A kinase-independent function of AKT promotes cancer cell survival
- Memorial Sloan-Kettering Cancer Center, United States
- Spanish National Cancer Research Centre, Spain
- Weill-Cornell Graduate School of Biomedical Sciences, United States
- Memorial Sloan-Kettering Cancer, United States
Figures
Figure 1 with 9 supplements
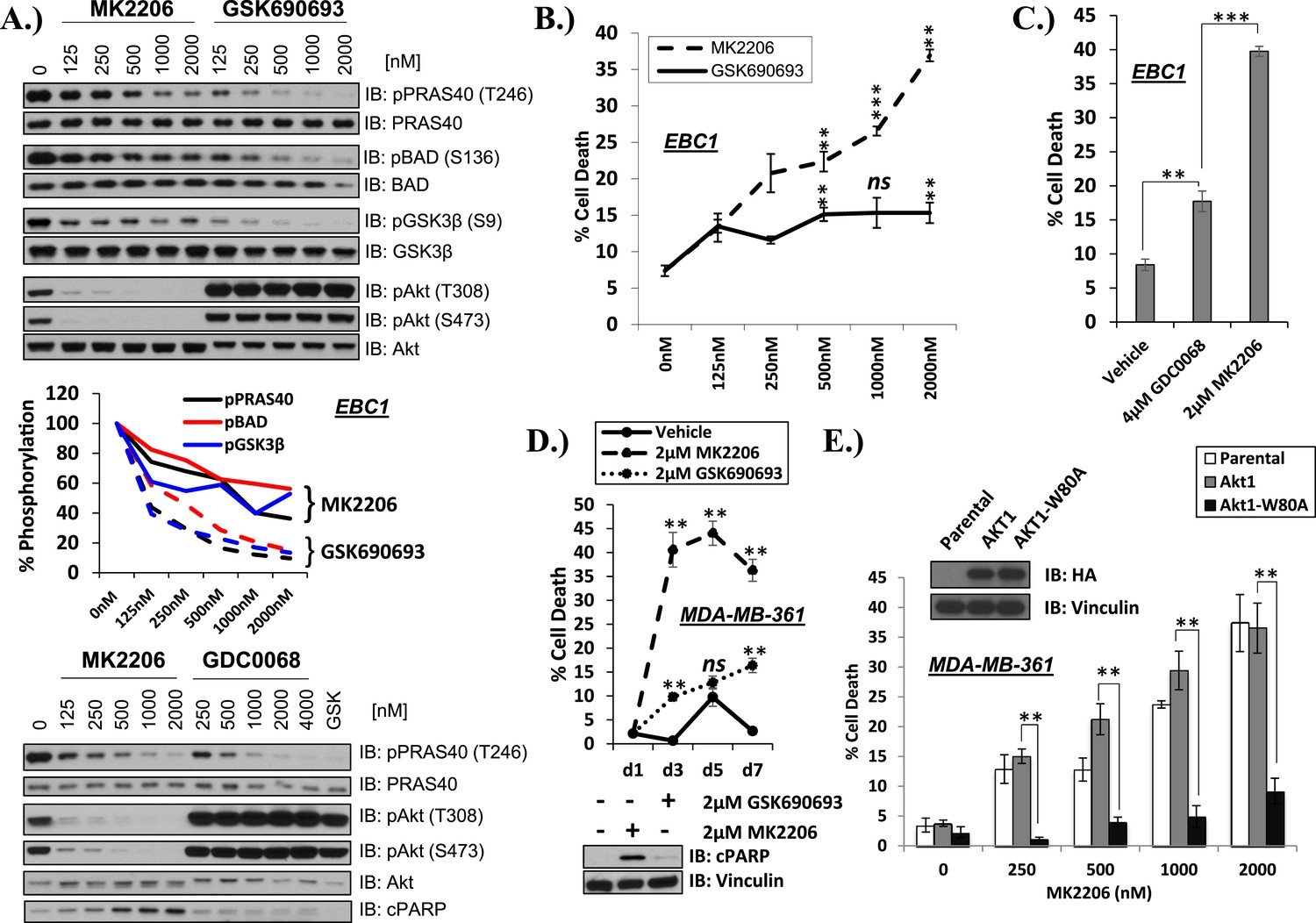
Differential sensitivity of human cancer cells to different classes of AKT inhibitors.
(A) Effect of AKT inhibitors on phosphorylation of AKT and AKT protein substrates. EBC1 cells were treated with the indicated doses of MK2206 and GSK690693 (top), or GDC0068 (bottom) for 24 hr. Treated cells were lysed and analyzed by immunoblot with the indicated antibodies. Phosphorylation of PRAS40, BAD, and GSK3β was quantified by image densitometry (middle). (B) Allosteric AKT inhibitor MK2206 induces more cell death than ATP-competitive inhibitor GSK690693 in EBC1 human lung cancer cells. Cells were treated with drug or vehicle for 96 hr. Cell death was determined by the trypan blue method. Error bars denote standard error of the mean (t-test *p ≤ 0.05, **p ≤ 0.01, treatment vs vehicle). (C) Allosteric AKT inhibitor MK2206 induces more cell death than ATP-competitive inhibitor GDC0068. Experimental conditions were as in Figure 1B. (D) Allosteric AKT inhibitor MK2206 induces more cell death than the ATP-competitive inhibitor GSK690693 in MDA-MB-361 human breast cancer cells. Cell death was assessed on day 1, day 3, day 5, and day 7 following treatment with vehicle or 2 µM of MK2206 or 2 µM of GSK690693(top) (t-test *p ≤ 0.05,**p ≤ 0.01, treatment vs vehicle). An additional plate from each treatment group was lysed 24 hr after drug treatment and analyzed by Western blot with the indicated antibodies (bottom). (E) Suppression of MK2206-induced cell death by a drug-resistant allele of AKT1. MDA-MB-361 cells were stably transduced with HA-tagged wild-type AKT1 or AKT1-W80A and treated with 2 µM MK2206 for 96 hr. Cell death was assessed as above. Expression of the transgenes was confirmed by immunoblot using an HA antibody, and loading was controlled with a vinculin antibody (inset).
-
Figure 1—source data 1
Contains source data for Figure 1 and all accompanying Figure 1—figure supplements.
- https://doi.org/10.7554/eLife.03751.004
Figure 1—figure supplement 1
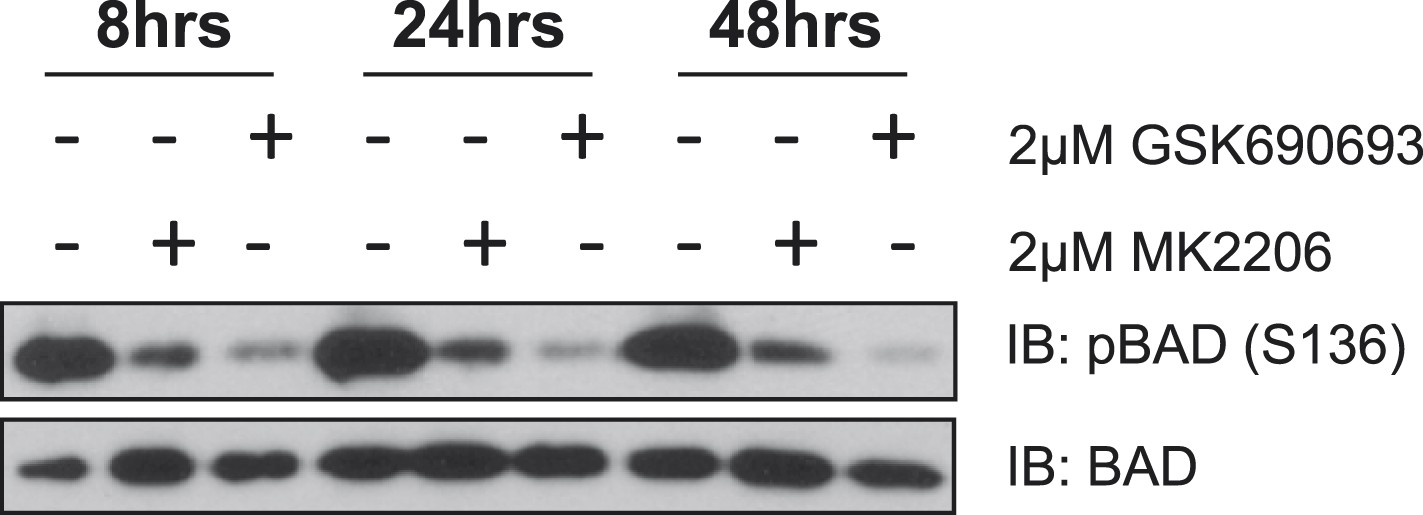
MK2206 and GSK690693 cause sustained suppression of AKT-dependent BAD phosphorylation.
EBC1 cells were treated with either 2 µM MK2206 or 2 µM GSK690693 and lysates were collected at the indicated times. Ser136 BAD phosphorylation was analyzed by immunoblot as indicated.
Figure 1—figure supplement 2
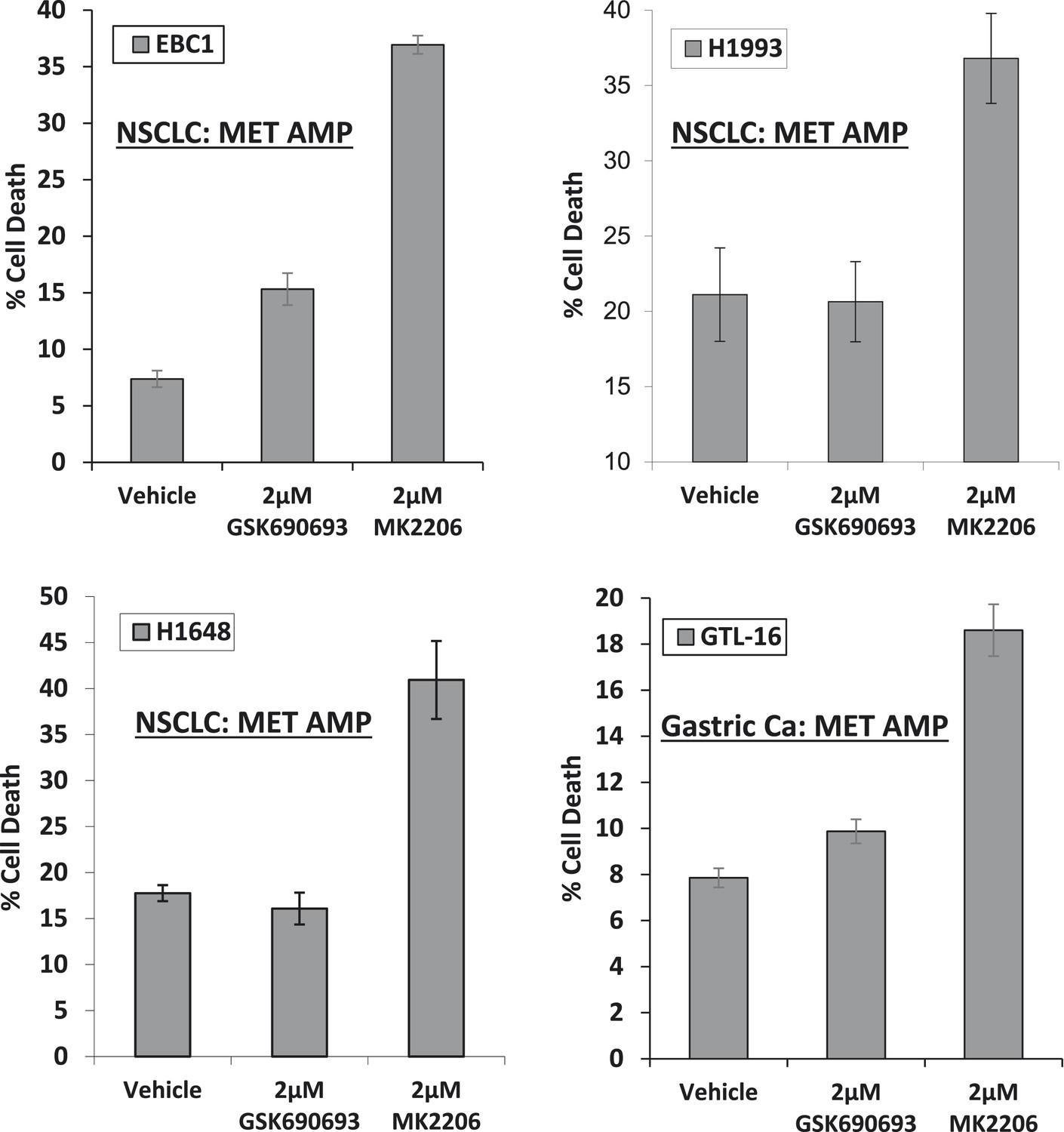
MET-amplified cancer cell lines die in response to an allosteric but not an ATP-competitive AKT inhibitor.
