Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB
- Goethe University, Germany
- University of Zurich, Switzerland
- National Institutes of Health, United States
- University of Konstanz, Germany
Figures
Figure 1 with 2 supplements

Topology and secondary structure representations of the AcrB protomers.
(A) Topology of the AcrB protomers in the transmembrane (TM) and porter domains. Our analysis indicates that the TM domain consists of two 5-helix parallel repeats, referred to as R1 and R2, and two flanking helices, TM2 and TM8. R1 (blue) is connected to R2 (red) through helix Iα (white), which lies parallel to the cytoplasmic face of the membrane. The porter domain also consists of two repeats of two subdomains each, namely PC1 and PN2 (blue), and PN1 and PC2 (red). TM2 (cyan) connects R1 to PN2, whereas TM8 (orange) links R2 to PC2. Other connections among porter sub-domains and R1/R2 are indicated by open/closed squares/circles. Hexagons indicate the connections between the porter domain and the funnel domain (FD), not drawn in this scheme. (B) Cartoon representation of the secondary structure of an AcrB protomer, in the context of the complete trimer. The other two protomers are drawn as a molecular surface (gray). Different sub-domains are colored as in (A), with inter-connecting loops and the FD shown in black.
Figure 1—figure supplement 1
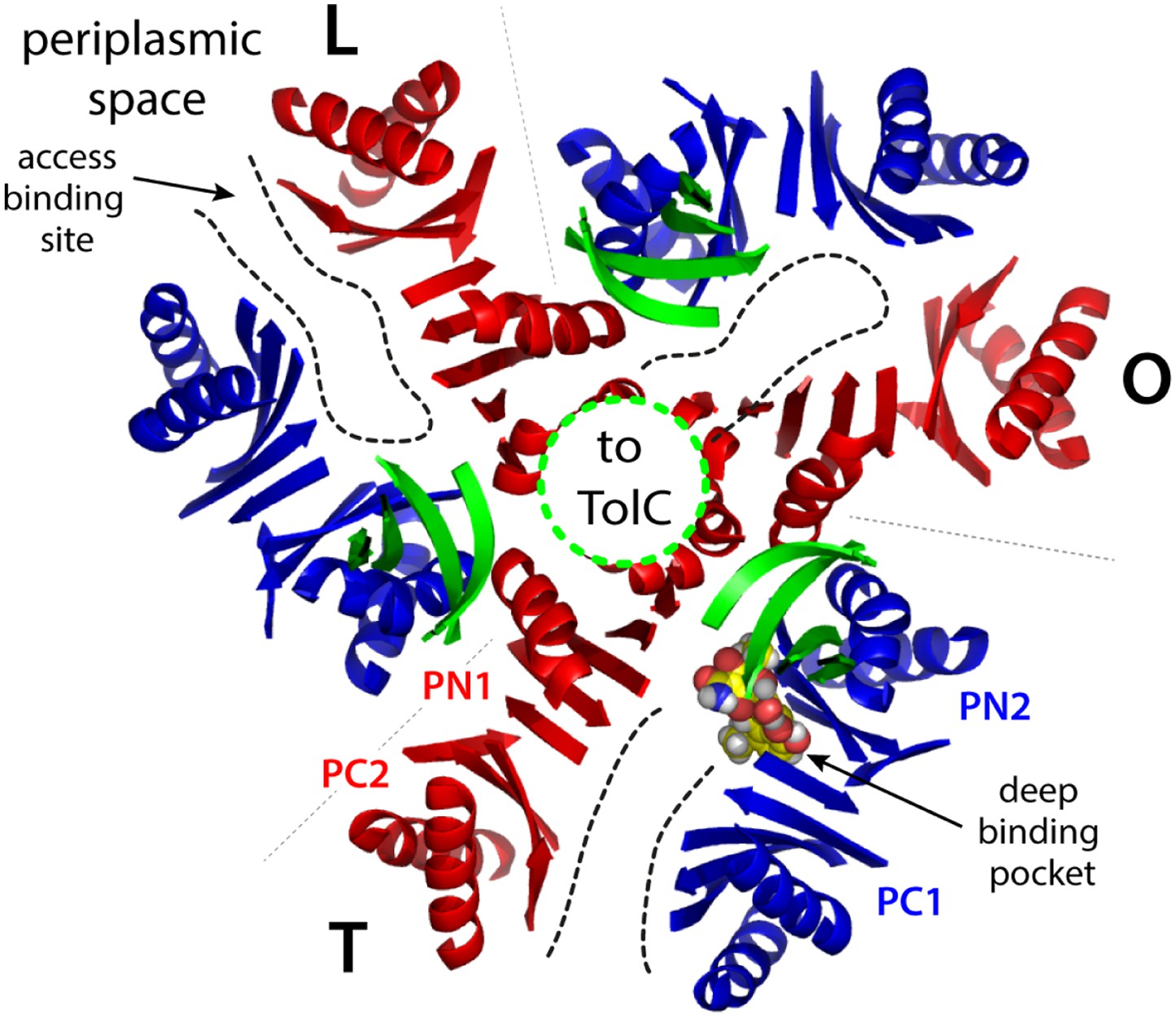
Cartoon representation of the porter domain of AcrB in the asymmetric state, viewed from the outer membrane, along the perpendicular to the inner membrane.
Each of the protomers adopt a distinct conformation, namely L, T and O; gray dashed lines indicate the protomer interfaces. Subdomains PC1/PN2 and PN1/PC2 are colored in blue and red, respectively, as in Figure 1. A minocycline molecule is bound in the T state, in the deep binding pocket between PC1 and PN2. Substrates can also bind more superficially, in the L state. Black dashed lines indicate access pathways from the periplasmic space into the drug-binding sites, in the L and T states, and from the binding site to the central funnel (green dashed line) within the funnel domain, in the O state. A fragment of this funnel domain is shown with green cartoons; this funnel is open to the lumen of the TolC channel, which traverses the outer membrane of E. coli.
