Forward genetics in C. elegans reveals genetic adaptations to polyunsaturated fatty acid deficiency
- Department of Chemistry and Molecular Biology, University of Gothenburg, Sweden
- Department of Molecular and Clinical Medicine/Wallenberg Laboratory, Institute of Medicine, University of Gothenburg, Sweden
eLife Assessment
This fundamental study investigates the role of polyunsaturated fatty acids (PUFAs) in physiology and membrane biology, using a unique model to perform a thorough genetic screen that demonstrates that PUFA synthesis defects cannot be compensated for by mutations in other pathways. These findings are supported by compelling evidence from a high quality genetic screen, functional validation of their hits, and lipid analyses. This study will appeal to researchers in membrane biology, lipid metabolism, and C. elegans genetics.
https://doi.org/10.7554/eLife.104181.4.sa0Significance of the findings:
Fundamental: Findings that substantially advance our understanding of major research questions
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Compelling: Evidence that features methods, data and analyses more rigorous than the current state-of-the-art
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
Polyunsaturated fatty acids (PUFAs) are essential for mammalian health and function as membrane fluidizers and precursors for signaling lipids, though the primary essential function of PUFAs within organisms has not been established. Unlike mammals who cannot endogenously synthesize PUFAs, C. elegans can de novo synthesize PUFAs starting with the Δ12 desaturase FAT-2, which introduces a second double bond to monounsaturated fatty acids to generate the PUFA linoleic acid. FAT-2 desaturation is essential for C. elegans survival since fat-2 null mutants are non-viable; the near-null fat-2(wa17) allele synthesizes only small amounts of PUFAs and produces extremely sick worms. Using fluorescence recovery after photobleaching (FRAP), we found that the fat-2(wa17) mutant has rigid membranes and can be efficiently rescued by dietarily providing various PUFAs, but not by fluidizing treatments or mutations. With the aim of identifying mechanisms that compensate for PUFA-deficiency, we performed a forward genetics screen to isolate novel fat-2(wa17) suppressors and identified four internal mutations within fat-2 and six mutations within the HIF-1 pathway. The suppressors increase PUFA levels in fat-2(wa17) mutant worms and additionally suppress the activation of the daf-16, UPRer and UPRmt stress response pathways that are active in fat-2(wa17) worms. We hypothesize that the six HIF-1 pathway mutations, found in egl-9, ftn-2, and hif-1, all converge on raising Fe2+ levels and in this way boost desaturase activity, including that of the fat-2(wa17) allele. We conclude that PUFAs cannot be genetically replaced and that the only genetic mechanism that can alleviate PUFA-deficiency do so by increasing PUFA levels.
Introduction
The fluidity of cellular membranes is heavily influenced by the saturation level of the phospholipids composing the membrane. Phospholipids containing saturated fatty acid (SFA) tails are more tightly packed and, therefore, form more rigid membranes, while phospholipids with an abundance of unsaturated fatty acids (UFAs) are more loosely packed and result in fluid membranes (Barelli and Antonny, 2016; Antonny et al., 2015). PUFAs themselves can also affect many different cellular processes and are precursors to both anti- and pro-inflammatory PUFA-derived signaling molecules called eicosanoids (Bazinet and Layé, 2014; Simopoulos, 1999; James et al., 2000; Bell et al., 1986). Imbalances between SFAs and PUFAs are associated with chronic diseases including coronary heart disease, diabetes, hypertension, and renal disease (Simopoulos, 1999).
C. elegans PAQR-2, and its mammalian ortholog AdipoR2, promote the production and incorporation of PUFAs into phospholipids to restore membrane homeostasis (Ruiz et al., 2023). Whether this is the primary function of PUFAs in cells or in organismal physiology is still not resolved. In particular, no unbiased forward genetic screens for suppressors of PUFA deficiency have been reported. Mammals are not able to endogenously synthesize PUFAs and must obtain omega-3 and omega-6 PUFAs from the diet, a fact known since 1930; Murff and Edwards, 2014; Burr and Burr, 1930; linoleic acid (LA, 18:2n6) and alpha-linolenic acid (ALA, 18:3n3) must be dietarily supplied and can be further desaturated and elongated into ≥20-carbon PUFAs used structurally or as precursors of signaling molecules (Watts and Browse, 2002). An exception to this exists during severe essential fatty acid deficiency when mammals can synthesize mead acid (20:3n9), though this is not a common occurrence (Ichi et al., 2014). In contrast, C. elegans expresses many desaturases and elongases that can convert dietary or de novo synthesized SFAs into a wide range of PUFAs: Δ9 desaturases are responsible for converting SFAs into monounsaturated fatty acids (MUFAs) by adding a first double bond, a Δ12 desaturase adds an additional double bond to transform MUFAs into LA, a PUFA with two double bonds, and Δ5 and Δ6 desaturases introduce additional double bonds to produce PUFAs with three, four, or five double bonds (Nakamura and Nara, 2004; Guillou et al., 2010; Zhang et al., 2016). To better understand the essential roles of PUFAs in cells and whole organisms, we leveraged a mutant allele of the C. elegans Δ12 desaturase FAT-2, whose function is to convert oleic acid (OA, 18:1n9) into linoleic acid (LA, 18:2n6) (Peyou-Ndi et al., 2000; Figure 1A). Because this is a critical step for PUFA production, worms devoid of FAT-2 activity (i.e. fat-2(-) null mutants) are not able to synthesize any PUFAs and are not viable. In contrast, the fat-2(wa17) allele produces a partially functional protein bearing a S101F substitution (Figure 1B–C): mutants homozygous for this allele produce <10% of the normal levels of PUFAs and are extremely slow growing and sickly, but, crucially, are viable (Watts and Browse, 2002).
Figure 1
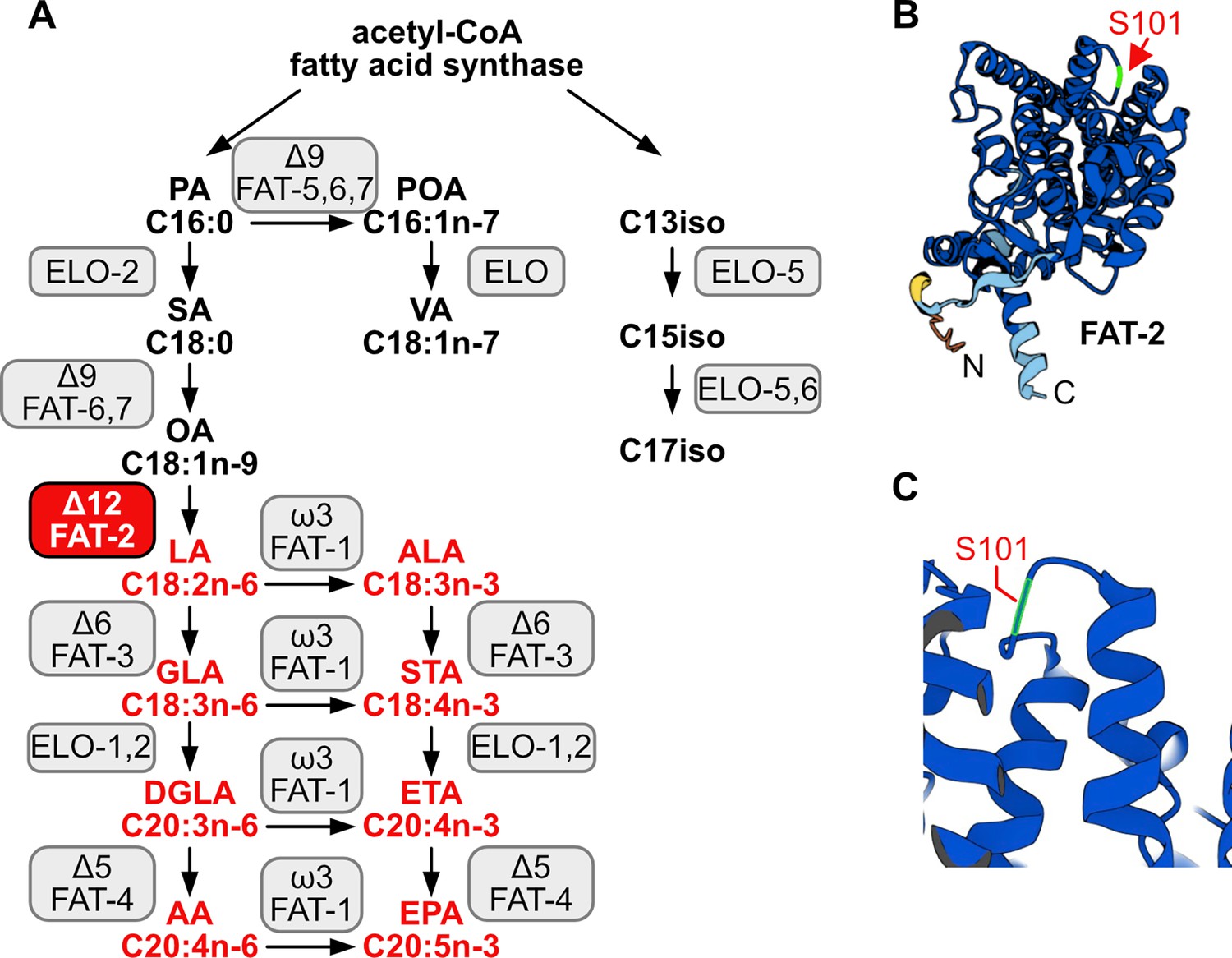
C.elegans fatty acid synthesis pathway and FAT-2 desaturase.
(A) Simplified pathway of fatty acid synthesis and desaturation in C. elegans. Boxes indicate the name of the enzymes, with the FAT-2 desaturase being indicated in a red box. Fatty acids whose synthesis is dependent on FAT-2 are indicated in red. Fatty acid abbreviations are as follow: palmitic acid (PA), palmitoleic acid (POA), vaccenic acid (VA), stearic acid (SA), oleic acid (OA), linoleic acid (LA), alpha-linolenic acid (ALA), gamma-linolenic acid (GLA), stearidonic acid (STA), dihomo-gamma-linolenic acid (DGLA), eicosatetraenoic acid (ETA), arachidonic acid (AA), and eicosapentaenoic acid (EPA). (B) AlphaFold2 predicted the FAT-2 structure with the serine at position 101 indicated with a red arrow. (C) Same structure as in B, zoomed in and angled to show that the S101 position that is mutated to phenylalanine in the fat-2(wa17) allele lies in a loop connecting two alpha helices.
Although there is no mammalian homolog of FAT-2 (Zhou et al., 2011), the fat-2(wa17) mutant can still serve as a useful genetic model to reveal evolutionarily conserved roles of PUFAs in organisms, and to help identify mechanisms that can compensate for PUFA deficiency. Here, we began by characterizing the fat-2(wa17) mutant in terms of its ability to be rescued with dietary PUFAs or with mutations previously identified as suppressors of paqr-2(tm3410) mutant phenotypes that are attributed to membrane rigidity. We then performed an exhaustive forward genetic screen for fat-2(wa17) suppressors in the hope that the critical functions of PUFAs could be discovered and to identify mechanisms that allow cells/organisms to cope with PUFA deficiencies. This screen yielded ten fat-2(wa17) suppressor alleles that fell into two groups: mutations within fat-2 itself, and mutations in the hif-1 pathway that converge on inhibition of ftn-2 expression.
Results
Characterization of the fat-2(wa17) mutant
The severe growth defect of fat-2(wa17) mutants can be suppressed by the wild-type fat-2(+) allele carried on an extrachromosomal array, confirming that this growth defect is due to reduced fat-2 activity (Figure 2A). As expected, given their low amounts of PUFAs, fluorescence recovery after photobleaching (FRAP) shows that the membranes of intestinal cells in the fat-2(wa17) mutant are excessively rigid and indeed appeared as rigid as those of the paqr-2(tm3410) mutant characterized by an excess of SFAs in its phospholipids (Svensk et al., 2013; Svensk et al., 2016; Figure 2B–C).