EBC1, H1993, H1648, and GTL6 cells were treated with vehicle, 2 µM GSK690693, or 2 µM MK2206 for 96 hr. Cell death was assessed by the trypan blue method as detailed in the ‘Methods’ section.
Figure 1—figure supplement 3
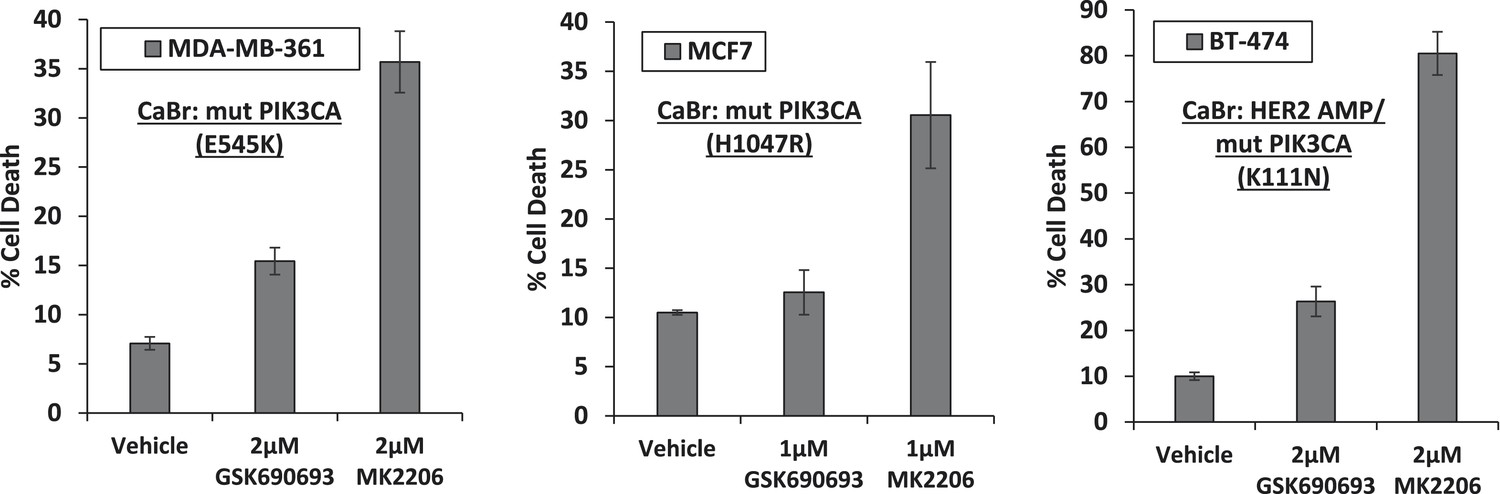
Breast cancer cell lines with HER2 amplification and/or activating PIK3CA-mutations die in response to an allosteric but not an ATP competitive AKT inhibitor.
MDA-MB-361, MCF7, and BT-474 breast cancer cells were treated with the indicated doses of GSK690693 or MK2206 for 96 hr. Cell death was assessed by the trypan blue method.
Figure 1—figure supplement 4
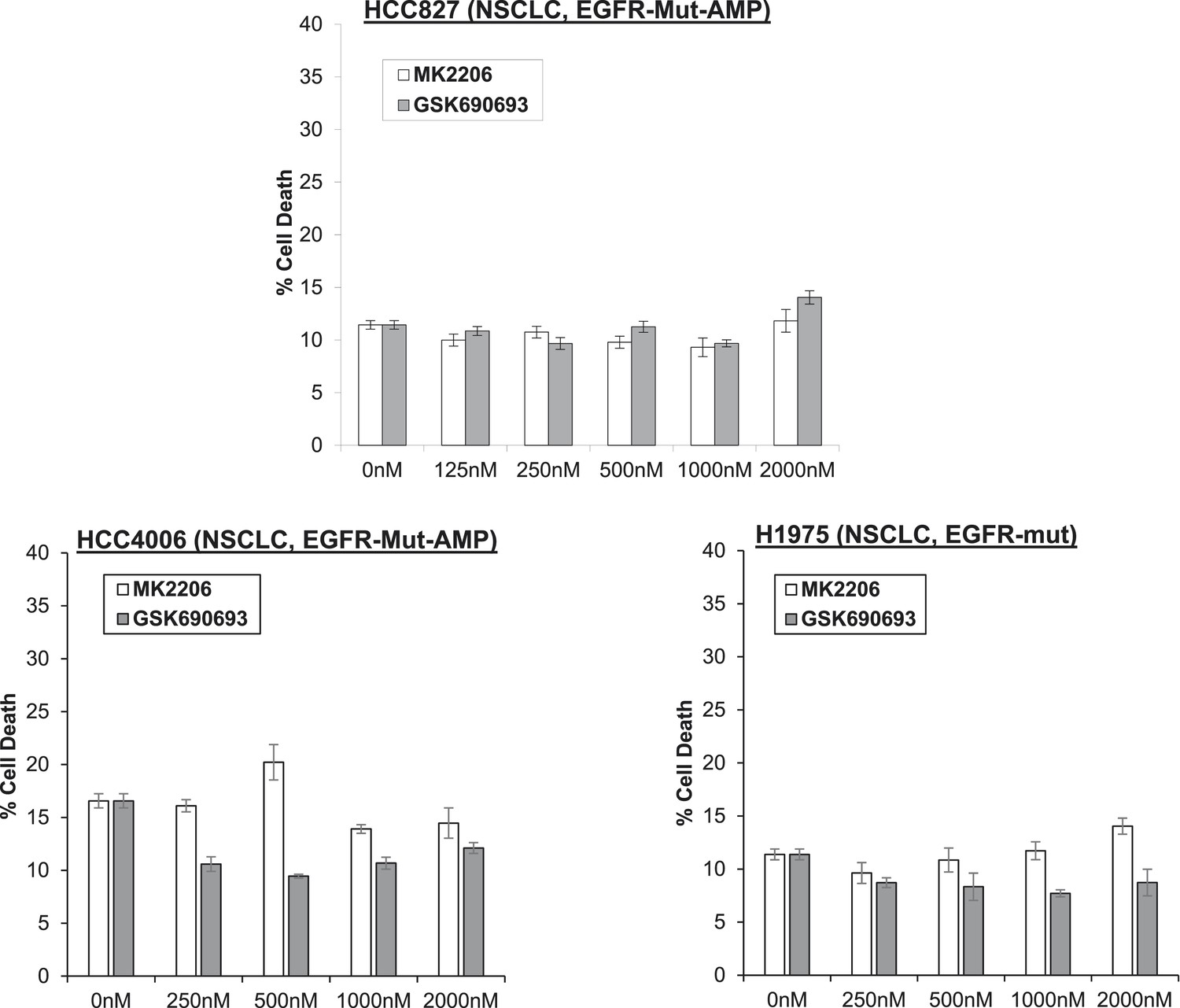
Non-small-cell lung cancer (NSCLC) cell lines with EGFR gene amplification and/or activating EGFR mutations are resistant to cell death induction by AKT inhibitors.
HCC827 (EGFR-Δ74-750/AMP) (top right), HCC4006 (EGFR-Δ747-749/AMP) (bottom left), and H1975 (EGFR-T790M-L858R) (bottom right) lung cancer cells were treated with the indicated doses of GSK690693 or MK2206 for 96 hr. Cell death was assessed by the trypan blue method.
Figure 1—figure supplement 5
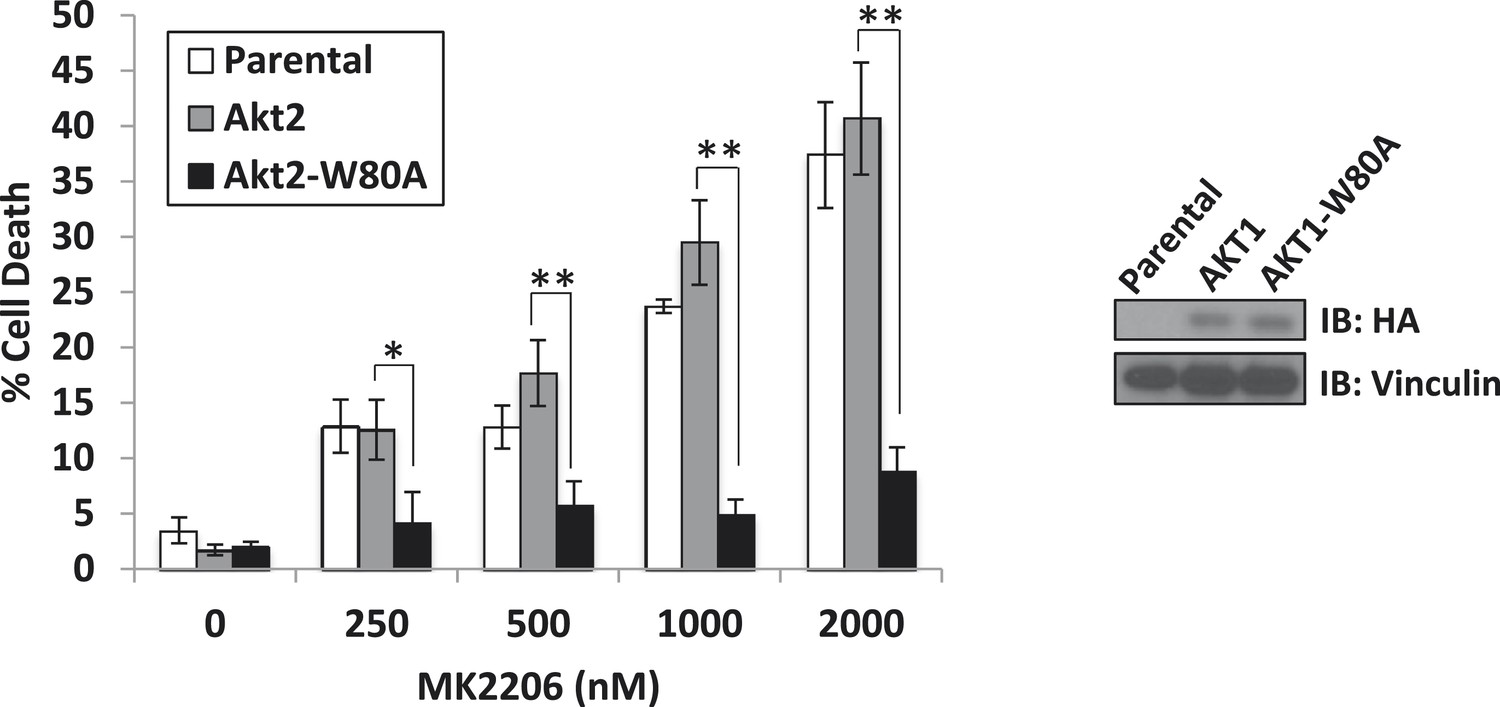
A drug-resistant allele of AKT2 suppresses MK2206-induced cell death.
