Promoter nucleosome dynamics regulated by signalling through the CTD code
- University of Namur, Belgium
- Universidad de Salamanca, Spain
- Universitat Pompeu Fabra, Spain
Figures
Figure 1 with 1 supplement
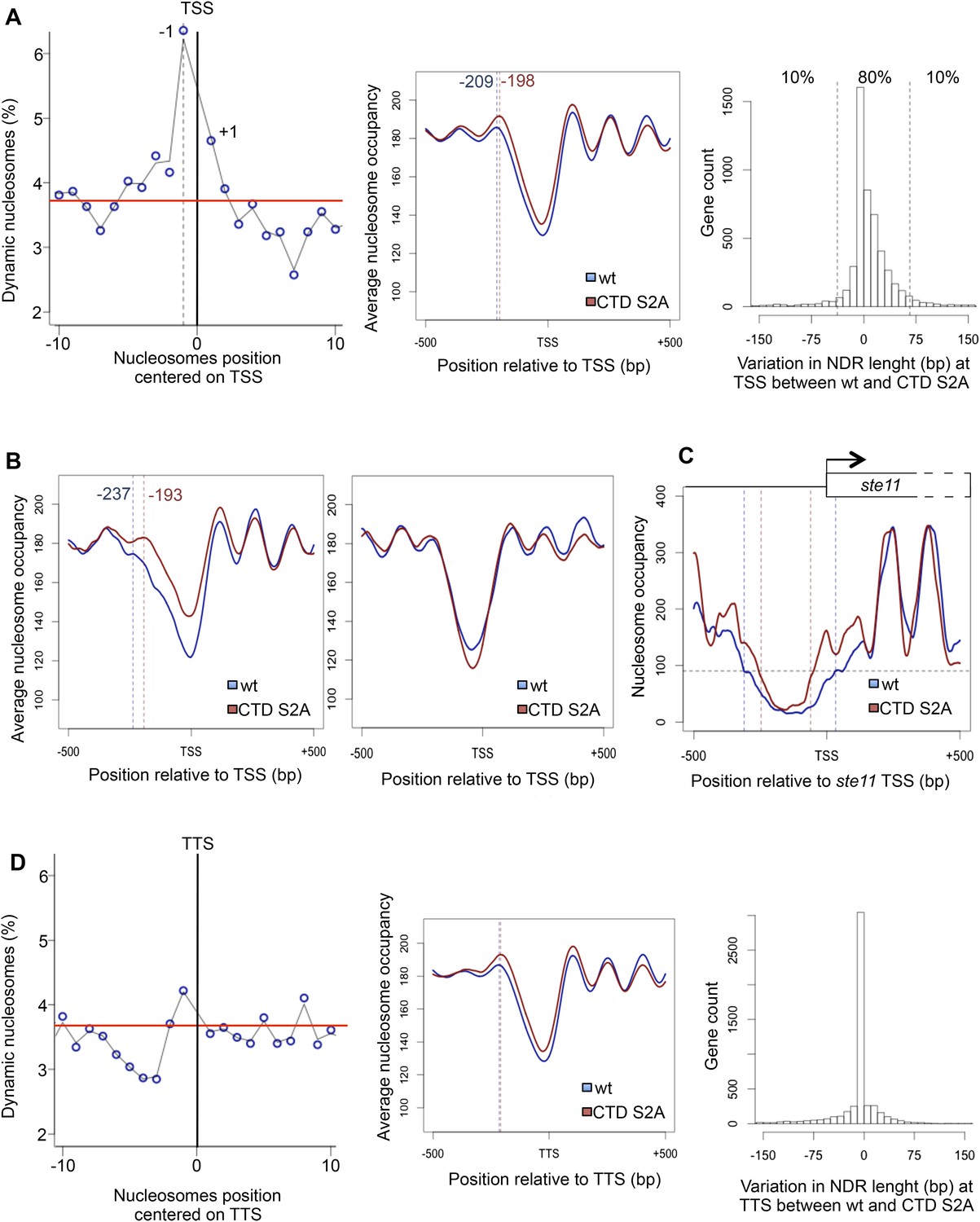
The RNA polymerase II S2P affects nucleosome dynamic at the promoter of a subset of genes.
(A) Left panel: the percentage of dynamic nucleosomes aligned relative to the transcription start site (TSS) in the fission yeast genome. The red line indicates the average percentage of dynamic nucleosome over the genome (3.7%). Middle panel: meta-gene analysis of the nucleosome occupancy signal for all protein-coding genes near the TSS. The distance between the TSS and the average −1 nucleosome midpoint position is indicated in blue for the wt and red for the S2A mutant. Right panel: distribution of the variation in nucleosome-depleted region (NDR) length at TSS between wt and the S2A mutant. The 10% genes showing the strongest increase or strongest decrease in NDR size are indicated. (B) Left panel: meta-gene analysis of the nucleosome occupancy signal for the 10% protein-coding genes showing the strongest decrease in NDR size at the TSS in the S2A mutant. The distance between the TSS and the average −1 nucleosome midpoint is indicated in blue for the wt and red for the S2A mutant. Right panel: meta-gene analysis of the nucleosome occupancy signal for the 10% protein-coding genes showing the strongest increase in NDR size at the TSS in the S2A mutant (blue: wt, red: S2A). (C) Nucleosomes occupancy nearby the promoter of ste11 (blue: wt, red: S2A). (D) Left panel: the percentage of dynamic nucleosomes aligned relative to the TTS in the fission yeast genome. The red line indicates the average percentage of dynamic nucleosome over the genome (3.7%). Middle panel: meta-gene analysis of the nucleosome occupancy signal for all protein coding genes near the TTS. Right panel: distribution of the variation in NDR length at TTS between wt and the S2A mutant (blue: wt, red: S2A).
-
Figure 1—source data 1
List of the synthetic lethal genetic interactions uncovered with the lsk1Δ and CTD S2A mutant strains.
- https://doi.org/10.7554/eLife.09008.004
-
Figure 1—source data 2
General features of the dynamic nucleosomes and the associated genes.
List of the genes showing dynamic changes in the −1 nucleosome (TSS) with false discovery rate < 0.5% and list of the 10% genes showing the strongest promoter NDR size decrease in the S2A mutant (see Figure 1A).
