Single-cell analysis of transcription kinetics across the cell cycle
- Baylor College of Medicine, United States
- Rice University, United States
- University of Illinois at Urbana-Champaign, United States
- Icahn School of Medicine at Mount Sinai, United States
Figures
Figure 1 with 5 supplements
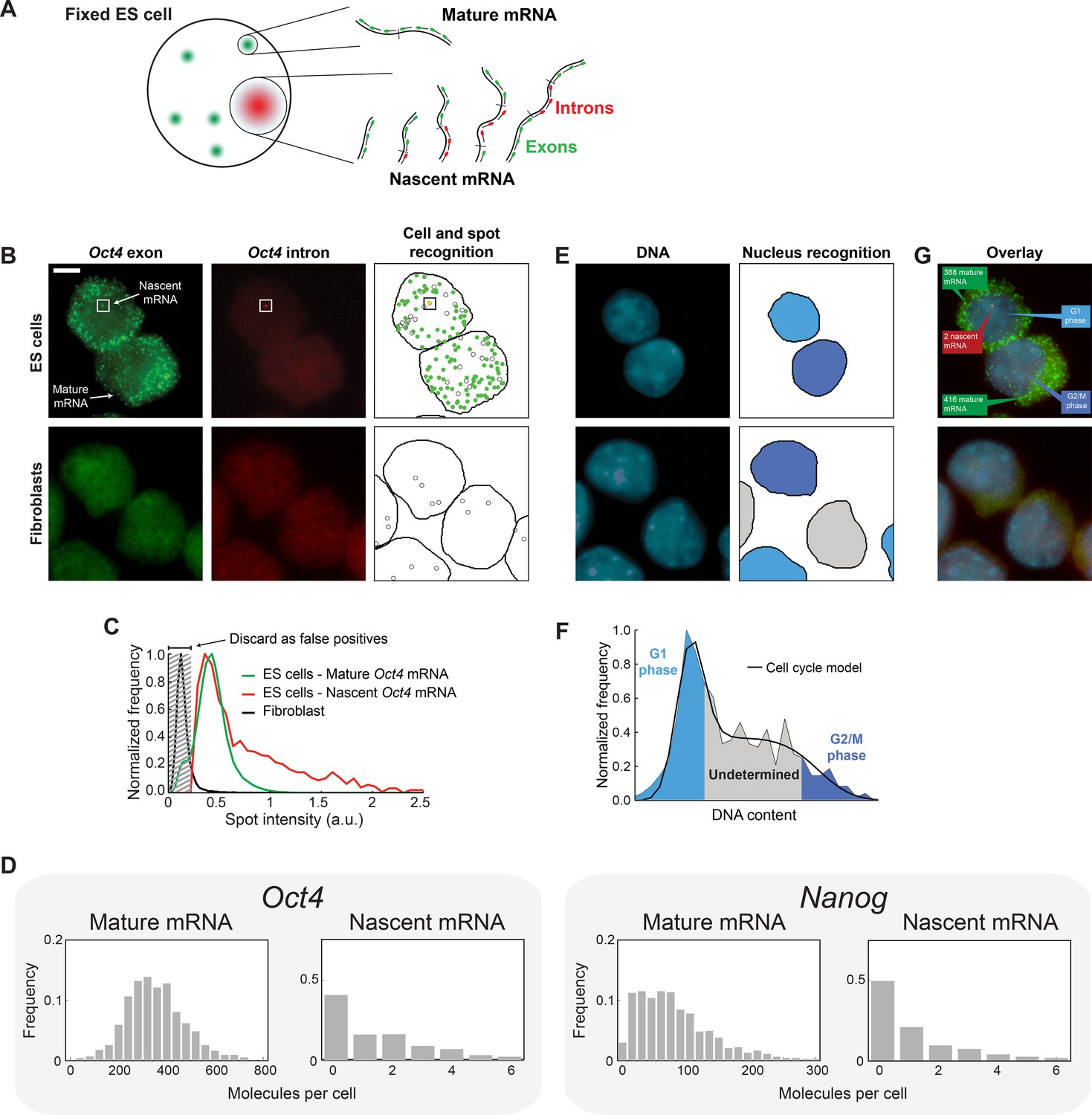
Quantifying mature mRNA, nascent mRNA and cell-cycle phase in individual mouse embryonic stem (ES) cells.