MDA-MB-361 cells were stably transduced with HA-tagged wild-type AKT2 or AKT2-W80A and treated with the indicated concentrations of MK2206 for 96 hr. Cell death was assessed by the trypan blue method (left). Expression of the transgenes was confirmed by immunoblot using an anti-HA antibody, and anti-vinculin was used to control for loading (right).
Figure 1—figure supplement 6
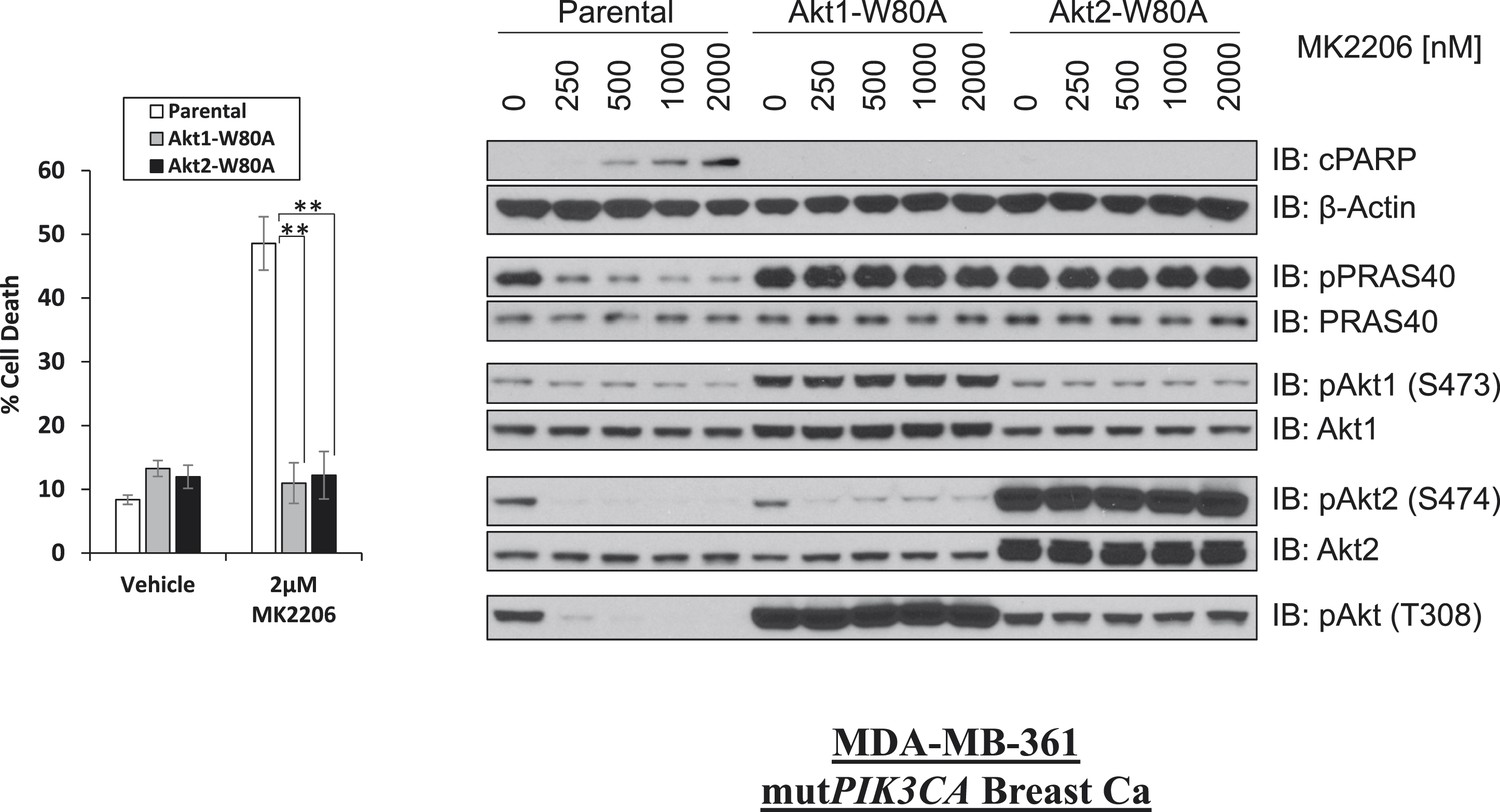
W80A mutation renders AKT1 and AKT2 resistant to MK2206.
MDA-MB-361 cells were stably transduced with HA-tagged AKT1-W80A or AKT2-W80A and treated with 2 µM MK2206 for 96 hr. Cell death was assessed by the trypan blue method (left). Parental MDA-MB-361 cells or MDA-MB-361 cells stably expressing AKT1-W80A or AKT2-W80A were treated with the indicated doses of MK2206 for 24 hr and lysed. To assess drug effects, lysates were subjected to immunoblot analysis with the indicated antibodies (right).
Figure 1—figure supplement 7
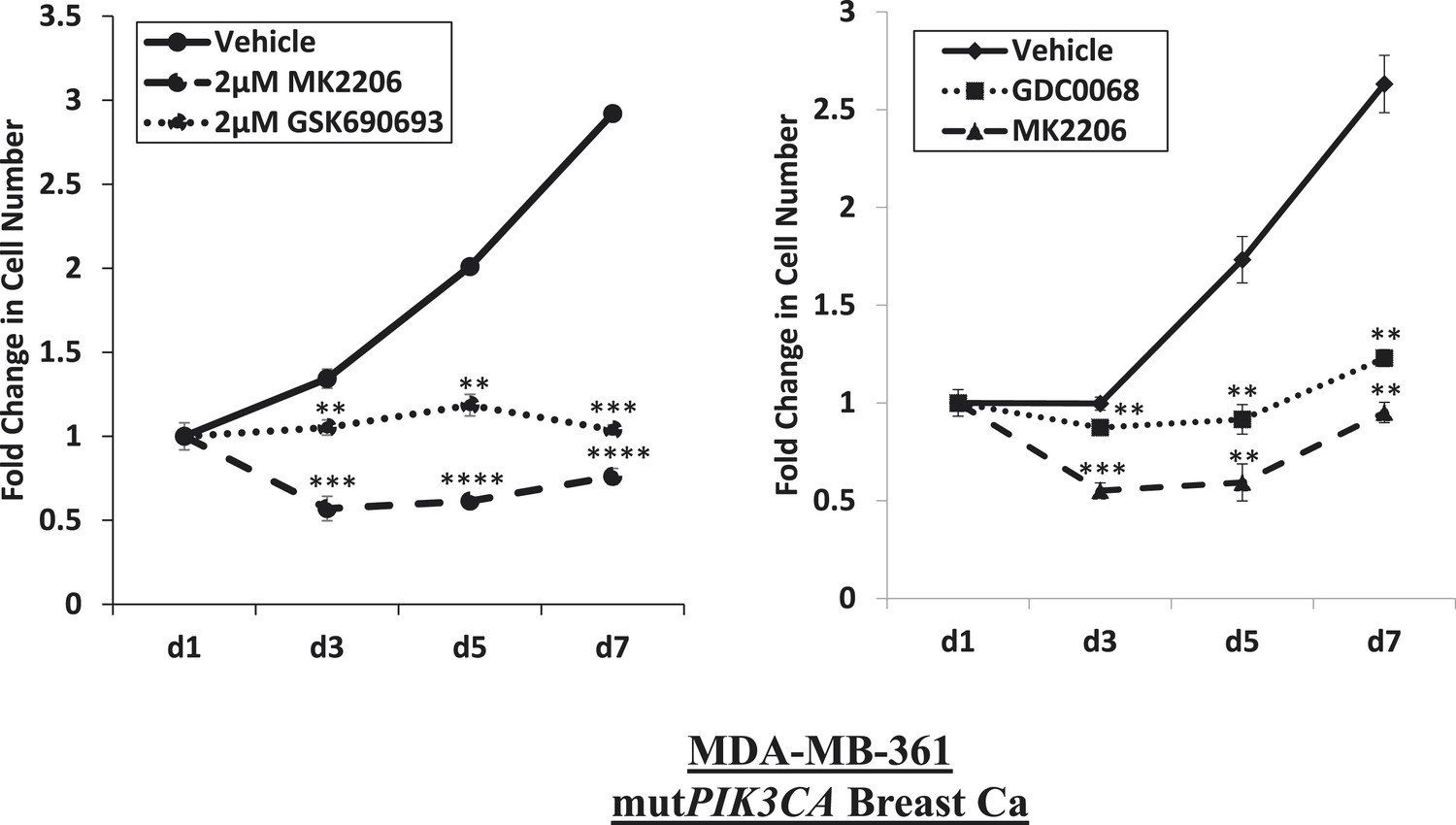
Near-complete inhibition of tumor cell proliferation by both allosteric and ATP-competitive inhibitors of AKT.
PIK3CA-mutant MDA-MB-361 cells were treated with vehicle, 2 µM MK2206, or 2 µM GSK690693 (left panel), or vehicle, 2 µM MK2206, or 4 µM GDC0068 (right panel). The number of viable cells was counted on day 1, day 3, day 5, and day 7.
Figure 1—figure supplement 8
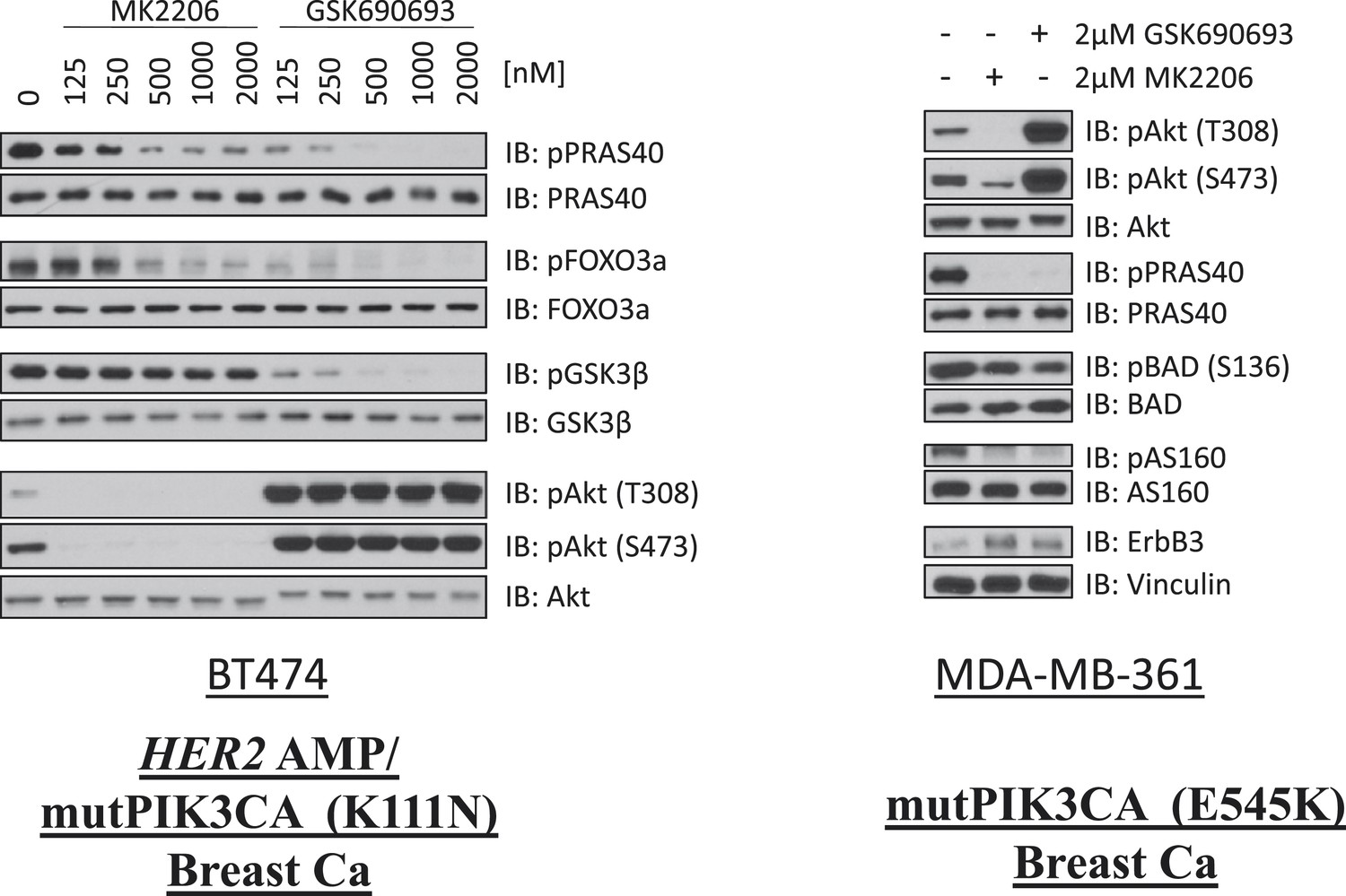
Comparison of AKT kinase inhibition by GSK690693 and MK2206.