Figure 1—figure supplement 2
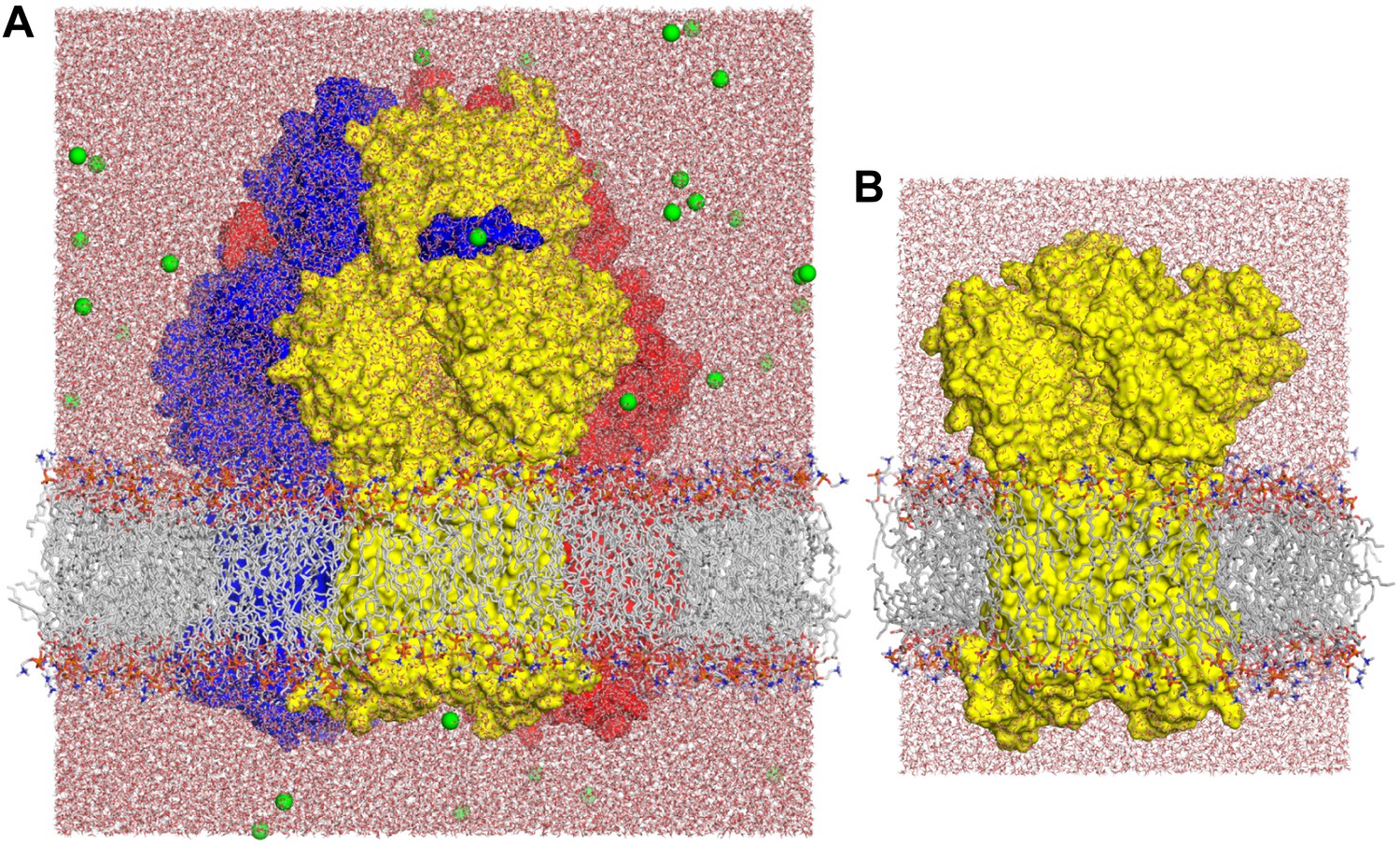
Molecular simulation systems employed in this study.
(A) Asymmetric AcrB trimer (residues 1 to 1044 per protomer) with one minocycline bound to the T state, embedded in a POPC lipid membrane and solvated by water. The system is enclosed in a periodic box of dimensions ∼150 × 150 × 160 Å. The molecular system includes ∼370,000 atoms. AcrB is shown as a van-der-Waals surface, with the L, T and O states in blue, yellow and red, respectively. Lipid and water molecules are represented with lines and sticks; free Na+ ions, added to neutralize the total charge of the system, are shown as green spheres. (B) Free-energy simulations were carried out on a construct of the individual AcrB protomers, comprising the transmembrane and porter domains only, embedded in a POPC lipid membrane. The figure shows the T state; L and O states were prepared similarly. Each of these systems consist of ∼101,000 atoms, enclosed in a rectangular box of dimensions ∼90 × 90 × 120 Å.
Figure 2 with 2 supplements

Identification of structural units within the TM domain of AcrB undergoing collective motions throughout the LTO conformational cycle.
We considered all possible combinations of 6, 5 and 4 transmembrane helices, within either TM1-TM6 or TM7-TM12, and compared the structure of each set of helices in the L vs T states (closed circles), T vs O (open circles) and O vs L (closed squares), through pairwise root-mean-square difference (RMSD) calculations. When these RMSD values are used as a rank, it becomes clear that combinations that exclude TM2 or TM8 (red) feature systematically smaller differences across the LTO cycle than those that include TM2 or TM8 (black). All other exclusions show no clear correlation. For example, comparing TM1-TM6 (left panel) in L vs T, this increasing ranking is ΔTM2, ΔTM4, ΔTM1, ΔTM5, ΔTM3, and ΔTM6; in T vs O: ΔTM2, ΔTM5, ΔTM4, ΔTM3, ΔTM1, and ΔTM6; and in O vs L: ΔTM2, ΔTM5, ΔTM1, ΔTM4, ΔTM3, and ΔTM6. Furthermore, the data shows that combinations of five helices excluding TM2 and TM8 are not significantly more divergent across the LTO conformers than combinations of four helices. Thus, the asymmetric crystal structure of AcrB captures collective displacements of two 5-helix bundles within each protomer, formed by TM1/TM3-TM6 and TM7/TM9-TM12 (referred to as repeats R1 and R2), relative to flanking helices TM2 and TM8, respectively. Note that comparisons of combinations of seven or more helices result in larger RMSD values than those shown here, and are therefore omitted.
Figure 2—figure supplement 1

Structural variations in the R1 and R2 repeats of the transmembrane domain of wildtype AcrB, in the L, T and O states.
The off-diagonal elements are comparisons (in terms of RMSD, as in Supplementary file 1) of the same repeat in different conformational states of the AcrB protomer. The diagonal elements are comparisons between R1 and R2 in the same state (all values in Å). Both R1 and R2 consist of a five-helix bundle. R1 includes TM1 and TM3 to TM6, while R2 comprises TM7 and TM9 to TM12. The definition of each transmembrane segment is as follows: TM1, residues 10–28; TM2, 329–358; TM3, 362–386; TM4, 393–423; TM5, 428–457; TM6, 463–496, TM7, 539–557; TM8, 872–892; TM9, 895–919; TM10, 925–955; TM11, 960–989; TM12, 998–1031.
Figure 2—figure supplement 2

Relative displacements of structural elements in the transmembrane and porter domain of wildtype AcrB across the LTO conformational cycle.
In each case, the L, T and O states are compared pair-wise. For example, to compute the displacement of R2 between the L and T states, the structures of R1+R2 in these two states are overlaid by least-square fitting R1, and the RMSD of R2 is computed (all values in Å). An analogous procedure is followed for TM2, relative to R1, and TM8, relative to R2, as well as for repeat PN1+PC2 in the porter domain relative to PN2+PC1. In all cases it is apparent that the computed displacements are significantly greater than the changes in the internal structure of these elements, quantified in Supplementary file 1 and Figure 2—figure supplement 1, indicating collective motions take place.
Figure 3 with 1 supplement

Collective motions of the structural repeats in TM domain of AcrB.
