Predicting effective microRNA target sites in mammalian mRNAs
- Howard Hughes Medical Institute, Whitehead Institute for Biomedical Research, United States
- Massachusetts Institute of Technology, United States
- Whitehead Institute for Biomedical Research, United States
- Hanyang University, Korea
Figures
Figure 1 with 5 supplements
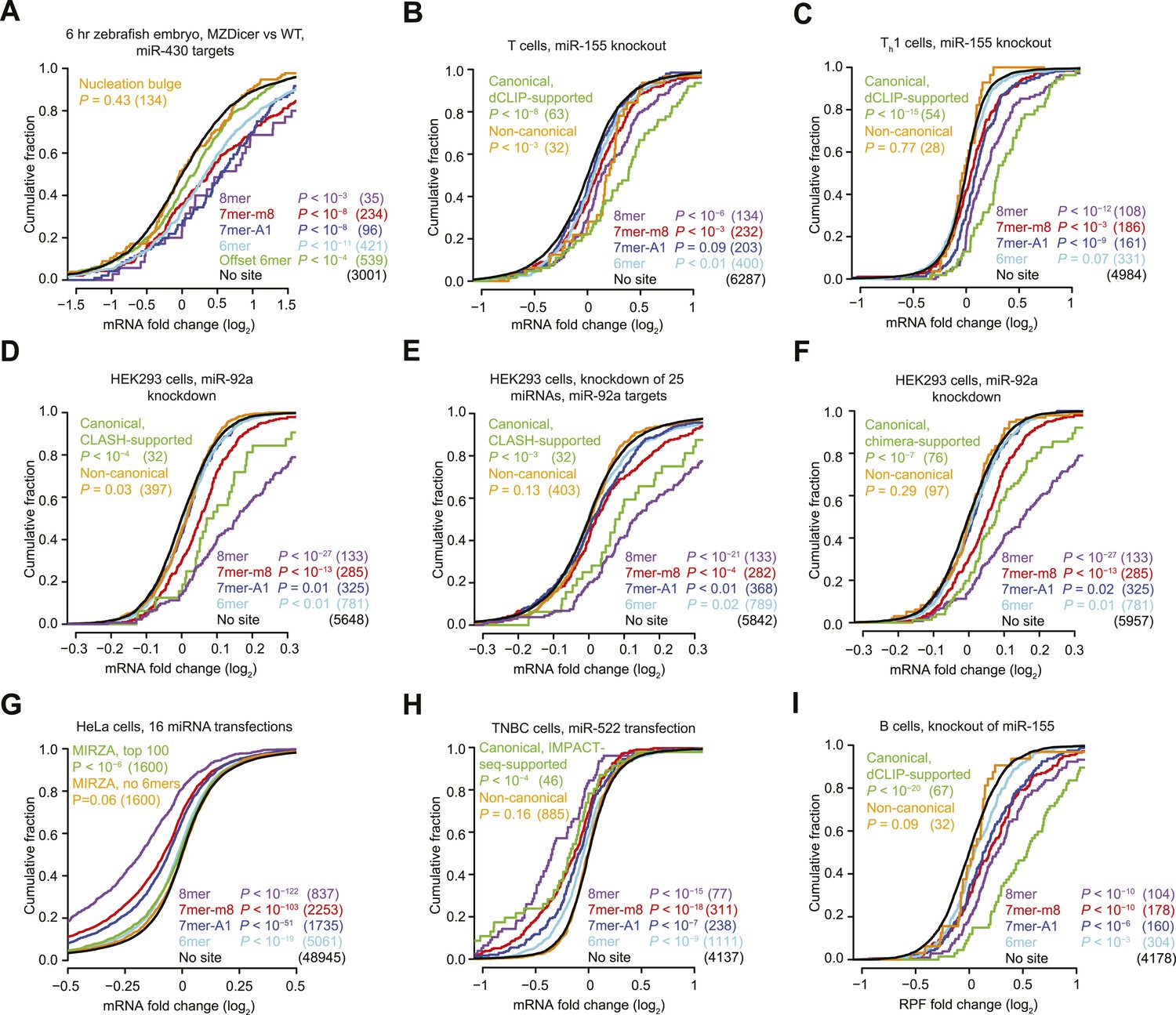
Inefficacy of recently reported non-canonical sites.
(A) Response of mRNAs to the loss of miRNAs, comparing mRNAs that contain either a canonical or nucleation-bulge site to miR-430 to those that do not contain a miR-430 site. Plotted are cumulative distributions of mRNA fold changes observed when comparing embryos that lack miRNAs (MZDicer) to those that have miRNAs (WT), focusing on mRNAs possessing a single site of the indicated type in their 3′ UTR. Similarity of site-containing distributions to the no-site distribution was tested (one-sided Kolmogorov–Smirnov [K–S] test, P values); the number of mRNAs analyzed in each category is listed in parentheses. See also Figure 1—figure supplement 1C and Figure 1—figure supplement 4A. (B and C) Response of mRNAs to the loss of miR-155, focusing on mRNAs that contain either a single canonical or ≥1 CLIP-supported non-canonical site to miR-155. These panels are as in (A), but compare fold changes for mRNAs with the indicated site type following genetic ablation of mir-155 in either T cells (B) or Th1 cells (C). See also Figure 1—figure supplement 2. (D and E) Response of mRNAs to the knockdown of miR-92a, focusing on mRNAs that contain either a single canonical or ≥1 CLASH-identified non-canonical site to miR-92a. These panels are as in (A), except CLASH-supported non-canonical sites were the same as those defined previously (Helwak et al., 2013) and thus were permitted to reside in any region of the mature mRNA, and these panels compare fold changes for mRNAs with the indicated site type following either knockdown of miR-92a (D) or combined knockdown of miR-92a and 24 other miRNAs (E) in HEK293 cells. See also Figure 1—figure supplement 3A,B. (F) As in (D), but focusing on mRNAs that contain ≥1 chimera-identified site. See also Figure 1—figure supplement 3C–E and Figure 1—figure supplement 4B. (G) Response of mRNAs to the transfection of 16 miRNAs, focusing on mRNAs that contain either a canonical or MIRZA-predicted non-canonical site. This panel is as in (A), but compares the fold changes for mRNAs with the indicated site type after introducing miRNAs, aggregating results from 16 individual transfection datasets. Fold changes are plotted for the top 100 non-canonical predictions for each of 16 miRNAs compiled either before (MIRZA, top 100) or after (MIRZA, no 6mers) removing mRNAs containing 6mer or offset-6mer 3′-UTR sites. (H) Response of mRNAs to a transfection of miR-522, focusing on mRNAs that contain either a single canonical or ≥1 IMPACT-seq–supported non-canonical site to miR-522. These panels are as in (A), except IMPACT-seq–supported non-canonical sites were the same as those defined previously (Tan et al., 2014) and thus were permitted in any region of the mature mRNA. (I) Response of ribosomes to the loss of miR-155, focusing on mRNAs that contain either a single canonical or ≥1 CLIP-supported non-canonical site to miR-155. This panel is as in (B and C) but compares the response of mRNAs using ribosome-footprint profiling (Eichhorn et al., 2014) after genetic ablation of mir-155 in B cells. Ribosome-footprint profiling captures changes in both mRNA stability and translational efficiency through the high-throughput sequencing of ribosome-protected mRNA fragments (RPFs).
Figure 1—figure supplement 1
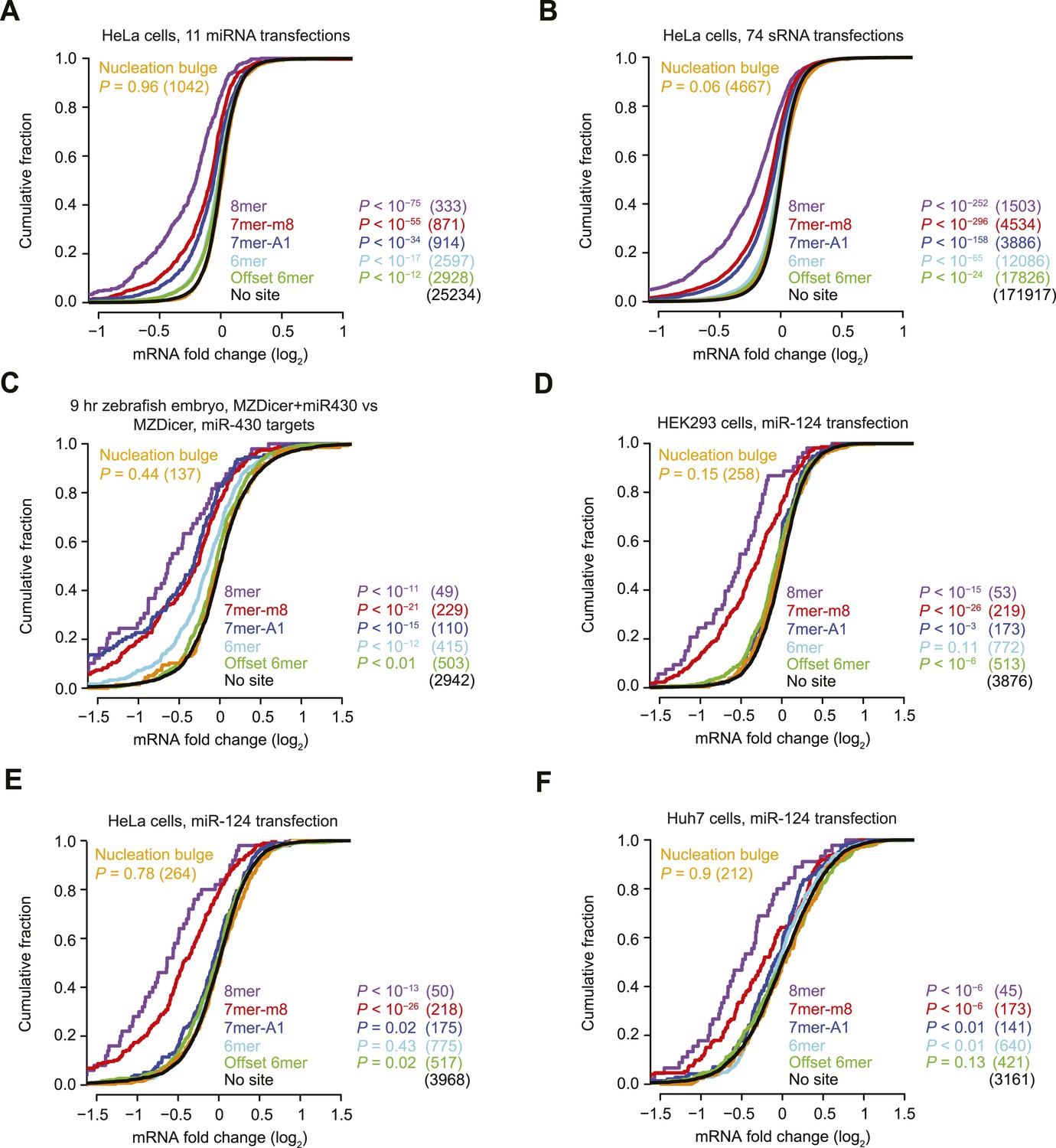
Inefficacy of nucleation-bulge sites.