- https://doi.org/10.7554/eLife.09008.005
Figure 1—figure supplement 1
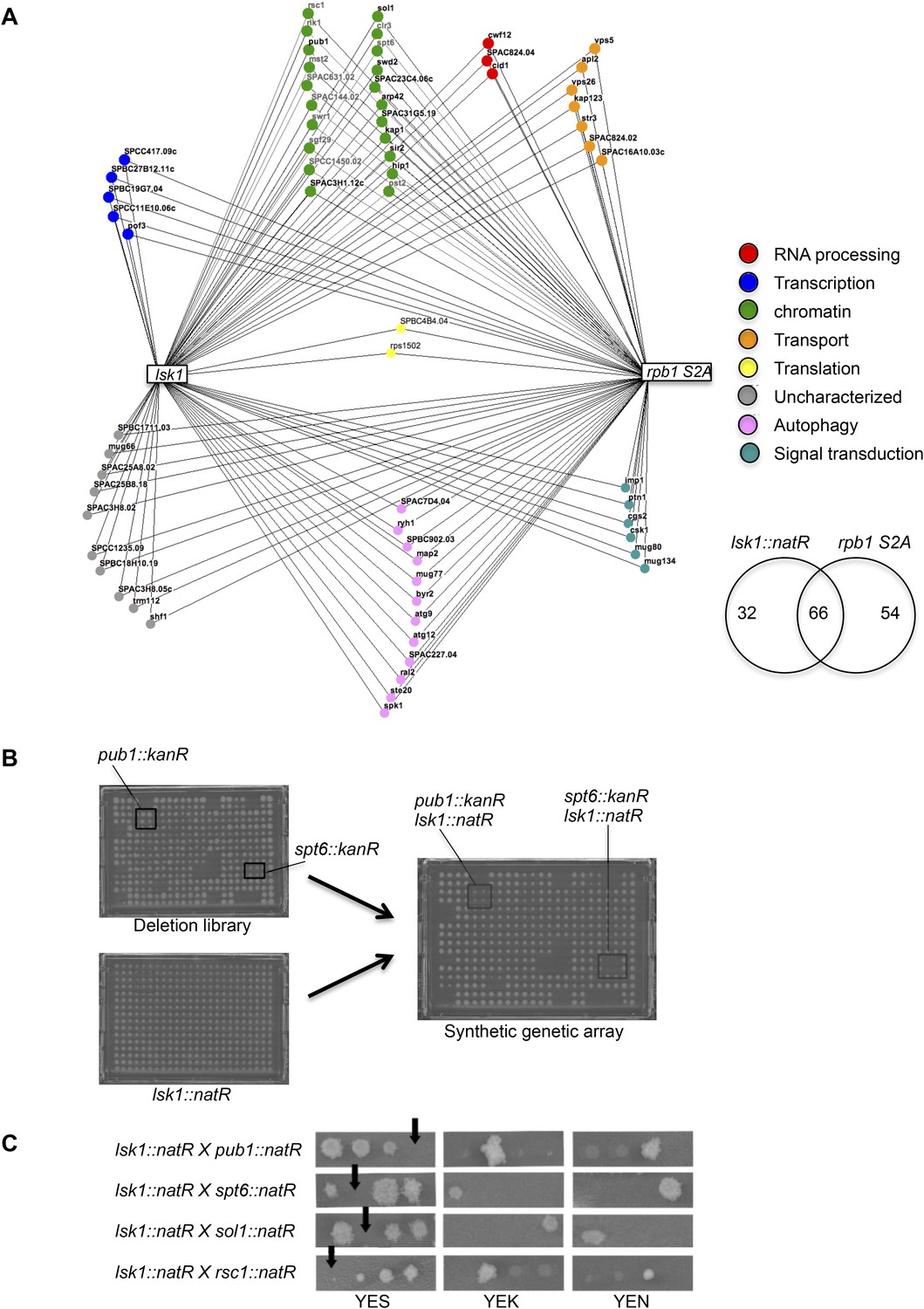
Genome-wide synthetic lethal interaction mappings of the lsk1Δ and rpb1 S2A mutants link Rpb1 CTD S2P to chromatin biology.
(A) The Bioneer deletion library was screened in quadruplicate for synthetic lethal interactions with either the lsk1 deletion or the rpb1 CTD S2A strains. 98 interactions were detected for lsk1, and 120 interactions were detected with rpb1 CTD S2A. The 66 shared interactions were classified by gene ontology in functional categories and displayed in the Osprey software (see ‘Material and methods’). (B) One plate of the Bioneer deletion library containing the pub1::kanR and spt6::kanR deletion mutants (in quadruplicate) is shown together with the mating partner plate that only contains lsk1::natR strains. After crossing and selection (see ‘Material and methods’), recombinants are selected on medium supplemented with kanamycin and nourseothricin. The co-lethal interactions are highlighted. (C) Confirmation of the genetic interactions by tetrad analyses followed by growth assay on unsupplemented rich medium, or rich medium supplemented with kanamycin or nourseothricin. Tetratype segregation was typically observed and reveals that the double mutants are not viables. Note that tetrad analysis was performed for about half (10) of the genes falling in the «chromatin regulation» GO functional category.
Figure 2 with 1 supplement

The CTD S2P regulates histone occupancy and acetylation at the promoter of the ste11 gene.
(A) Left panel: the occupancy of H3 measured by ChIP using the indicated amplicons in the 5′ proximal region of the ste11 and adh1 genes in the indicated strains. Right panel: similar to the left panel except that more amplicons were used and the data obtained in the lsk1Δ and S2A strains were normalized to the wt set as 1. (B) The wt and lsk1Δ strains were starved for nitrogen at the indicated time points (hours). The occupancy of acetylated H3 at the indicated places was determined by ChIP using anti-H3-K14-ac normalized against unmodified H3. (C) The occupancy of H3 was measured by ChIP using the indicated amplicon in a lsk1-as strain grown in the presence or absence of 10 μM 3-MB-PP1 for 1 hr. (D) A lsk1-as strain was cultured during vegetative growth (T0) and starved for nitrogen for 3 hr (T3). At T0, 10 μM 3-MB-PP1 (+I) or an identical volume of DMSO (−I) was added as indicated. The occupancy of acetylated H3 at the indicated places was determined by ChIP using anti-H3-K14-ac normalized against unmodified H3. (E) The occupancy of RNA polymerase II (8WG16 antibody) measured by ChIP using the indicated amplicons in the 5′ proximal region of the ste11 gene in the indicated strains. (F) Relative quantification (RQ) of the ste11 mRNA determined by quantitative Q-RT-PCR in the indicated strains. a.u.: arbitrary units. (G) The occupancy of H3 measured by ChIP using the indicated amplicons in the 5′ proximal region of the ste11 gene in the indicated strains.
Figure 2—figure supplement 1

Nucleosome scanning of the ste11 5′ region reveals increased occupancy in the CTD S2A mutant.
′(A) Nucleosome scanning analysis of the wt and rpb1 CTD S2A strains. Nucleosomal DNA enrichment at the indicated positions of the ste11 locus was determined by ChIP experiment on MNase-digested chromatin. Data are presented as the average of three independent experiments along with the SEM. Inferred nucleosome location is indicated and the position of the amplicons (−1 and 1) used in other experiments is indicated. (B) Schematic of amplicons used in the nucleosome scanning experiments. Green and blue arrows represent 17 overlapping amplicons (named a to q) covering a 1222 bp region encompassing the NDR and 255 bp of the ste11 transcript. The +1 site and its genomic coordinate are indicated. The red arrows represent the amplicons used in ChIP experiments (referred as −1, 1, 2). (C) The wt and lsk1Δ strains were cultured during vegetative growth (T0) and nitrogen starvation at the indicated time points (hour). The occupancy of methylated histone H3 was determined by ChIP using an antibody specific for the methylated H3-K36 residue normalized against a ChIP against the unmodified histone H3. The location of the amplicons used in Q-PCR is indicated. Each column represents the mean percentage immunoprecipitation value ± SEM (n = 3). (D) RQ of the ste11 mRNA determined by Q-RT-PCR using the ΔΔct method in the wt and the set2 strains during vegetative growth (T0) and nitrogen starvation at the indicated time points (hour). The error bars were calculated from duplicates. a.u.: arbitrary units. (E) The occupancy of acetylated histone H3 was determined by ChIP using an antibody specific for the acetylated H3-K36 residue normalized against a ChIP against the unmodified histone H3. The location of the amplicons used in Q-PCR is indicated. Each column represents the mean percentage immunoprecipitation value ± SEM (n = 2).