Figure 2
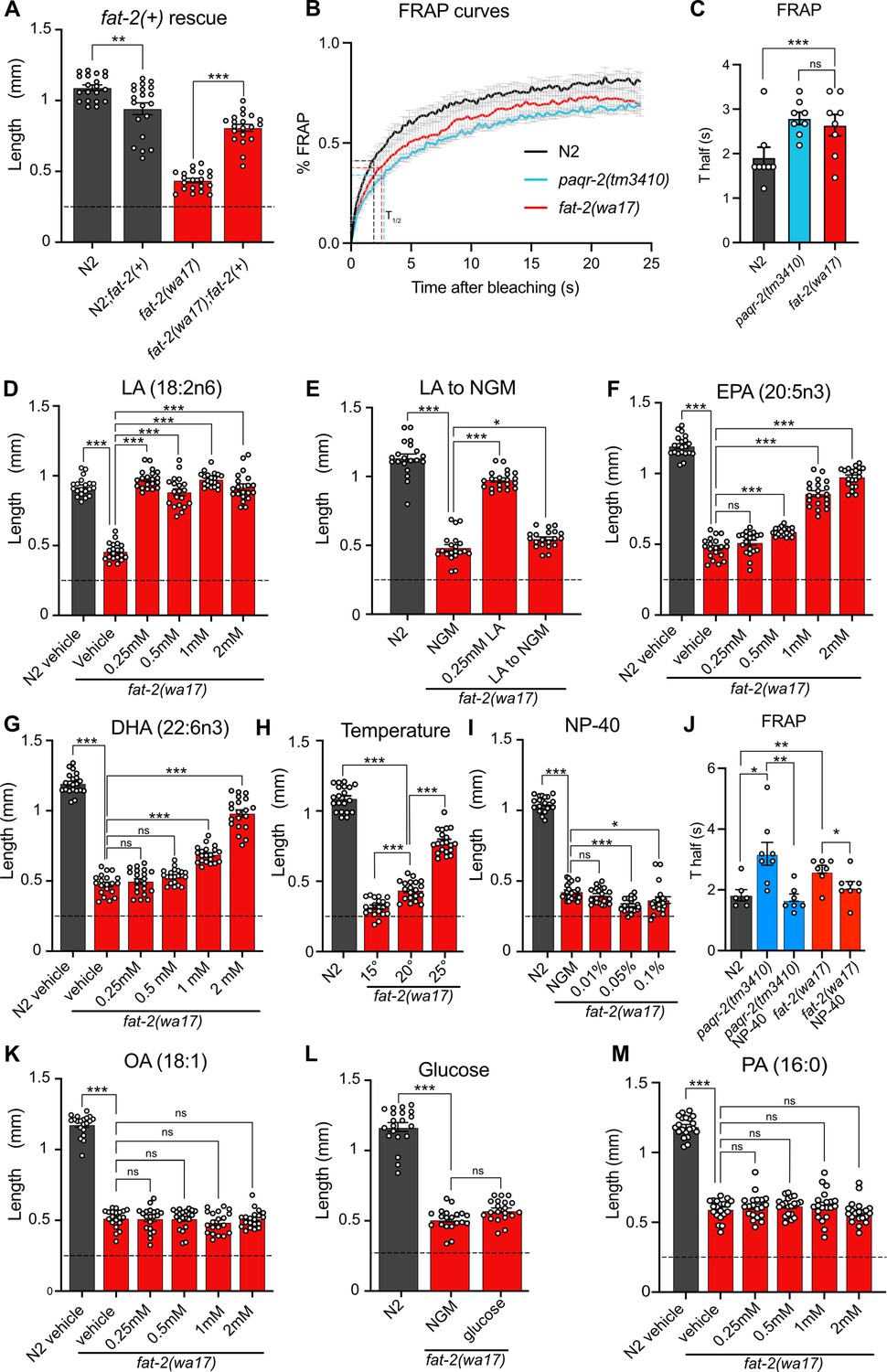
Characterization and rescue of fat-2(wa17).
(A) Introduction of the wild-type fat-2(+) allele on an extrachromosomal array rescues the fat-2(wa17) growth defect. n=20 for each genotype. (B–C) Fluorescence recovery after photobleaching (FRAP) curve and Thalf value show that fat-2(wa17) has rigid membranes similar to paqr-2(tm3410) control. n=8 for each genotype. (D–I, K–M) The lengths of fat-2(wa17) worms grown from L1 stage for 72 hr in the indicated conditions; horizontal dashed lines indicate the approximate lengths of the synchronized L1s at the start of the experiments. n=20 for each genotype/condition. (J) FRAP Thalf values show that NP-40 rescues fat-2(wa17) rigid membranes similarly to paqr-2(tm3410). From left to right, n=6, 8, 7, 7, 8. Error bars show the standard error of the mean. *p<0.05, **p<0.01, ***p<0.001 indicate significant differences compared to the fat-2(wa17) control (ordinary one-way ANOVA with Tukey multiple comparisons test).
Also, as expected, low doses of dietary linoleic acid (LA, 18:2n6) fully rescued the fat-2(wa17) growth defect (Figure 2D). This rescue by LA is transient and does not last when the following generation is transferred back to NGM plates (Figure 2E), which is consistent with the rapid turnover of fatty acids in C. elegans (Dancy et al., 2015). Eicosapentaenoic acid (EPA, 20:5n3) is the longest and most unsaturated PUFA produced in C. elegans (Figure 1A) and is also able to rescue the fat-2(wa17) mutant, albeit requiring higher concentrations than LA (Figure 2F). Surprisingly, docosahexaenoic acid (DHA, 22:6n3), which is not produced by C. elegans, is also able to rescue the fat-2(wa17) mutant (Figure 2G). Temperature has a direct effect on membrane fluidity: given a constant phospholipid composition, lower temperatures cause rigidification while higher temperatures promote fluidity (Tanaka et al., 1996). We found that the fat-2(wa17) mutant is growth-arrested when cultivated at membrane-rigidifying 15 °C, and conversely shows improved growth at 25 °C, which again is similar to earlier findings with the paqr-2(tm3410) mutant (Devkota et al., 2021; Figure 2H). However, cultivating the fat-2(wa17) mutant in the presence of the non-ionic detergent NP-40, which improves the growth of the paqr-2(tm3410) mutant (Svensk et al., 2013), did not suppress the poor growth phenotype of the fat-2(wa17) mutant even though it did improve membrane fluidity as measured using FRAP (Figure 2I–J). Similarly, supplementing the fat-2(wa17) mutant with the MUFA oleic acid (OA, 18:1), which also suppresses paqr-2(tm3410) phenotypes (Svensk et al., 2013), did not suppress the poor growth phenotype of the fat-2(wa17) mutant (Figure 2K). Conversely, membrane-rigidifying glucose (which causes a high SFA/UFA ratio in the dietary E. coli; Devkota et al., 2017) or the SFA palmitic acid (PA, 16:0) did not exacerbate the growth defect of fat-2(wa17) (Figure 2L–M). The observations that NP-40 and OA do not rescue fat-2(wa17), and that membrane rigidifying conditions (dietary glucose or PA) were not detrimental to fat-2(wa17), suggest that membrane rigidification may not be the main cause of the fat-2(wa17) growth defects.
Lipidomic analysis of phosphatidylcholines (PCs) and phosphatidylethanolamines (PEs) in the fat-2(wa17) mutant confirmed its reduced levels of PUFAs relative to wild-type worms (Figure 3A; Figure 3—figure supplement 1A), with the largest loss observed in longer PUFAs, namely dihomo-γ-linolenic acid (DGLA; C20:3), arachidonic acid or eicosatetraenoic acid (AA or ETA, C20:4) and, most strikingly, EPA (C20:5) (Figure 3B–C; Figure 3—figure supplement 1B–C). Consistent with previous publications (Watts and Browse, 2002), the levels of 18:1 fatty acids were greatly increased in the fat-2(wa17) mutant. Even though the lipid analysis methods used here are not able to distinguish between different 18:1 species, a previous study showed that the majority of the 18:1 fatty acids in the fat-2(wa17) mutant is actually 18:1n9 (OA) (Watts and Browse, 2002) and not 18:1n7 (vaccenic acid) as in most other strains (Watts and Browse, 2002; Hutzell and Krusberg, 1982); this is because OA is the substrate of FAT-2 and thus accumulates in the mutant. As expected, exogenous addition of LA to these worms resulted in an increase of PUFAs, and in particular, resulted in elevating EPA levels to more than 12% compared to less than 2% of total fatty acids in the PCs of fat-2(wa17). Transferring fat-2(wa17) from LA to NGM 6 hr prior to harvesting did not lessen this increase, suggesting minimal LA depletion during this time (Figure 3A–C; Figure 3—figure supplement 1A–C). Cultivation at 25°C did not result in any significant changes in the fat-2(wa17) mutant, indicating that the growth rescue seen at 25°C is not due to increased PUFA levels in the lipidome (Figure 3A–C; Figure 3—figure supplement 1A–C).
Figure 3 with 1 supplement see all
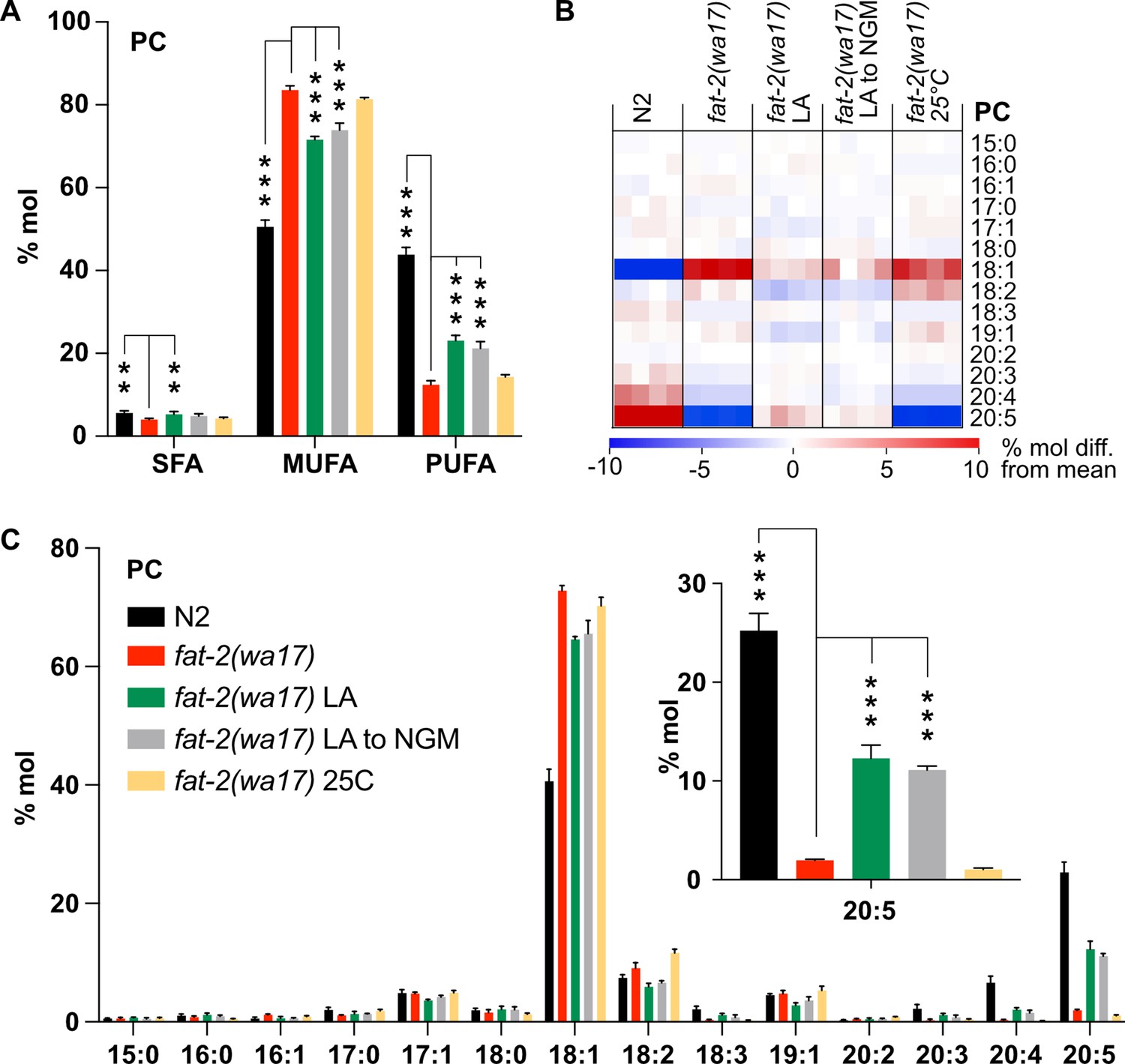
Lipidomic analysis of fat-2(wa17) mutant.
(A) Saturated fatty acid (SFA), monounsaturated fatty acid (MUFA), and polyunsaturated fatty acid (PUFA) levels in phosphatidylcholines (PCs) of fat-2(wa17) grown in various conditions. Note that cultivation on 2 mM LA boosts PUFA levels. Linoleic acid (LA) to NGM worms were grown on 2 mM LA before being transferred to NGM 6 hr prior to harvesting. (B) Heatmap of phosphatidylcholine (PC) species in fat-2(wa17) in all conditions. (C) Levels of individual FA species in PCs for all conditions. The inset shows that levels of 20:5 FA are increased by providing fat-2(wa17) with linoleic acid. n=4 populations for each genotype/condition. For A and C (inset), *p<0.05, **p<0.01, ***p<0.001 indicate significant differences compared to the fat-2(wa17) control and using one-way ANOVA followed by a Dunnett’s multiple comparison test.
