucleolus) versus MERFISH are highest for HFF TSA-seq data as compared to TSA-seq data from other cell lines (H1, K562, HCT116). Comparison of measured distances to nuclear locale (y-axis) versus TSA-seq scores (x-axis) from different cell lines labeled in red. Left to right: SON, LMNB1, and MKI67IP. Top to bottom: SON TSA-seq versus MERFISH for two TSA-seq replicates; TSA-seq from HFF, H1, K562, and HCT116 versus MERFISH.
Second, it predicts that the quantitative relationship between TSA-seq signal and mean distance from a nuclear compartment will depend on the convolution of the predicted exponential decay of spreading of the TSA signal produced by a point source with the more complicated staining distribution of nuclear compartments such as the nuclear lamina or nucleoli. We successfully used this concept to explain the differences emerging between LMNB1 DamID and TSA-seq signals for flat nuclei and to recognize the polarized distribution of different LADs over the nuclear periphery.
(4) After our genomic data production and during our data analysis, a valuable resource from the Zhuang lab was published, using MERFISH to visualize hundreds of genomic loci in IMR90 cells. We acknowledge that the much more extensive validation of TSA-seq by the multiplexed immuno-FISH MERFISH data is dependent on the degree to which the nuclear genome organization is similar between IMR90 and HFF fibroblasts. However, the correlation between distances to nuclear speckles, nucleoli, and the nuclear lamina measured in IMR90 fibroblasts and the nuclear speckle, nucleolar, and nuclear lamina TSA-seq measured in HFF fibroblasts is already striking (See Author response image 1). With regard to SON TSA-seq, the MERFISH versus HFF TSA-seq correlation is close to what we observe using published IMR90 SON TSA-seq data (correlation coefficients of 0.89 (IMR90 TSA-seq) versus 0.86 (HFF TSA-seq). Moreover, this correlation is highest using TSA-seq data from HFF cells as compared to the three other cell lines. (see Author response image 2). We believe these correlations can be considered a lower bound on the actual correlations between the FISH distances and TSA-seq that we would have observed if we had performed both assays on the same cell line.
(5) Currently, we still require tens of millions of cells to perform each TSA-seq assay. This requires significant expansion of cells and a resulting increase in passage numbers of the IMR90 cells before we can perform the TSA-seq. During this expansion we observe a noticeable slowing of the IMR90 cell growth as expected for secondary cell lines as we approach the Hayflick limit. We still do not know to what degree nuclear organization relative to nuclear locales may change as a function of cell cycle composition (ie percentage of cycling versus quiescent cells) and cell age. Thus, even if we performed TSA-seq on IMR90 cells we would be comparing MERFISH from lower passages with a higher percentage of actively proliferating cells with TSA-seq from higher passages with a higher percentage of quiescent cells.
We are currently working on a new TSA-seq protocol that will work with thousands of cells. We believe it is better investment of time and resources to wait until this new protocol is optimized before we repeat TSA-seq in IMR90 cells for a better comparison with multiplexed FISH data.
Specific Comments in response to Reviewer 2:
(1) As we acknowledge in our Response summary, we were limited in the degree to which we could actually follow-up our findings with experiments designed to test specific hypotheses generated by our data. However, we do want to point out that our comparison of wild-type K562 cells with the LMNA/LBR double knockout was designed to test the long-standing model that nuclear lamina association of genomic loci contributes to gene silencing. This experiment was motivated by our surprising result that gene expression differences between cell lines correlated strongly with differences in positioning relative to nuclear speckles rather than the nuclear lamina. Despite documenting in these double knockout cells a decreased nuclear lamina association of most LADs, and an increased nuclear lamina association of the “p-w-v” fiLADs identified in this manuscript, we saw no significant change in gene expression in any of these regions as compared to wild-type K562 cells. Meanwhile, distances to nuclear speckles as measured by TSA-seq remained nearly constant.
We would argue that this represents a specific example in which new insights generated by our genomics comparison of cell lines led to a clear and specific hypothesis and the experimental testing of this hypothesis.
In response to Reviewer 2, we are modifying the text to make this clearer and to explicitly describe how we were testing the hypothesis that distance to nuclear lamina is correlated with but not causally linked to gene expression and how to test this hypothesis we used a DKO of LMNA and LBR to change distances relative to the nuclear lamina and to test the effect on gene expression.