Figure 3 with 2 supplements
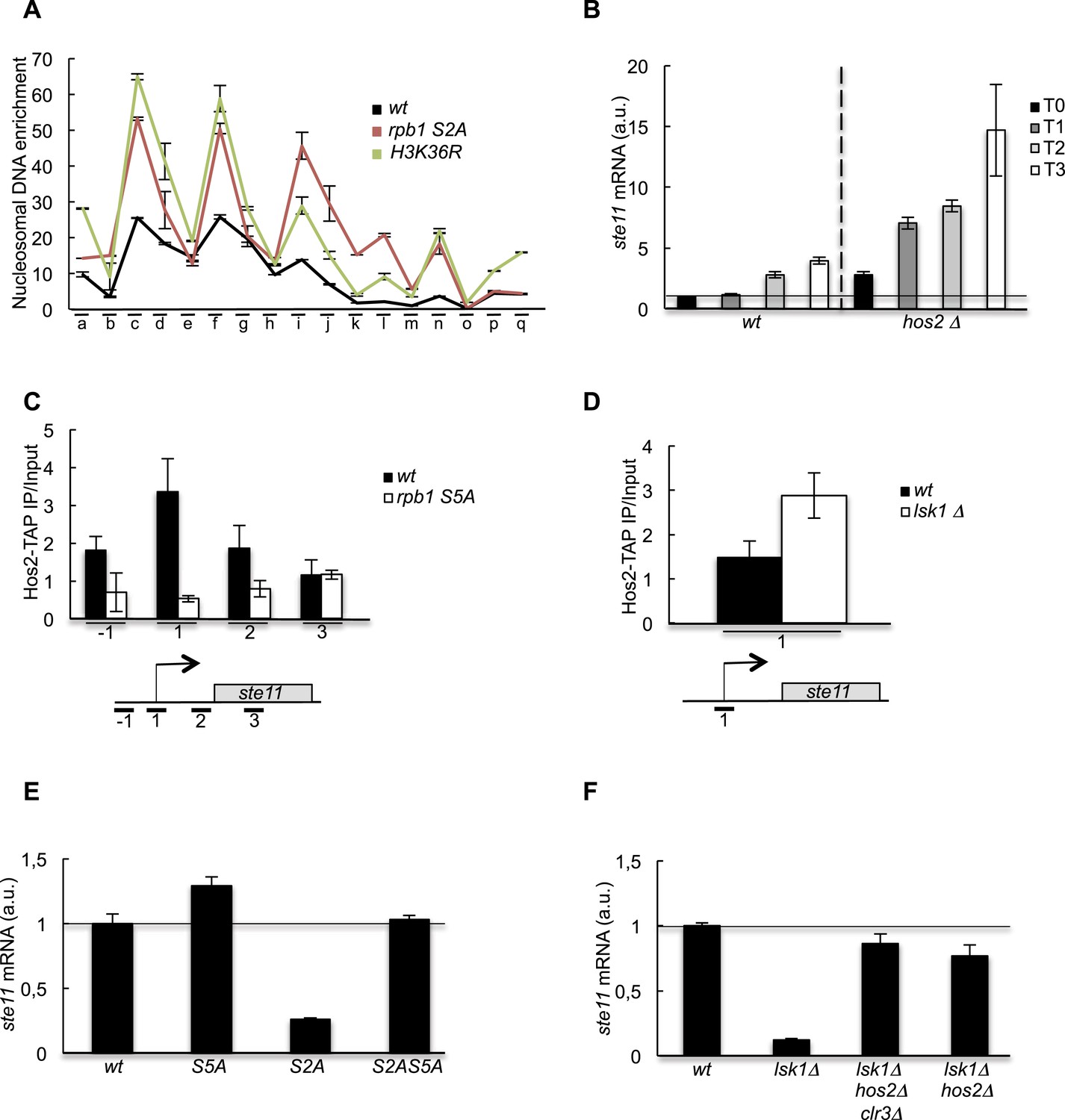
The increase of CTD S2P nearby the promoter region is necessary to reverse the CTD S5P-dependent deacetylation of nucleosomes.
(A) Nucleosome scanning analysis of the S2A and H3K36R strains. Nucleosomal DNA enrichment at the indicated positions of the ste11 locus was determined by ChIP experiment on MNase-digested chromatin. Data are presented as the average of two independent experiments along with the SEM. (B) RQ of the ste11 mRNA determined by Q-RT-PCR in the wt and hos2Δ strains during nitrogen starvation at the indicated time points (hour). a.u.: arbitrary units. (C) The occupancy of Hos2-TAP was measured by ChIP using the indicated amplicons at the ste11 locus in a wt strain and a CTD S5A mutant. Each column represents the averaged value ± SEM (n = 4). (D) Identical to C with a wt strain and an lsk1Δ mutant. (E–F) RQ of the ste11 mRNA determined by quantitative Q-RT-PCR in the indicated strains. a.u.: arbitrary units.
Figure 3—figure supplement 1

HDAC-dependent control of ste11 expression by CTD S2P independently of Gcn5.
(A) The occupancy of Gcn5-TAP was measured by ChIP at indicated loci in a wt strain and a lsk1Δ mutant. Each column represents the mean percentage immunoprecipitation value ± SEM (n = 3) normalized to an untagged control strain. (B) RQ of the ste11, adh1, and sam1 mRNAs determined by Q-RT-PCR using the ΔΔct method in the indicated strains grown in the presence of Trichostatin A (50 μg/ml) or an equivalent volume of DMSO for 2 hr. The number indicated in each column is the fold increase of expression in the TSA vs DMSO condition. The data were normalized to the wt set as 1 in order to highlight the fold increase of expression in the lsk1Δ and the rpb1 CTD S2A mutants. All experiments were performed in duplicate. (C) Schematic of the construction of the histone mutants, based on previous work by the Allshire laboratory. Two of the loci harbouring the histones H3 and H4 genes are ultimately deleted by the natR and kanR markers, while the third locus (h3.2) harbours the mutation. (D) The occupancy of histone H3 was measured by ChIP using the indicated amplicon in the indicated strains. Each column represents the mean percentage immunoprecipitation value ± SEM (n = 2). (E) RQ of the ste11 mRNA determined by quantitative RT-PCR using the ΔΔct method in the indicated strains during vegetative growth (T0) and nitrogen starvation at the indicated time points (hour). a.u.: arbitrary units. (F) RQ of the ste11 mRNA determined by quantitative RT-PCR using the ΔΔct method in the indicated strains lacking one of the fission yeast HDAC. Note that for clr6, a ts strain (clr6-1) and the isogenic wild type were used and cultured 4 hr at 36°C. a.u.: arbitrary units.