-
Figure 3—source data 1
Data from the targeted lipidomics analysis, related to Figures 3 and 7, Figure 3—figure supplement 1, Figure 7—figure supplement 1.
- https://cdn.elifesciences.org/articles/104181/elife-104181-fig3-data1-v1.xlsx
Testing the effect of paqr-2(tm3410) suppressors
The previously characterized paqr-2(tm3410) null mutant has excess SFAs within phospholipids and many of the resulting defects, including membrane rigidity, can be suppressed by mutations that activate fatty acid desaturases or promote the incorporation of UFAs into phospholipids (Svensk et al., 2013; Ruiz et al., 2018; Ruiz et al., 2019; Busayavalasa et al., 2020). Given the phenotypic similarities between paqr-2(tm3410) and fat-2(wa17), such as cold intolerance and rigid membranes, we hypothesized that previously characterized paqr-2(tm3410) suppressors may be able to suppress fat-2(wa17) as well. However, the paqr-2(tm3410) suppressors tested (mdt-15(et14), nhr-49(et8), fld-1(et46), paqr-1(et52), acs-13(et54); Svensk et al., 2013; Ruiz et al., 2018; Ruiz et al., 2019; Busayavalasa et al., 2020) resulted in either no or only slight rescue of fat-2(wa17) growth (Figure 4A–D). Additionally, Oil Red O staining of fat-2(wa17) mutants suggests that they have an excessive lipid content, and this too was not normalized by the tested paqr-2(tm3410) suppressors (Figure 4E–G). These results suggest that membrane rigidity is at most only a minor cause of the fat-2(wa17) defects since fluidizing treatments (NP-40 or OA) or mutations (the tested paqr-2 suppressors) provide only minimal or no suppression.
Figure 4
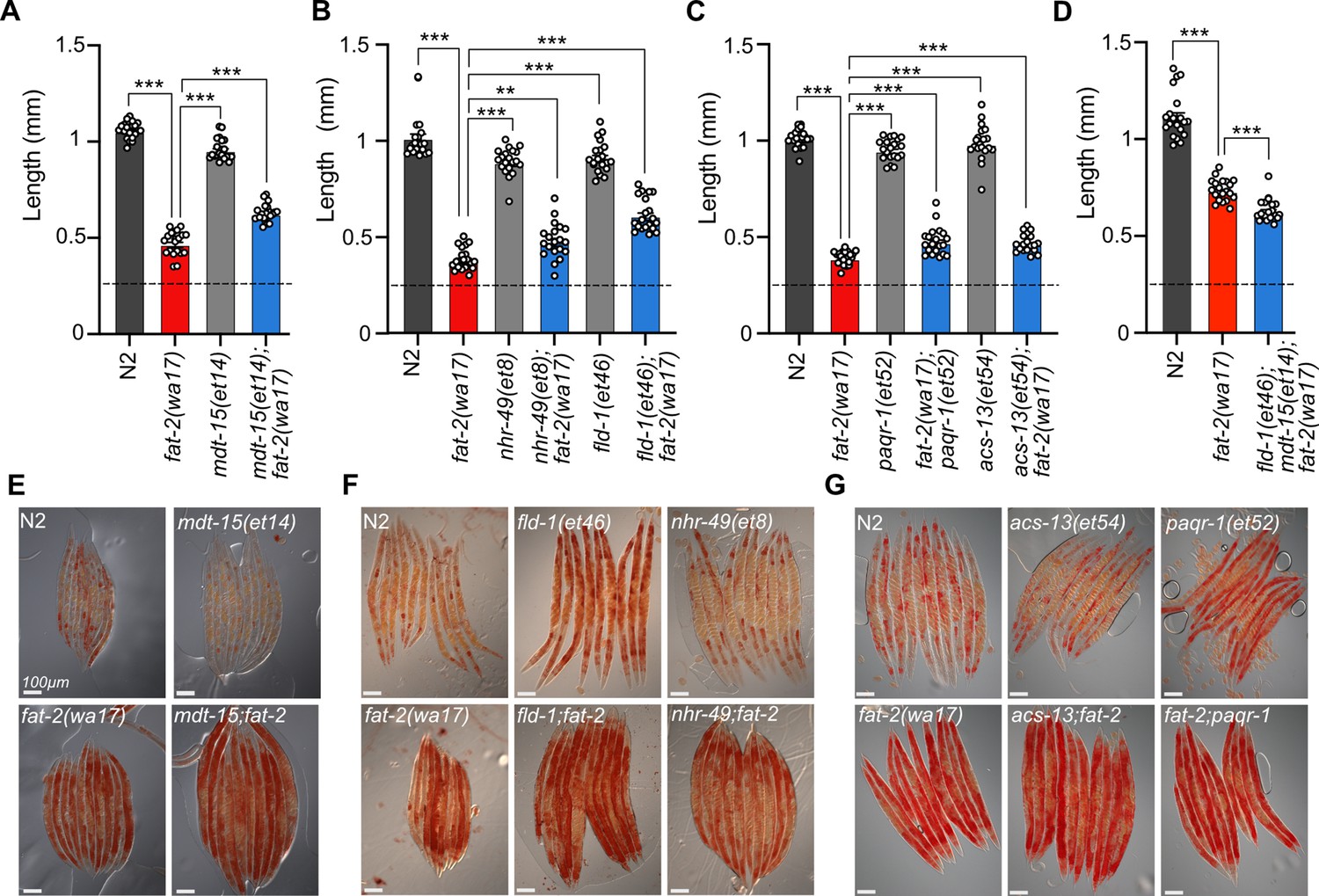
Membrane fluidizing mutations partially rescue fat-2(wa17).
(A–D) Fluidizing paqr-2(tm3410) suppressor mutations only slightly rescue fat-2(wa17) growth. Dashed horizontal lines indicate the approximate length of L1s at the start of the experiments; length was measured 72 hr post-synchronization. n=20 for each genotype. Error bars show the standard error of the mean. *p<0.05, **p<0.01, ***p<0.001 indicate significant differences compared to the fat-2(wa17) control (ordinary one-way ANOVA with Tukey multiple comparisons test). (E–G) Oil Red O staining of day 1 adults shows that the high lipid abundance in fat-2(wa17) is not suppressed by paqr-2(tm3410) fluidizing mutations.
A forward genetics screen for fat-2(wa17) suppressors
With the aim of identifying essential roles of PUFAs and molecular mechanisms that can compensate for PUFA deficiency, we performed a forward genetic screen for fat-2(wa17) suppressors that allow growth to adulthood within 72 hr, as opposed to the ~120 hr needed for the parental strain (Figure 5A). Approximately 40,000 EMS-mutagenized haploid genomes were screened, leading to the isolation of ten fat-2(wa17) suppressors, which fell into two groups: mutations within the fat-2 locus itself and mutations within genes of the HIF-1 pathway (Figure 5B–C). The fat-2(wa17) suppressors all reached adulthood within 72 hr (criteria for the screen) and improved the growth of the mutant when assessed by measuring worm length at 72 hr (Figure 5D–E). The hif-1(et69) mutation was recreated by CRISPR-Cas9 within the fat-2(wa17) background to confirm its fat-2(wa17) suppressor activity (Figure 5—figure supplement 1A); multiple independent isolations of essentially the same alleles within egl-9 (alelles et60-et62), fat-2 (alleles et63-et66), and ftn-2(et67-68) also serve as confirmation for these loci.
Figure 5 with 1 supplement see all
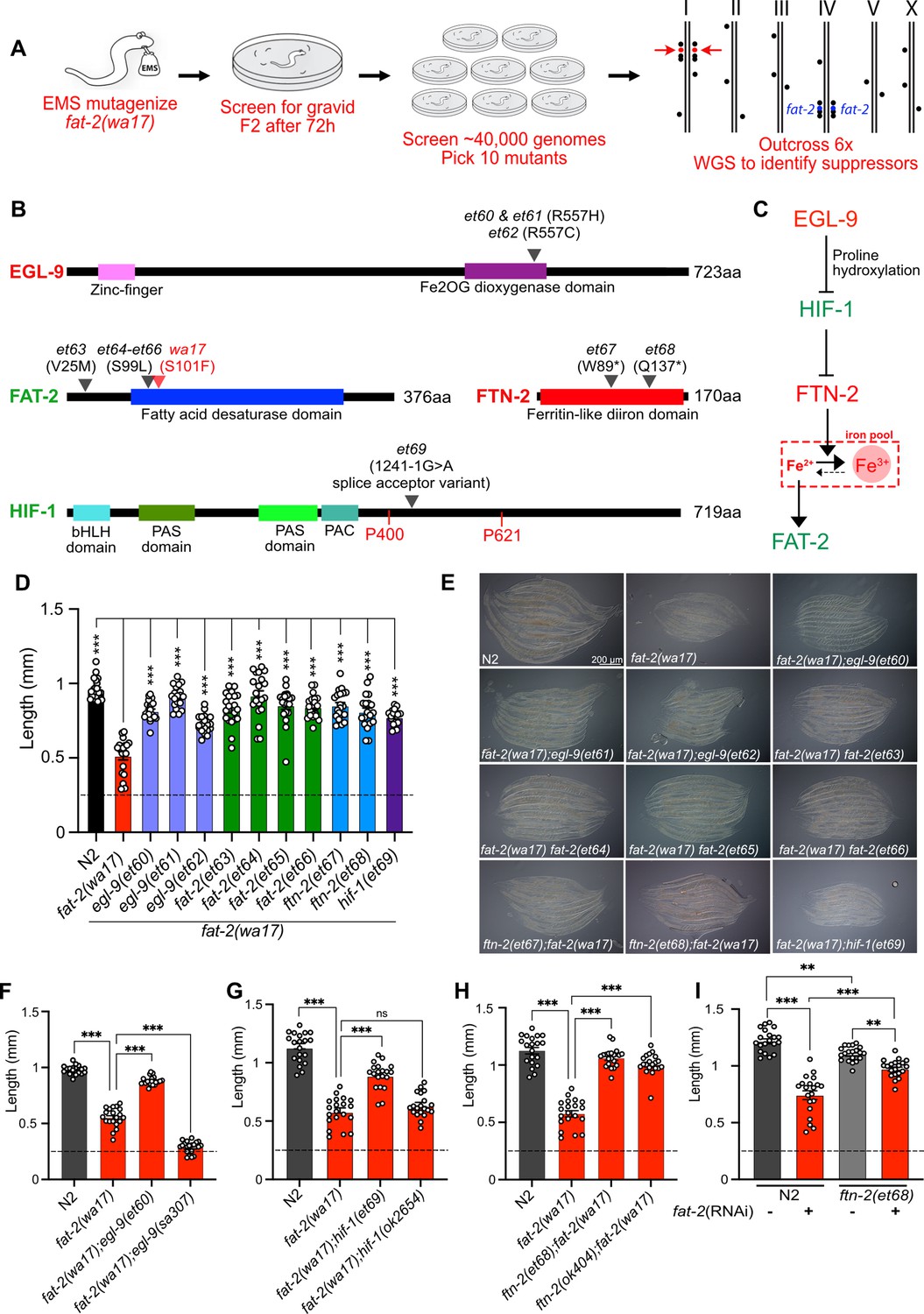
A forward genetic screen reveals that fat-2(wa17) is suppressed by mutations in the HIF-1 pathway.
(A) Overview of the forward genetics screen strategy to isolate fat-2(wa17) suppressors. (B) Identity and position of the fat-2(wa17) suppressors as well as the positions of functional domains. Novel mutations are marked by a black triangle with the corresponding allele name and mutation effect; the red triangle in FAT-2 indicates the original wa17 allele. Gene names in red represent loss- or reduction-of-function mutations; gene names in green represent gain-of-function mutations. (C) Proposed pathway of fat-2(wa17) suppression by mutations in the HIF-1 pathway. Reduction of EGL-9 constitutively activates HIF-1, and HIF-1 activation inhibits FTN-2. The loss of FTN-2 increases the levels of Fe2+, thus boosting FAT-2 desaturase activity. Gain-of-function mutations are labeled in green, loss- or reduction-of-function mutations are labeled in red. (D) Length of all fat-2(wa17) suppressors measured 72 hr after the L1 stage. (E) Representative images of fat-2(wa17) suppressors after 72 h of growth. (F–H) Null alleles of egl-9 and hif-1 do not rescue fat-2(wa17), but the null allele of ftn-2 does, confirming that ftn-2(et67) and ftn-2(et68) are loss-of-function alleles. Lengths were measured 72 hr after L1 synchronization. (I) ftn-2(et68) rescue of fat-2(RNAi) worms, confirming that the suppressors are not wa17 specific. The horizontal dashed line indicates the approximate length of L1s at the start of each experiment. n=20 for each genotype/condition. Error bars show the standard error of the mean. *p<0.05, **p<0.01, ***p<0.001 indicate significant differences compared to the fat-2(wa17) control (ordinary one-way ANOVA with Tukey multiple comparisons test).