BT474 cells (left) and MDA-MB-361 cells (right) were treated with the indicated doses of either MK2206 or GSK690693 for 24 hr and lysed. Lysates were analyzed by immunoblot with the indicated antibodies.
Figure 1—figure supplement 9
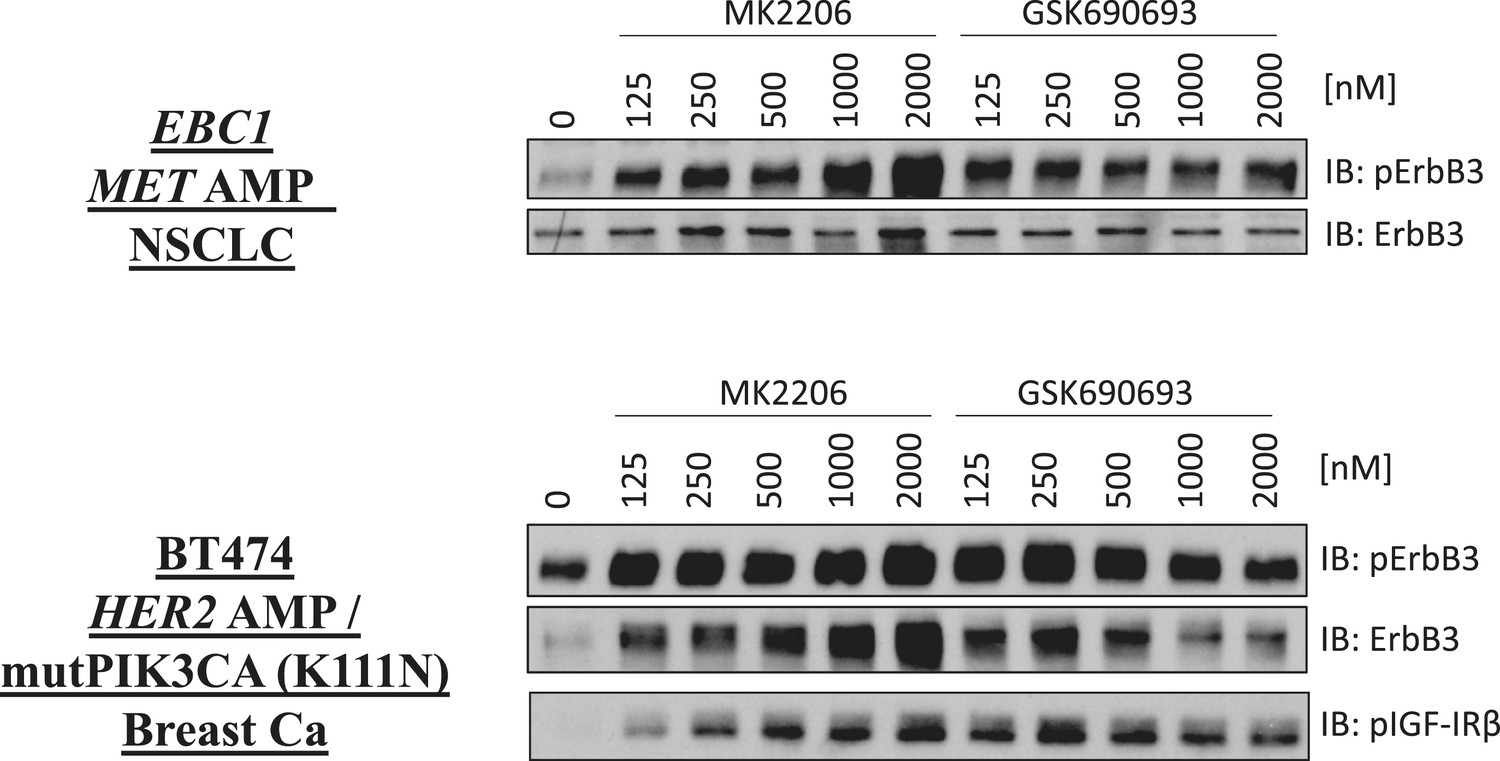
De-inhibition of ErbB3 and p-IGF-IRβ by GSK690693 and MK2206.
EBC1 and BT474 cells were treated with the indicated doses of MK2206 and GSK690693 for 24 hr and lysates were analyzed by Western blot with the indicated antibodies.
Figure 2 with 1 supplement
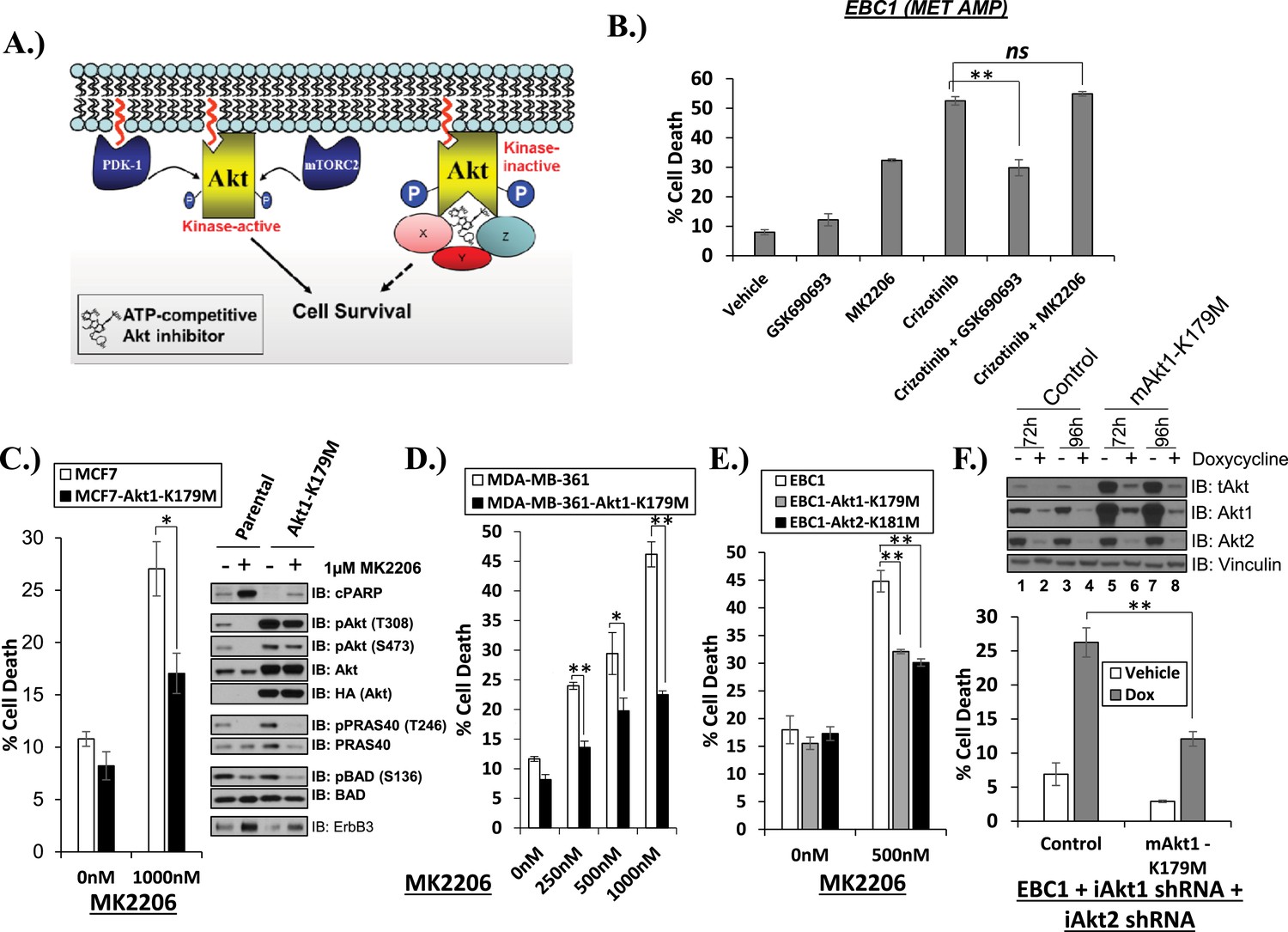
Catalytically inactive AKT supports the survival of AKT-dependent human cancer cells.
(A) Cartoon illustrating how ATP-competitive AKT inhibitors may promote a kinase-independent pro-survival function of AKT. In this model, ATP-competitive inhibitors of AKT block kinase activity, induce AKT hyper-phosphorylation, and induce the formation of a pro-survival complex. (B) EBC1 cells were treated with either 25 nM crizotinib, 1 µM GSK690693, 1 µM MK2206, or the indicated combinations for 96 hr. Cell death was assessed following treatment by the trypan blue assay. (C) MCF7 cells or a sub-line generated by stable transduction with kinase-deficient AKT1-K179M were treated with either vehicle or 1 µM MK2206 for 96 hr. Cell death was assessed by the trypan blue method (left). A duplicate set of plates was lysed and analyzed by immunoblot with the indicated antibodies (right) to confirm kinase inhibition and transgene expression. (D) MDA-MB-361 cells or a sub-line stably expressing AKT1-K179M were treated with the indicated doses of MK2206 for 96 hr. Cell death was assessed by the trypan blue assay. (E) Parental EBC1 cells of EBC1 cells stably expressing AKT1-K179M or AKT2-K181M were treated with MK2206 as indicated. After 96 hr, cell death was assessed as elsewhere. (F) EBC1 cells expressing doxycycline-inducible hairpins targeting both hAKT1 and hAKT2 and a sub-line expressing a hairpin-resistant kinase-deficient mAKT1-K179M allele were grown in the presence or absence of 2.5 µg/ml doxycycline for 7 days. Cell death was assessed on day 7 by the trypan blue assay (bottom). Replicate plates were cultured with or without doxycycline for the indicated times and lysed. Lysates were analyzed by immunoblot (top) using the indicated antibodies.
-
Figure 2—source data 1
Contains source data for Figure 2 and Figure 2—figure supplement 1.