Figure 3—figure supplement 2
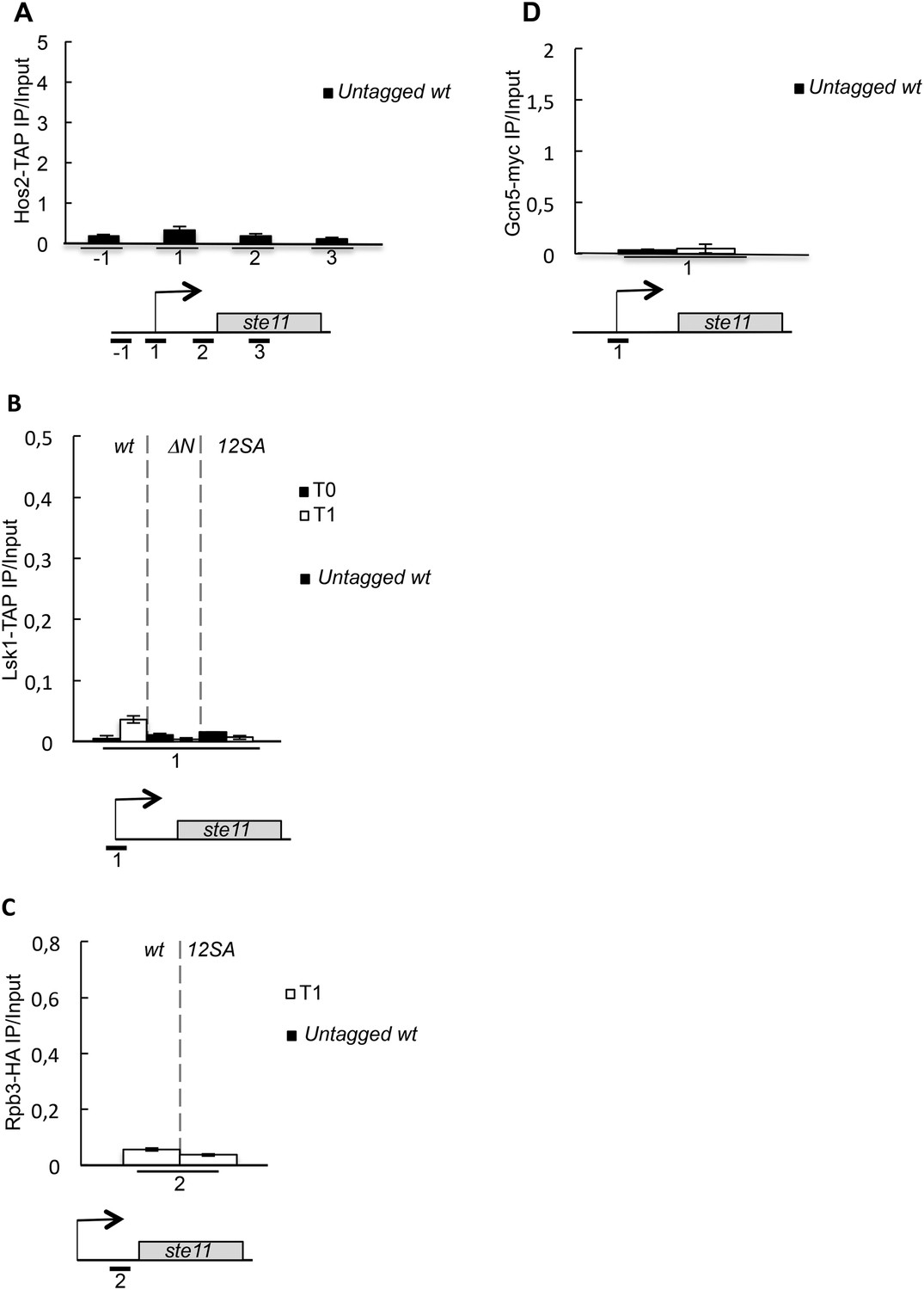
Untagged control experiments for the chromatin immunoprecipitations.
(A) Similar to Figure 3C except that an untagged wild-type strain was used. (B) Similar to Figure 5C except that an untagged wild-type strain was used. (C) Similar to Figure 5D except that an untagged wild-type strain was used. (D) Similar to Figure 3—figure supplement 1A except that an untagged wild-type strain was used.
Figure 4
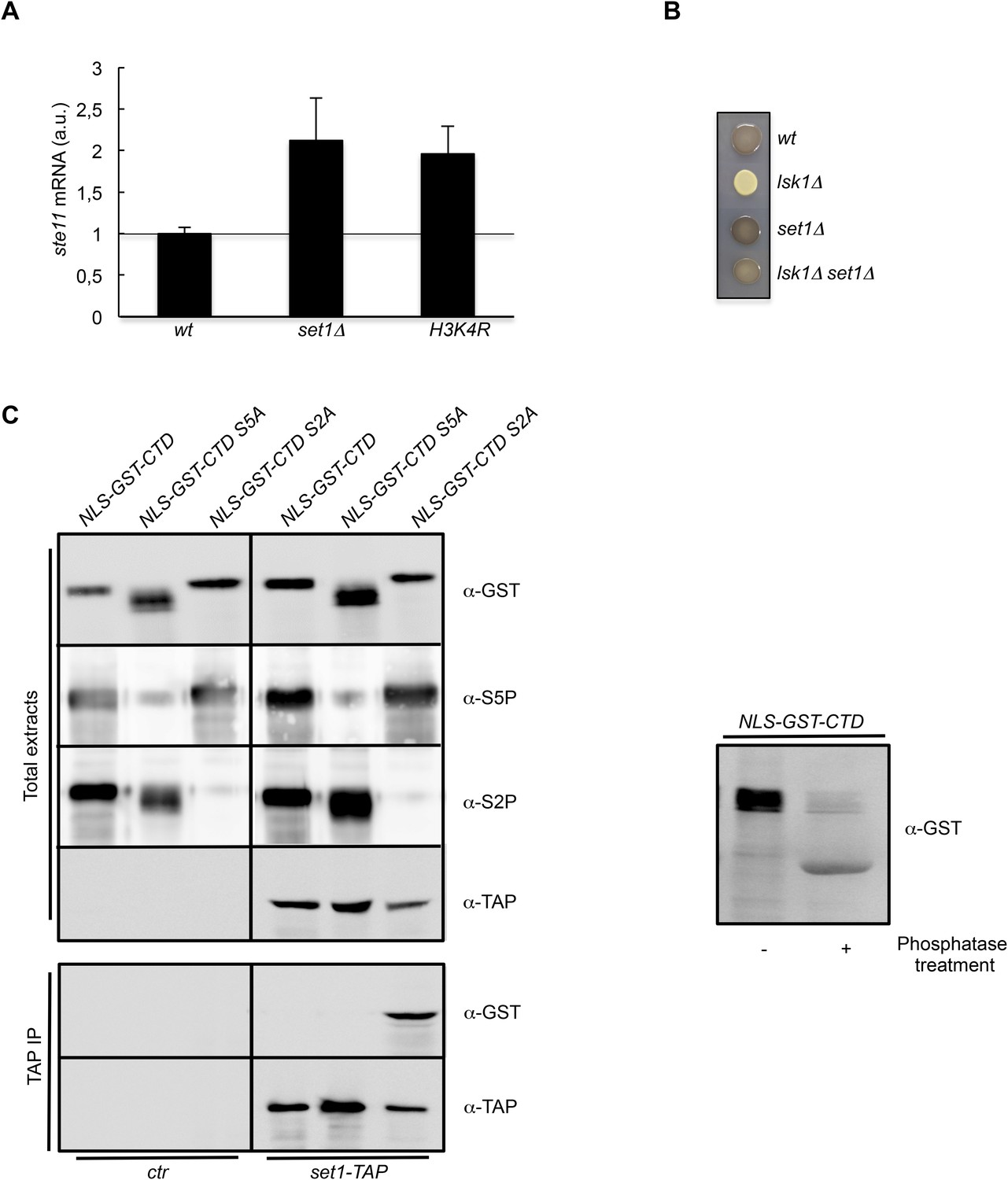
The S2P antagonizes Set1 binding to the CTD.