Three of the four fat-2 intragenic alleles (et64-et66) carried a substitution of serine to leucine at position 99 (S99L), only two amino acids away from the S101F mutation in fat-2(wa17); the fourth, et63, is a missense mutation substituting valine with methionine at position 25 (V25M; Figure 5B). These four internal fat-2 alleles likely compensate structurally for the S101F mutation in fat-2(wa17) and thus improve its activity.
Three independent fat-2(wa17) suppressor mutations were found to affect the same arginine at position 557 of EGL-9 (et60 and et61 resulted in a R557H missense while et62 caused a R557C missense; Figure 5B). EGL-9 is a proline hydroxylase (PHD) that interacts via its R557 with Fe2+/2-oxoglutarate, a required cofactor for its oxygenase activity (Epstein et al., 2001). EGL-9 regulates the response to iron depletion and hypoxia: in the presence of sufficient Fe2+ and oxygen, EGL-9 can hydroxylate HIF-1 (hypoxia-inducible factor 1), leading to its ubiquitination and degradation. When either Fe2+ or oxygen are unavailable, HIF-1 is stable and can bind DNA to regulate adaptive transcriptional responses (Epstein et al., 2001; Bracken et al., 2006; Huang et al., 1996). It is surprising that all three fat-2(wa17) suppressor alleles affected precisely the same amino acid within EGL-9, and we surmise that a special property is conferred to EGL-9 by this specific mutation. For example, this mutant version of EGL-9 may be unable to inactivate HIF-1 by hydroxylation but still retain other important functions. In agreement with this interpretation, we found that the egl-9(sa307) null mutant cannot act as a fat-2(wa17) suppressor (Figure 5F).
One of the fat-2(wa17) suppressors corresponds to a splice acceptor mutation in the fifth intron of HIF-1, which would result in a frameshift after the first 413 amino acids if splicing instead occurs with the following sixth intron splice acceptor site (Figure 5B). This hif-1(et69) allele is dominant: heterozygosity for hif-1(et69)/+ provides better fat-2(wa17) suppression than hif-1(et69)/hif-1(et69) homozygosity (Figure 5—figure supplement 1B). This suggests that the hif-1(et69) allele is a gain-of-function allele, which may be because the frameshift occurs just after the first of potentially two prolines that are hydroxylated by EGL-9 when oxygen and Fe2+ levels are sufficient (Epstein et al., 2001). This is also consistent with the observation that the hif-1(ok2654) null allele is not a fat-2(wa17) suppressor (Figure 5G). Usually, hydroxylation of the prolines P400 and P621 causes recruitment of a ubiquitin ligase, leading to HIF-1 degradation (Epstein et al., 2001). In the case of the hif-1(et69) allele, such regulation is likely impossible, and a constitutive HIF-1 may act as a fat-2(wa17) suppressor in several ways: promote overexpression of lipid metabolism genes including fat-2 (Xie and Roy, 2012), inhibit fatty acid beta-oxidation (Huang et al., 2014; Papandreou et al., 2006), which may help PUFAs to reach adequate levels even in the fat-2(wa17) mutant, or suppress the expression of the ferritin-encoding ftn-2, thus increasing the levels of ferrous ions required for desaturase activity (Romney et al., 2011; Shen et al., 2023).
Most informatively, the last two fat-2(wa17) suppressor mutations introduced premature STOP codons within the ftn-2 gene (alleles et67 and et68; Figure 5B). Additionally, the ftn-2(ok404) null allele also acted as a potent fat-2(wa17) suppressor (Figure 5H), which is consistent with inhibition of ftn-2 being the key outcome from HIF-1 pathway activation. C. elegans ftn-2 encodes a ferritin that is expressed in the intestine, muscle and several neurons (Romney et al., 2008). The FTN-2 protein is constitutive and 10 X faster as a ferroxidase (oxidising the reactive ferrous Fe2+ to the harmless ferric Fe3+) than FTN-1, which is an inducible intestine-specific ferritin in C. elegans (Romney et al., 2011; Kim et al., 2004; Romero et al., 2020; Mubarak et al., 2023; Cha’on et al., 2007). Additionally, FTN-2 is the major binder of iron in worms, and ftn-2 mutants, therefore, contain much less iron than wild-type worms, though the Fe2+/Fe3+ ratio is increased among the remaining iron (James et al., 2015). This is likely the mechanism by which the ftn-2(et67) and ftn-2(et68) alleles act as fat-2(wa17) suppressors: increasing the availability of ferrous ions is a potent way to activate desaturases (Shen et al., 2023) and thus likely increases the activity of the near null fat-2(wa17) allele leading to the production of more/sufficient PUFAs. Importantly, the ftn-2(et68) allele was also able to suppress the growth defect resulting from fat-2 knockdown (using RNAi; Figure 5I); this shows that ferritin mutations compensate for reduced fat-2 activity generally rather than suppressing specifically only the fat-2(wa17) allele. Additionally, the ftn-2(et68) allele was not able to rescue the fat-2(syb7458) null allele (Figure 5—figure supplement 1C), suggesting that some fat-2 activity must exist for ftn-2(et68) to act upon. Lastly, ftn-2(et68) is still a potent fat-2(wa17) suppressor when hif-1 is knocked out (Figure 5—figure supplement 1D), suggesting that no other HIF-1-dependent functions are required as long as ftn-2 is downregulated; this conclusion is supported by the observation that the potency of the ftn-2(ok404) null allele to act as a fat-2(wa17) suppressor is not increased by including the hif-1(et69) allele (compare Figure 5H and Figure 5—figure supplement 1E). Altogether, the genetic interaction studies suggest that the suppressor mutations in ftn-2 and hif-1 are acting via the same mechanism to rescue fat-2(wa17) and that ftn-2 is downstream of hif-1 in the fat-2 suppression pathway.
Effect of fat-2(wa17) suppressors on HIF-1 and PUFA levels
The results of the fat-2(wa17) suppressor screen support a model where the egl-9 R557 substitution alleles have an impaired ability to suppress HIF-1, while the gain-of-function hif-1(et69) allele constitutively suppresses ftn-2 expression and the ftn-2 null alleles are unable to sequester ferrous ions of which elevated levels increase fat-2 activity (Figure 5C). A 3xFLAG-tagged version of the endogenous HIF-1 allowed us to monitor HIF-1 levels in different conditions using Western blots (Figure 6A–B). As expected, hypoxia caused elevated levels of HIF-1 in wild-type worms. HIF-1 levels are abnormally low in fat-2(wa17) during normoxia but restored to normal levels by the internal fat-2(et65) mutation, suggesting that low PUFA levels cause HIF-1 downregulation. Nevertheless, HIF-1 levels are also increased by hypoxia in the fat-2(wa17) mutant, indicating that the hif-1 locus is still responsive to oxygen levels in the fat-2(wa17) mutant. As expected, egl-9(et60) drastically increases HIF-1 expression in fat-2(wa17), which is consistent with the R557 substitution impairing the ability of EGL-9 to inhibit HIF-1. Finally, the null ftn-2(et68) allele caused near-loss (a faint HIF-1 band is occasionally seen) of detectable HIF-1 in fat-2(wa17), suggesting feedback regulation between ftn-2 and hif-1 (Figure 6A–B, Figure 6—figure supplement 1A–B).
Figure 6 with 1 supplement see all
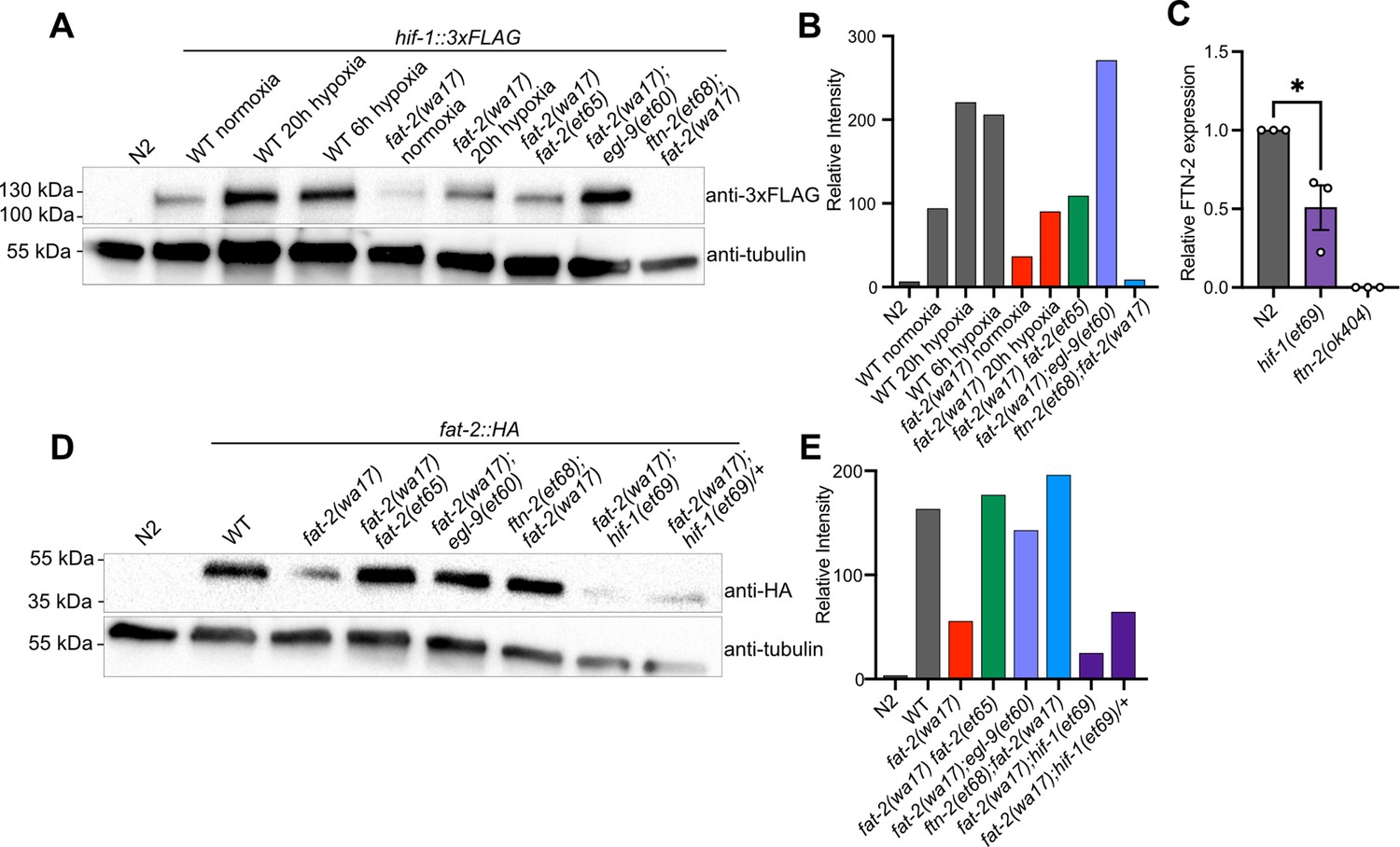
fat-2(wa17) suppressors belong in the HIF-1 pathway and influence HIF-1 levels.
(A) Western blot confirming that hif-1::3xFLAG levels in fat-2(wa17) are increased by egl-9(et60), but not by ftn-2(et68). Hypoxia treatment increases HIF-1 levels in wild-type (WT) and fat-2(wa17), confirming successful protein tagging. (B) Quantification of Western blot in A showing relative intensity of the HIF-1 signal normalized to that of tubulin. (C) mRNA expression of FTN-2, confirming that hif-1(et69) reduces FTN-2 levels. n = the mean of 3 independent normalized replicates for each genotype. *p<0.05 (unpaired t-test). (D) Western blot confirming that fat-2::HA levels in fat-2(wa17) are greatly reduced but increased in suppressor strains. (E) Quantification of Western blot in D showing relative intensity of the FAT-2 signal normalized to that of tubulin.
-
Figure 6—source data 1
PDF file containing original western blots for Figure 6, indicating the relevant bands and treatments.
- https://cdn.elifesciences.org/articles/104181/elife-104181-fig6-data1-v1.zip
-
Figure 6—source data 2
Original files for western blot analysis displayed in Figure 6.
- https://cdn.elifesciences.org/articles/104181/elife-104181-fig6-data2-v1.zip
Inhibition of egl-9 promotes HIF-1 activity (Shao et al., 2009), which we here verified for the egl-9(et60) allele using western blots (Figure 6A). Additionally, we found by qPCR that ftn-2 mRNA levels are as expected reduced by the proposed gain-of-function hif-1(et69) allele (Figure 6C). We conclude that the egl-9 and hif-1 suppressor mutations likely converge on inhibiting ftn-2 and thus act similarly to the ftn-2 loss-of-function alleles.