For clarity, the figure shows only the transmembrane α-helices at the interface between repeats R1 (blue) and R2 (red), alongside the two α-helices flanking these repeats, TM2 (cyan), TM8 (orange), and the helix that links the two repeats, Iα (gray). Each panel depicts two conformations in the LTO cycle, overlaid optimally on TM4-TM6; thus the figure highlights the motions of R2 relative to R1. Initial and final states (e.g., L vs T) are shown in light and bright colors, respectively. Note that the L vs T overlay is viewed from a different angle than T vs O and O vs L, for clarity. Interpolations between these conformational states using the complete TM domain are shown in Video 4, Video 5 and Video 6.
Figure 3—figure supplement 1

Arrangement of helix Iα across the conformational cycle of wildtype AcrB.
Iα is an additional α-helix inserted between TM6 and TM7, which lies parallel to the membrane domain (residues 512 to 536). RMSD analysis (shown in Å) of R2+Iα indicates that this helix moves alongside the R2 repeat. RMSD analysis of TM2+I also shows that the relative orientation of these two helices is also approximately constant in the L, T, and O states. Altogether, these data indicate that Iα functions as a connecting rod coupling the displacements of TM2 and R2.
Figure 4

Configuration of the proton-dependent conformational switch in the core of the TM domain of AcrB, in the L, T and O states, viewed from the periplasm.
The structure corresponds to the asymmetric crystal structure of AcrB at a resolution of 1.9 Å (PDB entry 4DX5). The top panels show a close-up of the proton-binding sites, D407 and D408, along with neighboring side-chains, including K940 in the foreground and R971 in the background. The lower panels are close-ups of R971 and interacting side-chains in its proximity. The highlighted side-chains are shown along with 2Fo-Fc electron density maps contoured at 1.0σ. Note this proton-relay network is precisely at the interface between the two transmembrane repeats, R1 and R2 (blue and red).
Figure 5 with 1 supplement
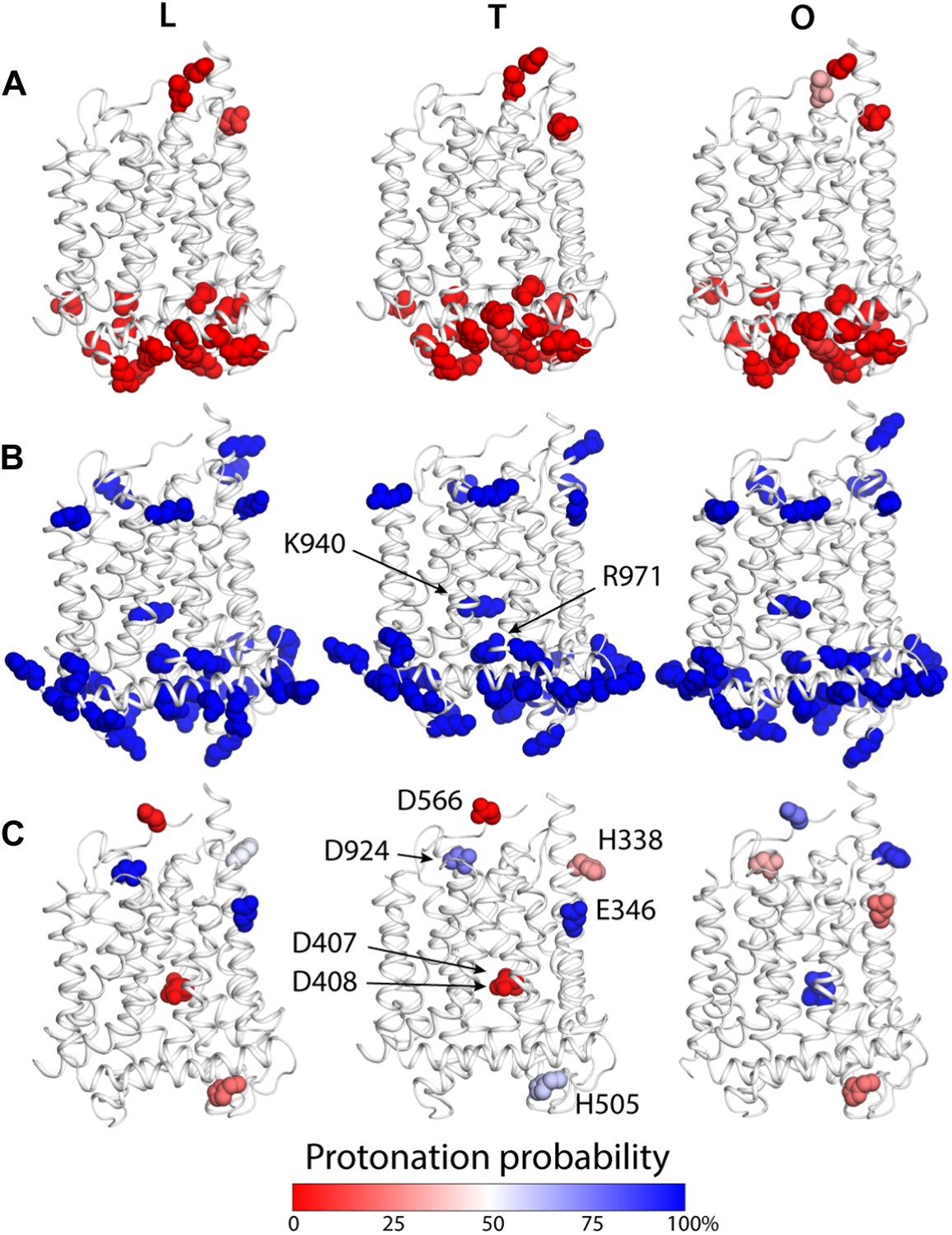
Changes in the computed protonation probability of ionizable residues in the TM domain, across the LTO conformational cycle.
The three protomers in the asymmetric crystal structure of AcrB are shown side by side, in a cartoon representation (white), in the same orientation as in Figure 1. Ionizable residues are represented as van-der-Waals spheres, and colored according to their protonation probability, computed via Monte-Carlo Poisson–Boltzmann calculations (‘Materials and methods’). (A) Glu, Asp and His residues predicted to be in the deprotonated state in all conformations of the protomer. (B) K940 and R971 are predicted to be protonated in the L, T and O states; other lysine and arginine residues are assumed to be protonated in the calculation. (C) Glutamate, aspartate and histidine residues whose protonation state is predicted to be inter-dependent with the conformation of the protomer.
Figure 5—figure supplement 1

Electrostatic switch involving K940 and R971 upon protonation of D407 and D408.
The plot shows net electrostatic interactions between K940, R971 and D407/D408, in each conformational state of AcrB; ‘other’ indicates the net interaction with all residues in the protein except for K940, R971, D407 and D408. D407 and D408 are deprotonated in states L (blue) and T (yellow), and protonated in state O (red). This analysis shows that in the transition from T to O, K940 and R971 lose favorable electrostatic interactions with D407 and D408, as a result of their becoming protonated; however, by re-orienting and retracting towards their own repeat, that is, R2 (see Figure 3), they gain much of the lost electrostatic energy, by increasing their favorable interactions with other residues in the protein, by minimizing the repulsion between each other, and by increasing their exposure to solvent. In turn, this reconfiguration results in a lateral displacement of the repeats relative to each other. Electrostatic interaction energies were extracted from a Monte-Carlo Poisson–Boltzmann simulation of the protonation equilibrium of AcrB (‘Materials and methods’); the plot shows average values over 500,000 steps of the Monte-Carlo simulation; error bars correspond to the standard-deviation in each case (not visible in all cases).