- https://doi.org/10.7554/eLife.03751.015
Figure 2—figure supplement 1
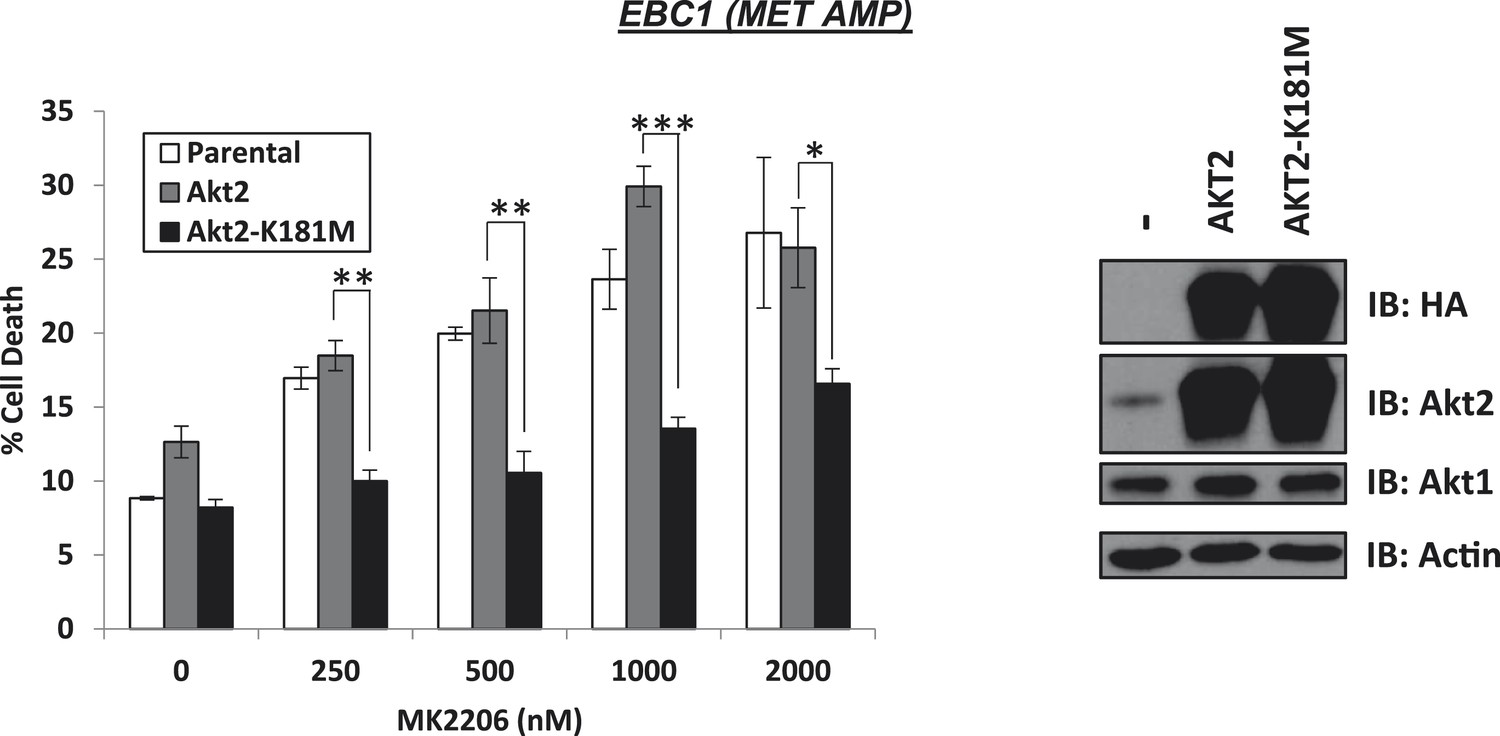
Kinase-dead AKT2-K181M but not wild-type AKT2 protects from MK2206-induced cell death.
EBC1 cells were stably transduced with either HA-tagged wild-type AKT2 or AKT2-K181M. Cells were treated with the indicated doses of MK2206 for 96 hr and cell death was analyzed by the trypan blue method (left). Expression of the transgenes was confirmed by immunoblot with the indicated antibodies (right).
Figure 3 with 4 supplements
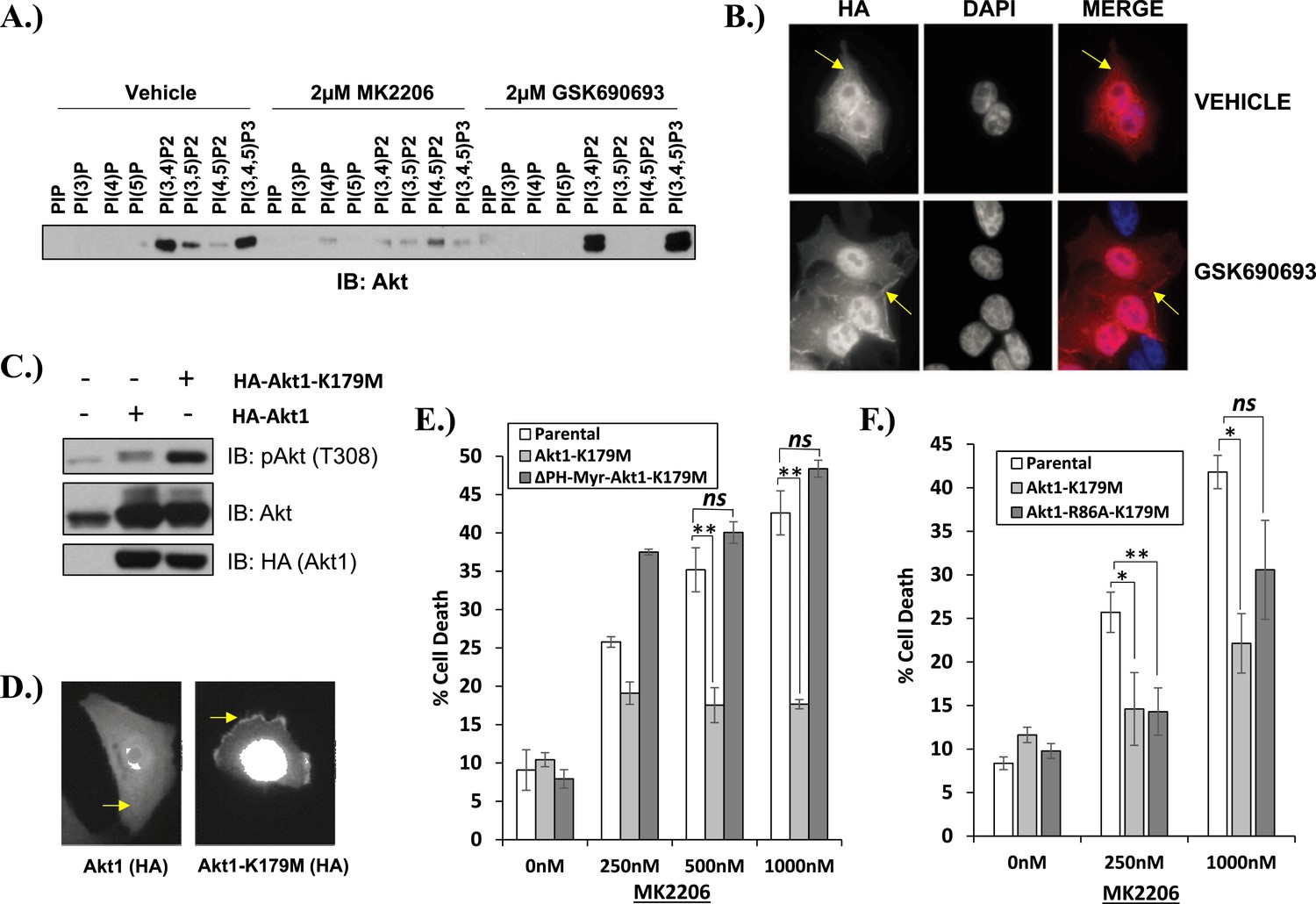
Kinase-dead AKT protects from drug-induced cell death in a PH-domain-dependent manner.
(A) HCT116 AKT1−/− AKT2 −/− cells stably transduced with human influenza hemagglutinin (HA) epitope-tagged AKT2 were treated with vehicle, MK2206, or GSK690693 for 4 hr and lysed. Lysates were subjected to a PIP-binding assay to assess phosphoinositide binding preference. AKT binding was assessed by immunoblot using an AKT-specific antibody. (B) The localization of AKT2 was assessed in the same cells as in A by HA immunofluorescence following 24 hr treatment with the indicated drugs. Arrows indicate HA staining. (C) Parental MCF10A cells and MCF10A cells stably transduced with HA-tagged wild-type (WT) AKT1 (lane 2) or AKT1-K179M (lane 3) were analyzed by Western blot with the indicated antibodies. (D) The localization of WT and K179M-AKT1 in MCF10A cells was assessed by HA immunostaining. Shown are representative immunofluorescence images. Arrows indicate HA staining. (E) Parental MDA-MB-361 cells or sub-lines stably expressing AKT1-K179M or ΔPH-Myr-AKT1-K179M were treated with the indicated doses of MK2206 for 96 hr. Cell death was assessed by the trypan blue assay. (F) Parental MDA-MB-361 cells, or sub-lines stably expressing kinase-dead AKT1-K179M, or the PIP-binding-deficient variant AKT1-R86A-K179M were treated with the indicated doses of MK2206 for 96 hr. Cell death was measured by the trypan blue assay.
-
Figure 3—source data 1
Contains source data for Figure 3 and Figure 3—figure supplement 2.
- https://doi.org/10.7554/eLife.03751.018
Figure 3—figure supplement 1
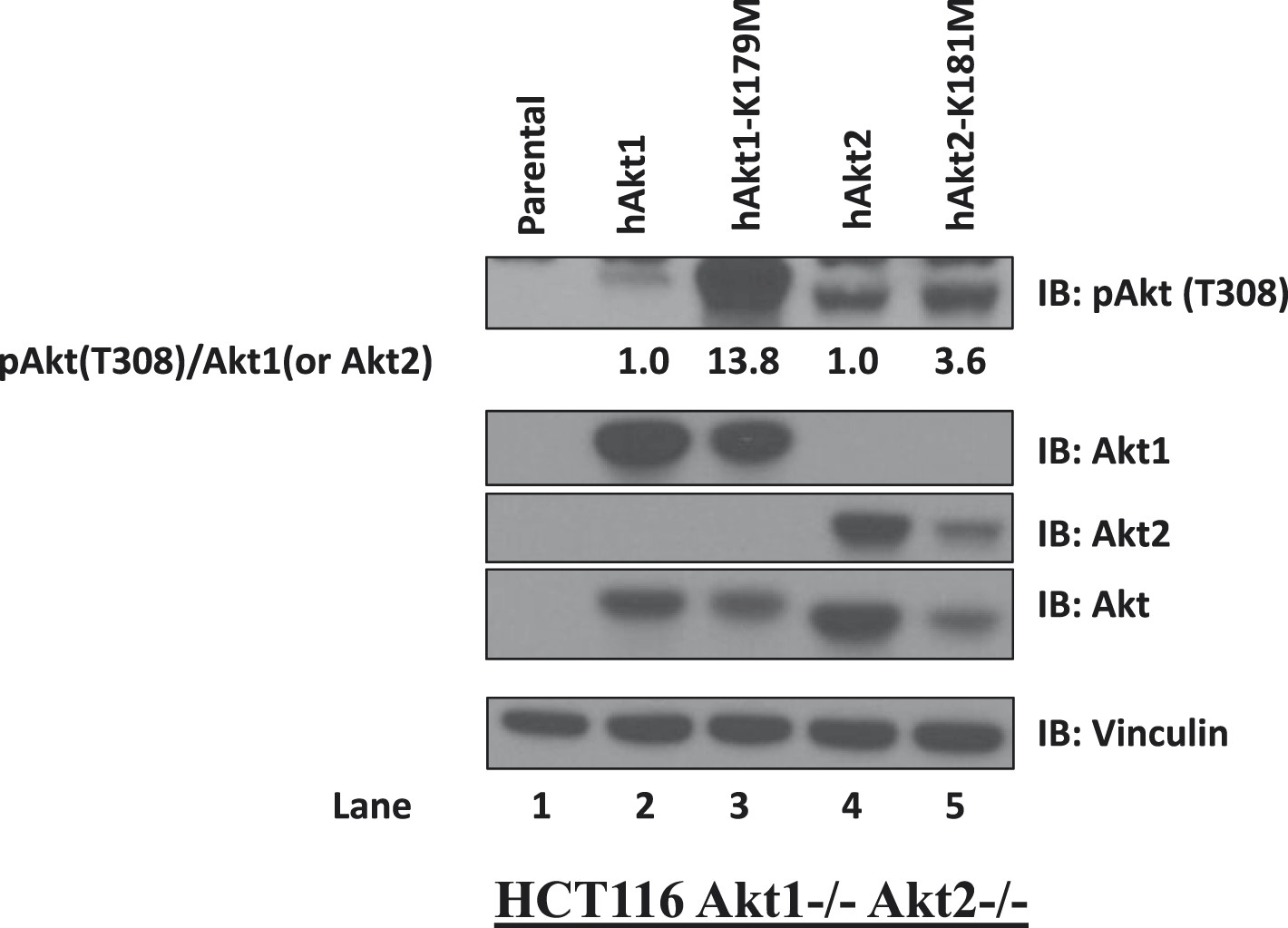
Kinase-deficient AKT mutants display enhanced activation loop phosphorylation at threonine 308.