We also used Western blots to evaluate the abundance of the FAT-2 protein expressed from endogenous wild-type or mutant loci, but to which a HA tag was fused using CRISPR/Cas9. We found that the FAT-2::HA levels are severely reduced when the locus contains the S101F substitution present in the wa17 allele, but are restored close to wild-type levels by the fat-2(et65) suppressor mutation (Figure 6D–E, Figure 6—figure supplement 1C–D). The levels of FAT-2 in the HIF-1 pathway suppressors varied between experiments, with the suppressors sometimes restoring FAT-2 levels and sometimes not, even when the worms were growing well (Figure 6D–E, Figure 6—figure supplement 1C–D). The fat-2(wa17) suppressors, except for the intragenic fat-2 alleles, likely do not act by increasing FAT-2 protein levels.
As already mentioned, ferrous ions (Fe2+) are potent activators of desaturases (Shen et al., 2023). Given that each of the fat-2(wa17) suppressor mutants within the HIF-1 pathway are predicted to ultimately inhibit ftn-2, thus increasing the ferrous ion pool, we hypothesized that PUFA levels should be at least partially normalized in the fat-2(wa17) suppressors. This was confirmed by lipidomic analysis of phosphatidylcholines (Figure 7A) and phosphatidylethanolamines (Figure 7—figure supplement 1A). In particular, while levels of 18:2 (LA) were not significantly increased in the suppressor strains, the levels of 20:5 (EPA) were significantly increased by more than three folds and to levels near those obtained earlier by supplementing with LA (Figure 7B–C, Figure 7—figure supplement 1B–C), likely because the suppressor mutations allow fat-2(wa17) to produce more LA that is converted by other elongases and desaturases into EPA, the end product.
Figure 7 with 1 supplement see all
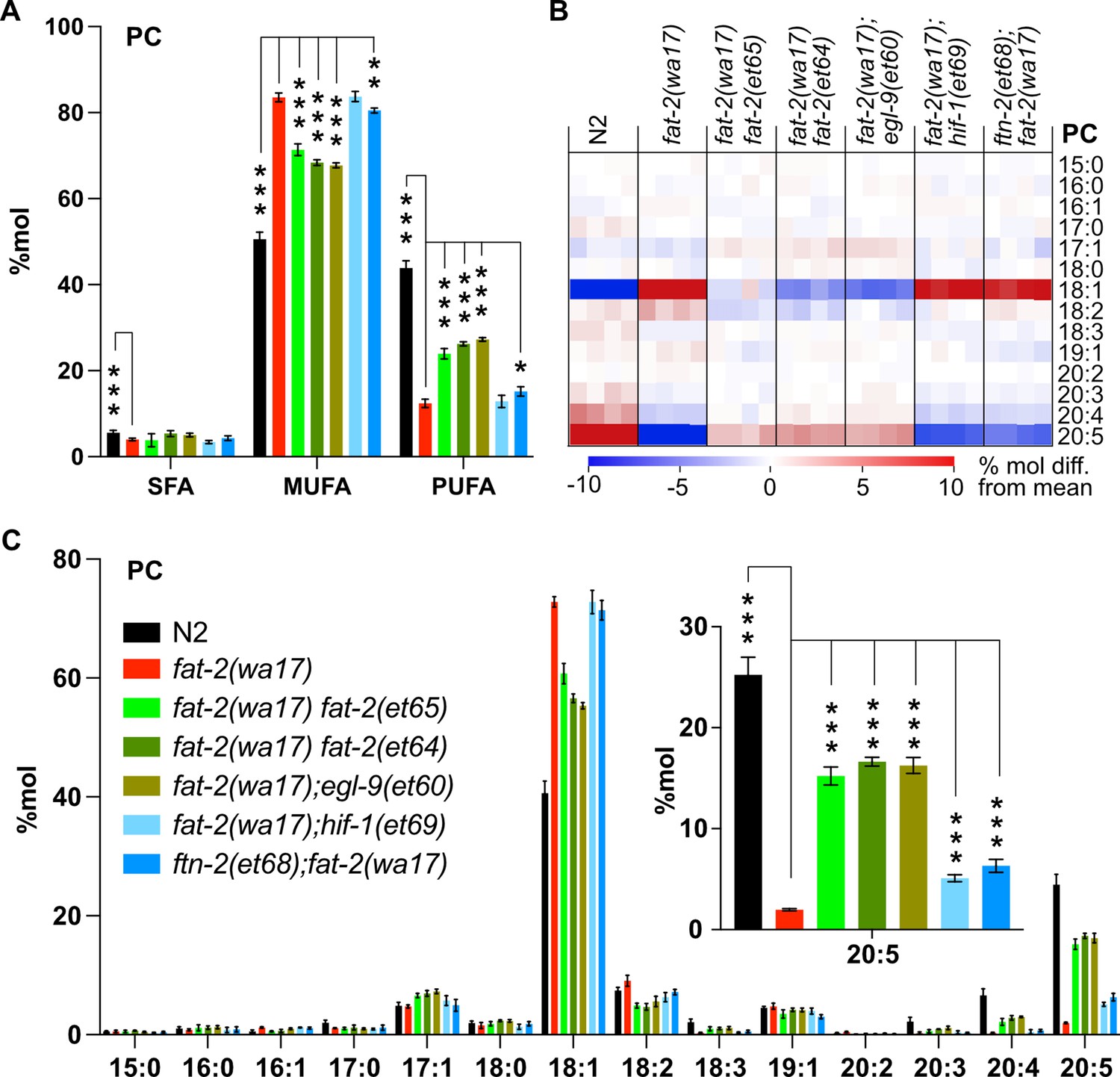
Lipidomic analysis of fat-2(wa17) suppressors reveals that polyunsaturated fatty acid (PUFA) levels are increased.
(A) Levels of saturated fatty acids (SFAs), monounsaturated fatty acids (MUFAs), and PUFAs in phosphatidylcholines (PCs) measured in fat-2(wa17) suppressors confirm that the suppressors increase PUFA levels in fat-2(wa17). Worms were homozygous for all indicated genotypes, but note that the hif-1(et69) allele suppresses fat-2(wa17) best in a heterozygous state. (B) Heat map analysis of PC species in suppressor mutants. (C) Levels of individual FA species in PCs in fat-2(wa17) suppressors, the insert shows that levels of C20:5 are significantly increased in all double mutant strains. n=4 populations for each genotype. For A and C (inset), *p<0.05, **p<0.01, ***p<0.001 indicate significant differences compared to the fat-2(wa17) control and using one-way ANOVA followed by a Dunnett’s multiple comparison test. Note that the N2 and fat-2(wa17) samples are the same as in Figure 3.
Multiple stress response pathways are active in fat-2(wa17) and suppressed by ftn-2(et68)
The increase in EPA, and PUFA levels in general, likely explains the improved development and growth of fat-2(wa17). We examined other traits that may be rescued by the fat-2 suppressors, using the ftn-2(et68) mutant as a representative because the egl-9 and hif-1 alleles converge on it. We found that the membrane fluidity defects in fat-2(wa17) were suppressed by ftn-2(et68) (Figure 8A–C). Additionally, several stress response pathways that are constitutively activated in the fat-2(wa17) mutant were also rescued by ftn-2(et68). The mitochondrial UPR visualized with a hsp-60::GFP reporter (Yoneda et al., 2004) is activated in fat-2(wa17) at a level similar to that in afts-1(et15), a known activator of mitochondrial stress (Rauthan et al., 2013), and this is suppressed by ftn-2(et68) (Figure 8D–E). Similarly, the metabolic stress reporter DAF-16::GFP (Libina et al., 2003) is constitutively nuclear-localized in fat-2(wa17), and this is also suppressed by ftn-2(et68) (Figure 8F–G). Using a hsp-4::GFP reporter (Calfon et al., 2002), we found that the ER UPR is only slightly activated in fat-2(wa17) relative to WT (especially in spermatheca), and that this stress response too is partially suppressed by ftn-2(et68) (Figure 8H–I). Altogether, these results show that the PUFA-deficient fat-2(wa17) mutant engages multiple stress response pathways and that these are abated by ftn-2(et68).
Figure 8
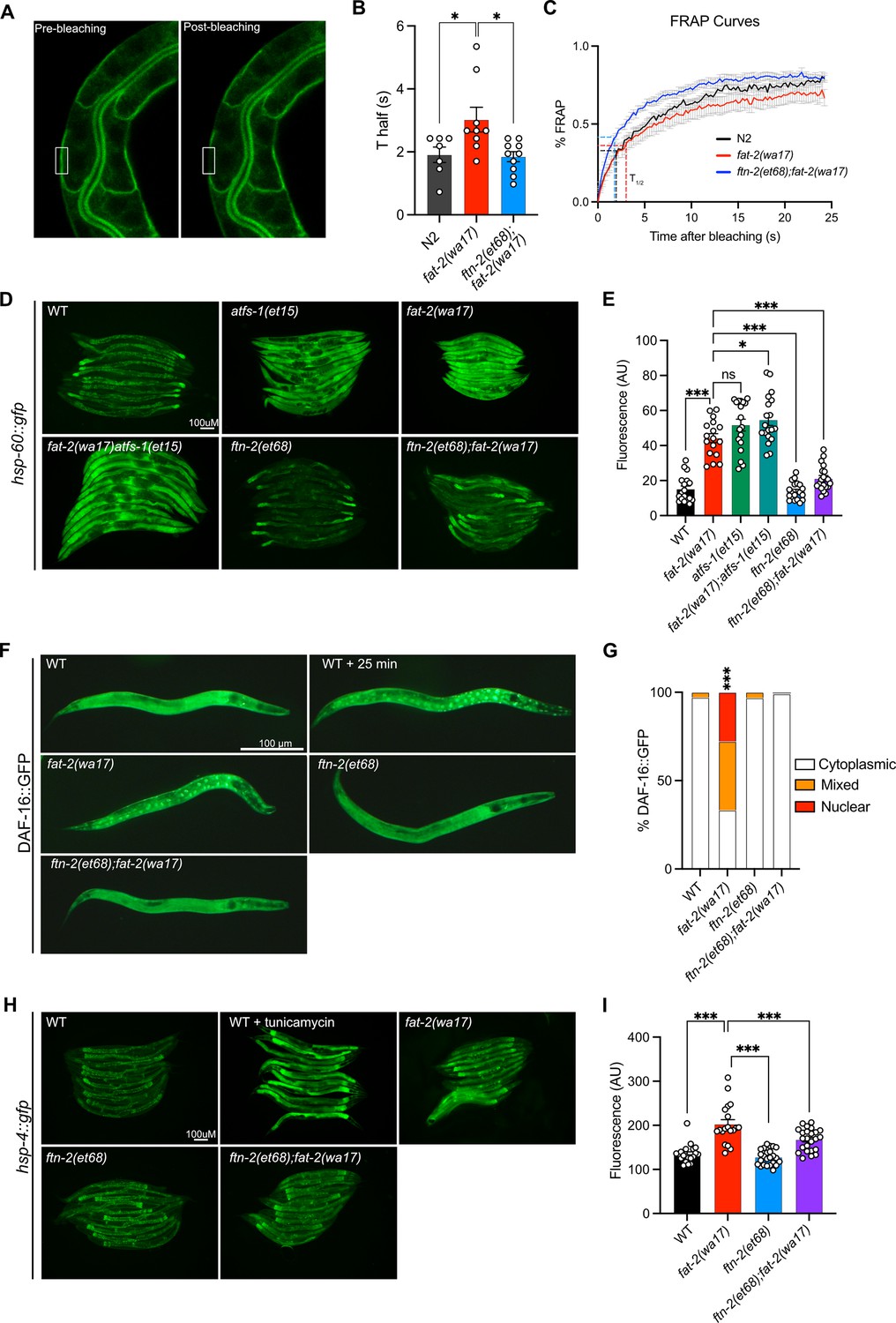
ftn-2(et68) rescues fat-2(wa17)’s stress responses.
(A) Representative image of a fluorescence recovery after photobleaching (FRAP) experiment, showing pGLO-1::GFP-CAAX-positive intestinal membranes. The rectangle indicates the bleached area. (B–C) Thalf values and FRAP curves show that ftn-2(et68);fat-2(wa17) has less rigid membranes than fat-2(wa17). From left to right, n=7, 9, 10. (D–E) Representative images and quantification of ftn-2(et68) rescue of fat-2(wa17) mitochondrial stress with a hsp-60::gfp reporter. atfs-1(et15) serves as a control for high mitochondrial UPR activation. n=20 for each genotype. (F–G) Representative images and quantification of DAF-16::GFP localization showing that the DAF-16 stress response is constitutively active in the fat-2(wa17) mutant but normalized by ftn-2(et68). Chi-squared test shows that fat-2(wa17) is significantly different from wild-type (WT). n=100 for each genotype. (H–I) Representative images and quantification of mild ER stress in fat-2(wa17) that is slightly rescued by ftn-2(et68) using a hsp-4::gfp reporter. n=20 for each genotype. *p<0.05, **p<0.01, ***p<0.001 indicate significant differences compared to the fat-2(wa17) control (ordinary one-way ANOVA with Tukey multiple comparisons test).