(A and B) These panels are as in Figure 1A but compare the response of cognate site-containing mRNAs in a compendium of either 11 miRNA transfection datasets (A) or 74 sRNA transfection datasets (B). The datasets were pre-processed (Figure 3) and are provided in Supplementary file 1. (C) This panel is as in Figure 1A but compares the response of mRNAs in MZDicer embryos in which miR-430 has been injected. (D–F) These panels are as in Figure 1A but compare the response of mRNAs with the indicated miR-124 site types after transfecting miR-124 into either HEK293 cells (D), HeLa cells (E), or Huh7 cells (F).
Figure 1—figure supplement 2
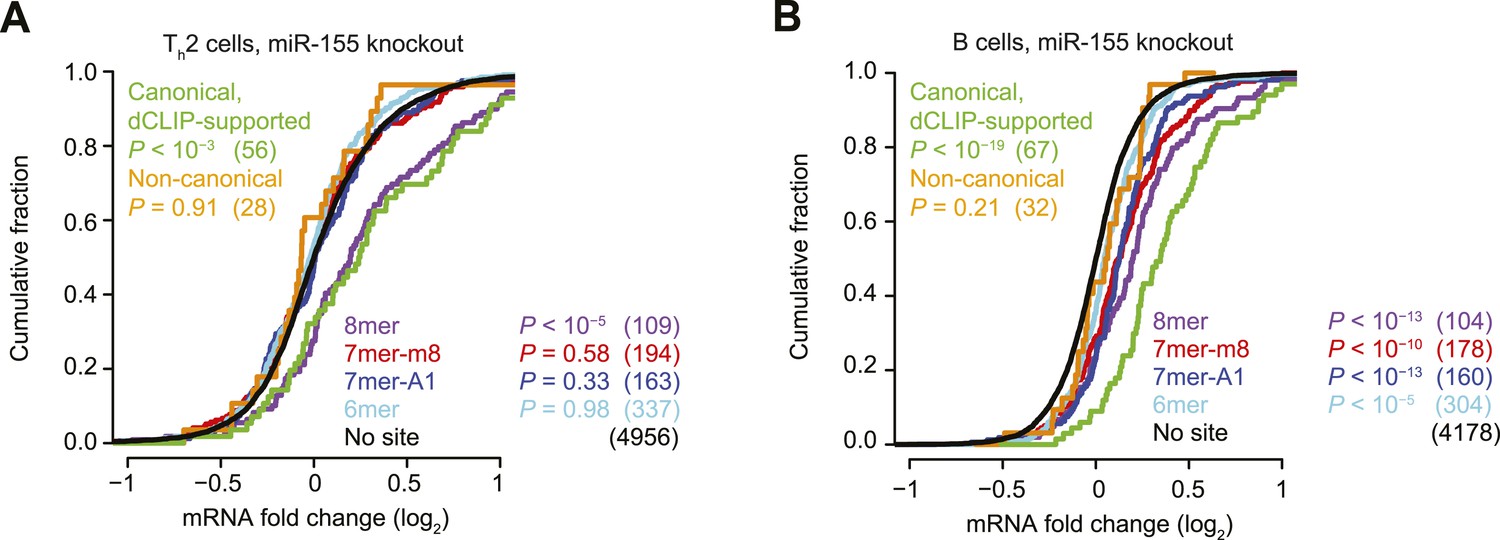
Inefficacy of CLIP-supported non-canonical miR-155 sites.
(A and B) These panels are as in Figure 1B but compare the response of mRNAs after genetic ablation of miR-155 in Type 2 helper T cells (Th2, A) or B cells (B).
Figure 1—figure supplement 3
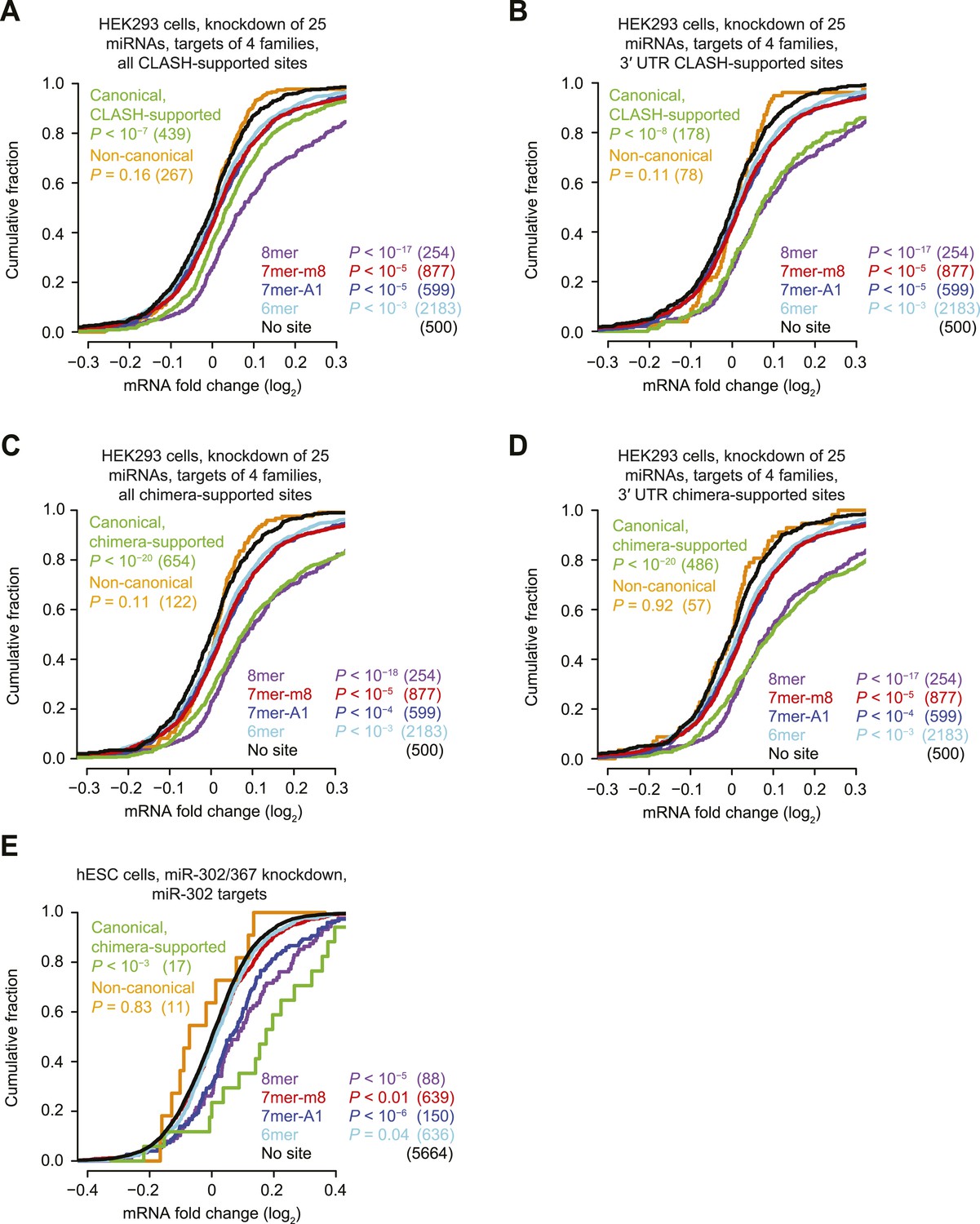
Inefficacy of CLASH- and chimera-supported non-canonical sites.
(A–D) These panels are as in Figure 1D but compare the response of mRNAs with sites cognate to any one of four miRNA families (miR-15/16, miR-19, miR-17/20/93/106, or miR-25/92), for either all CLASH-supported targets (A), mRNAs with CLASH-supported 3′-UTR sites (B), all chimera-supported targets (C), or mRNAs with chimera-supported 3′-UTR sites (D). These four miRNA families were chosen because their predicted targets were the most responsive to knockdown of the 25 miRNAs. p values reflect the median p value (as evaluated by a K–S test) across 100 trials in which a no-site control cohort with matched 3′-UTR lengths was chosen for each site-containing distribution. Length-matched no-site controls were required for this analysis because longer 3′ UTRs had a greater chance of containing additional sites to at least one of the many miRNAs that were knocked down, and thus had a greater chance of being derepressed as a result of interactions otherwise not considered in the analysis. To populate each control cohort, 500 different no-site mRNAs were chosen, considering the 3′-UTR length of each site-containing mRNA and selecting (without replacement) control mRNAs from among the 10 no-site mRNAs with the most similar 3′-UTR lengths. Shown is the response of a control cohort for mRNAs containing non-canonical sites. mRNAs with 3′ UTRs >2000 nt were excluded from the analysis because so many of the 3′ UTRs >2000 nt had a site to at least one of the four miRNA families, making it impossible to select appropriate length-matched controls. (E) This panel is as in Figure 1F but compares the response of mRNAs with the indicated miR-302 site types after knocking down miR-302/367 in hESCs.