Figure 6 with 1 supplement
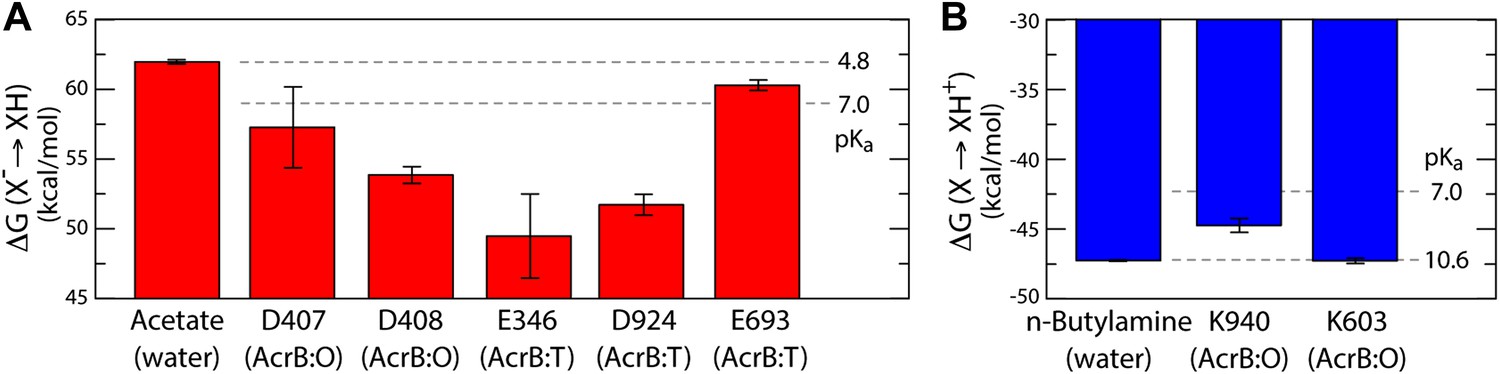
Free energy of protonation for selected glutamate, aspartate and lysine residues in the L, T and O states.
All values were computed via all-atom molecular simulations in a phospholipid membrane (‘Materials and methods’). Computed values for side-chain analogs in solution, whose pKa is known experimentally, provide a means to translate the computed free-energy values into a pKa scale (dashed lines). (A) The calculations indicate upward shifts in the pKa of E346 and D924 in the T state, and of D407 and D408 in the O state, relative to acetate in water, whose magnitude indicate that in those conformational states these residues are protonated at physiological pH. By contrast, the pKa of E693 is hardly shifted, consistent with its location on a water-exposed, electrostatically neutral area of the protein surface. (B) The pKa of K940 in the O state is also shifted downwards relative to n-butylamine in solution, on account of its being largely unexposed, but does not become deprotonated owing to polar contacts with N941 and T978 (Figure 4).
Figure 6—figure supplement 1
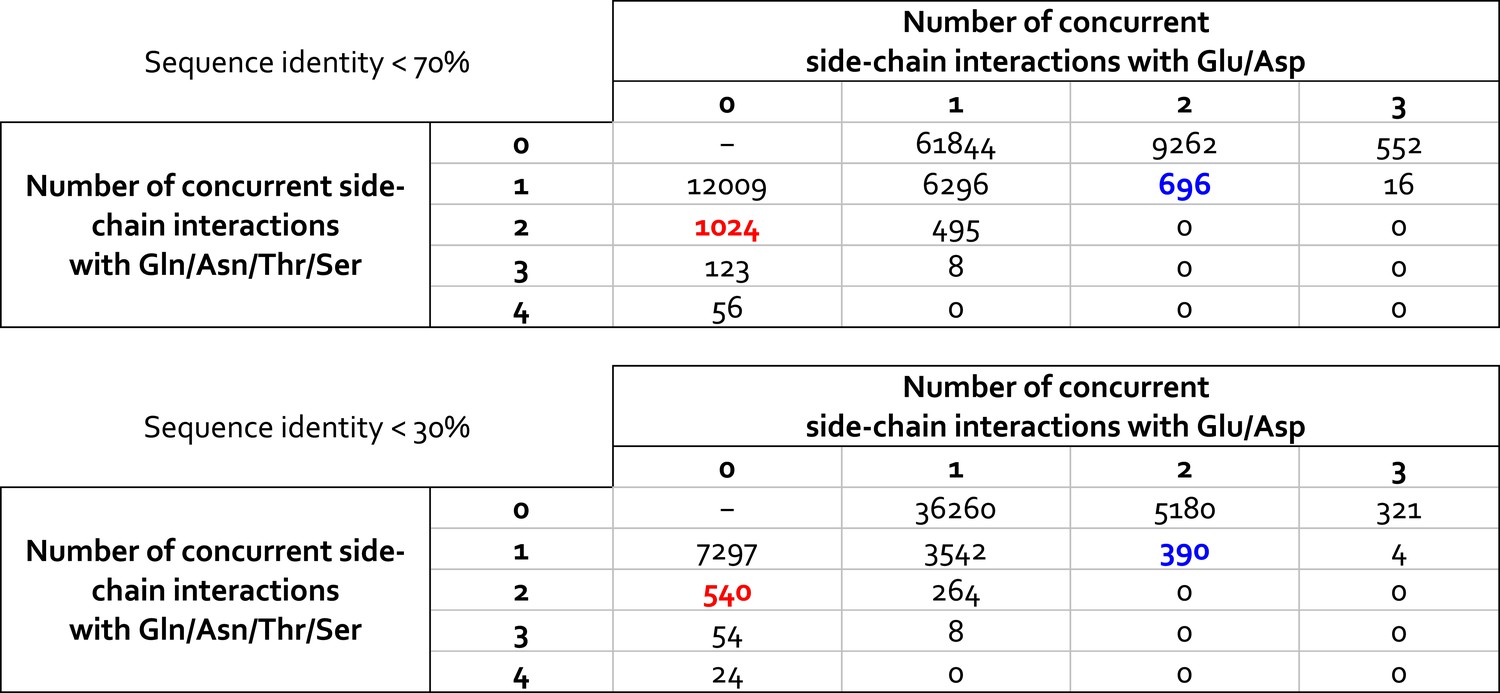
Statistical analysis of ionic and non-ionic side-chains interactions formed by lysine side-chains in high-resolution crystal structures deposited in the Protein Data Bank.
The datasets analyzed comprise crystal structures with non-redundant amino-acid sequences, based on either a 70% or a 30% sequence-identity threshold (21,780 and 13,735 structures, respectively). For each lysine side-chain in each structure, we identify the number of polar and/or acidic side-chains with which the lysine side-chain interacts directly, that is, via a direct hydrogen-bond. This analysis shows that the interaction patterns observed for K940 in AcrB (highlighted in blue/red) are frequently observed in protein structures; interestingly, the frequency with which two concurrent Glu/Asp interactions are observed, as in the L and T states of AcrB (blue) is comparable (slightly smaller) to that with which two non-ionic interactions are observed, as in the O state of AcrB (red).