Mimicking fat-2(wa17) suppressors using hypoxia or iron supplements
Attempts to mimic the effects of the fat-2(wa17) suppressor mutations by hypoxia or supplement treatments were only partially successful. Providing fat-2(wa17) with ferric ammonium citrate (FAC), which increases the levels of ferric ions that can be converted into ferrous ions as well as overall iron levels in worms (Valentini et al., 2012), provided only a slight rescue of the fat-2(wa17) mutant (Figure 9A). Additionally, providing fat-2(wa17) with ferrous ions in the form of ferrous chloride did not provide any rescue (Figure 9—figure supplement 1A). Reducing the levels of ferrous ions with the iron chelator deferoxamine, which we hypothesized would further hinder fat-2(wa17) growth, had no effect (Figure 9—figure supplement 1B); however, given that the fat-2(syb7458) null mutant grows at the same rate as fat-2(wa17) in 72 hr (Figure 9—figure supplement 1C) but never develops into an adult, we theorize that 72 hr may be too short to see a negative effect from deferoxamine on fat-2(wa17). HIF-1-activating paraquat (PQ; Hwang et al., 2014) likewise conferred only a small rescue (Figure 9B), while the combination of FAC and PQ did not provide any additional growth rescue, both at 20°C and 25°C (Figure 9C; Figure 9—figure supplement 1D). The HIF-1 activator hydrogen peroxide (Xie and Roy, 2012) also only mildly rescued fat-2(wa17) (Figure 9D), while two separate hypoxia mimetics (cobalt chloride [Padmanabha et al., 2015] and sodium sulfite [Jiang et al., 2011]) did not suppress the poor growth of fat-2(wa17) (Figure 9—figure supplement 1E–F). Additionally, exposing fat-2(wa17) to multiple short hypoxia treatments slightly increased growth, but longer hypoxia treatments had no effect (Figure 9E). Finally, we also tested a cocktail of eicosanoids, which are derived from PUFAs such as EPA and could be limiting in the fat-2(wa17) mutant, but found that they had no rescuing effect when added as a supplement to the culture plates; their half-life and uptake by the worms are unknown (Figure 9—figure supplement 1G). Taken together, these results suggest that increasing iron and activating HIF-1 are beneficial to fat-2(wa17), but that achieving physiologically optimal dosing via experimental treatments is difficult.
Figure 9 with 1 supplement see all
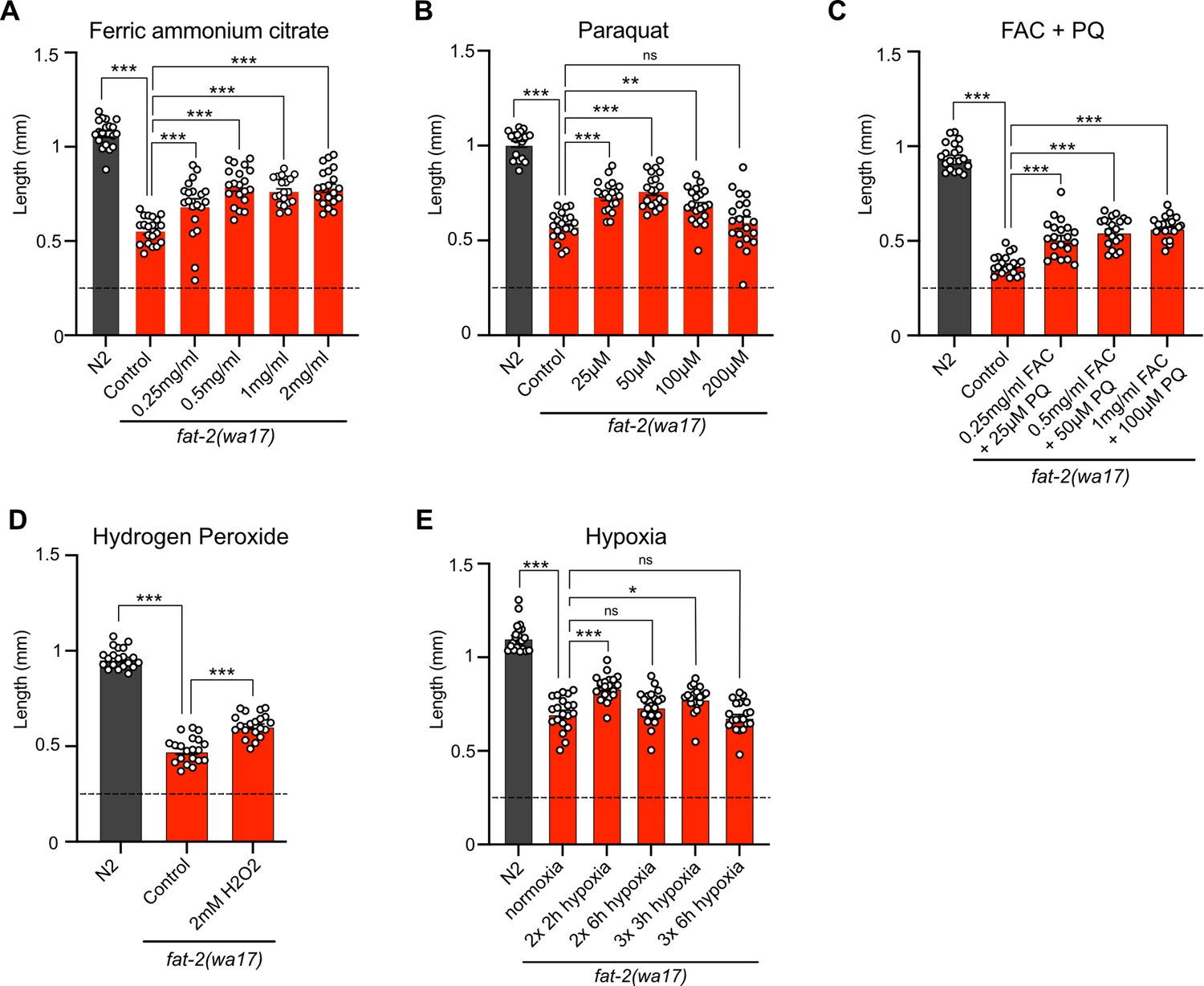
Exogenous treatments that mimic fat-2(wa17) suppressors partially rescue fat-2(wa17).
(A–E) Length assay of fat-2(wa17) cultivated with different treatments for 72 hr after L1 stage synchronization. The horizontal dashed line represents the approximate length of L1 worms at the start of each experiment. n=20 for each genotype/condition. *p<0.05, **p<0.01, ***p<0.001 indicate significant differences compared to the fat-2(wa17) control (ordinary one-way ANOVA with Tukey multiple comparisons test).
Discussion
That dietary PUFAs are essential for mammalian health, with LA and ALA acting as precursors for the synthesis of other PUFAs, is known since the 1930s (Burr and Burr, 1930). PUFAs have been linked to several important cellular and physiological processes (reviewed in Calder, 2012; Vrablik and Watts, 2013; Harayama and Shimizu, 2020), including cell membrane properties and organelle dynamics (Antonny et al., 2015), autophagy (O’Rourke et al., 2013), mitochondria function (Stanley et al., 2012), ferroptosis (Yang et al., 2016; Lee et al., 2020; Perez et al., 2020), regulation of the daf-2/insulin, mTOR and p38-MAPK pathways (Horikawa and Sakamoto, 2010; Chamoli et al., 2020; Liu et al., 2020), SREBP stability and signaling (Worgall et al., 1998; Yahagi et al., 1999), lipid droplet fusion (Wang et al., 2022), neuronal signaling and neurotransmission (Kahn-Kirby et al., 2004; Marza and Lesa, 2006; Vásquez et al., 2014), TRPV-dependent sensory signaling (Kahn-Kirby et al., 2004), oocyte development (Chen et al., 2016), and telomere length (Wu et al., 2024). Which of these, if any, is the specific essential role of PUFAs in animal physiology? And are there molecular mechanisms that can compensate for PUFA deficiency? In the present study, we approached these questions using forward genetics in C. elegans. While C. elegans can de novo synthesize PUFAs, mutations that impair the production of certain PUFAs can lead to developmental defects or lethality (Watts and Browse, 2002; Dancy et al., 2015; Perez and Van Gilst, 2008), offering opportunities for suppressor screens. Here, we showed that defects in the fat-2(wa17) mutant, which has limited Δ12 desaturase activity and only produces trace amounts of PUFAs, are suppressed by either compensatory intragenic mutations within fat-2 itself or by mutations within the HIF-1 pathway. The fact that screening approximately 40,000 haploid genomes for fat-2(wa17) suppressors and finding mutations only within fat-2 itself or within the HIF-1 pathway suggests that the screen has reached near-saturation and that we may have identified most, if not all, possible genetic ways to compensate for the fat-2(wa17) mutation. Importantly, none of the fat-2(wa17) suppressor mutations that we identified compensate for the PUFA shortage itself. Instead, the fat-2(wa17) suppressors act by boosting desaturase activity to allow the fat-2(wa17) mutant to synthesize more PUFAs; the fat-2(wa17) suppressors, therefore, cannot suppress the defects of the fat-2 null mutant, as we specifically showed for ftn-2(et68). We draw the important conclusion that PUFAs are not only essential but also that their essential functions cannot be genetically replaced.
The fat-2(wa17) suppressor mutations within the HIF-1 pathway converge on the inhibition of ftn-2. The primary function of ferritin is to provide a harmless storage of iron within cells: ferritin promotes the oxidation of ferrous ions and stores the resulting ferric ions in a mineralized form (Plays et al., 2021). Thus, ftn-2 inhibition results in reduced total cellular iron but increased levels of ferrous ions, i.e., Fe2+ (Jenkins et al., 2020; Pekec et al., 2022). Importantly, ferrous ions are required for desaturase reactions, and increasing ferrous ion concentration is a potent way to increase activity because it accelerates the rate at which the desaturase cycles from the inactive post-reaction Fe3+-bound state to the active Fe2+-bound state (Shen et al., 2023; Shen et al., 2020). Because eukaryotic desaturases are all evolutionarily closely related (Sperling et al., 2003) and act in essentially the same way, ferrous ions must also be potent FAT-2 activators and thus boost the output from the mutant FAT-2(S101F) protein produced by the fat-2(wa17) allele or from the reduced FAT-2 protein levels in fat-2 RNAi-treated worms. Our findings suggest an elegant explanation for the observation that HIF-1 inhibits ftn-2 expression in C. elegans (Romney et al., 2011): this is likely an adaptive response to boost desaturase activity when oxygen or iron is limiting, ensuring a maximum output under adverse conditions. Fe2+ and HIF may contribute to desaturase boost also in human since CytB5 (which supplies Fe2+ to desaturases) promotes SFA tolerance while VHL (which causes HIF degradation) prevents SFA tolerance in cultured cells (Zhu et al., 2019). Other mechanisms are also possible. For example, mutations in the HIF-1 pathway could somehow reduce EPA turnover rates in the fat-2(wa17) mutant and allow its levels to rise above an essential threshold. This hypothesis is consistent with the observation that the suppressors can rescue both the fat-2(wa17) mutant and fat-2 RNAi-treated worms but not the fat-2 null mutant. It is even possible, though deemed unlikely, that the fat-2(wa17) suppressors act by compensating for the PUFA shortage via some undefined separate process downstream of the lipid changes and that they only indirectly result in elevated EPA levels.