Figure 1—figure supplement 4
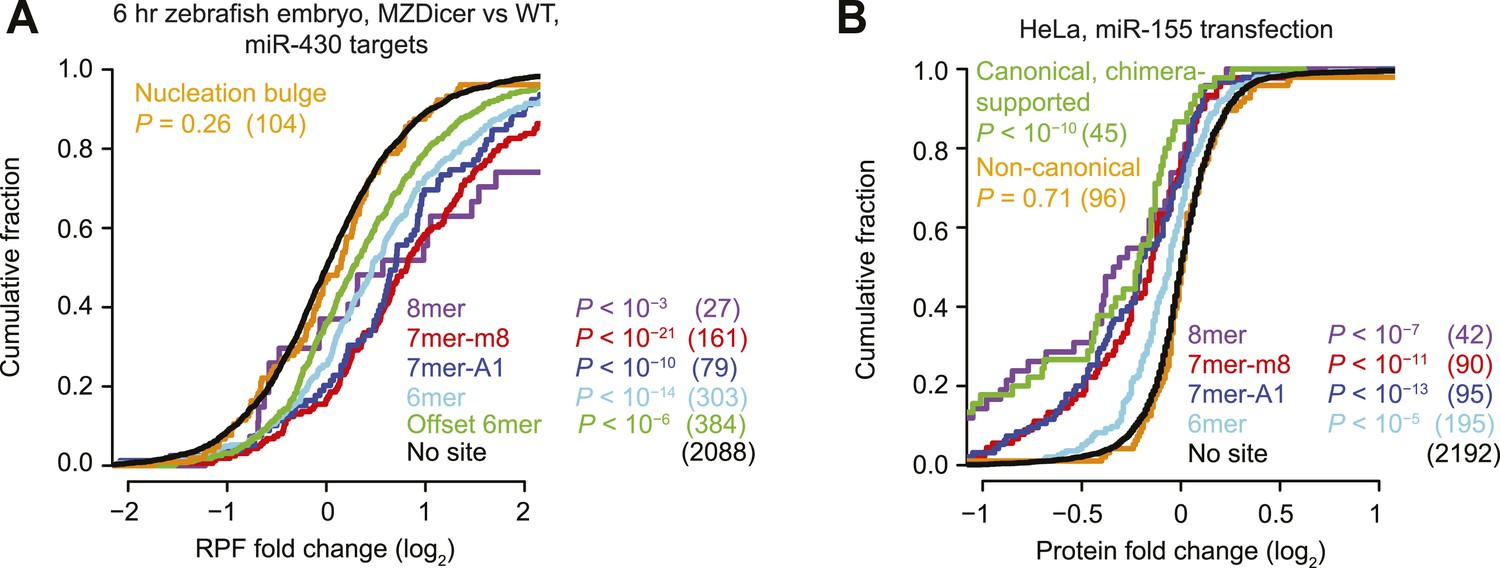
Inefficacy of non-canonical sites in mediating translational repression.
(A) This panel is as in Figure 1A but compares the response of mRNAs using ribosome footprint profiling (Bazzini et al., 2012), which captures changes in both mRNA stability and translational efficiency through the high-throughput sequencing of ribosome-protected mRNA fragments (RPFs). (B) This panel is as in (Figure 1I) but compares protein fold changes for chimera-supported targets, as evaluated by pulsed SILAC (Selbach et al., 2008) after transfection of miR-155 in HeLa cells.
Figure 1—figure supplement 5

Re-evaluating conservation of chimera-supported non-canonical sites.
(A) Conservation of chimera-supported non-canonical sites detected in an analysis modeled after that of Grosswendt et al. (2014) but modified to control for background conservation. Plotted for the indicated miRNAs is the average conservation of chimera-supported non-canonical sites, as measured by branch-length score (BLS), compared to the average conservation of 100 equally sized cohorts of controls; error bars, standard deviation of cohort averages; **, p < 0.01; *, p < 0.05, one-sided Z test. We considered chimera-supported non-canonical sites that mapped within 3′ UTRs and contained a single mismatch to the 6 nt seed of the miRNA. This set of sites mirrored that analyzed previously (Grosswendt et al., 2014), and excluded offset 6mers, which as a class was already known to mediate repression and exhibit preferential conservation (Friedman et al., 2009). Cohorts of control sites were generated such that for each chimera-supported site, each control cohort contained a single example of the identical 6 nt motif that was present in the indicated region (either an AGO cluster or 3′ UTR) but not supported by chimeric reads. To control for local background conservation and thereby avoid treating sites within slowly evolving 3′ UTRs the same as those within rapidly evolving 3′ UTRs, we used the binning procedure developed for calculating PCT scores (Friedman et al., 2009); 3′ UTRs were partitioned into 10 conservation bins (based on the median BLS of the nucleotides of the human sequence), and control sites were randomly selected (with replacement) from 3′ UTRs in the same bin as the actual site. Control AGO clusters were collected as was done previously (Grosswendt et al., 2014), using genome-wide data downloaded from clipz.unibas.ch and derived from multiple AGO PAR-CLIP experiments performed in HEK293 cells (Kishore et al., 2011). The union of AGO clusters for all experiments was computed and filtered for overlap with Ensembl-annotated 3′ UTRs, using the ‘merge’ and ‘intersectBED’ utilities, respectively, found in BEDTools v2.20.1 (parameter ‘-s’) (Quinlan and Hall, 2010). (B) Attribution of the conservation signal to the confounding effects of conserved regions. Considered are 1443 non-canonical chimera-supported sites selected as in (A) but including sites of all miRNA families. For each chimera-supported site, a z score was generated using the distribution of BLSs for 100 control sites chosen as in panel (A) from either AGO clusters or 3′ UTRs, as indicated. Each z score reflected how the conservation of the actual site differed from that of its controls. Compared are cumulative distributions of the z scores for sites of broadly conserved miRNAs and those of less conserved miRNAs, using the previously defined sets of broadly and less conserved miRNAs (Friedman et al., 2009). If the chimera-supported non-canonical sites were preferentially conserved because of their function in mediating repression, then sites of broadly conserved miRNAs would be expected to have a right-shifted distribution compared to sites of less conserved miRNAs. However, no significant difference was discerned between each pair of z-score distributions. The remainder of this legend outlines the rationale for the analysis of this panel. One way to reconcile the conservation signal observed in panel (A) with our conclusion that a large majority if not all of these sites bind miRNA but do not mediate repression is to consider the potentially confounding biochemical properties of conserved regions, which are illustrated by the observation that artificial siRNAs preferentially target sites that are evolutionarily conserved over those that are not (Nielsen et al., 2007). Because these siRNAs are not natural (and do not share a seed with conserved miRNAs) the evolutionary conservation of these preferred sites could not have arisen because they function to mediate sRNA-guided repression. Instead, some other function of these 3′-UTR regions, such as greater accessibility to RNA-binding factors, must explain their preferential conservation and also endow them with properties that favor sRNA binding (Nielsen et al., 2007). To examine whether confounding properties of conserved 3′-UTR regions might similarly explain the elevated conservation of chimera-supported sites, we compared the z scores for sites bound by broadly conserved miRNAs (miRNAs in families conserved beyond mammals, as listed in TargetScan7) with those bound by less conserved miRNAs. MicroRNAs conserved among mammals but not more broadly were grouped with the less conserved miRNAs because canonical 6mer and 7mer sites to these miRNAs have no conservation signal above background, presumably because these miRNAs have not been present long enough for the number of preferentially conserved 6mer and 7mer sites to rise above the background (Friedman et al., 2009); we reasoned that the same would be true of non-canonical sites, to the extent that any are preferentially conserved. If the conservation signal observed in panel (A) were related to miRNA binding, we would have expected a difference between the scores for the sites of broadly and less conserved miRNAs. The lack of a significant difference supports the idea that chimera-supported non-canonical sites tend to be conserved for the same reason that functional sites to artificial siRNAs tend to be conserved.
Figure 2 with 2 supplements
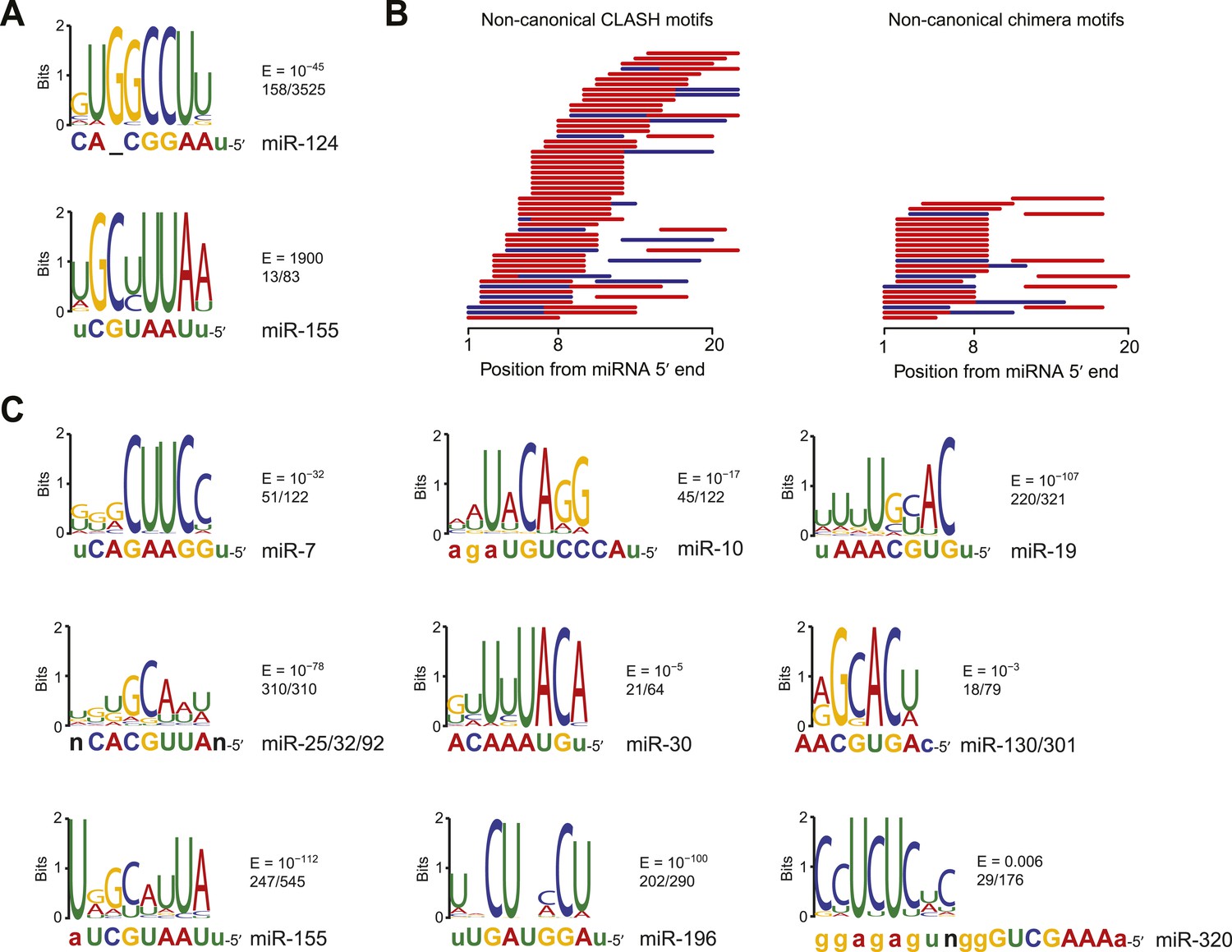
Confirmation of experimentally identified non-canonical miRNA binding sites.