(A) Identifying nascent and mature mRNA. Introns (red) and exons (green) were labeled using different colors of smFISH probes. In the cell, pre-spliced nascent mRNA at the site of active transcription are bound by both probe sets, whereas mature mRNA are only bound by the exon probe set. (B) Mouse embryonic stem (ES) cells (top row) labeled for Oct4 exons (left column, green) and introns (center column, red). Automated image analysis (right column) was used to identify the cell boundaries (black line), intron (red) and exon (green) spots, as well as false-positive spots (black circles, see Panel C). Co-localized exon and intron spots (yellow) were identified as nascent mRNA (square), whereas spots found only in the exon channel were identified as mature mRNA. Fibroblasts (bottom row) were used as negative control. Scale bar, 5 µm. (C) The distribution of Oct4 mRNA spot intensities for mature mRNA (green, >100000 spots), nascent mRNA (red, >1000 spots), and spots found in Fibroblasts (black, >1000 spots). The histograms were used to discard false positive spots (gray region) and to identify the signal intensity corresponding to a single mRNA. (D) The distributions of mature and nascent mRNA numbers per cell for Oct4 (>700 cells) and Nanog (>1000 cells). (E) The same cells as in panel B, labeled for DNA using DAPI (left column, blue). Automated image analysis (right column) was used to identify the nuclear boundary (black line). The DNA content of each nucleus was used to estimate the phase of the cell cycle (cyan, grey, and blue shading; see Panel F). (F) The distribution of DNA content per cell (>700 cells), estimated from the nuclear DAPI signal (panel E). The histogram of DNA content per cell was fitted to a theoretical model of the cell cycle (black line), and used to identify which cells are in G1 phase (cyan) and which in G2 (blue). (G) Overlay of the smFISH and DAPI channels for mouse embryonic stem cells (top) and fibroblasts (bottom). The estimated number of mature (green) and nascent (red) mRNA, as well as the phase of the cell cycle (blue), are indicated for the two stem cells.