Lipidomic analysis showed that among all PUFAs, it was the EPA levels that were best restored by the fat-2(wa17) suppressors. It is likely that any LA molecule produced in the mutants is quickly acted upon by downstream desaturases and elongases, leading to increased levels of the end product, namely EPA. EPA may be a sufficient or particularly important PUFA for sustaining C. elegans health, given that the fat-2(wa17) mutant is well rescued by EPA supplements. Indeed, DHA, which is not produced by C. elegans, is also able to rescue the fat-2(wa17) mutant. Others have shown that supplementing nearly completely EPA-deficient fat-3 C. elegans mutants with DHA significantly restored their EPA levels, suggesting that DHA supplements reduce EPA turnover (Lesa et al., 2003). EPA and DHA, being long chain PUFAs should have similar fluidizing effects on membrane properties (though in vitro experiments challenge this view; Sherratt et al., 2021), and both can serve as precursors of eicosanoids or docosanoids, particularly inflammatory ones (Chiang and Serhan, 2020). Abundant literature indicates that EPA is a particularly important PUFA in C. elegans. Phosphatidylcholines containing two attached EPA molecules are very abundant in C. elegans membranes, and their abundance increases the most in response to a temperature shift from 25–15°C, suggesting an important role in fluidity homeostasis (Tanaka et al., 1999). Long chain PUFAs such as EPA are required for efficient neurotransmission in C. elegans: mutants unable to produce them have depleted levels of synaptic vesicles accompanied by poor motility, and these defects are rescued by exogenous PUFAs, including DHA (Lesa et al., 2003). C. elegans can also convert EPA to eicosanoids in a cytochrome P450-dependent manner (Kulas et al., 2008; Mokoena et al., 2020); inhibiting this process results in reduced pharyngeal pumping rate, suggesting that regulation of muscular contraction by eicosanoids is conserved from nematodes to mammals (Kosel et al., 2011). EPA-derived eicosanoids are also required for guiding some cell migrations in C. elegans, including that of sperm (Hoang et al., 2013). EPA, and other PUFAs, can inhibit the nuclear localization of DAF-16 in fat-2-RNAi-treated worms, suggesting that they mediate signaling via this insulin receptor homolog and thus generally promote growth in C. elegans rather than stress resistance and fat storage (Horikawa and Sakamoto, 2010). In conclusion, EPA is clearly an important PUFA in C. elegans, and our work suggests that its multifaceted functions cannot be replaced by mutations in any one gene.
Finally, the case of the three novel egl-9 alleles isolated in our screen deserves special attention. All three alleles specifically affect the arginine at position 557 of the EGL-9 protein (it is converted to a histidine in two of the alleles, and to a cysteine in the third). This Arg557 in EGL-9 is specifically required for its ability to hydroxylate HIF-1, thus marking it for ubiquitin-dependent degradation (Epstein et al., 2001). Null alleles of egl-9 were not picked in our screen, and directly testing such a null allele revealed it to be ineffective as a fat-2(wa17) suppressor. We conclude that the EGL-9 proteins bearing a mutation at position Arg557 retain important functions while being unable to hydroxylate HIF-1. Others have previously demonstrated that EGL-9 could inhibit HIF-1 even when unable to hydroxylate it (Shao et al., 2009). Clearly, there is more to EGL-9 than its function as a HIF-1 hydroxylase, and it would be interesting in the future to detail this further.
We conclude that PUFA-deficient fat-2(wa17) mutants benefit only slightly from membrane-fluidizing treatments, and that there is likely no genetic way to compensate for PUFA deficiency. fat-2(wa17) mutants can only be rescued by boosting the activity of its defective desaturase, and restoring EPA levels are likely sufficient to suppress most fat-2(wa17) phenotypes suggesting a particularly important role for this PUFA in C. elegans. In the future, it will be interesting to determine if boosting desaturase activity by inhibition of ferritin expression via HIF-1 is also a beneficial response to hypoxia in worms and humans.
Materials and methods
C. elegans strains and cultivation
Request a detailed protocolThe wild-type C. elegans reference strain N2, fat-2(wa17), nhr-49(et8), mdt-15(et14), paqr-1(et52), paqr-2(3410), acs-13(et54), fld-1(et46), hif-1(ok2564), ftn-2(ok404), ftn-1(ok3625), egl-9(sa307), atfs-1(et15), zcIs4 [hsp-4::GFP], zcIs9 [hsp-60::GFP +lin-15(+)] and zIs356 [daf-16p::daf-16a/b::GFP +rol-6(su1006)] are available from the C. elegans Genetics Center (CGC; USA). The PHX7548 (fat-2(syb7458)/nT1[qIs51](IV;V)) strain was created by Suny Biotech Co using CRISPR/Cas9 and carries a deletion of 1387 bp between flanking sequences 5’-aaacttggcccccgacgaagatg-3’ and 5’-gtgataatgacgagaataagtcct-3’. fat-2(syb7458) worms were maintained in an unbalanced state on non-peptone plates containing OP50 grown overnight in LB containing 2 mM linoleic acid.
Unless otherwise stated, experiments were performed at 20 °C, using the E. coli strain OP50 as a food source, which was re-streaked every 6–8 wk and maintained on LB plates at 4°C. Single colonies were cultivated overnight at 37 °C in LB medium before being used to seed NGM plates. Stock solutions of supplements were filter-sterilized and added to cooled NGM after autoclaving to produce supplement plates.
Construction of fat-2(+)
Request a detailed protocolThe pfat-2(+) construct was generated with the NEB PCR Cloning Kit for amplification of fat-2(+) with the following primers: 5’-gagctcaagaagcgtttcca-3’ and 5’-gggcaagaatttgtagtgtca-3’ using N2 genomic DNA. Plasmids were prepared with a GeneJet Plasmid Miniprep Kit and injected at the following concentrations: pfat-2(+) of 20 µg/µl, pRF4(rol-6) of 40 µg/µl, and pBSKS of 35 µg/µl into fat-2(wa17) and N2 worms.
Pre-loading of E. coli with fatty acids or eicosanoids
Request a detailed protocolStock solutions of fatty acids (Merck) or eicosanoids (Primary Eicosanoid HPLC Mixture; Cayman Chemical) dissolved in ethanol (EPA, DHA, OA), DMSO (LA), or methyl acetate (eicosanoids) were diluted in LB media to the appropriate final concentration, inoculated with OP50 bacteria, and shaken overnight at 37 °C. The bacteria were washed twice in M9 to remove fatty acids, concentrated 10 X by centrifugation, dissolved in LB, and seeded onto NGM plates lacking peptone.
Growth assays
Request a detailed protocolFor length measurement studies, synchronized L1s were plated onto test plates seeded with OP50, and worms were mounted and photographed 72 hr later. Experiments performed at 15 °C were photographed after 144 hr. The length of 20 worms was measured using ImageJ.
For hydrogen peroxide treatment, synchronized worms were incubated in 2 mM hydrogen peroxide for 2 hr at the L1 stage before being plated on NGM plates for 72 hr. For hypoxia treatment, synchronized L1s were incubated for 2–6 hr in a hypoxia chamber, returned to normoxia for 24 hr, and hypoxia exposure was repeated as stated in the figure.
Oil Red O staining
Request a detailed protocolSynchronized day 1 adults were washed three times with PBST and fixed for 3 min in 60% isopropanol. Worms were then rotated for 2 hr in filtered 60% Oil Red O staining solution. The stained worms were washed three times in PBST before being mounted on agarose pads and imaged with a Zeiss Axioscope microscope.
Mutagenesis and screen for fat-2(wa17) suppressors
Request a detailed protocolfat-2(wa17) worms were mutagenized for 4 hr by incubation in the presence of 0.05 M ethyl methane sulfonate (EMS) according to the standard protocol (Sulston and Hodgkin, 1988). The worms were then washed and spotted onto an NGM plate. After 2 hr, L4 hermaphrodite animals were transferred to new plates. 8–10 d later, F1 progeny were bleached, washed, and their eggs allowed to hatch overnight in M9. The resulting L1 larvae were spotted onto new plates, cultivated at 20 °C, then screened after 72 hr for gravid F2 worms, which were then picked to new plates for further analysis. In total, approximately 40, 000 independently mutagenized haploid genomes were screened. The isolated suppressor mutants were outcrossed 4–6 times prior to whole genome sequencing, and 8–10 times prior to characterization. Outcrossing was done by mating N2 males to a suppressor, then crossing male progeny to fat-2(wa17) mutant worms. Progeny from this cross were picked to individual plates and kept at 20 °C, then screened for fat-2(wa17) homozygosity using PCR, followed by testing the F2 progeny for ability to grow to adults in 72 hr. Genotyping primers for the suppressor mutants are included in Supplementary file 1.
Whole genome sequencing
Request a detailed protocolThe genomes of the ten suppressor mutants that had been outcrossed 4 or 6 times were sequenced by Eurofins (Constance, Germany) with a mean coverage varying from 40.68X to 63.05X and their genomes assembled using the C. elegans genome version cel235 from Ensembl (REF: PMID: 37953337). Eurofins applied customised filters to the variants to filter false positives using GATK’s Variant Filtration module (DePristo et al., 2011; McKenna et al., 2010). Variants detected were annotated based on their gene context using snpEff (Cingolani et al., 2012). For each suppressor mutant, one or two hot spots, i.e., small genomic area containing several mutations, were identified and candidate mutations tested experimentally as described in the text.
CRISPR-Cas9 genome editing
Request a detailed protocolThe recreation of the candidate suppressor mutations and insertion of the 3xFLAG tag into the hif-1 gene was performed using CRISPR-Cas9 gene editing as previously described (Ghanta and Mello, 2020; Dokshin et al., 2018). The insertion of the ssDNA oligos was performed utilizing the homology-directed repair (HDR) mechanisms. The protospacer-adjacent motif (PAM) site of the ssDNA oligo template was flanked by 40 bp homology arms. Design and synthesis of the ssDNA and CRISPR RNA (crRNA) was performed using the Alt-R HDR Design Tool from IDT (Integrated DNA Technologies, Inc; Coralville, IA, USA), including proprietary modifications that improve oligo stability. To recreate the hif-1(et69) allele, we used the crRNA sequence 5’- UUUCUUAACGUGUGUAUUUCGUUUUAGAGCUAUGCU-3’ and the DNA oligo donor sequence 5’-AGTTCCATACATTTAGCAAGTGATTTCTTAACGTGTGTATTTCAAGAGCACGTAAGAACAGCTACGATGACGTTTTGCAATGGCT-3. To introduce the 3xFLAG at the N-terminus coding end of hif-1, we used the crRNA sequence 5’- GAAAAUAAUCAAGAGAGCAUGUUUUAGAGCUAUGCU-3’ and the DNA oligo donor sequence 5’-AAATGAACAACAGCCTAGTTCTTATTCCCCATTTCCAATGCTCTCTGACTACAAGGACCACGACGGCGATTATAAGGATCACGACATCGACTACAAAGACGACGATGACAAGTGATTATTTTCTACCCCCTCTCAAACTGTTCATTGTTTTG-3’. The injection mixes were prepared using 10 μg/μl of the Cas9 enzyme (IDT), 0.4 μg/μl tracrRNA (IDT), 2.8 0.4 μg/μl crRNA (IDT), 1 μg/μl of ssDNA (IDT), and 40 ng/μl of PRF4(rol-6) or Pmyo-2(GFP) plasmid. The mixture was microinjected into the posterior gonad of the worm and the F1 generation was screened for animals expressing the reporter plasmid. Genotypes were tested by PCR and successfully edited genes were confirmed by Sanger sequencing (Eurofins).
Fluorescence recovery after photobleaching (FRAP)
Request a detailed protocolFRAP experiments were carried out using a membrane-associated prenylated GFP reporter expressed in intestinal cells as previously described (Devkota and Pilon, 2018), using a Zeiss LSM700inv laser scanning confocal microscope with a 40 X water immersion objective. Briefly, the GFP-positive membranes were photobleached over a rectangular area (30×4 pixels) using 30 iterations of the 488 nm laser with 50% laser power transmission. Images were collected at a 12-bit intensity resolution over 256×256 pixels (digital zoom 4 X) using a pixel dwell time of 1.58 µs, and were all acquired under identical settings. The recovery of fluorescence was traced for 25 s. Fluorescence recovery and Thalf were calculated as previously described (Svensk et al., 2016).
Stress response assay
Request a detailed protocolWorms were imaged with a Zeiss Axioscope and fluorescence intensity was quantified with ImageJ (n≥20 for all experiments). Worm strains carrying hsp-60::GFP were imaged as day 1 adults, and the fluorescence values were taken from a 39 μm circumference circle in the brightest part of the anterior part of the worm. Worm strains carrying DAF-16::GFP were imaged as L4s and the percentage of worms with cytoplasmic or nuclear localization was quantified. Worm strains carrying hsp-4::GFP were imaged as day 1 adults and the fluorescence of the whole worm was quantified.
Lipidomics
Request a detailed protocolSamples were composed of synchronized L4 larvae (one 9 cm diameter plate/sample; each treatment/genotype was prepared in four independently grown replicates) grown on NGM or non-peptone plates seeded with linoleic acid. In the case of LA to NGM samples, worms were grown until late L3/early L4 stage on linoleic acid seeded non-peptone plates before being transferred to NGM plates for 6 hr before collection. Worms were washed three times in M9, pelleted and stored at –80 °C until analysis. For lipid extraction, the pellet was sonicated for 10 min in methanol;butanol [1:3] and then extracted according to published methods (Löfgren et al., 2016). Lipid extracts were evaporated and reconstituted in chloroform:methanol [1:2] with 5 mM ammonium acetate. This solution was infused directly (shotgun approach) into a QTRAP 5500 mass spectrometer (Sciex) equipped with a TriVersa NanoMate (Advion Bioscience) as described previously (Jung et al., 2011). Phospholipids were measured using precursor ion scanning in negative mode using the fatty acids as fragments (Ekroos et al., 2003; Ejsing et al., 2009). To generate the phospholipid composition (as mol%), the signals from individual phospholipids (area under the m/z peak in the spectra) were divided by the signal from all detected phospholipids of the same class. The data were evaluated using the LipidView software (Sciex). The data were further analyzed using Qlucore Omics Explorer n.n (Qlucore AB) for analysis. The data were normalized for the purpose of the heat map visualization (mean = 0; variance = 1).