(A) RQ of the ste11 mRNA determined by quantitative RT-PCR using the ΔΔct method in the wt, set1Δ, and H3K4R strains. Each column represents the averaged value ± SEM (n = 2). a.u.: arbitrary units. Note that the wild-type control is different in the case of the set1Δ and H3K4R strains because the histone mutant is constructed in a background where only one copy of the histone H3 is retained (Figure 3—figure supplement 1C). Isogenic strains are used as control and set as 1. (B) Heterothallic wild-type, lsk1Δ, set1Δ, and set1Δ lsk1Δ strains were plated for 48 hr on mating medium before iodine staining to reveal sterility. (C) NLS-GST-CTD fusions containing wt, S2A, or S5A repeats were expressed in fission yeast from a pREP-3 plasmid. Protein extracts from the ctr and set1-TAP strains expressing these fusions were probed by Western blot using the anti-GST, anti-S5P, anti-S2P, or anti-TAP antibodies as indicated (Top panel-Total extracts). After TAP immunoprecipitation, the resulting beads were probed by Western blot using anti-GST or anti-TAP antibodies (Bottom panel-TAP IP). Right panel: a protein extract from the ctr strain expressing the NLS-GST-CTD fusion was treated or not by a phosphatase and probed by Western blot using anti-GST antibodies.
Figure 5 with 1 supplement

The phosphorylation of Lsk1 by Sty1 is required for CTD S2 phosphorylation in the 5′ proximal region of the ste11 gene.
(A) The Sty1-TAP protein was precipitated and used on beads for in vitro kinase assays using wt and mutated (4SA or 12SA) forms of the GST-Lsk1 protein as indicated. The MAP kinase was activated by nitrogen starvation. The amount of precipitated proteins was estimated by Western blot analysis using peroxidase–antiperoxidase (PAP). (B) RQ of the ste11 mRNA determined by Q-RT-PCR in the indicated strains during nitrogen starvation at the indicated time points (hour). a.u.: arbitrary units. lsk1 4SE refers to a mutant where the four residues fitting the perfect MAPK consensus were mutated to glutamic acid to mimic phosphorylation. (C) Left panel, the occupancy of Lsk1-TAP and the Lsk1ΔN-TAP and Lsk112SA-TAP mutants was measured by ChIP using the indicated amplicons. Middle panel, same experiment in wt or sty1Δ strains. Right panel, the occupancy of S2P was measured by ChIP using the indicated amplicons in the indicated strains. These experiments were performed during vegetative growth (T0) and early nitrogen starvation (T1). (D) The occupancy of RNA Pol II was measured by ChIP in the indicated strains using the indicated amplicons. The experiment was performed during early nitrogen starvation (T1).
Figure 5—figure supplement 1
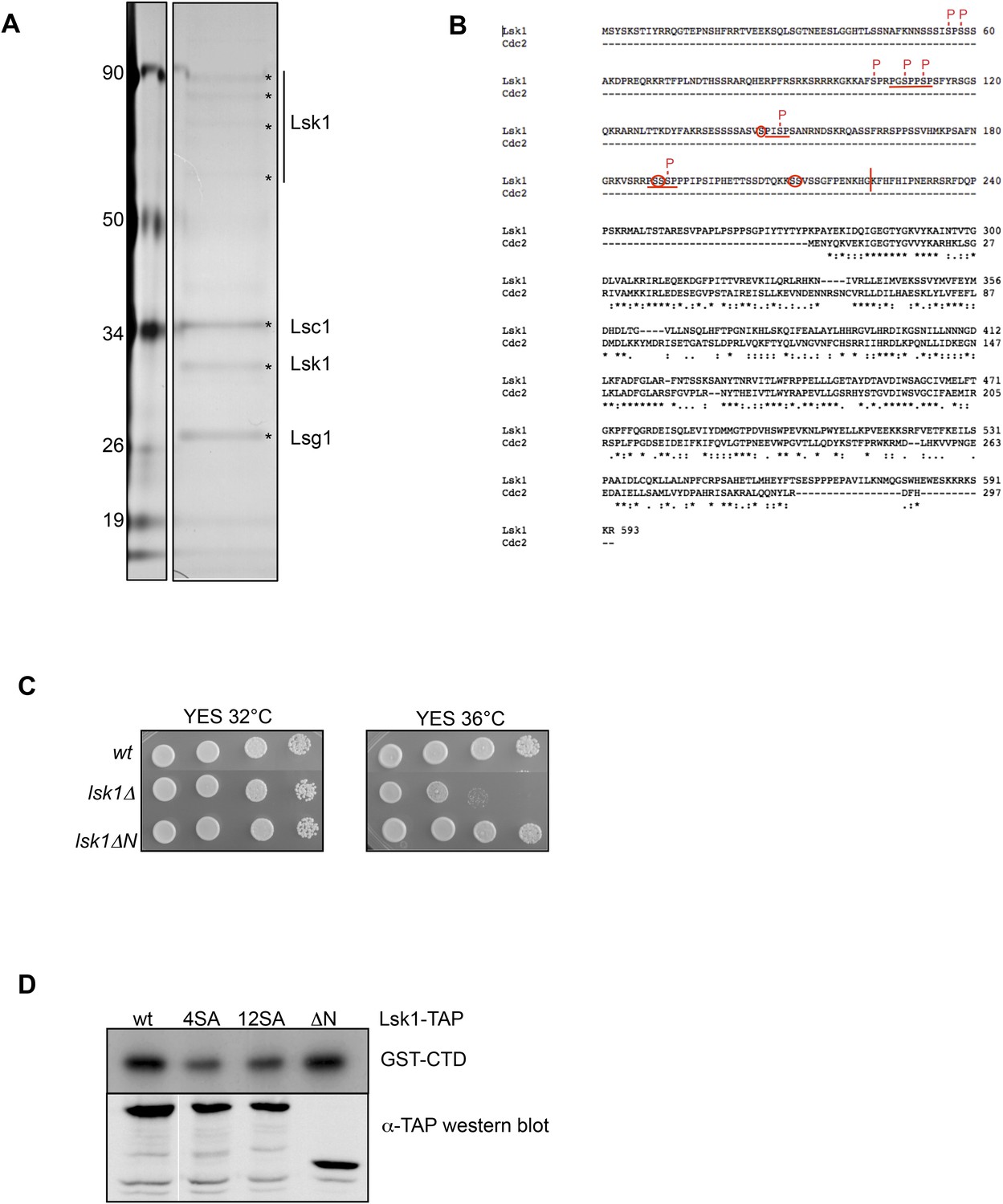
The phosphorylation of the CTD S2 kinase Lsk1 on 12 sites in vivo is not required for its activity.