(A) Sequence logos corresponding to motifs enriched in dCLIP clusters that either appear following transfection of miR-124 into HeLa cells (Chi et al., 2009) (top) or disappear following knockout of miR-155 in T cells (Loeb et al., 2012) (bottom). Shown to the right of each logo is its E-value among clusters lacking a seed-matched or offset-6mer canonical site and the fraction of these clusters that matched the logo. Shown below each logo are the complementary regions of the cognate miRNA family, highlighting nucleotides 2–8 in capital letters. (B) Position of the top-ranked motif corresponding to non-canonical sites enriched in CLASH (Helwak et al., 2013) (left) or chimera (Grosswendt et al., 2014) (right) data for each human miRNA family supported by at least 50 interactions without a seed-matched or offset-6mer canonical site. For each family the most enriched logo was aligned to the reverse complement of the miRNA. In cases in which a logo mapped to multiple positions along the miRNA, the positions with the best and second best scores are indicated (red and blue, respectively). (C) Sequence logos of motifs enriched in chimera interactions that lack canonical sites. As in (A), but displaying sequence logos identified in the chimera data of panel (B) for a sample of nine human miRNAs. Logos identified for the other human miRNAs are also provided (Figure 2—figure supplement 1B). A nucleotide that differs between miRNA family members is indicated as a black ‘n’.
Figure 2—figure supplement 1
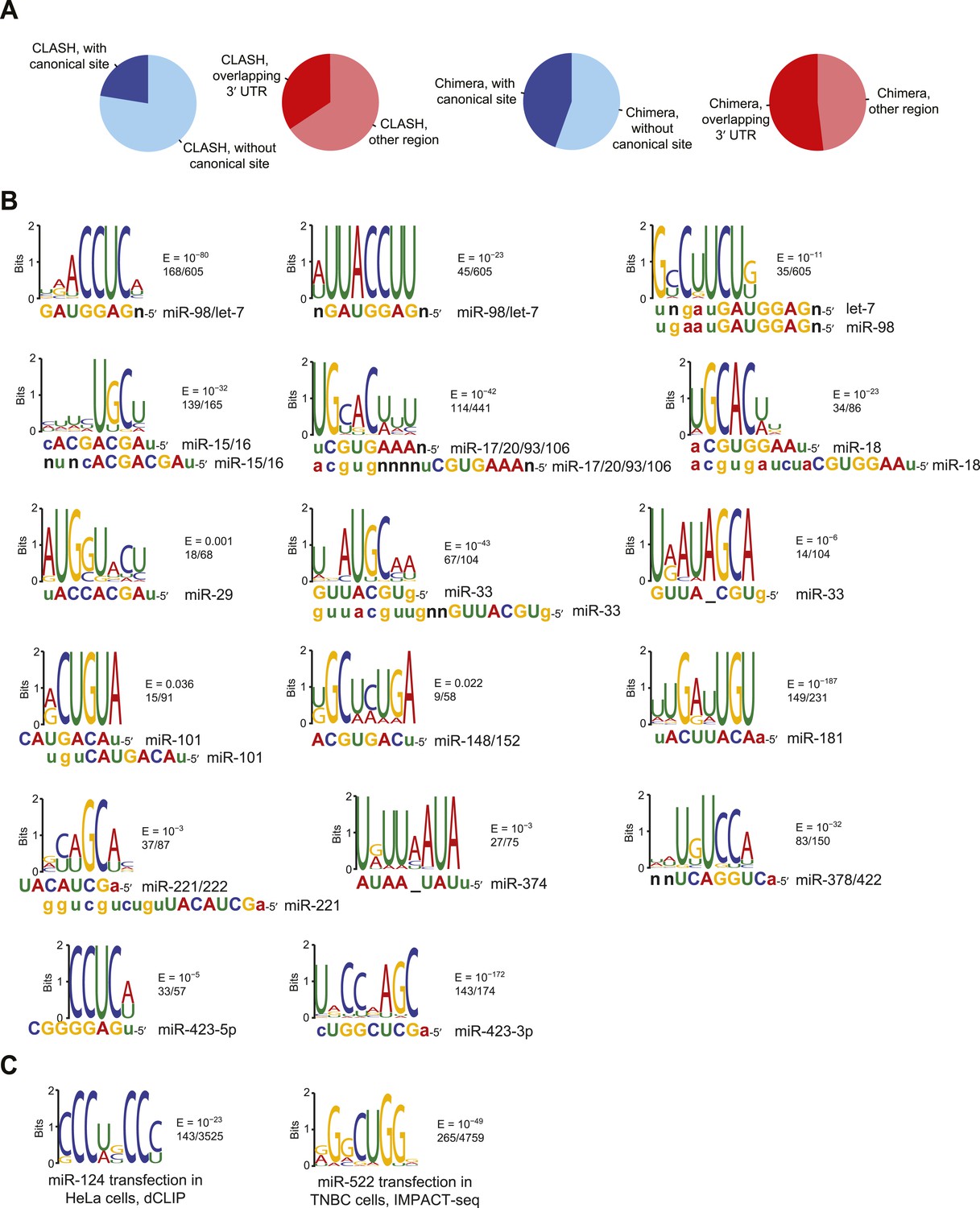
Comparison of CLASH and chimera data and identification of motifs enriched in human chimera interactions that lack canonical sites.
(A) Comparison of CLASH (left) and chimera (right) reads from human cells, showing the proportion possessing a canonical site (blue) and overlapping 3′ UTRs (red). In total, 18,514 CLASH and 10,567 chimera interactions were analyzed. (B) Sequence logos of motifs enriched in chimera interactions that lack canonical sites. This panel is as in Figure 2C but displays the remaining motifs identified from the chimera data analyzed in Figure 2B. In cases of alignment ambiguity, both alignments are shown below the logo. For some miRNA families, multiple motifs were significantly enriched (E ≤ 0.001) and are shown separately. Significantly enriched motifs (or a top-ranked motif matching the miRNA) were not found for miR-21, and miR-3168 was excluded from the analysis due to poor support for its authenticity as a miRNA. (C) Sequence logos of motifs that do not match the cognate miRNA but are nonetheless enriched in miR-124 dCLIP (Chi et al., 2009) and miR-522 IMPACT-seq (Tan et al., 2014) clusters that lack canonical sites to the miRNA. The miR-124 logo was nearly identical to a non-specific motif previously identified as enriched in CLIP data from the mouse brain (Chi et al., 2012). The miR-522 logo was found instead of the previously reported miRNA-matching logo (Tan et al., 2014).
Figure 2—figure supplement 2
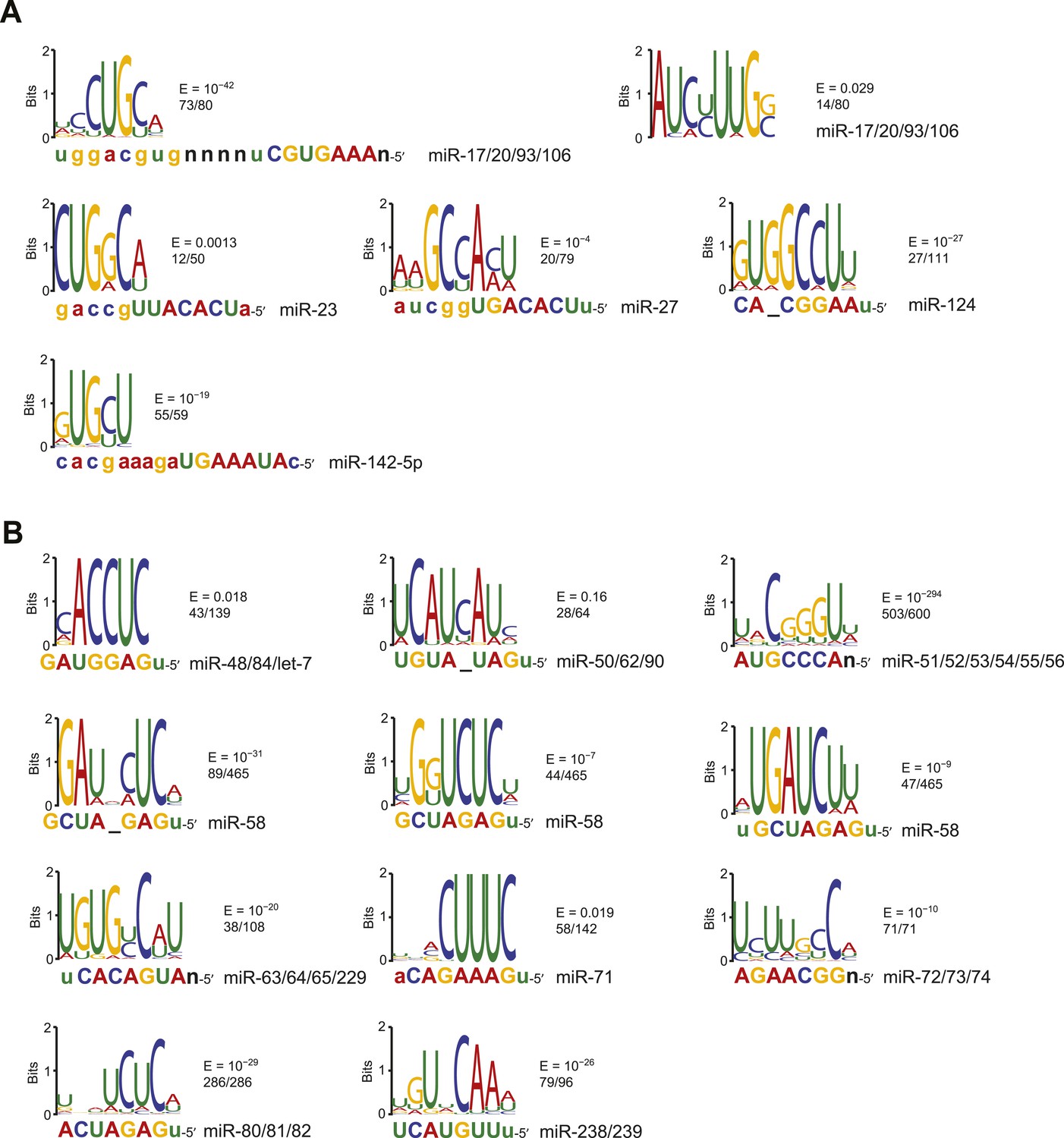
Identification of motifs enriched in mouse and nematode chimera interactions that lack canonical sites.