Figure 7 with 1 supplement

Configuration of the proton-binding site in asymmetric crystal structures of four AcrB variants inactive in drug efflux, compared to the wildtype protein.
The binding site is viewed and represented as in Figure 4, omitting the electron-density map. The most probable protonation states of D407 and D408, based on Poisson–Boltzmann Monte-Carlo simulations (wildtype and mutants) as well as all-atom free-energy perturbation molecular dynamics simulations (wildtype), are indicated in each case with ‘H+’. K940 and R971 are protonated in all cases, according to these calculations. Hydrogen-bonds identified on the basis of the distance and orientation of potential donors and acceptors, are indicated with dashed lines.
Figure 7—figure supplement 1
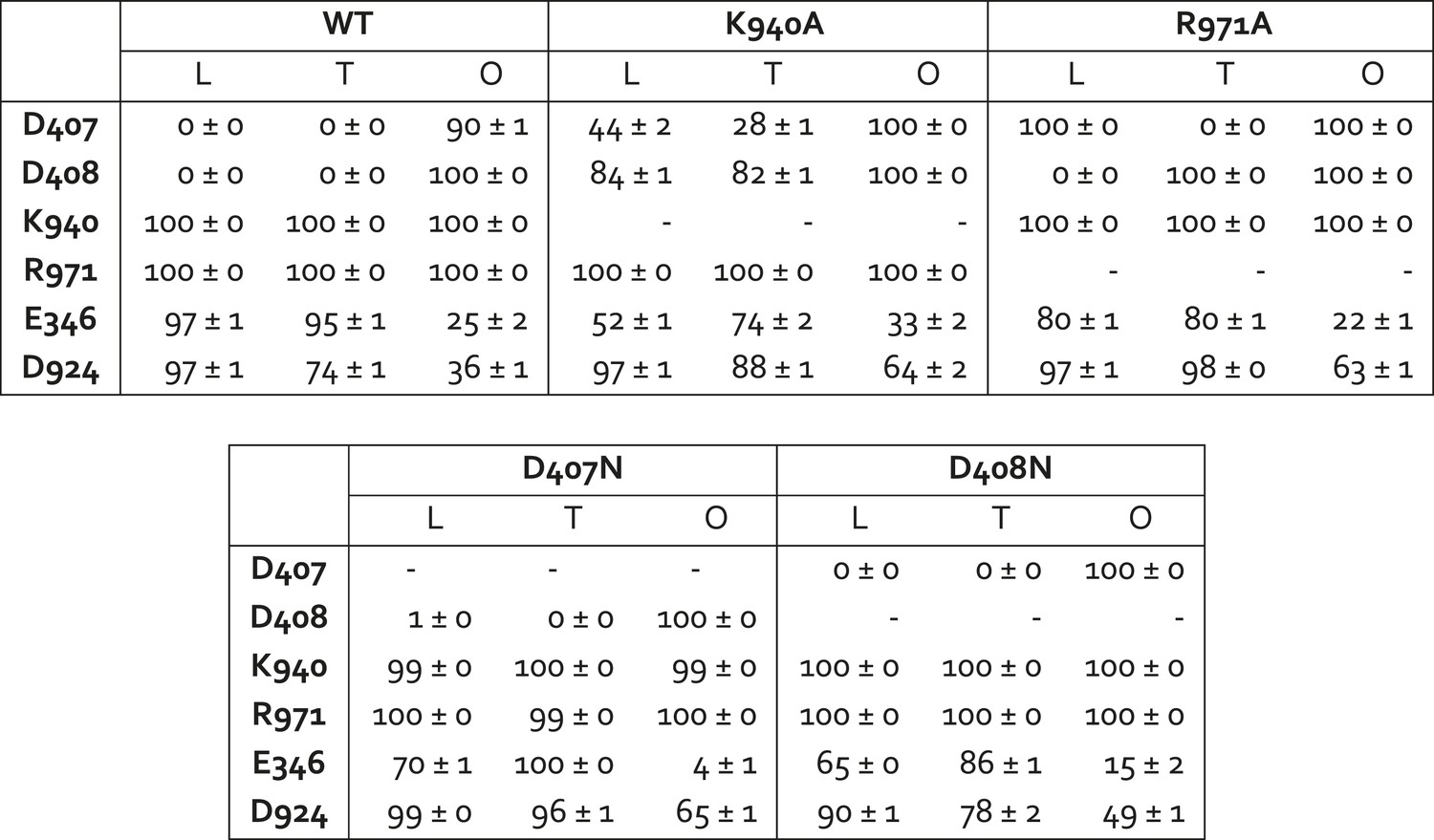
Protonation probability (%) of selected side-chains in the transmembrane domain of AcrB, in crystal structures of wildtype and mutagenized variants of the transporter.
Each of the protonation probability values shown is an average of the results of 10 independent Monte-Carlo/Poisson–Boltzmann simulations of the coupled protonation equilibrium of all glutamate, aspartate and histidine side-chains, plus K940 and R971, in full-length AcrB trimers (see ‘Materials and methods’ for further details). Standard deviations are also shown.