(A) Lsk1-TAP was purified from 1 g of soluble extract and the final eluted fraction was separated on SDS–PAGE, silver stained, and analyzed by mass spectrometry. (B) Protein sequence alignment of the fission yeast Lsk1 and Cdc2 cyclin-dependent kinases. * indicates identity, indicates high similarity, indicates similarity. MAPK consensus motifs (PXS/TP) are underlined, phosphorylated residues identified by mass spectrometry are marked with P, and additional residues phosphorylated in vitro are circled. The vertical bar refers to the truncation in the lsk1ΔN mutant. (C) Spot dilution assay of wt, lsk1Δ, and lsk1Δ strains grown 2 days at 32°C or 36°C on rich medium. (D) Strains harbouring the indicated tagged proteins were lysed, and immunoprecipitation was performed on IgG beads followed by kinase assay on GST-CTD. A Western blot analysis was performed using the PAP antibody. 4SA refers to the lsk1 mutant where only the four residues fitting the perfect MAPK consensus were mutated. 12SA refers to the lsk1 mutant where 12 residues were mutated. ΔN refers to the truncated mutant.
Figure 6

A chimeric HMG-Lsk1-HA protein induces ste11 expression independently of nitrogen starvation.
(A) A schematic of the chimeric HMG-Lsk1-HA protein. The wild-type Ste11 and Lsk1 proteins are depicted and the size of various regions indicated in amino acids. (B) Western blot analysis (anti-HA) of a wild-type strain containing the pREP-1 HMG-lsk1-HA plasmid or the corresponding empty vector and grown for 22 hr in the presence or absence of thiamine (that represses expression) as indicated. (C) RQ of the ste11 mRNA determined by quantitative RT-PCR using the ΔΔct method in a wild-type strain containing the indicated plasmids and grown for 22 hr in the presence or absence of thiamine as indicated. a.u.: arbitrary units. (D) The occupancy of HMG-Lsk1-HA at the ste11 and the adh1 promoters was measured by ChIP (anti-HA) using the indicated amplicon in a wild-type strain containing the pREP-1 HMG-lsk1-HA plasmid and grown for 22 hr in the presence or absence of thiamine as indicated. Each column represents the mean percentage immunoprecipitation value ± SEM (n = 2). (E) Similar to D except that the immunoprecipitation was performed with an anti-S2P antibody.
Figure 7 with 1 supplement
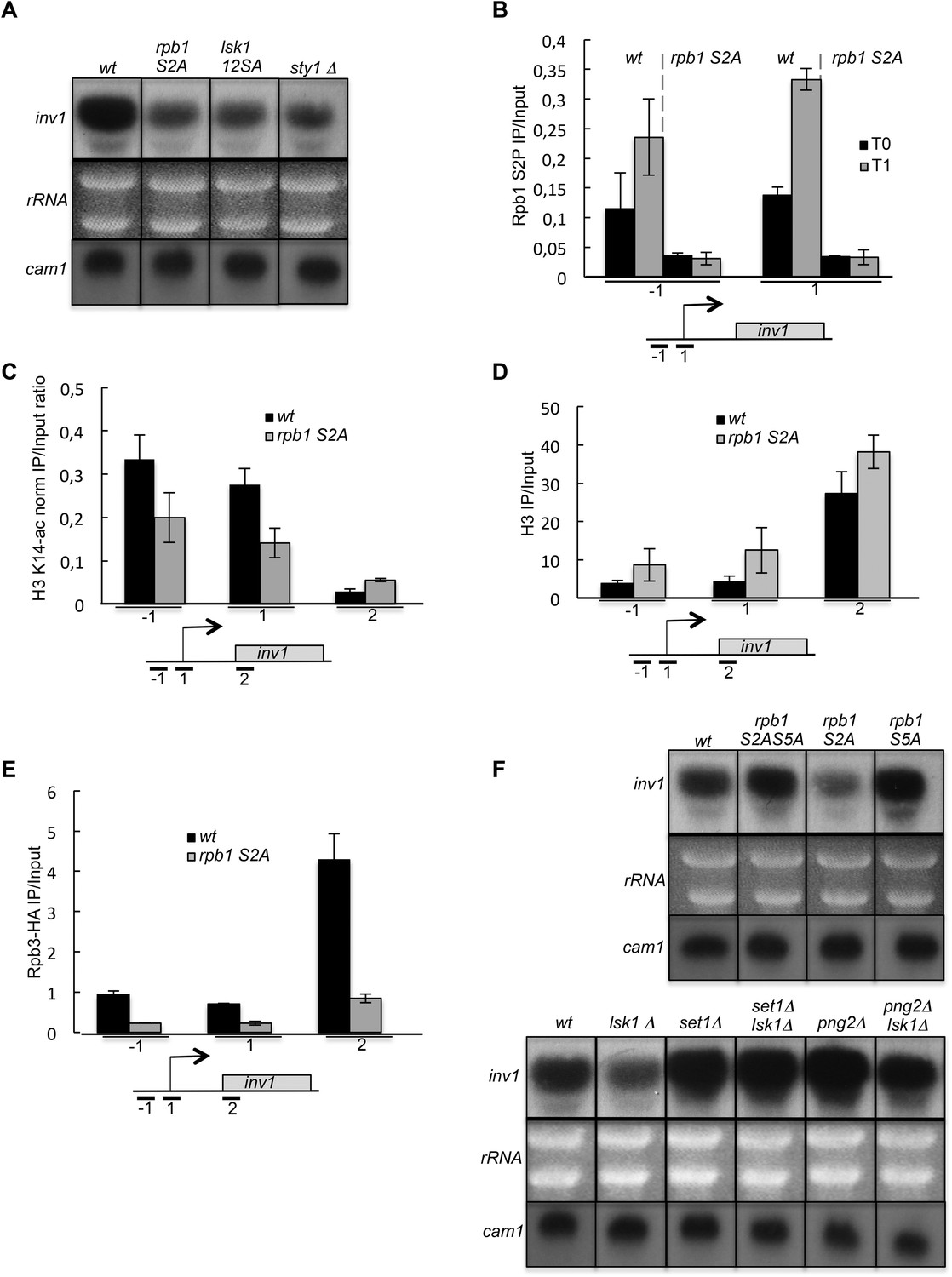
The induction of inv1 upon glucose deprivation is regulated by S2P-dependent control of promoter nucleosome acetylation.