(A) Sequence logos of motifs enriched in M. musculus chimera interactions that lack canonical sites; otherwise as in Figure 2C. Significantly enriched motifs (or a top-ranked motif matching the miRNA) were not found for let-7 and miR-142-3p. (B) Sequence logos of motifs enriched in C. elegans chimera interactions that lack canonical sites; otherwise as in Figure 2C. Significantly enriched motifs (or a top-ranked motif matching the miRNA) were not found for miR-1.
Figure 3 with 1 supplement

Pre-processing the microarray datasets to minimize nonspecific effects and technical biases.
(A) Example of the correlated response of mRNAs after transfecting two unrelated sRNAs (sRNA 1 and 2, respectively). Results for mRNAs containing at least one canonical 7–8 nt 3′-UTR site for either sRNA 1, sRNA 2, or both sRNAs are highlighted in red, blue, and green, respectively. Values for mRNAs without such sites are in grey. All mRNAs were used to calculate the Spearman correlation (rs). (B) Correlated responses observed in a compendium of 74 transfection experiments from six studies (colored as indicted in the publications list). For each pair of experiments, the rs value was calculated as in panel (A), colored as indicated in the key, and used for hierarchical clustering. (C) Study-dependent relationships between the responses of mRNAs to the transfected sRNA and either 3′-UTR length or 3′-UTR AU content, focusing on mRNAs without a canonical 7–8 nt 3′-UTR site to the sRNA. Boxplots indicate the median rs (bar), 25th and 75th percentiles (box), and the minimum of either 1.5 times the interquartile range or the most extreme data point (whiskers), with the width of the box proportional to the number of datasets used from each study. The studies are colored as in panel (B), abbreviating the first author and year. (D) Reduced correlation between the responses of mRNAs to unrelated sRNAs after applying the PLSR technique. This panel is as in (A) but plots the normalized mRNA fold changes. (E) Reduced correlations in results of the compendium experiments after applying the PLSR technique. This panel is as in (B) but plots the correlations after normalizing the mRNA fold changes. (F) Reduced study-dependent relationships between mRNA responses and either 3′-UTR length or 3′-UTR AU content. This panel is as in (C) but plots the correlations after normalizing the mRNA fold changes. (G and H) Cumulative distributions of fold changes for mRNAs containing at least one canonical 7–8 nt 3′-UTR site or no site either before normalization (raw) or after normalization (normalized). Panel (G) plots the results from experiments shown in (A) and (D), and (H) plots results from all 74 datasets.
Figure 3—figure supplement 1
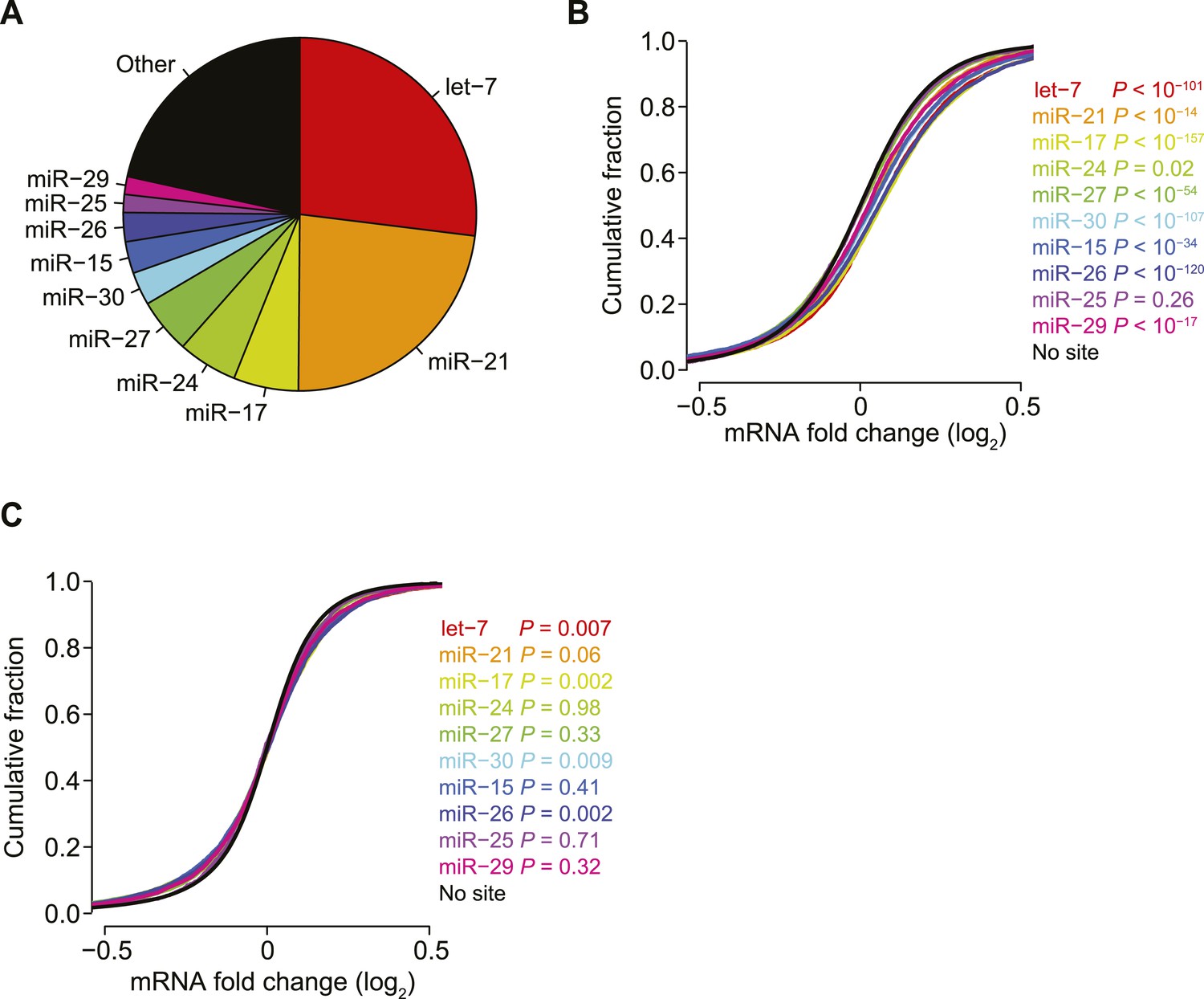
Reduced biases from derepression of endogenous miRNA targets.
(A) Pie chart reflecting the relative proportions of reads for the indicated miRNA families observed when sequencing small RNAs from HeLa cells. Relative miRNA levels were quantified as described previously (Denzler et al., 2014). (B and C) Cumulative distributions of fold changes for mRNAs with at least one canonical 7–8 nt 3′-UTR site to the indicated miRNA family in the compendium of 74 sRNA transfection datasets, either before (B) or after (C) normalization. p values were computed using a one-sided Wilcoxon rank-sum test, comparing each of the site-containing distributions to the no-site distribution. This test was a more stringent alternative to the K–S test, which led to highly significant p values for very slight differences, due to the large number of mRNAs in each distribution. To account for multiple hypotheses, an appropriate Bonferroni-corrected significance threshold would be p < 0.005, which was not achieved for most comparisons in panel (C).
Figure 4
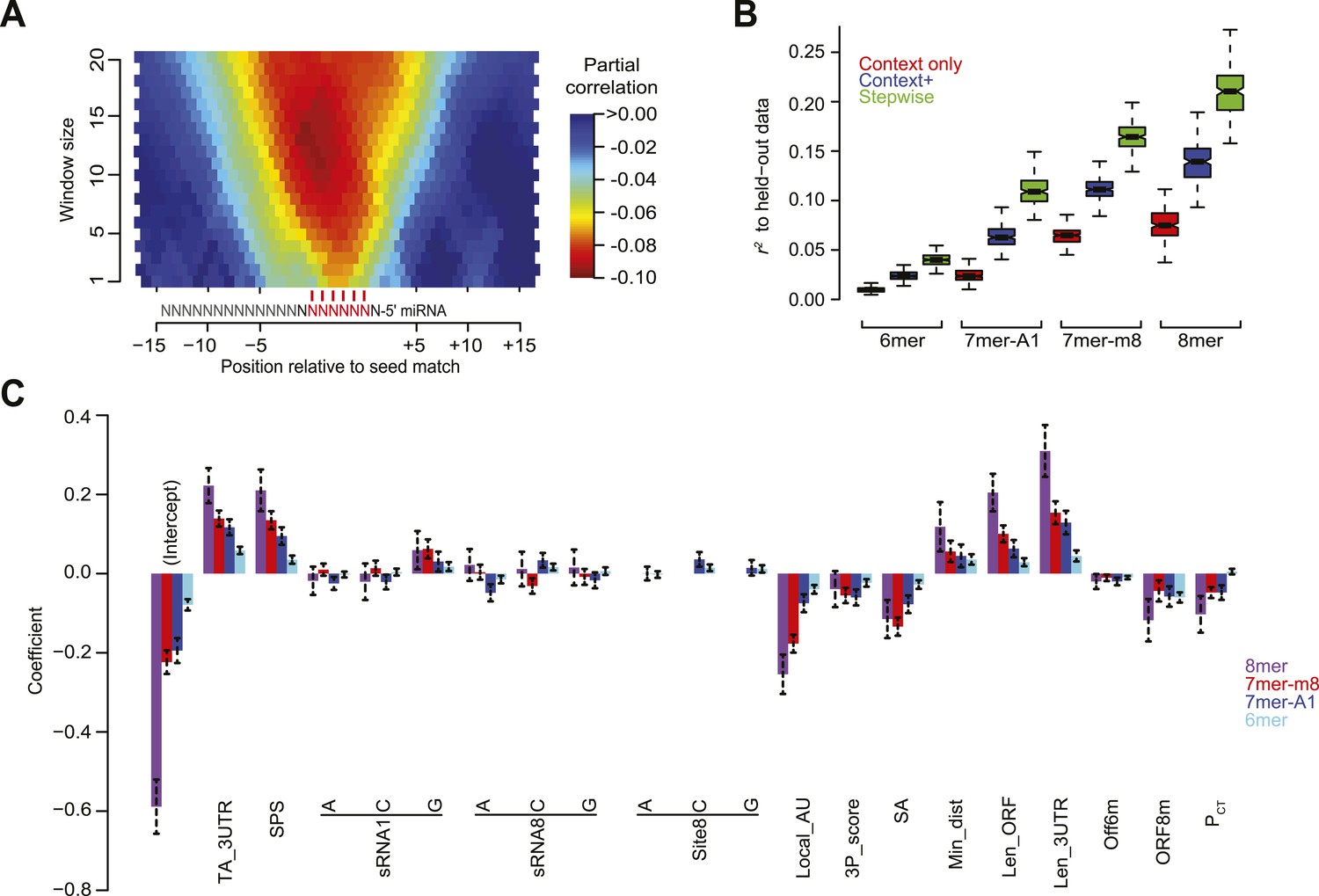
Developing a regression model to predict miRNA targeting efficacy.