Figure 8
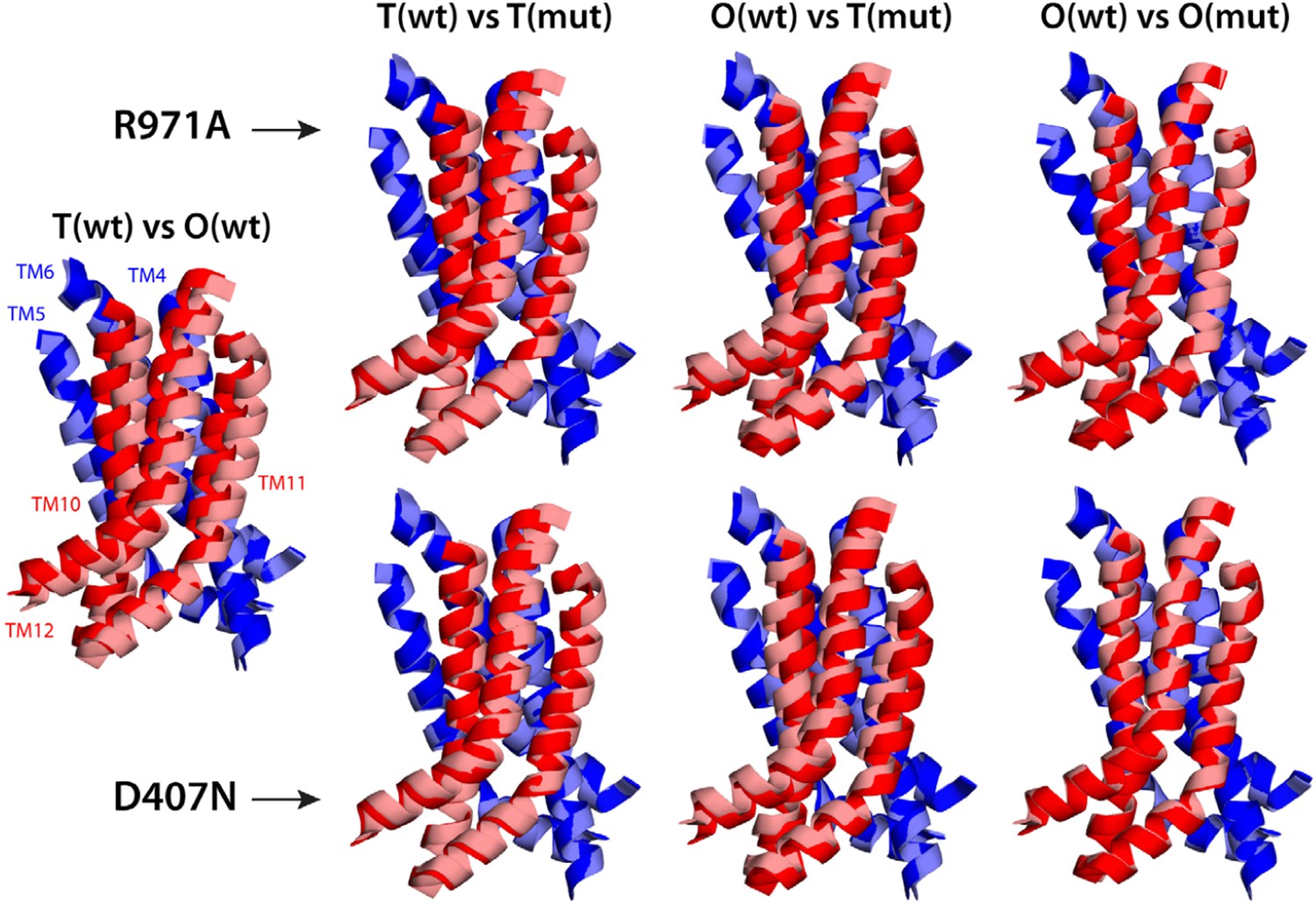
Relative motions of transmembrane repeats R1 and R2 in wildtype AcrB and in the R971A and D407N variants, in the T to O transition.
For clarity, only the three transmembrane helices at the interface between repeats are shown, namely TM4-TM6 (R1, blue cartoons) and TM10-TM12 (R2, red cartoons), respectively. The structural superimpositions shown are optimal overlays of TM4-TM6, and thus highlight the differences in the orientation of TM10-TM12 relative to TM4-TM6. This comparison shows that the T state in the crystal structures of the R971A and D407N mutants is an intermediate between the T and O conformations in wildtype AcrB. The conformation of the O state, however, is largely unaffected by these mutations.
Figure 9
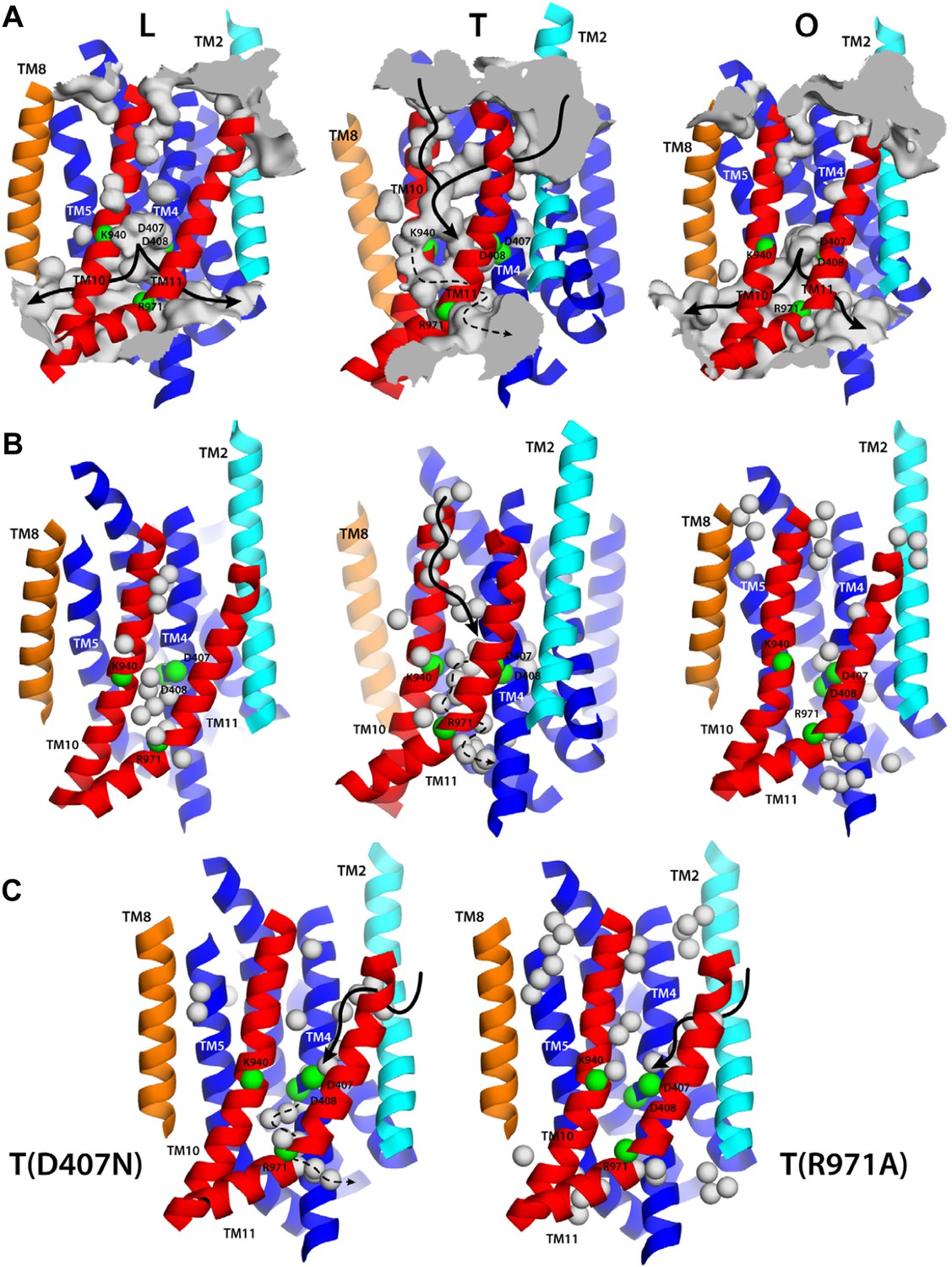
Alternating access to the proton-binding site in AcrB.
(A) Conformation-dependent water channels within the TM domain of AcrB, observed in all-atom simulations in a phospholipid membrane. Transmembrane helices are represented as cartoons and colored as in Figure 1; for clarity, TM7, TM9 and TM12 in repeat R2 are omitted. The locations of D407, D408, K940, R971, E346 and D924 are indicated with green spheres representing their Cα atoms. The water channels are represented by average density maps computed from the molecular dynamics trajectory, and depicted as an iso-density surface (gray). Based on this analysis, the proton-binding site is open to the cytoplasm in the L and O states, via two possible channels (solid black lines), whereas in the T state it is open to the periplasm, also via two water wires (solid black lines). A third narrow wire in the T state seems to connect the proton-binding site to the cytoplasm (dashed black lines), but the positively charged R971, which traverses this water wire, likely blocks H+ leakage. (B and C) Crystallographic water molecules (gray spheres) detected within the TM domain of AcrB, in each of the protomer states of the wildtype structure, as well as in the T protomer of the D407N and R971A variants.