HCT116 AKT1−/− AKT2 −/− cells were stably transduced with wild-type and kinase-deficient alleles (K179M for AKT1 and K181M for AKT2) of either AKT1 or AKT2. Stably transduced cells were lysed and analyzed by immunoblot with the indicated antibodies. The ratio of phosphorylated AKT (Thr308)/total AKT was determined for each AKT isoform by image quantification densitometry and is indicated below each lane.
Figure 3—figure supplement 2
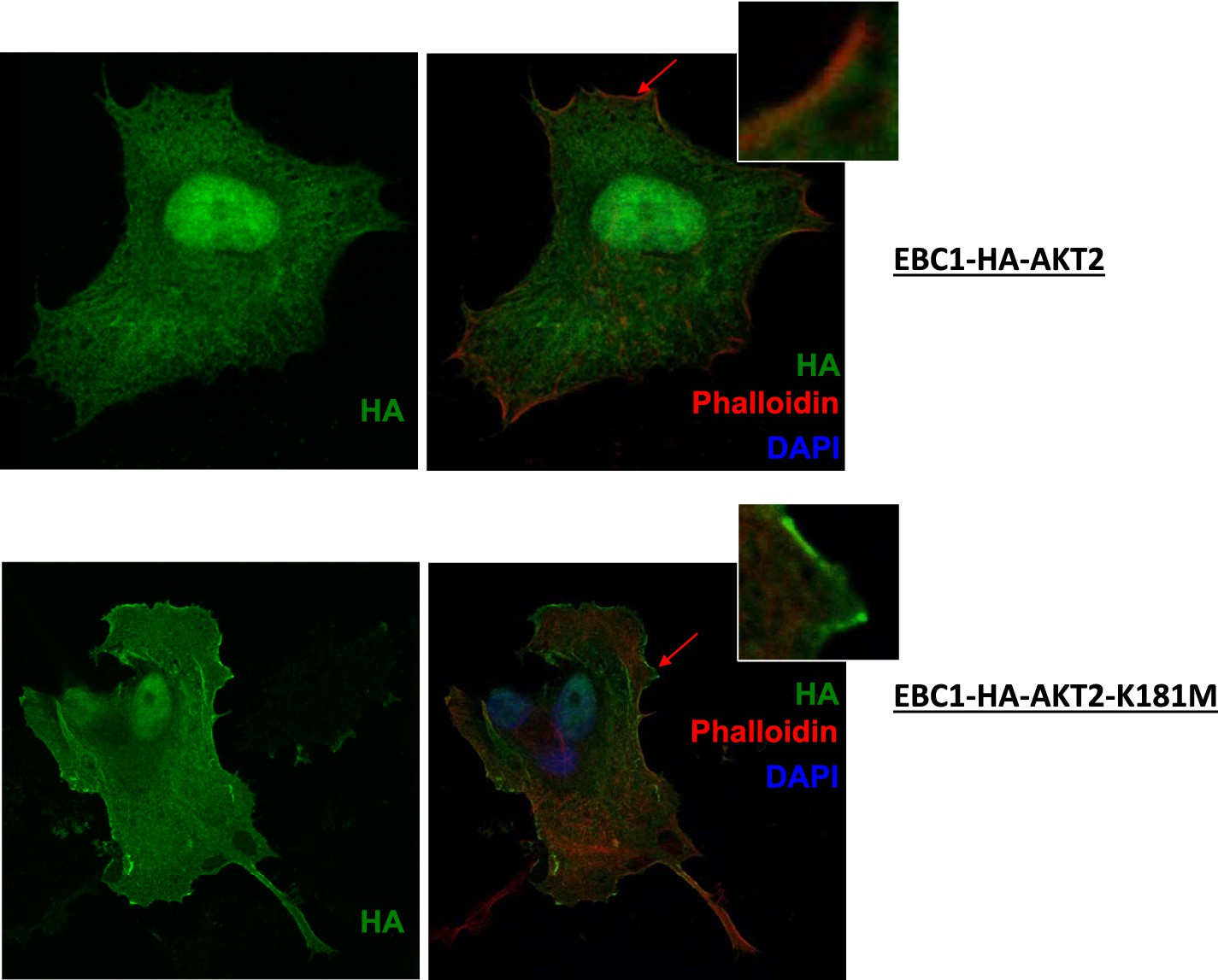
Kinase-deficient mutant AKT2 exhibits enhanced membrane localization.
EBC1 cells stably transduced with HA-tagged wild-type AKT2 (top) or kinase-dead AKT2-K181M (bottom) were stained with antibodies against HA. Cells were also stained with DAPI and phalloidin, which binds F-actin and marks membrane ruffles. Note, increased membrane localization of mutant AKT2 even in the absence of membrane ruffles. Shown are representative confocal images of each sample.
Figure 3—figure supplement 3

Biochemical effects of PH-domain removal in kinase-dead AKT.
Parental MDA-MB-361 cells or MDA-MB-361 cells stably expressing AKT1-K179M or ΔPH-Myr-AKT1-K179M were treated with the indicated doses of MK2206 for 24 hr and lysed. Lysates were subjected to immunoblot analysis with the indicated antibodies.
Figure 3—figure supplement 4
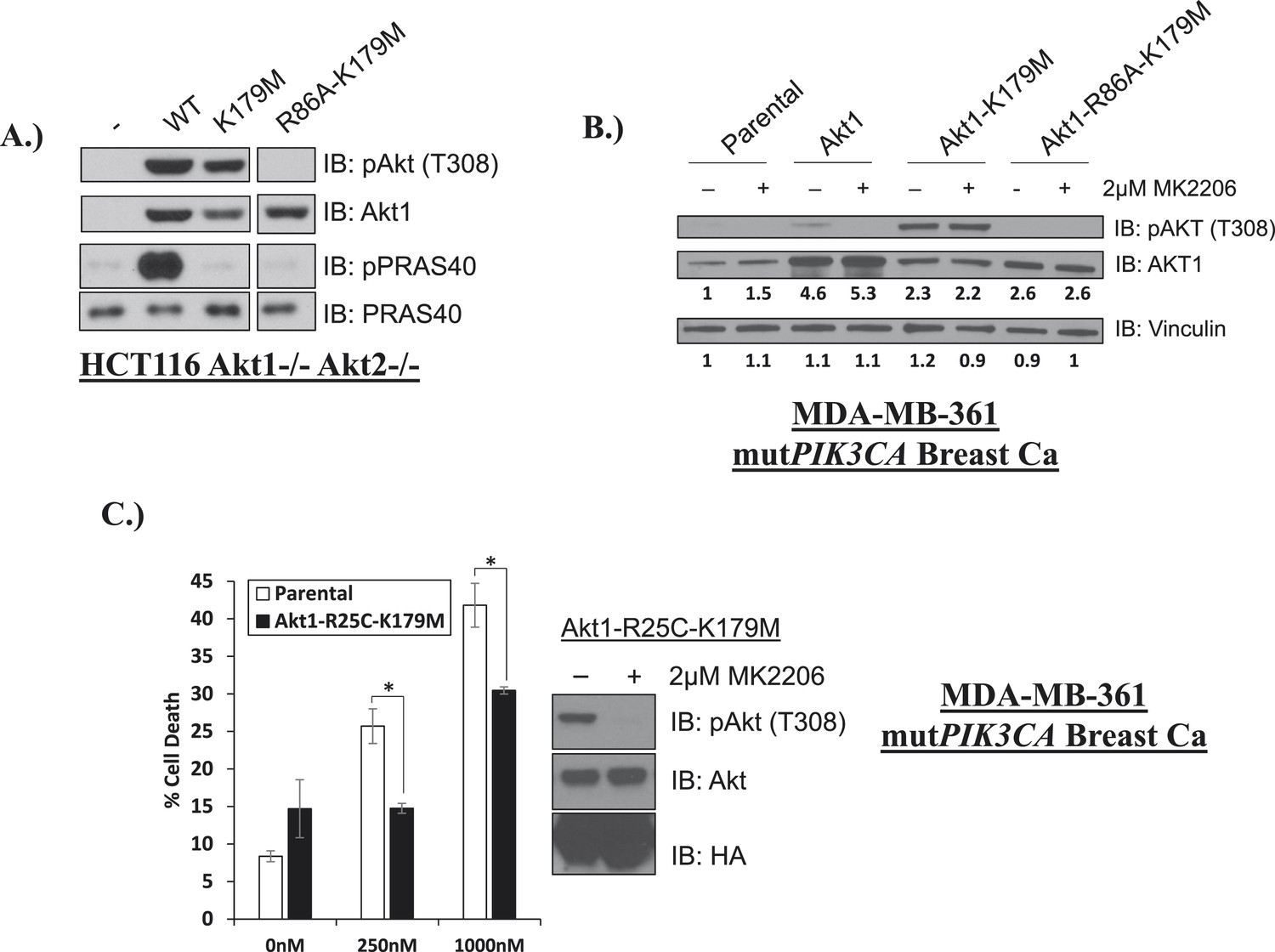
Phosphoinositide binding deficient kinase-dead AKT can protect from drug-induced cell death.
(A) To assess AKT and AKT substrate phosphorylation of mutant AKT1 alleles, parental HCT116 AKT1−/− AKT2 −/− cells, or HCT116 AKT1−/− AKT2 −/− cells stably expressing WT AKT1, AKT1-K179M, or AKT1-R86A-K179M were lysed and analyzed by Western blot with the indicated antibodies. (B) Parental MDA-MB-361 cells or MDA-MB-361 cells stably expressing AKT1-K179M or AKT1-R86A-K179M were lysed and analyzed by Western blot with the indicated antibodies. Relative levels of AKT1 and vinculin were quantified by image densitometry and are indicated under each lane. (C) Parental MDA-MB-361 cells or sub-lines stably expressing kinase-dead AKT1-K179M or the PIP-binding-deficient variant AKT1-R25C-K179M were treated with the indicated doses of MK2206 for 96 hr. Cell death was measured by the trypan blue assay. Lysates from MDA-MB-361 cells expressing AKT1-R25C-K179M were collected after treatment with vehicle or 2 µM MK2206 for 24 hr.