(A) Northern blot analysis of inv1 expression in the indicated strains after an hour shift from high glucose (2%) to low glucose (0.1%). Note that the mRNA is not detected in high glucose (not shown). Ethidium bromide strained ribosomal RNAs and the level of expression of the cam1 gene are shown as loading controls. (B) The occupancy of S2P was measured by ChIP using the indicated amplicons in the 5′ proximal region of the inv1 gene in the indicated strains. These experiments were performed in high-glucose medium (T0) and after 1 hr in low-glucose medium (T1). (C) The wt and S2A strains were cultured in low glucose for 1 hr to induce inv1. The occupancy of acetylated H3 was determined by ChIP using anti-H3-K14-ac normalized against unmodified H3 (D). The location of the amplicons used in Q-PCR is indicated. (E) The occupancy of RNA Polymerase II was measured by ChIP in the indicated strains using the indicated amplicons in the 5′ proximal region of the inv1 gene. The strains were cultured in low glucose for 1 hr to induce inv1. (F) Northern blot analyses of inv1 expression in the indicated strains after an hour shift from high glucose (2%) to low glucose (0.1%). Ethidium bromide strained ribosomal RNAs and the level of expression of the cam1 gene are shown as loading controls.
Figure 7—figure supplement 1
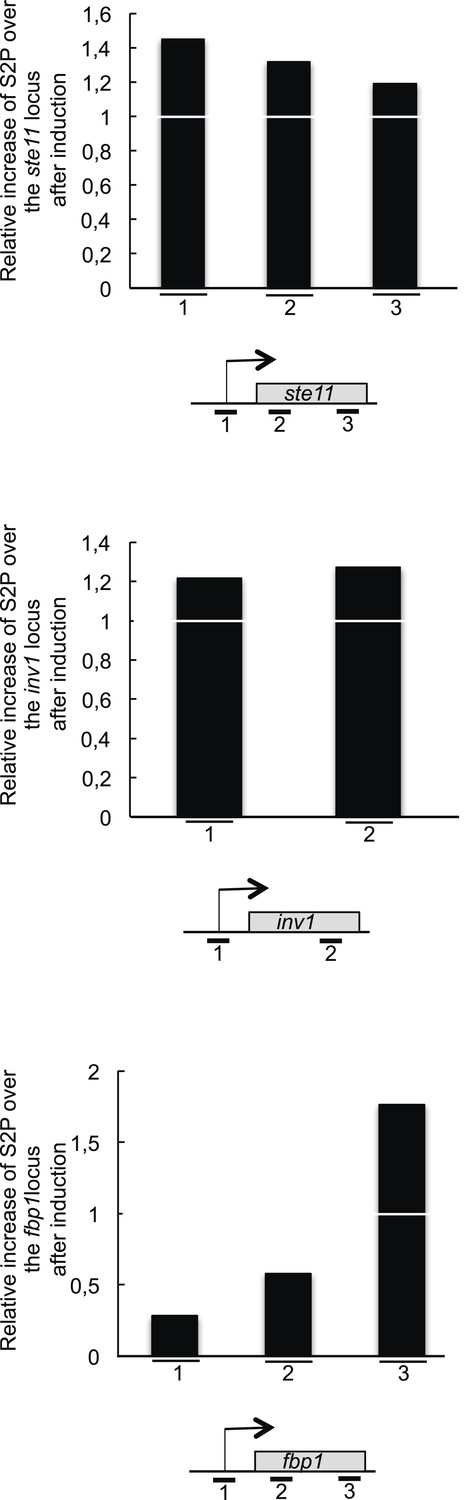
The RNA polymerase II CTD S2P reaches its maximal level early during the induction of the ste11 and inv1 genes.
The level of S2P (based on a ChIP using the 3E10 antibody) was determined and normalized on the total level of polymerase (based on a ChIP using the 8WG16 antibody). The ratio of the normalized S2P signal between T0 on T1 (1 hr of induction) is shown for the ste11, inv1, and fbp1 loci. Therefore, a value of 1 indicates that there is no increase in the level of S2P after 1 hr of induction.
Figure 8 with 1 supplement
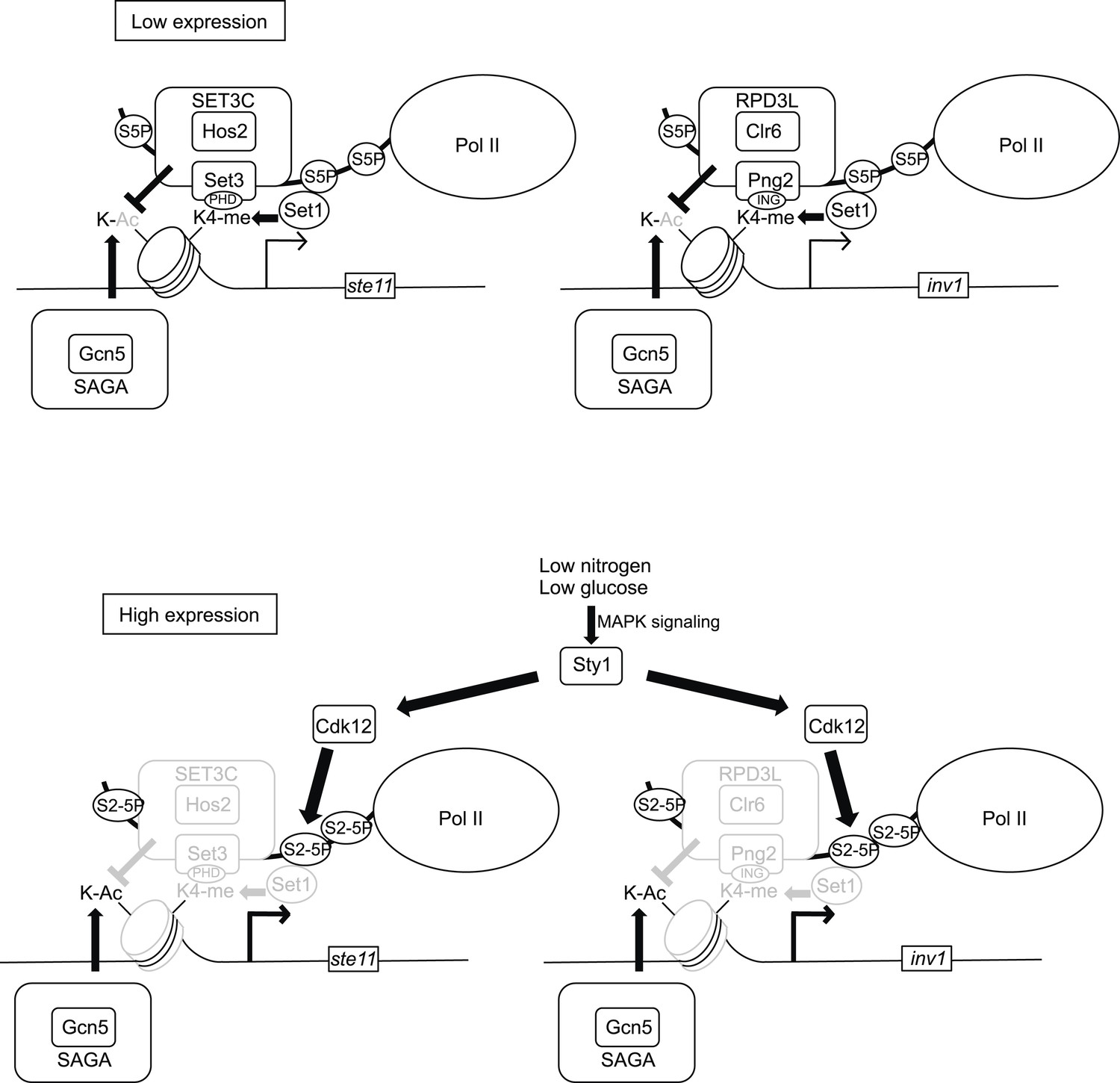
Model of the control of promoter nucleosome dynamics by CTD S2 phosphorylation.