Figure 10

Dynamic exchange of water molecules between the proton-binding site in the core of the TM domain of AcrB and the bulk solution at either the periplasmic or cytoplasmic sides of the membrane and proposed role of R971.
(A) A random selection of single-water trajectories extracted from the molecular dynamics simulation are shown as blue lines, cut-off at 30 Å from the proton-binding site. The location of residues D407, D408, K940 and R971 is indicated by their Cα atoms, shown as green spheres. Transmembrane helices in each protomer/state are represented as cartoons (white); for clarity, TM7, TM9 and TM12 in repeat R2 are omitted. (B) Proposed role of R971 as a selectivity filter against cytoplasmic H+ leakage in the T state of AcrB. The plot quantifies the frequency of water exchanges between the proton-binding site within the TM domain of AcrB and the bulk solution at the cytoplasmic side of the membrane, as a function of the proximity of the individual molecular trajectories to R971. In the T state, almost 100% of the water trajectories approach R971 within 10 Å, and therefore this water wire (dashed lines in Figure 9) is highly unlikely to sustain H+ conduction, for electrostatic reasons. In the L and O states, by contrast, 40% of the exchanging water molecules follow trajectories that do not approach R971 (solid lines in Figure 9).
Figure 11 with 1 supplement
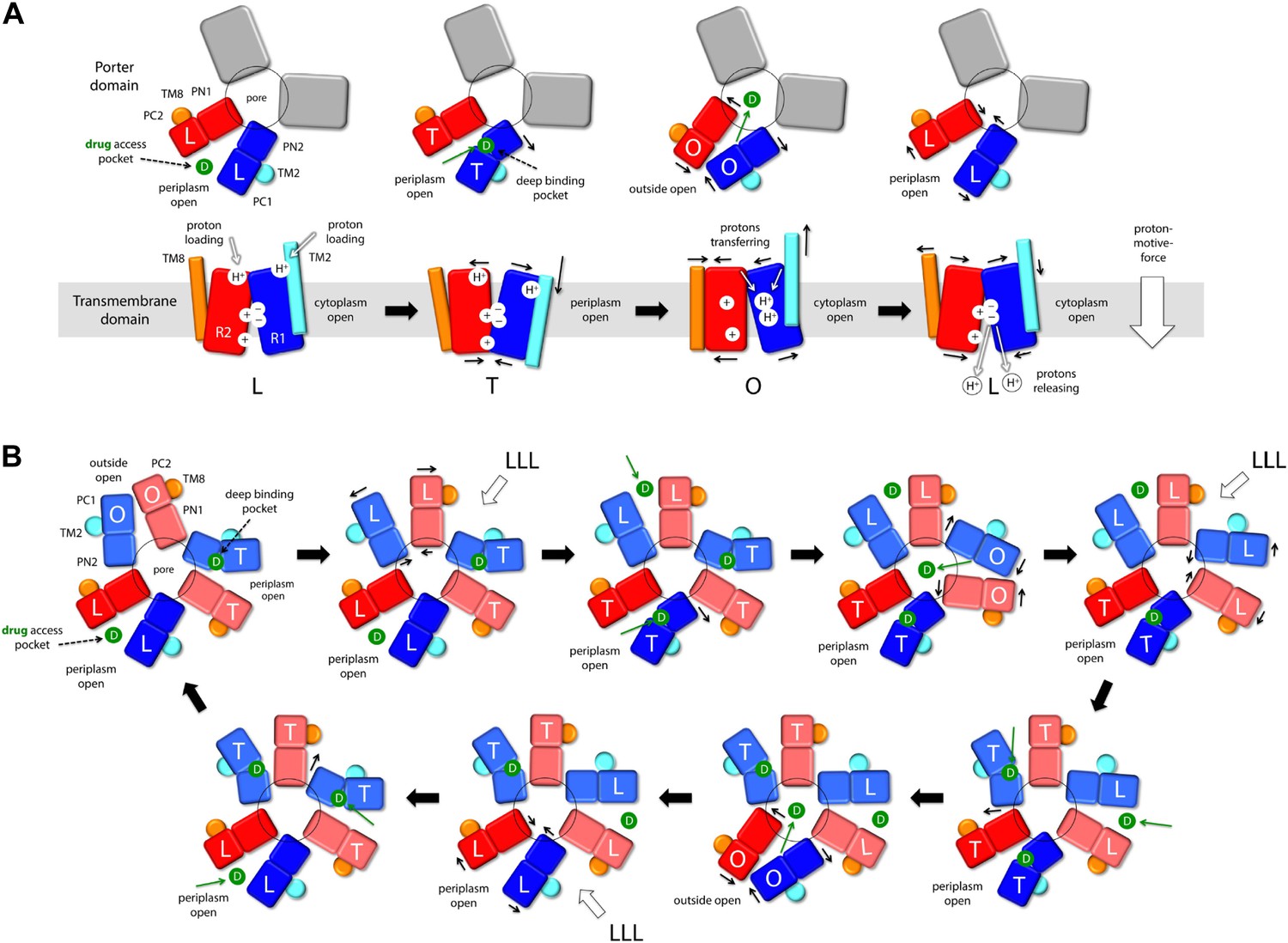
Proposed mechanism of drug-proton antiport in AcrB.
(A) The scheme shows the conformational cycle of one of the protomers; the porter domain is viewed from the outer membrane, and the TM domain is viewed along the membrane plane. The structural repeats in the TM and porter domains, R1 and R2, and PC1/PN2 and PN1/PC2, respectively, are colored as in Figure 1. Likewise for the TM2 and TM8, which flank R1 and R2 and couple them to the repeats in the porter domain. The conformational state (L/T/O) of each protomer is indicated alongside the proposed location of the substrate (D) and the protons (H+) in each state. Residues in the proton relay network formed by K940, R971, D407 and D408, which controls the relative orientation of R1 and R2, are indicated with ‘+’ and ‘−’ symbols. The series of states proposed for the TM domain is based on the analysis of protonation states and water accessibility of wildtype AcrB and its variants, reported above. The model postulates that TM2 and TM8 mediate the strict coupling between these two conformational cycles. (B) The scheme shows the conformational cycle of the porter-domain trimer, viewed from the outer-membrane. The specific series of states proposed for the porter domain derives from analysis of potential steric clashes at the protomer–protomer interface in all possible combinations of L, T and O conformations (Figure 11—figure supplement 1). Note that the frequency of protomers in the O state is minimized, relative to other possible cycles, while that of protomers in the L and T state is maximized; we propose this kinetically advantageous, in that it would result in minimum back-flow of substrates from the exit pore, and maximal capture from the periplasmic space.