(A) Optimizing the scoring of predicted structural accessibility. Predicted RNA structural accessibility scores were computed for variable-length windows within the region centered on each canonical 7–8 nt 3′-UTR site. The heatmap displays the partial correlations between these values and the repression associated with the corresponding sites, determined while controlling for local AU content and other features of the context+ model (Garcia et al., 2011). (B) Performance of the models generated using stepwise regression compared to that of either the context-only or context+ models. Shown are boxplots of r2 values for each of the models across all 1000 sampled test sets, for mRNAs possessing a single site of the indicated type. For each site type, all groups significantly differ (P < 10−15, paired Wilcoxon sign-rank test). Boxplots are as in Figure 3C. (C) The contributions of site type and each of the 14 features of the context++ model. For each site type, the coefficients for the multiple linear regression are plotted for each feature. Because features are each scored on a similar scale, the relative contribution of each feature in discriminating between more or less effective sites is roughly proportional to the absolute value of its coefficient. Also plotted are the intercepts, which roughly indicate the discriminatory power of site type. Dashed bars indicate the 95% confidence intervals of each coefficient.
-
Figure 4—source data 1
Coefficients of the trained context++ model corresponding to each site type.
Using these coefficients and corresponding scaling factors (Table 3), context++ scores can be computed essentially as illustrated in Supplementary Figure 5 of Garcia et al. (2011).
- https://doi.org/10.7554/eLife.05005.016
Figure 5 with 1 supplement

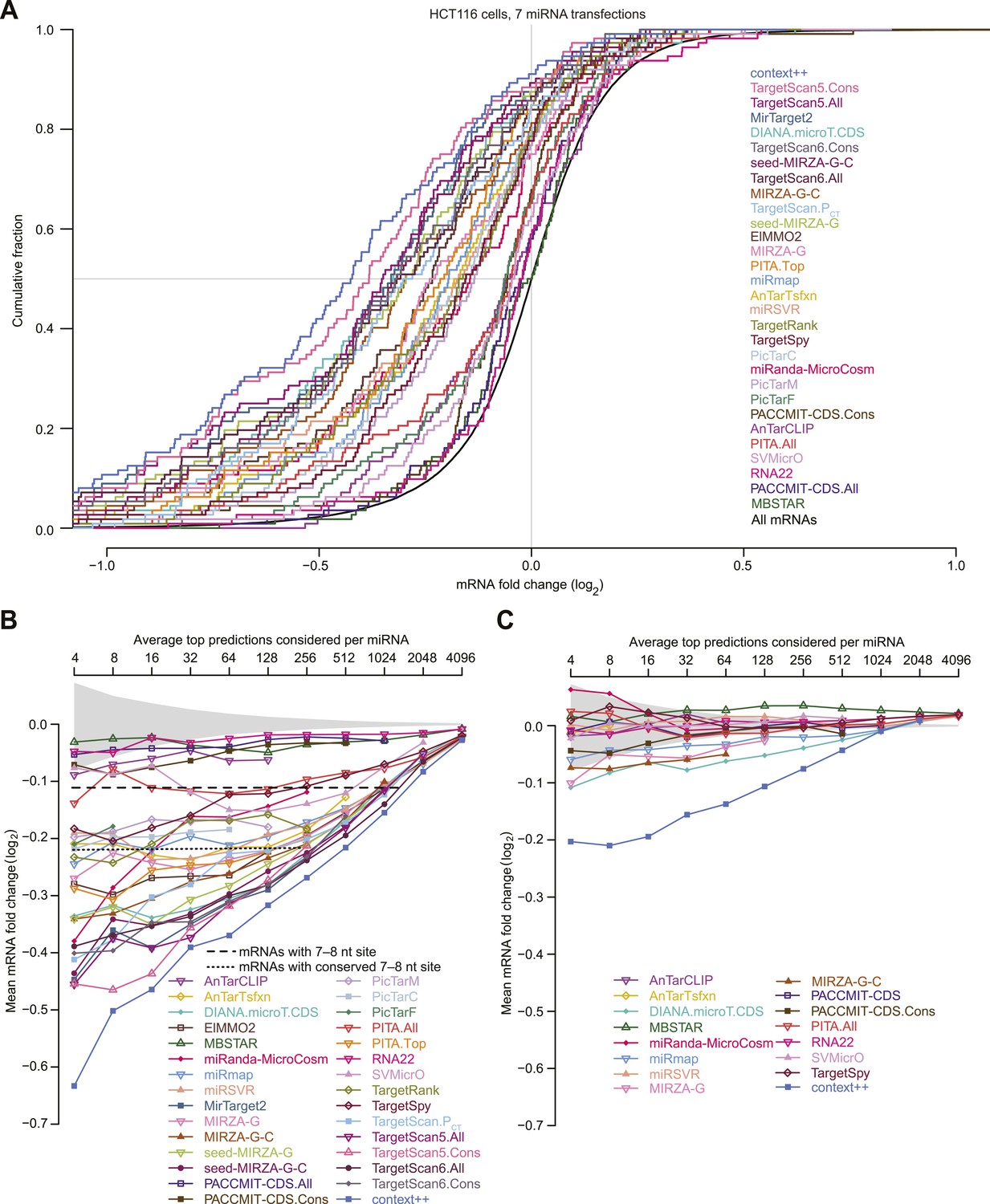
Performance of target prediction algorithms on a test set of seven experiments in which miRNAs were individually transfected into HCT116 cells.
(A) Average number of targets predicted by the indicated algorithm for each of the seven miRNAs in the test set (let-7c, miR-16, miR-103, miR-106b, miR-200b, miR-200a, and miR-215). The numbers of predictions with at least one canonical 7–8 nt 3′-UTR site to the transfected miRNA (dark blue) are distinguished from the remaining predictions (light blue). Names of algorithms are colored according to whether they consider only sequence or thermodynamic features of site pairing (grey), only site conservation (orange), pairing and contextual features of a site (red), or pairing, contextual features, and site conservation (purple). The most recently updated predictions were downloaded, with year that those predictions were released indicated in parentheses. (B and C) Extent to which the predictions explain the mRNA fold changes observed in the test set. For predictions tallied in panel (A), the explanatory power, as evaluated by the r2 value for the relationship between the scores of the predictions and the observed mRNA fold changes in the test set, is plotted for either mRNAs with 3′ UTRs containing at least one canonical 7–8 nt 3′-UTR site (B) or other mRNAs (C). Algorithms designed to evaluate only targets with seed-matched 7–8 nt 3′-UTR sites are labeled ‘N/A’ in (C). (D) Repression of the top predictions of the context++ model and of our previous two models, focusing on an average of 16 top predicted targets per miRNA in the test set. The dotted lines indicate the median fold-change value for each distribution, otherwise as in Figure 1A. (E and F) Median mRNA fold changes observed in the test set for top-ranked predicted targets, considering either all predictions (E) or only those with 3′ UTRs lacking at least one canonical 7–8 nt site (F). For each algorithm listed in panel (A), all reported predictions for the seven miRNAs were ranked according to their scores, and the indicated sliding threshold of top predictions was implemented. For example, at the threshold of 4, the 28 predictions with the top scores were identified (an average of 4 predictions per miRNA, allowing miRNAs with more top scores to contribute more predictions), mRNA fold-change values from the cognate transfections were collected, and the median value was plotted. When the threshold exceeded the number of reported predictions, no value was plotted. Also plotted is the median mRNA fold change for all mRNAs with at least one cognate canonical 7–8 nt site in their 3′ UTR (dashed line; an average of 1366 mRNAs per miRNA), the median fold change for all mRNAs with at least one conserved cognate canonical 7–8 nt site in their 3′ UTR (dotted line; an average of 461 mRNAs per miRNA), and the 95% interval for the median fold change of randomly selected mRNAs, determined using 1000 resamplings (without replacement) at each cutoff (shading). Conserved sites were defined as in TargetScan6, with conservation cutoffs for each site type set at different branch-length scores (cutoffs of 0.8, 1.3, and 1.6 for 8mer, 7mer-m8, and 7mer-A1 sites, respectively).
Figure 5—figure supplement 1
Performance of miRNA prediction algorithms on the test set.
(A) This panel is as in Figure 5D, but shows the results for all algorithms evaluated in Figure 5A. Algorithm names are listed in the order of the median fold change for their top predictions, with each name colored using the color used for its cumulative distribution. (B and C) These panels are as in Figure 5E–F, respectively, but compare mean fold changes instead of median fold changes.