Figure 4
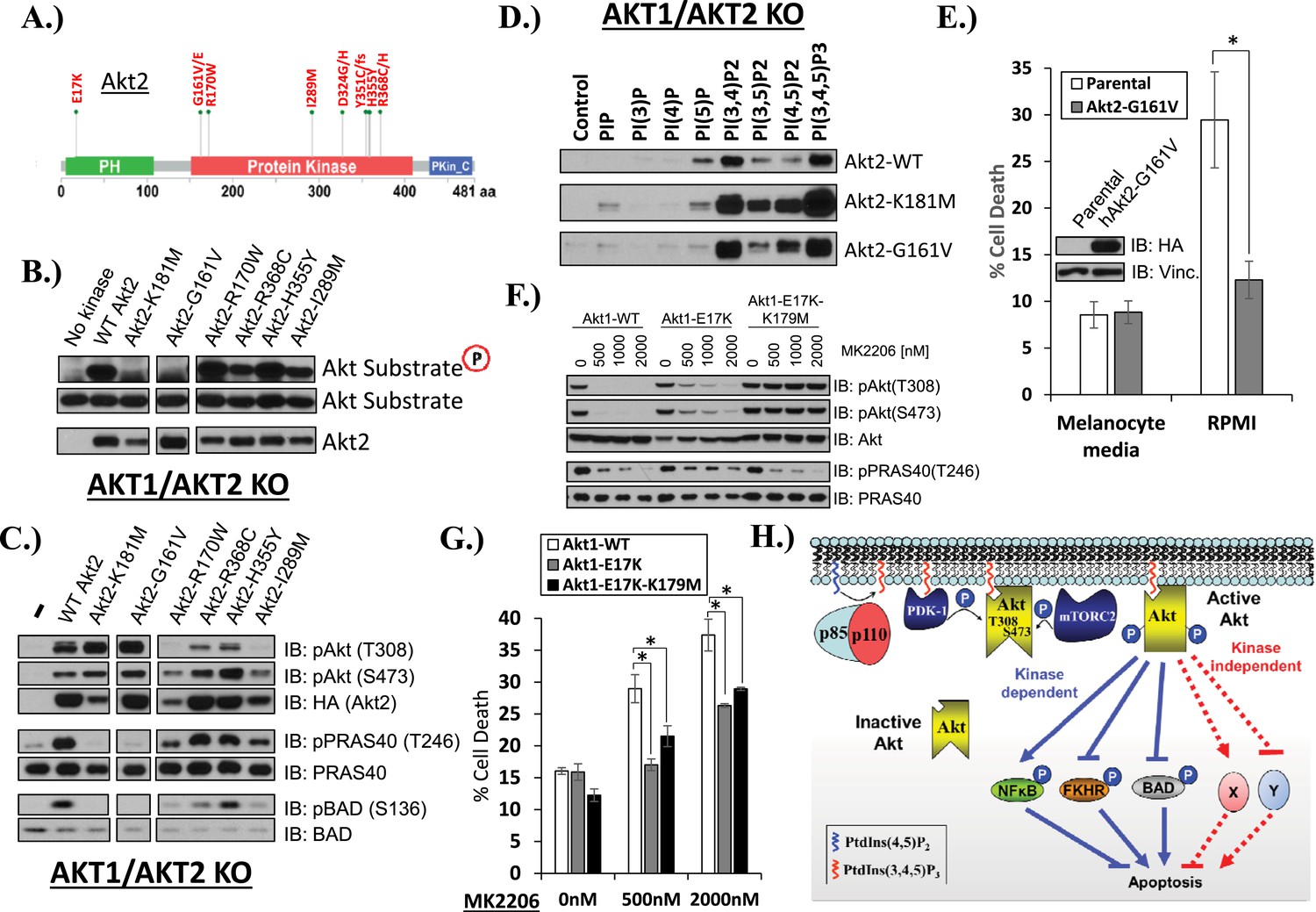
AKT mutants found in human cancer can promote cell survival independently of kinase activity.
(A) Distribution of AKT2 mutations that occur in human cancers in 2 or more independent samples. (B) HA-tagged wild-type and the indicated AKT2 mutant proteins were immunoprecipitated with an HA antibody from stably transduced HCT116 AKT1−/− AKT2 −/− cells and subjected to non-radioactive in vitro kinase assay. ‘No kinase’ control consists of an HA-immunoprecipitate from parental HCT116 AKT1−/− AKT2 −/− cells. Substrate phosphorylation, substrate loading, and AKT2 loading were all measured by immunoblot (for further details see ‘Methods’). (C) To evaluate the in vivo kinase activity of various AKT2 alleles, cells described in B were also lysed and analyzed by immunoblot with the indicated antibodies. (D) To determine the PIP-binding preference of WT and mutant AKT2, HCT116 AKT1−/− AKT2 −/− cells expressing WT, K181M, or G161V alleles of AKT2 were subjected to PIP-binding assay. AKT binding was assessed by immunoblot using an HA antibody. (E) Parental or AKT2-G161-expressing immortalized human epidermal melanocytes were plated on melanocyte media and allowed to attach overnight. Cells were then given fresh melanocyte media or switched to RMPI media containing 10% fetal bovine serum. 96 hr following media switch, cell death was assessed as elsewhere. Expression of the transgene was confirmed by immunoblot (inset). Vinculin was used as a loading control. (F) Parental EBC1 cells or EBC1 cells stably expressing exogenous WT AKT1, AKT1-E17K or AKT1-E17K-K179M were treated with the indicated doses of MK2206 for 24 hr and lysed. To asses target inactivation, lysates were analyzed by Western blot with the indicated antibodies. (G) Cells described in F were treated with the indicated doses of MK2206 for 96 hr. Cell death was assessed as before. (H) Model of AKT-dependent protection from apoptosis. AKT becomes fully activated following PI3K activation and subsequent phosphorylation at the T308 and S473 regulatory sites. Fully active AKT negatively regulates pro-apoptotic signals such as BAD and FKHR and positively regulates anti-apoptotic signals such as NFκB through phosphorylation (kinase-dependent functions). Fully AKT also regulates survival signals through kinase-independent activities.
-
Figure 4—source data 1
Contains source data for Figure 4.
- https://doi.org/10.7554/eLife.03751.024
Figure 5
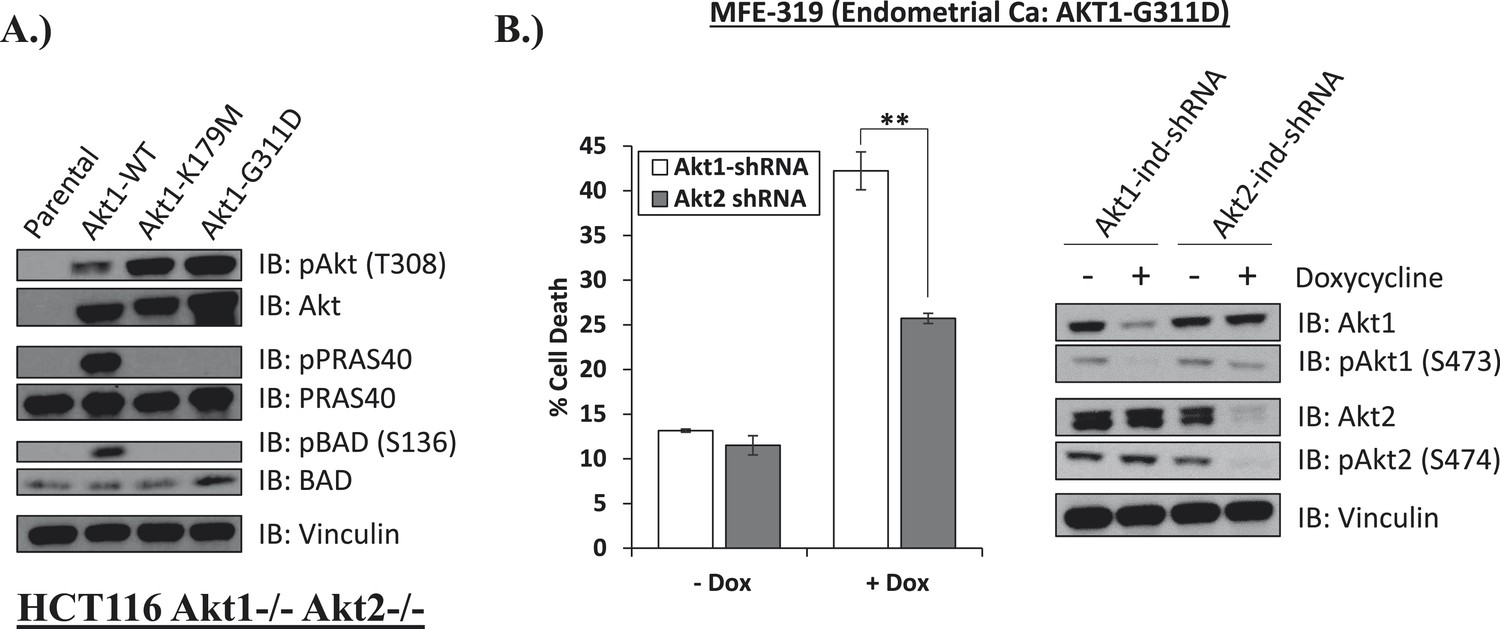
Endometrial cancer cells expressing endogenous kinase-deficient AKT1 are sensitive to AKT1 but not AKT2 knockdown.
(A) AKT-deficient HCT116 cells were stably transduced with wild-type or the indicated mutant AKT1 alleles. In cell AKT kinase activity was determined by assessing the phosphorylation of the AKT substrates PRAS40 and BAD by immunoblot as shown. Expression of vinculin was used as a loading control. Note that AKT1-G311D does not promote AKT substrate phosphorylation. (B) MFE-319 endometrial cancer cells which carry and endogenous AKT1-G311D mutation (http://www.broadinstitute.org/ccle/home) were engineered to express tetracycline-inducible hairpins targeting either AKT1 or AKT2. Cells were grown in the presence or absence of 2.5 µg/ml doxycycline for 6 days. Cell death was assessed on day 7 as before (left). To confirm knockdown of AKT and selectivity of the hairpins, a separate set of plates was grown with or without doxycycline for the indicated times and lysed. Vinculin expression was used as a loading control.
-
Figure 5—source data 1
Contains source data for Figure 5.
- https://doi.org/10.7554/eLife.03751.026
Author response image 1

A) and B) MDA‐MB‐361 cells were stably transduced with wild type or MK‐2206‐resistant AKT1 (AKT1‐W80A) or AKT2 (AKT2‐W80A) and assessed for MK2206 response as shown. Note that overexpression of wild type AKT alleles does not change the response to MK2206. C) EBC1 cells were stably transduced with wild type or kinase‐dead AKT2 (AKT2‐K181M) and assessed for MK2206 response as in A. Note that mutant, but not wild type AKT overexpression, confers resistance to MK2206.