Protein extraction and western blots
Request a detailed protocolWorms were lysed using lysis buffer containing 25 mM Tris (pH 7.5), 300 mM NaCl, 0.1% NP40, and 1 X protease inhibitor on ice with a motorized pestle. Samples were centrifuged at 20,000 g for 15 min at 4°C, and protein sample concentration was quantified using the BCA protein assay kit. 15 µg of protein were mixed with Laemmli sample loading buffer containing β-mercaptoethanol, boiled for 10 min, and loaded in 4–20% gradient precast SDS gel. After electrophoresis, the proteins were transferred to nitrocellulose membranes using Trans-Blot Turbo Transfer Packs and a Trans-Blot Turbo apparatus/predefined mixed-MW program. Blots were blocked in 5% nonfat dry milk in PBST for 1 hr at room temperature. Blots were incubated with primary antibodies overnight at 4 °C (mouse monoclonal anti-FLAG antibody (M2, Sigma Aldrich) 1:5000 dilution) or 1 hr at room temperature (mouse monoclonal anti-alpha-Tubulin (B512, Sigma Aldrich) 1:5000 dilution). Blots were then washed with PBST and incubated with swine-anti-rabbit HRP 1:3000 dilution or goat anti-mouse HRP 1:3000 dilution for 1 hr at room temperature and washed again with PBST. Detection of the hybridized antibody was performed using an ECL detection kit (Immobilon Western, Millipore), and the signal was visualized with a digital camera (VersaDoc, Bio-Rad).
Quantitative PCR (qPCR)
Request a detailed protocolTotal cellular worm RNA was isolated using the RNeasy Plus Kit according to the manufacturer’s instructions (Qiagen) and quantified using a NanoDrop spectrophotometer (ND-1000; Thermo Scientific). cDNA was obtained using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific) with random hexamers. qPCR was performed with a CFX Connect thermal cycler (Bio-Rad) using HOT FIREpol EvaGreen qPCR SuperMix (Solis Biodyne) and standard primers. Samples were measured in triplicates. The relative expression of each gene was calculated according to the delta-delta CT method. Expression of the housekeeping gene tba-1 was used to normalize for variations in RNA input. Primers used were as follows: ftn-2, forward (TACCACTCCGAGGTTGAAGC) and reverse (TGGAAGGGCAACATCGTCAC); tba-1, forward (TCTCGCAGGTTGTGTCTTCC) and reverse (AGCCTCATGGTAAGCCTGAA).
Statistics
Error bars for the worm length measurements show the standard error of the mean, and one-way ANOVA tests were used to identify significant differences from fat-2(wa17) control, unless otherwise stated. All experiments were independently repeated at least twice with similar results, and the statistics shown apply to the presented experimental results.
Materials availability statement
Request a detailed protocolStrains generated in this study will be deposited at the Caenorhabditis Genetics Center (CGC). Strains, and other reagents, can also be requested from the corresponding author.
Data availability
The targetted lipidomics data for Figures 3 and 7 (and their associated figure supplements) is provided as Source Data.
References
-
From zero to six double bonds: phospholipid unsaturation and organelle functionTrends in Cell Biology 25:427–436.https://doi.org/10.1016/J.TCB.2015.03.004
-
Lipid unsaturation and organelle dynamicsCurrent Opinion in Cell Biology 41:25–32.https://doi.org/10.1016/J.CEB.2016.03.012
-
Polyunsaturated fatty acids and their metabolites in brain function and diseaseNature Reviews. Neuroscience 15:771–785.https://doi.org/10.1038/nrn3820
-
The role of polyunsaturated fatty acids in fishComparative Biochemistry and Physiology Part B 83:711–719.https://doi.org/10.1016/0305-0491(86)90135-5
-
Cell-specific regulation of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha stabilization and transactivation in a graded oxygen environmentThe Journal of Biological Chemistry 281:22575–22585.https://doi.org/10.1074/jbc.M600288200
-
On the nature and rôle of the fatty acids essential in nutritionJournal of Biological Chemistry 86:587–621.https://doi.org/10.1016/S0021-9258(20)78929-5
-
Mechanisms of action of (n-3) fatty acidsThe Journal of Nutrition 142:592S–599S.https://doi.org/10.3945/jn.111.155259
-
Disruption of iron homeostasis increases phosphine toxicity in Caenorhabditis elegansToxicological Sciences 96:194–201.https://doi.org/10.1093/toxsci/kfl187
-
Specialized pro-resolving mediator network: an update on production and actionsEssays in Biochemistry 64:443–462.https://doi.org/10.1042/EBC20200018
-
Charting molecular composition of phosphatidylcholines by fatty acid scanning and ion trap MS3 fragmentationJournal of Lipid Research 44:2181–2192.https://doi.org/10.1194/jlr.D300020-JLR200
-
Roles of polyunsaturated fatty acids, from mediators to membranesJournal of Lipid Research 61:20000800.https://doi.org/10.1194/JLR.R120000800
-
Polyunsaturated fatty acids are involved in regulatory mechanism of fatty acid homeostasis via daf-2/insulin signaling in Caenorhabditis elegansMolecular and Cellular Endocrinology 323:183–192.https://doi.org/10.1016/j.mce.2010.03.004
-
Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its alpha subunitThe Journal of Biological Chemistry 271:32253–32259.https://doi.org/10.1074/jbc.271.50.32253
-
Fatty acid compositions of Caenorhabditis elegans and C. BriggsaeComparative Biochemistry and Physiology Part B 73:517–520.https://doi.org/10.1016/0305-0491(82)90068-2
-
Dietary polyunsaturated fatty acids and inflammatory mediator productionThe American Journal of Clinical Nutrition 71:343S–348S.https://doi.org/10.1093/ajcn/71.1.343S
-
Sodium sulfite is a potential hypoxia inducer that mimics hypoxic stress in Caenorhabditis elegansJournal of Biological Inorganic Chemistry 16:267–274.https://doi.org/10.1007/s00775-010-0723-1
-
High throughput quantitative molecular lipidomicsBiochimica et Biophysica Acta 1811:925–934.https://doi.org/10.1016/j.bbalip.2011.06.025
-
Transcriptional regulation and life-span modulation of cytosolic aconitase and ferritin genes in C. elegansJournal of Molecular Biology 342:421–433.https://doi.org/10.1016/j.jmb.2004.07.036
-
Eicosanoid formation by a cytochrome P450 isoform expressed in the pharynx of Caenorhabditis elegansThe Biochemical Journal 435:689–700.https://doi.org/10.1042/BJ20101942
-
Cytochrome P450-dependent metabolism of eicosapentaenoic acid in the nematode Caenorhabditis elegansArchives of Biochemistry and Biophysics 472:65–75.https://doi.org/10.1016/j.abb.2008.02.002
-
Long chain polyunsaturated fatty acids are required for efficient neurotransmission in C. elegansJournal of Cell Science 116:4965–4975.https://doi.org/10.1242/jcs.00918
-
Polyunsaturated fatty acids and neurotransmission in Caenorhabditis elegansBiochemical Society Transactions 34:77–80.https://doi.org/10.1042/BST0340077
-
Synthesis and function of fatty acids and oxylipins, with a focus on Caenorhabditis elegansProstaglandins & Other Lipid Mediators 148:106426.https://doi.org/10.1016/j.prostaglandins.2020.106426
-
Endogenous production of long-chain polyunsaturated fatty acids and metabolic disease riskCurrent Cardiovascular Risk Reports 8:1–9.https://doi.org/10.1007/S12170-014-0418-1/FULLTEXT.HTML
-
Structure, function, and dietary regulation of delta6, delta5, and delta9 desaturasesAnnual Review of Nutrition 24:345–376.https://doi.org/10.1146/annurev.nutr.24.121803.063211
-
Identification and characterization of an animal delta(12) fatty acid desaturase gene by heterologous expression in Saccharomyces cerevisiaeArchives of Biochemistry and Biophysics 376:399–408.https://doi.org/10.1006/abbi.2000.1733
-
An iron enhancer element in the FTN-1 gene directs iron-dependent expression in Caenorhabditis elegans intestineThe Journal of Biological Chemistry 283:716–725.https://doi.org/10.1074/jbc.M707043200
-
AdipoR2 recruits protein interactors to promote fatty acid elongation and membrane fluidityThe Journal of Biological Chemistry 299:104799.https://doi.org/10.1016/j.jbc.2023.104799
-
Structure and mechanism of a unique diiron center in mammalian stearoyl-CoA desaturaseJournal of Molecular Biology 432:5152–5161.https://doi.org/10.1016/j.jmb.2020.05.017
-
Free ferrous ions sustain activity of mammalian stearoyl-CoA desaturase-1The Journal of Biological Chemistry 299:104897.https://doi.org/10.1016/j.jbc.2023.104897
-
EPA and DHA containing phospholipids have contrasting effects on membrane structureJournal of Lipid Research 62:100106.https://doi.org/10.1016/j.jlr.2021.100106
-
Essential fatty acids in health and chronic diseaseThe American Journal of Clinical Nutrition 70:560s–569s.https://doi.org/10.1093/AJCN/70.3.560S
-
The evolution of desaturasesProstaglandins, Leukotrienes and Essential Fatty Acids 68:73–95.https://doi.org/10.1016/S0952-3278(02)00258-2
-
Update on lipids and mitochondrial function: impact of dietary n-3 polyunsaturated fatty acidsCurrent Opinion in Clinical Nutrition and Metabolic Care 15:122–126.https://doi.org/10.1097/MCO.0B013E32834FDAF7
-
Biosynthesis of 1,2-dieicosapentaenoyl-sn-glycero-3-phosphocholine in Caenorhabditis elegansEuropean Journal of Biochemistry 263:189–195.https://doi.org/10.1046/j.1432-1327.1999.00480.x
-
Manipulation of in vivo iron levels can alter resistance to oxidative stress without affecting ageing in the nematode C. elegansMechanisms of Ageing and Development 133:282–290.https://doi.org/10.1016/j.mad.2012.03.003
-
Polyunsaturated fatty acid derived signaling in reproduction and development: insights from Caenorhabditis elegans and Drosophila melanogasterMolecular Reproduction and Development 80:244–259.https://doi.org/10.1002/MRD.22167
-
Polyunsaturated fatty acids promote the rapid fusion of lipid droplets in Caenorhabditis elegansJournal of Biological Chemistry 298:102179.https://doi.org/10.1016/j.jbc.2022.102179
-
The relationship between n-3 polyunsaturated fatty acids and telomere: A review on proposed nutritional treatment against metabolic syndrome and potential signaling pathwaysCritical Reviews in Food Science and Nutrition 64:4457–4476.https://doi.org/10.1080/10408398.2022.2142196
-
A crucial role of sterol regulatory element-binding Protein-1 in the regulation of lipogenic gene expression by polyunsaturated fatty acidsJournal of Biological Chemistry 274:35840–35844.https://doi.org/10.1074/jbc.274.50.35840
-
Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperonesJournal of Cell Science 117:4055–4066.https://doi.org/10.1242/jcs.01275
-
Desaturase and elongase-limiting endogenous long-chain polyunsaturated fatty acid biosynthesisCurrent Opinion in Clinical Nutrition and Metabolic Care 19:103–110.https://doi.org/10.1097/MCO.0000000000000254
Article and author information
Author details
Funding
Cancerfonden (22 1984 Pj)
- Marc Pilon
Vetenskapsrådet (2024-04012)
- Marc Pilon
NIH Office of Research Infrastructure Programs (P40 OD010440)
- No recipients declared.
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
Some strains were provided by the CGC, which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440). This research was funded by Cancerfonden (22 1984 Pj) and Vetenskapsrådet (2024–04012).
Version history
- Sent for peer review:
- Preprint posted:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Reviewed Preprint version 3:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.104181. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2024, Kaper et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,500
- views
-
- 141
- downloads
-
- 2
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 2
- citations for umbrella DOI https://doi.org/10.7554/eLife.104181
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Forward genetics in C. elegans reveals genetic adaptations to polyunsaturated fatty acid deficiency
eLife 13:RP104181.
https://doi.org/10.7554/eLife.104181.4




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}