Figure 6
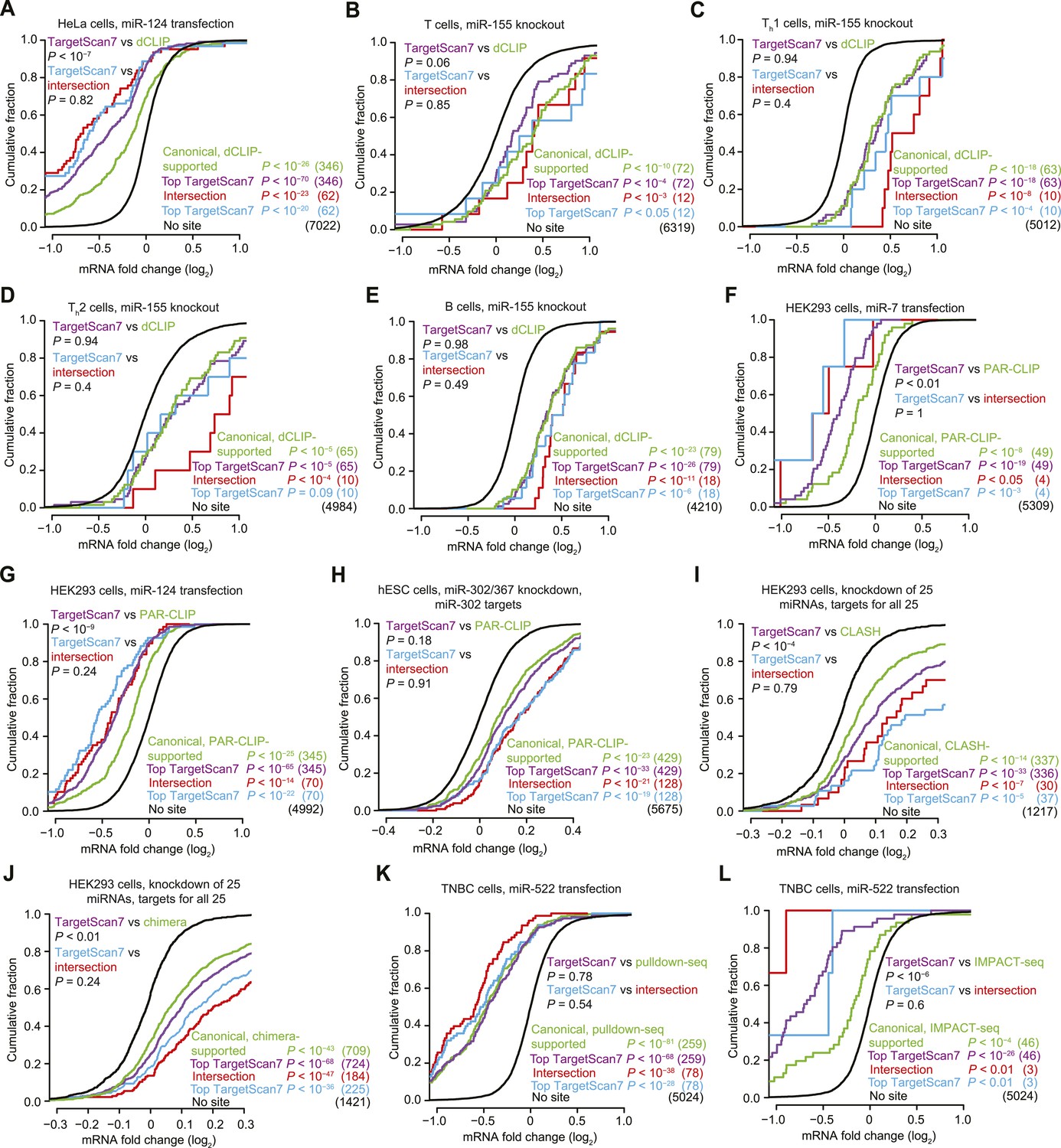
Response of predictions and mRNAs with experimentally supported canonical binding sites.
(A–E) Comparison of the top TargetScan7 predicted targets to mRNAs with canonical sites identified from dCLIP in either HeLa cells with and without transfected miR-124 (Chi et al., 2009) or lymphocytes with and without miR-155 (Loeb et al., 2012). Plotted are cumulative distributions of mRNA fold changes after transfection of miR-124 in HeLa cells (A), or after genetic ablation of miR-155 in either T cells (B), Th1 cells (C), Th2 cells (D), and B cells (E) (one-sided K–S test, P values). For genes with alternative last exons, the analysis considered the score of the most abundant alternative last exon, as assessed by 3P-seq tags (as is the default for TargetScan7 when ranking predictions). Each dCLIP-identified mRNA was required to have a 3′-UTR CLIP cluster with at least one canonical site to the cognate miRNA (including 6mers but not offset 6mers). Each intersection mRNA (red) was found in both the dCLIP set and top TargetScan7 set. Similarity between performance of the TargetScan7 and dCLIP sets (purple and green, respectively) and TargetScan7 and intersection sets (blue and red, respectively) was tested (two-sided K–S test, P values); the number of mRNAs analyzed in each category is in parentheses. TargetScan7 scores for mouse mRNAs were generated using human parameters for all features. (F–H) Comparison of top TargetScan7 predicted targets to mRNAs with canonical binding sites identified using photoactivatable-ribonucleoside-enhanced CLIP (PAR-CLIP) (Hafner et al., 2010; Lipchina et al., 2011). Plotted are cumulative distributions of mRNA fold changes after either transfecting miR-7 (F) or miR-124 (G) into HEK293 cells, or knocking down miR-302/367 in hESCs (H). Otherwise these panels are as in (A–E). (I) Comparison of top TargetScan7 predicted targets to mRNAs with canonical sites identified using CLASH (Helwak et al., 2013). Plotted are cumulative distributions of mRNA fold changes after knockdown of 25 miRNAs from 14 miRNA families in HEK293 cells. For each of these miRNA families, a cohort of top TargetScan7 predictions was chosen to match the number of mRNAs with CLASH-identified canonical sites, and the union of these TargetScan7 cohorts was analyzed. The total number of TargetScan7 predictions did not match the number of CLASH-identified targets due to slightly different overlap between mRNAs targeted by different miRNAs. Otherwise these panels are as in (A–E). (J) Comparison of top TargetScan7 predicted targets to mRNAs with chimera-identified canonical sites (Grosswendt et al., 2014). Otherwise this panel is as in (I). (K) Comparison of top TargetScan7 predicted targets to mRNAs with canonical binding sites within 3′ UTRs of mRNAs identified using pulldown-seq (Tan et al., 2014). Plotted are cumulative distributions of mRNA fold changes after transfecting miR-522 into triple-negative breast cancer (TNBC) cells. Otherwise this panel is as in (A–E). (L) Comparison of top TargetScan7 predicted targets to mRNAs with canonical sites identified using IMPACT-seq (Tan et al., 2014). Otherwise this panel is as in (K).
Figure 7 with 1 supplement
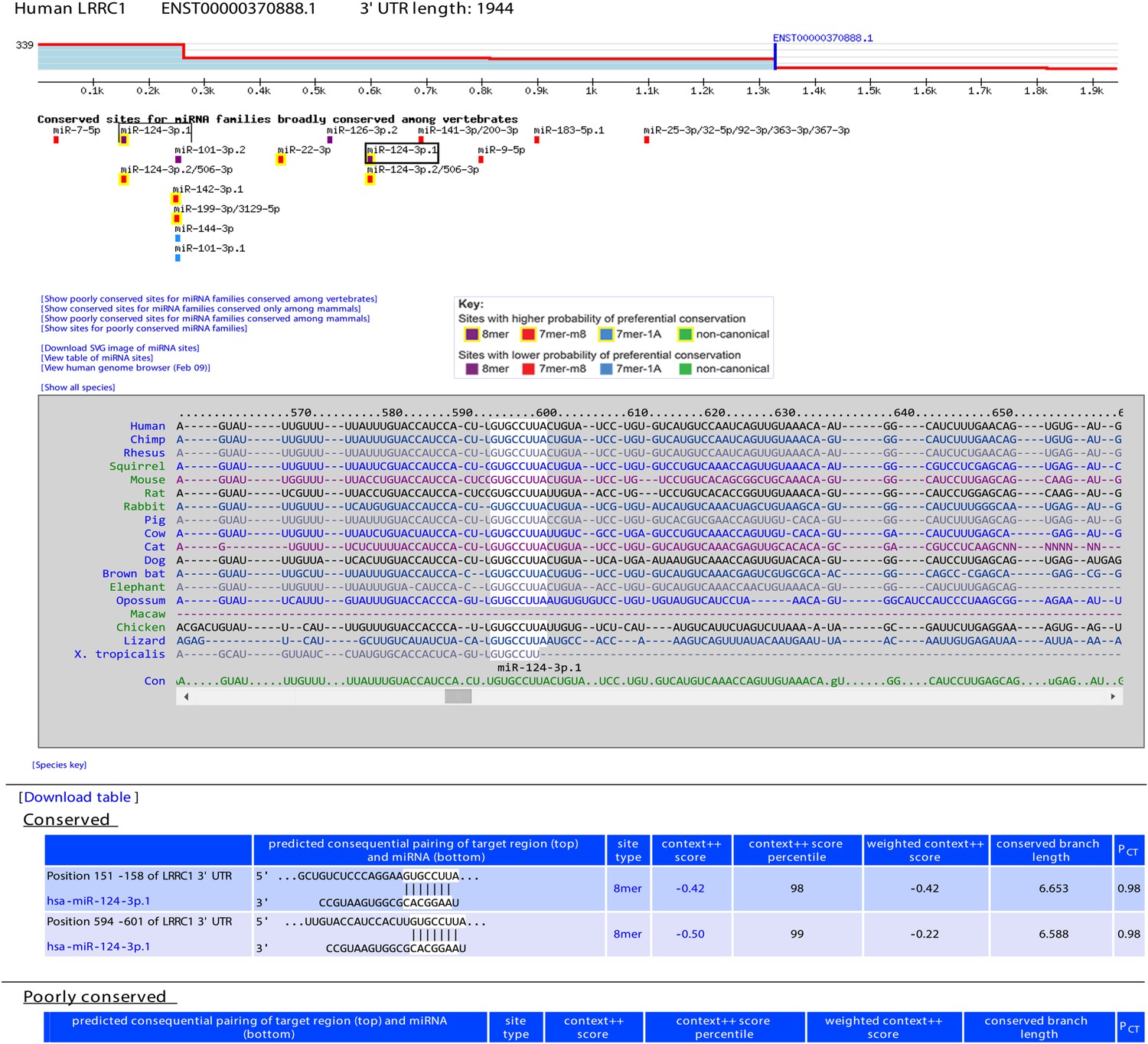
Example display of TargetScan7 predictions.
The example shows a TargetScanHuman page for the 3′ UTR of the LRRC1 gene. At the top is the 3′-UTR profile, showing the relative expression of tandem 3′-UTR isoforms, as measured using 3P-seq (Nam et al., 2014). Shown on this profile is the end of the longest Gencode annotation (blue vertical line) and the total number of 3P-seq reads (339) used to generate the profile (labeled on the y-axis). Below the profile are predicted conserved sites for miRNAs broadly conserved among vertebrates (colored according to the key), with options to display conserved sites for mammalian conserved miRNAs, or poorly conserved sites for any set of miRNAs. Boxed are the predicted miR-124 sites, with the site selected by the user indicated with a darker box. The multiple sequence alignment shows the species in which an orthologous site can be detected (white highlighting) among representative vertebrate species, with the option to display site conservation among all 84 vertebrate species. Below the alignment is the predicted consequential pairing between the selected miRNA and its sites, showing also for each site its position, site type, context++ score, context++ score percentile, weighted context++ score, branch-length score, and PCT score.