Author response image 2
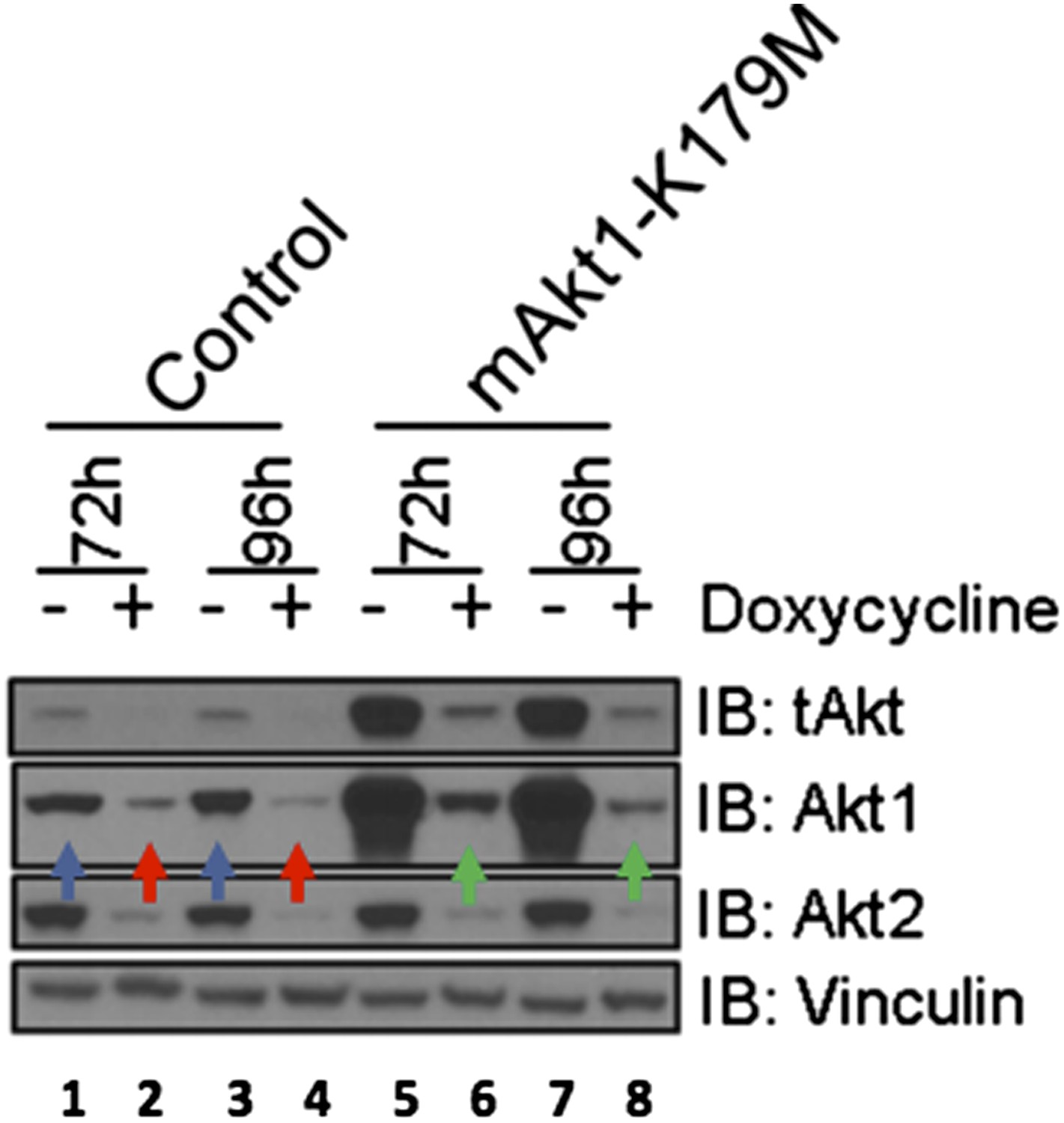
EBC1 cells were stably transduced with doxycycline‐inducible human‐specific AKT1 and AKT2 shRNAs (Control) and subsequently transduced with a murine kinase‐deficient AKT1 cDNA (mAKT1‐K179M). Cells were treated with doxycycline for the indicated times. AKT levels were measured at each time point by western blot as shown. Note that the levels of shRNA‐resistant AKT1 expression (green arrows) are equivalent to those seen in untreated control cells (blue arrows).
Author response image 3

EBC1 cells were treated with the pan‐class‐I PI3K inhibitor, and/or the AKT inhibitor GSK690693. Cells were allowed to grow for 96 hours and assessed for cell death following this incubation period by the trypan blue method (left). A duplicate set of samples was analysed by western blot to document drug activity. Note that GSK690693 does not protect (red arrow) from PI3K‐inhibitor‐induced cell death (green arrow).
Author response image 4

EBC1 cells were treated with the MET inhibitor crizotinib and/or AKT inhibitors GSK690693 and MK2206, and assessed for cell death. Panel (A) was shown in the original manuscript as Figure 2B and only contained data with crizotinib and GSK690693. Panel (B) has now replaced this figure in the revised manuscript and contains additional data using MK2206 (red arrows).
Author response image 5
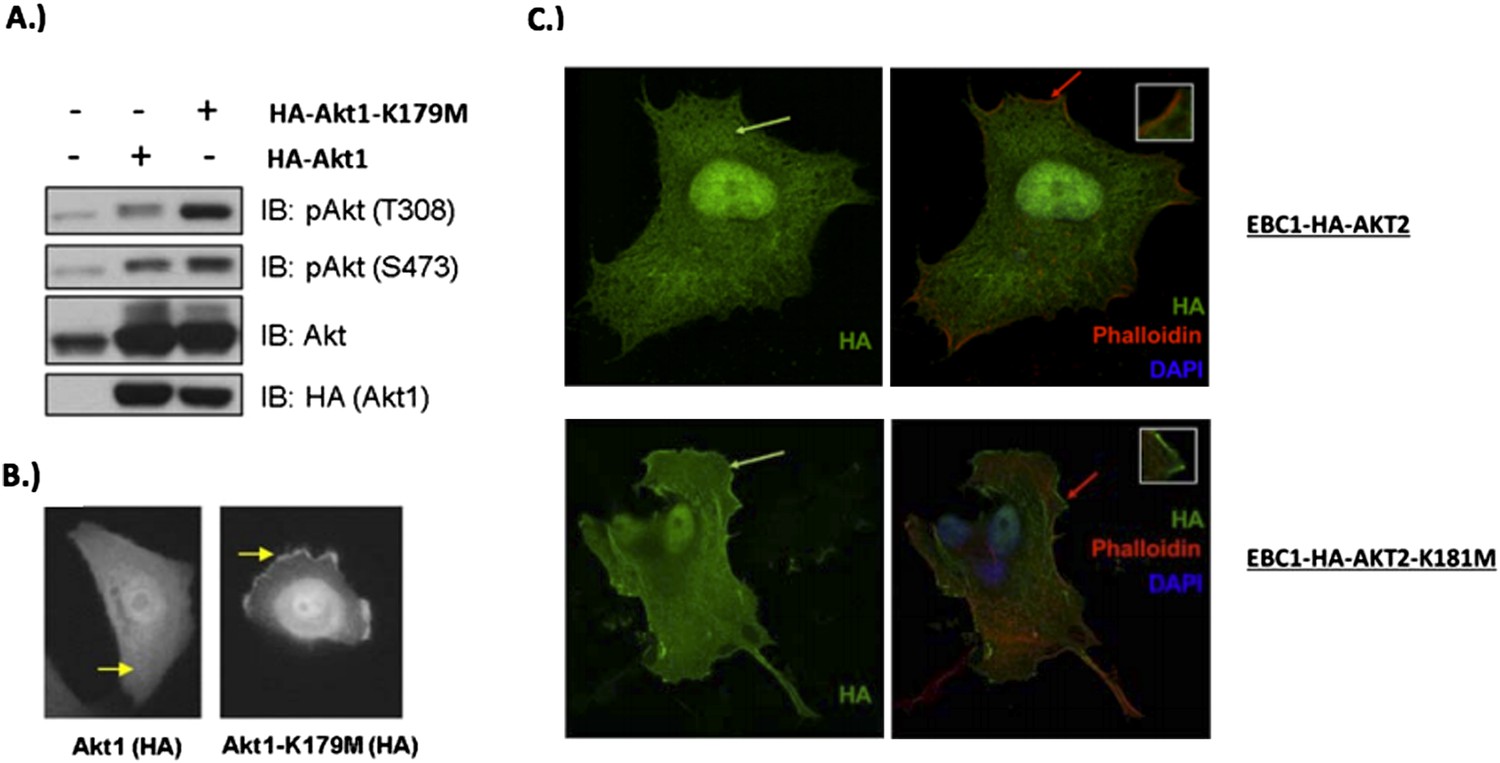
MCF10A cells were stably transduced with wild type or kinase‐dead (K179M) AKT1. Western blot analysis (A) (Figure 3C of revised manuscript) shows enhanced pAKT(T308) in mutant‐AKT‐expressing cells, and immunofluorescence analysis indicates this increase corresponds to enhanced membrane localization (B) (Figure 3D of revised manuscript). (C) EBC1 cells were stably transduced with WT or kinase‐dead AKT2 (K181M). Cells were stained as indicated and imaged using confocal microscopy. Note the enhanced membrane localization of mutant AKT2 (bottom left, green arrow), compared to wild type AKT2 (top left, green arrow). Insets are an enlarged images of the region indicated by the red arrows.
Author response image 6
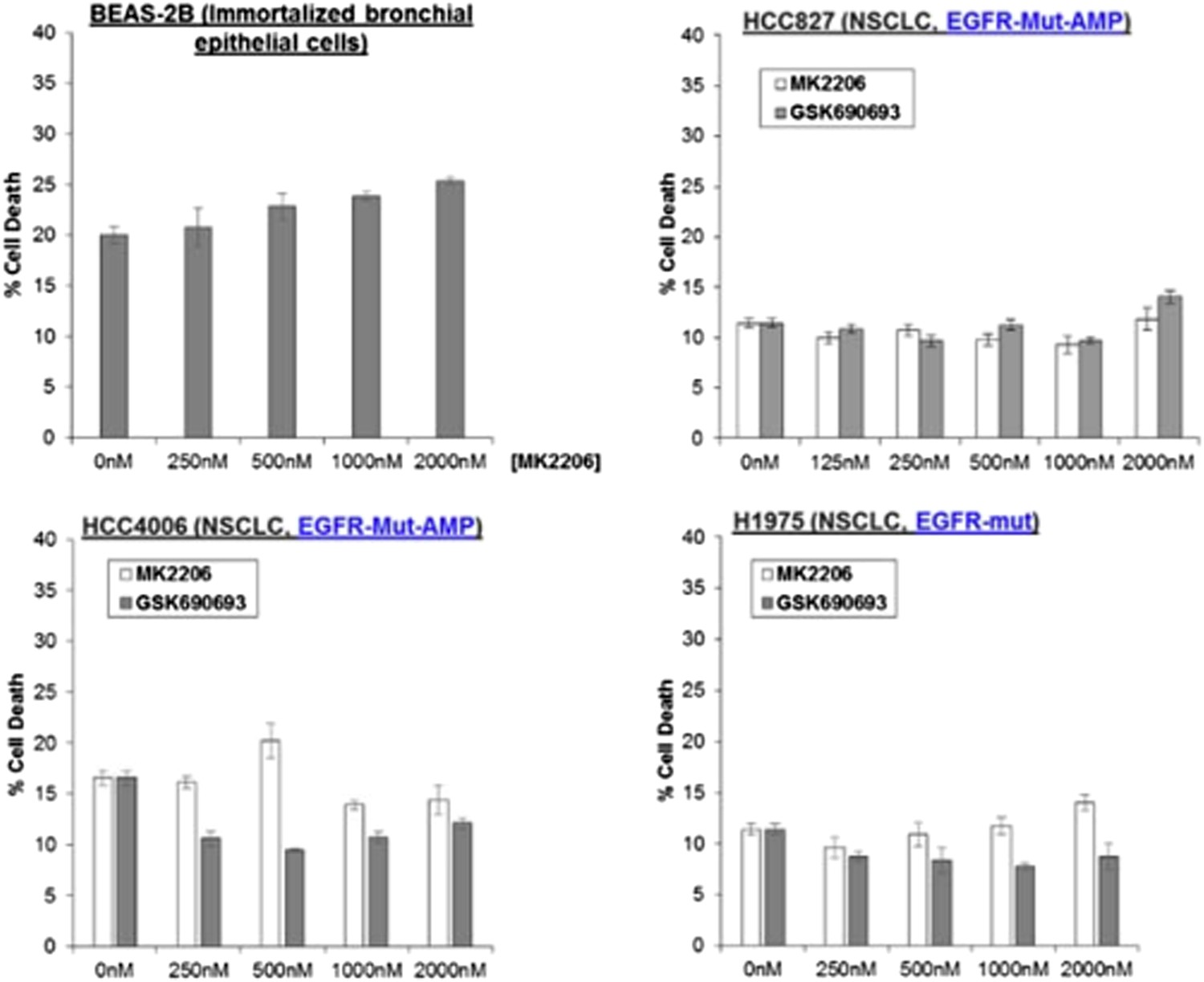
Author response image 7

Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A kinase-independent function of AKT promotes cancer cell survival
eLife 3:e03751.
https://doi.org/10.7554/eLife.03751




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}