When gene expression is low in non-induced conditions, the phosphorylation of the CTD on S5 recruits the Set1 methylase and the SET3C or the RPD3L HDAC complexes. The Set3 and Png2 subunits of these two complexes mediate interactions with the methylated (di or tri-) H3K4. Upon induction of gene expression, the Sty1 MAP kinase is activated and phosphorylates the CTD S2 kinase Lsk1, which is required for an increase in S2P nearby the promoter. We propose that the S2P-S5P CTD displaces the HDACs complexes, which leads to a local increase in nucleosome acetylation and dynamics. In the absence of CTD S2P, the nucleosomes occupancy over the NDR is high, which impedes the RNA pol II access and efficient transcription.
Figure 8—figure supplement 1
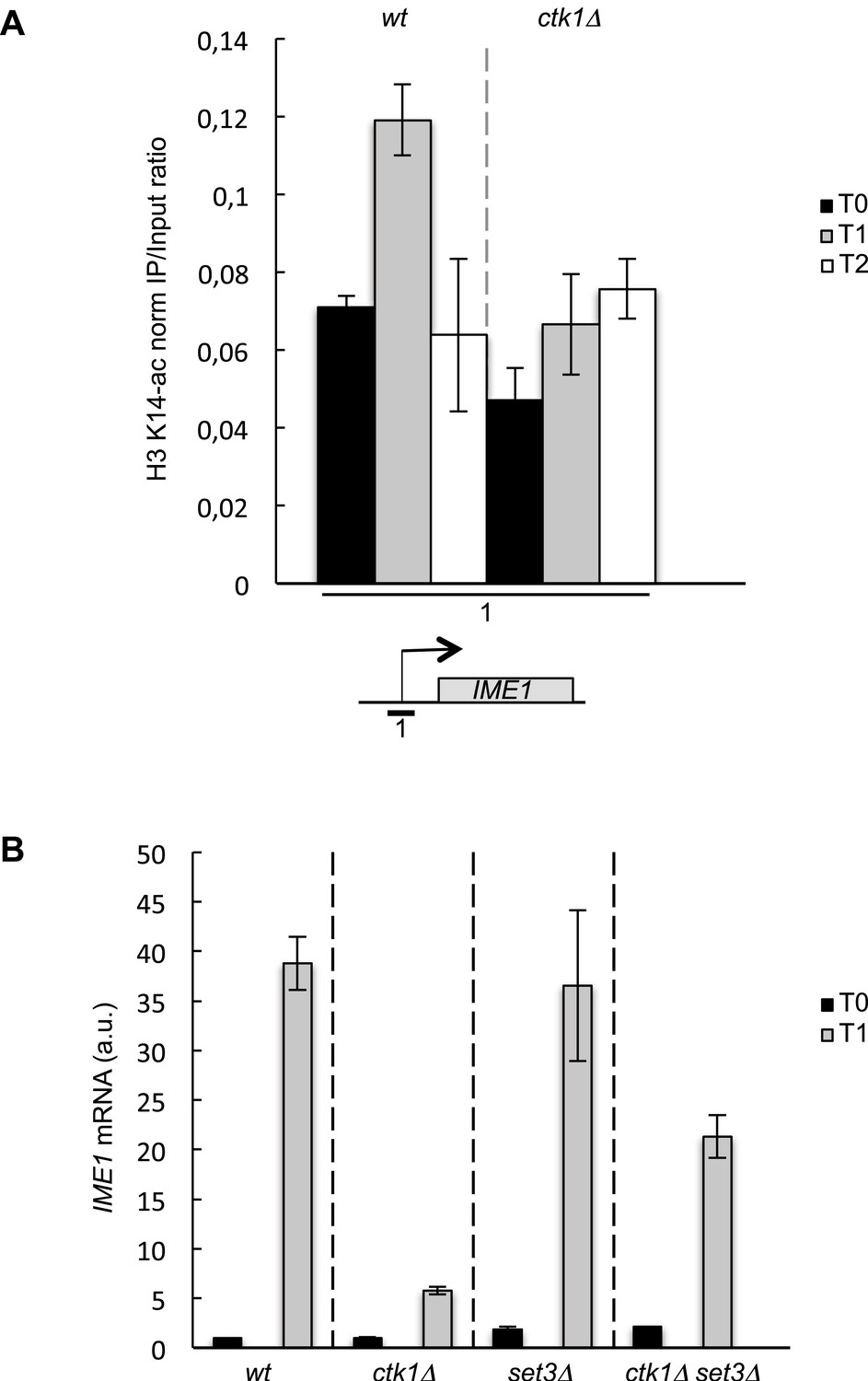
The role of CTD S2P in promoter histone acetylation is conserved in budding yeast.
(A) The wt and ctk1Δ strains were starved for nitrogen at the indicated time points (hours). The occupancy of acetylated H3K14 at the IME1 TSS was determined by ChIP using anti-H3-K14-ac normalized against unmodified H3. (B) RQ of the IME mRNA determined by Q-RT-PCR using the ΔΔct method in the indicated strains during vegetative growth (T0) and nitrogen starvation at the indicated time points (hour). a.u.: arbitrary units.
Additional files
-
Supplementary file 1
Oligonucleotides used in this study.
- https://doi.org/10.7554/eLife.09008.020
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Promoter nucleosome dynamics regulated by signalling through the CTD code
eLife 4:e09008.
https://doi.org/10.7554/eLife.09008




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}