Figure 7—figure supplement 1
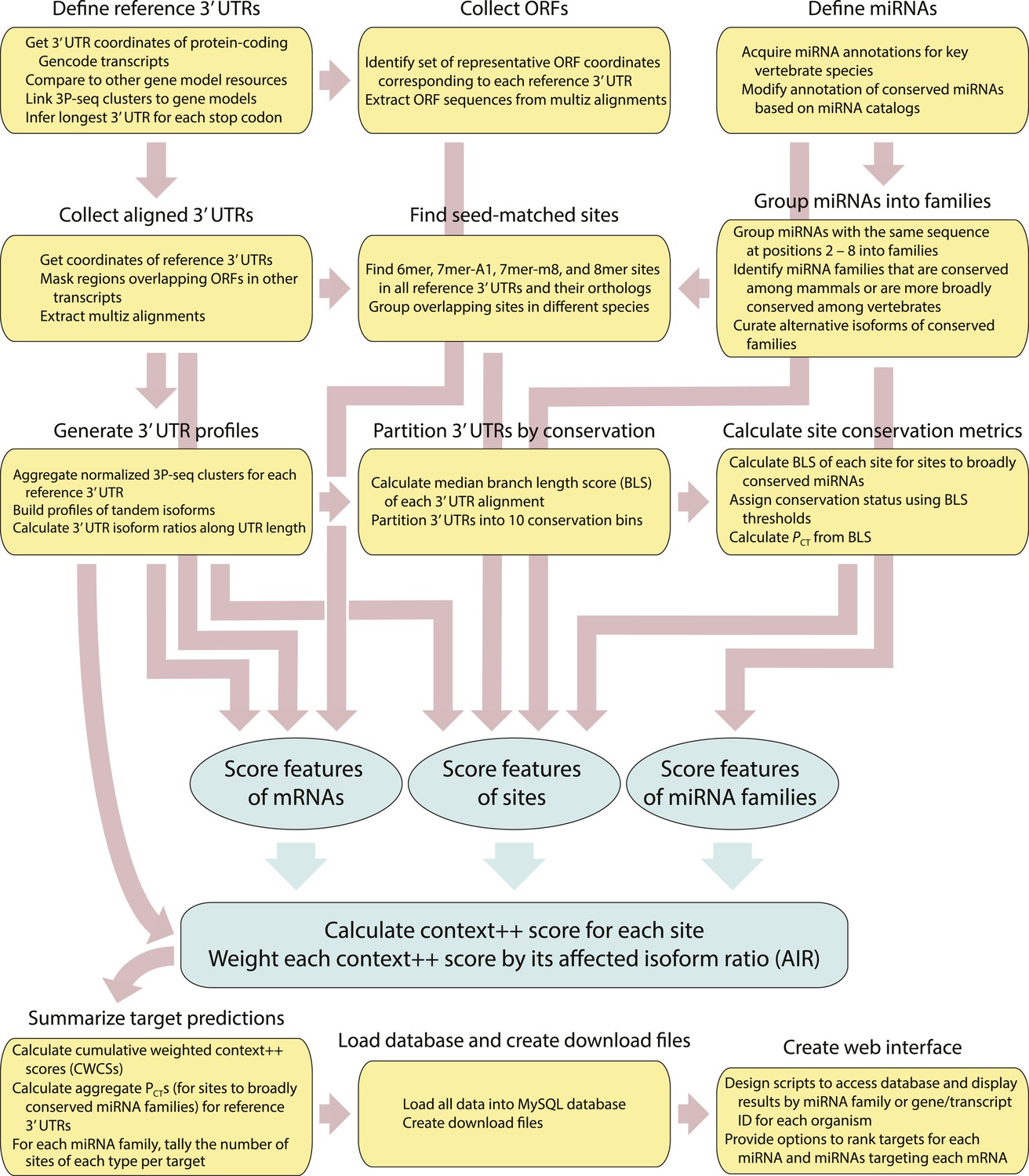
Flowchart of the computational pipeline used to build the TargetScan7 database.
https://doi.org/10.7554/eLife.05005.021Tables
Table 1
The 26 features considered in the models, highlighting the 14 robustly selected through stepwise regression (bold)
| Feature | Abbreviation | Description | Frequency chosen | |||
|---|---|---|---|---|---|---|
| 8mer | 7mer-m8 | 7mer-A1 | 6mer | |||
| miRNA | ||||||
| 3′-UTR target-site abundance | TA_3UTR | Number of sites in all annotated 3′ UTRs (Arvey et al., 2010; Garcia et al., 2011) | 100% | 100% | 100% | 100% |
| ORF target-site abundance | TA_ORF | Number of sites in all annotated ORFs (Garcia et al., 2011) | 9.4% | 0.7% | 68.1% | 93.4% |
| Predicted seed-pairing stability | SPS | Predicted thermodynamic stability of seed pairing (Garcia et al., 2011) | 100% | 100% | 100% | 100% |
| sRNA position 1 | sRNA1 | Identity of nucleotide at position 1 of the sRNA | 68% | 100% | 99.7% | 97.7% |
| sRNA position 8 | sRNA8 | Identity of nucleotide at position 8 of the sRNA | 0% | 0.8% | 100% | 100% |
| Site | ||||||
| Site position 1 | site1 | Identity of nucleotide at position 1 of the site | N/A | 57.1% | N/A | 2% |
| Site position 8 | site8 | Identity of nucleotide at position 8 of the site | 0.8% | 95.1% | 99.4% | 100% |
| Site position 9 | site9 | Identity of nucleotide at position 9 of the site (Lewis et al., 2005; Nielsen et al., 2007) | 15.4% | 7.1% | 0.9% | 93.7% |
| Site position 10 | site10 | Identity of nucleotide at position 10 of the site (Nielsen et al., 2007) | 0.1% | 100% | 8.5% | 26.3% |
| Local AU content | local_AU | AU content near the site (Grimson et al., 2007; Nielsen et al., 2007) | 100% | 100% | 100% | 100% |
| 3′ supplementary pairing | 3P_score | Supplementary pairing at the miRNA 3′ end (Grimson et al., 2007) | 42.5% | 100% | 100% | 100% |
| Distance from stop codon | dist_stop | log10(Distance of site from stop codon) | 62.4% | 10.8% | 8.7% | 25.7% |
| Predicted structural accessibility | SA | log10(Probability that a 14 nt segment centered on the match to sRNA positions 7 and 8 is unpaired) | 100% | 100% | 100% | 100% |
| Minimum distance | min_dist | log10(Minimum distance of site from stop codon or polyadenylation site) (Gaidatzis et al., 2007; Grimson et al., 2007; Majoros and Ohler, 2007) | 99.9% | 100% | 87.4% | 100% |
| Probability of conserved targeting | PCT | Probability of site conservation, controlling for dinucleotide evolution and site context (Friedman et al., 2009) | 100% | 100% | 100% | 20.8% |
| mRNA | ||||||
| 5′-UTR length | len_5UTR | log10(Length of the 5′ UTR) | 98.2% | 8.2% | 4.6% | 17.2% |
| ORF length | len_ORF | log10(Length of the ORF) | 100% | 100% | 100% | 100% |
| 3′-UTR length | len_3UTR | log10(Length of the 3′ UTR) (Hausser et al., 2009) | 100% | 100% | 100% | 100% |
| 5′-UTR AU content | AU_5UTR | Fraction of AU nucleotides in the 5′ UTR | 13% | 38.9% | 91.1% | 31.3% |
| ORF AU content | AU_ORF | Fraction of AU nucleotides in the ORF | 1.2% | 72.4% | 28.4% | 35.8% |
| 3′-UTR AU content | AU_3UTR | Fraction of AU nucleotides in the 3′ UTR (Robins and Press, 2005; Hausser et al., 2009) | 5.4% | 73.3% | 65.3% | 80.6% |
| 3′-UTR offset-6mer sites | off6m | Number of offset-6mer sites in the 3′ UTR (Friedman et al., 2009) | 65.9% | 89.6% | 99.8% | 100% |
| ORF 8mer sites | ORF8m | Number of 8mer sites in the ORF (Lewis et al., 2005; Reczko et al., 2012) | 99.5% | 99.1% | 100% | 100% |
| ORF 7mer-m8 sites | ORF7m8 | Number of 7mer-m8 sites in the ORF (Reczko et al., 2012) | 4.7% | 4.3% | 85.3% | 100% |
| ORF 7mer-A1 sites | ORF7A1 | Number of 7mer-A1 sites in the ORF (Reczko et al., 2012) | 68.4% | 34.2% | 97.8% | 98.4% |
| ORF 6mer sites | ORF6m | Number of 6mer sites in the ORF (Reczko et al., 2012) | 91% | 13.3% | 0.7% | 36.7% |
-
The feature description does not include the scaling performed (Table 3) to generate more comparable regression coefficients.
Table 2
Summary of datasets analyzed in this study, and corresponding figures using the datasets
Table 3
Scaling parameters used to normalize data to the (0, 1) interval
| Feature | 8mer | 7mer-m8 | 7mer-A1 | 6mer | ||||
|---|---|---|---|---|---|---|---|---|
| 5th % | 95th % | 5th % | 95th % | 5th % | 95th % | 5th % | 95th % | |
| 3P_score | 1.000 | 3.500 | 1.000 | 3.500 | 1.000 | 3.500 | 1.000 | 3.500 |
| SPS | −11.130 | −5.520 | −11.130 | −5.490 | −8.410 | −3.330 | −8.570 | −3.330 |
| TA_3UTR | 3.113 | 3.865 | 3.067 | 3.887 | 3.145 | 3.887 | 3.113 | 3.887 |
| Len_3UTR | 2.392 | 3.637 | 2.409 | 3.615 | 2.413 | 3.630 | 2.405 | 3.620 |
| Len_ORF | 2.788 | 3.753 | 2.773 | 3.729 | 2.773 | 3.730 | 2.775 | 3.731 |
| Min_dist | 1.415 | 3.113 | 1.491 | 3.096 | 1.431 | 3.117 | 1.477 | 3.106 |
| Local_AU | 0.308 | 0.814 | 0.277 | 0.782 | 0.342 | 0.801 | 0.295 | 0.772 |
| SA | −4.356 | −0.661 | −5.218 | −0.725 | −4.230 | −0.588 | −5.082 | −0.666 |
| PCT | 0.000 | 0.816 | 0.000 | 0.364 | 0.000 | 0.449 | 0.000 | 0.193 |
-
Provided are the 5th and 95th percentile values for continuous features that were scaled, after the values of the feature were appropriately transformed as indicated (Table 1).
Additional files
-
Supplementary file 1
Normalized values for fold changes (log2) of mRNAs detectable in the compendium of 74 sRNA transfection datasets.
- https://doi.org/10.7554/eLife.05005.024
-
Supplementary file 2
Normalized values for fold changes (log2) of mRNAs detectable in the seven datasets examining the response of transfecting miRNAs into HCT116 cells.
- https://doi.org/10.7554/eLife.05005.025
-
Supplementary file 3
Genomic coordinates of CLIP clusters that appeared in annotated 3′ UTRs after transfecting miR-124 into HeLa cells.
- https://doi.org/10.7554/eLife.05005.026
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Predicting effective microRNA target sites in mammalian mRNAs
eLife 4:e05005.
https://doi.org/10.7554/eLife.05005




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}