Figure 11—figure supplement 1
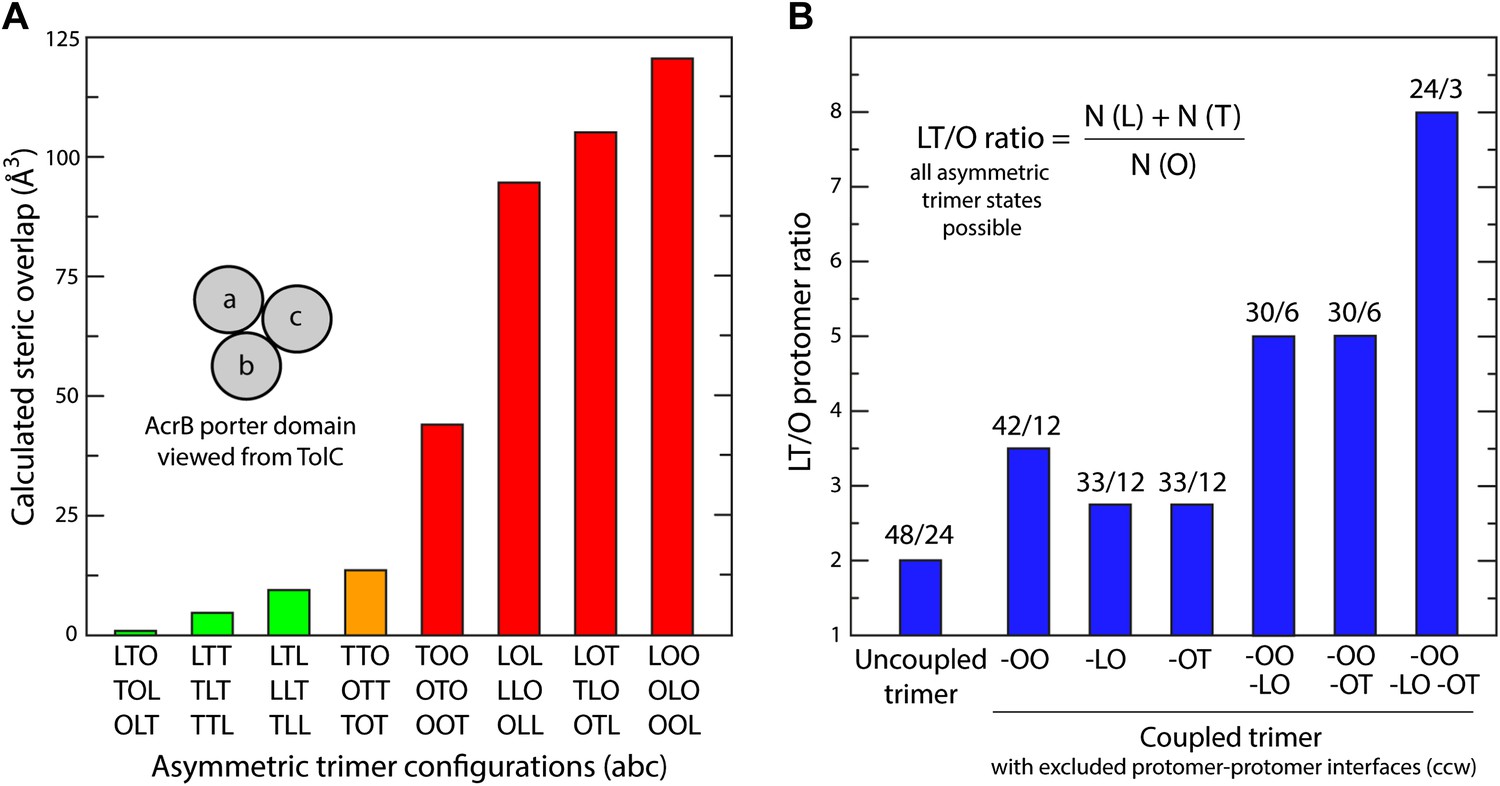
Mechanistic significance of the conformational coupling among protomers.
(A) Steric clashes in hypothetical models of the porter domain of AcrB, in all possible non-symmetric permutations of the L, T and O conformations. The steric clash in a given trimer configuration is defined as the volume of the overlapping atoms from different protomers at the three interfaces. The trimer models were generated via rigid-body rotations of the PC1/PN2 and PN1/PC2 sub-domains around the trimer axis, and optimized with a side-chain prediction algorithm (‘Materials and methods’). The combinations shown in green are the most feasible from a steric standpoint; these are also sufficient to draw a complete transport cycle that, arguably, would be also ideal from a kinetic standpoint, as it would minimize the chances of drug re-binding after extrusion into the TolC lumen (see manuscript). The conformations shown in red are likely to be energetically prohibited, because LO and OO interfaces would require extensive deformations in the protomers structure. (B) For alternative hypothetical transport cycles of the AcrB trimer, ratio of the number of instances in which a protomer adopts either the L or T state, relative to the O state. Only non-symmetric trimer states are considered. The conformational cycle of an uncoupled trimer features the largest number of possible trimer configurations, but also the largest proportion of O states. In the coupled trimer, many of those trimer configurations are excluded because speficic protomer–protomer interfaces are sterically incompatible, for example, OO and LO (A). The plot shows that these specific exclusions imply an optimization of the LT/O ratio in the conformational cycle, which is likely to translate into a more kinetically efficient efflux system.
Videos
Video 1
Structural repeats within the periplasmic porter domain of AcrB, PN1/PC2 and PN2/PC1, and their collective motions during the transport cycle.
L to T transition.
Video 2
Structural repeats within the periplasmic porter domain of AcrB, PN1/PC2 and PN2/PC1, and their collective motions during the transport cycle.
T to O transition.
Video 3
Structural repeats within the periplasmic porter domain of AcrB, PN1/PC2 and PN2/PC1, and their collective motions during the transport cycle.
O to L transition.
Video 4
Structural repeats within the transmembrane domain of AcrB, R1 and R2, and their collective motions during the transport cycle.
L to T transition.
Video 5
Structural repeats within the transmembrane domain of AcrB, R1 and R2, and their collective motions during the transport cycle.
T to O transition.
Video 6
Structural repeats within the transmembrane domain of AcrB, R1 and R2, and their collective motions during the transport cycle.
O to L transition.
Additional files
-
Supplementary file 1
Structural variations in the porter domain of the periplasmic region of wildtype AcrB, in the L, T and O states.
- https://doi.org/10.7554/eLife.03145.029
-
Supplementary file 2
Data collection and refinement statistics for the crystal structures of AcrB mutants D407N, D408N, R971A, and K940A.
- https://doi.org/10.7554/eLife.03145.030
-
Supplementary file 3
Differences in the structures of the repeats in the transmembrane and porter domains of AcrB, in wildtype vs. D407N, D408N, R971A, and K940A variants.
- https://doi.org/10.7554/eLife.03145.031
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Coupling of remote alternating-access transport mechanisms for protons and substrates in the multidrug efflux pump AcrB
eLife 3:e03145.
https://doi.org/10.7554/eLife.03145




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}