Figure 1—figure supplement 1
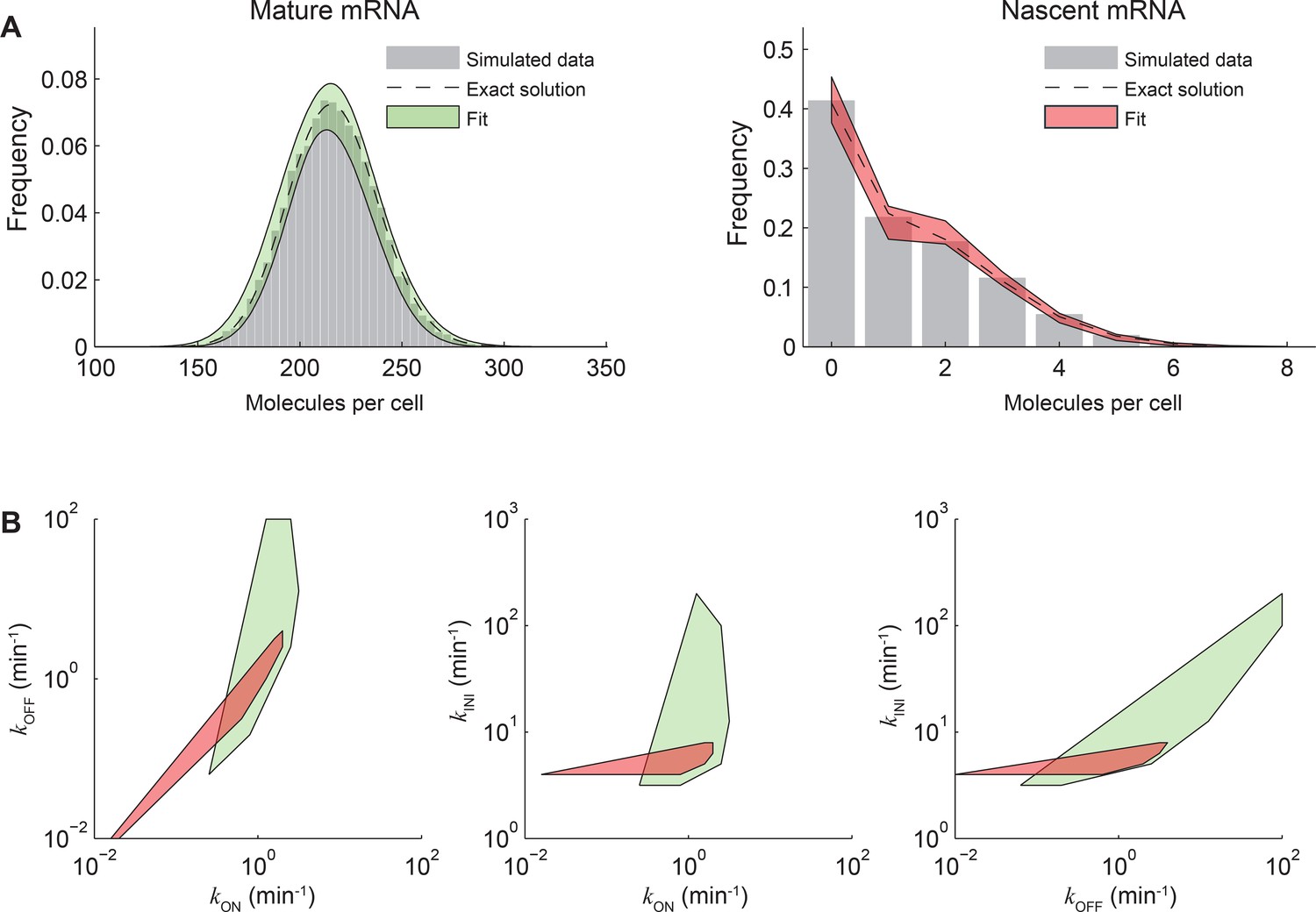
Fitting both nascent and mature mRNA constrains model parameters.
(A) The distributions of mature (left) and nascent (right) mRNA numbers were calculated (dashed line) and simulated using the Gillespie algorithm (Gillespie, 1977) (gray bars, 10000 simulations) for the stochastic model of transcription kinetics described in the main text (Figure 3A). The parameters used were: kON = 1 min-1, kOFF = 1 min-1, kINI = 5 min-1, τRES = 1 min, kD = log(2)/60 min-1. For simplicity, no cell-cycle effects were included. Each simulation was run for a total of 20000 min and an observation time tob was randomly selected from the last 10000 min. At tob, the number of mature mRNA was recorded, and the equivalent number of full-length transcripts of nascent mRNA was calculated from the timing of initiation events occurring between the times t = tob-τRES and t = tob. The simulated mature and nascent mRNA data were then each fitted back to the same model using maximum likelihood estimation (Neuert et al., 2013), with kON, kOFF and kINI as fitting parameters. The best fits (log-likelihood values differing from the maximum log-likelihood by <1%) are indicated on the plots in green and red shading. (B) Convex hull of the estimated parameters kON, kOFF, kINI that obey the quality criterion above for mature (green) and nascent (red) mRNA. It can be seen that parameters estimated from mature or nascent mRNA data independently span ~2 orders of magnitude, while using the fits from both species significantly constrains the parameter space.
Figure 1—figure supplement 2
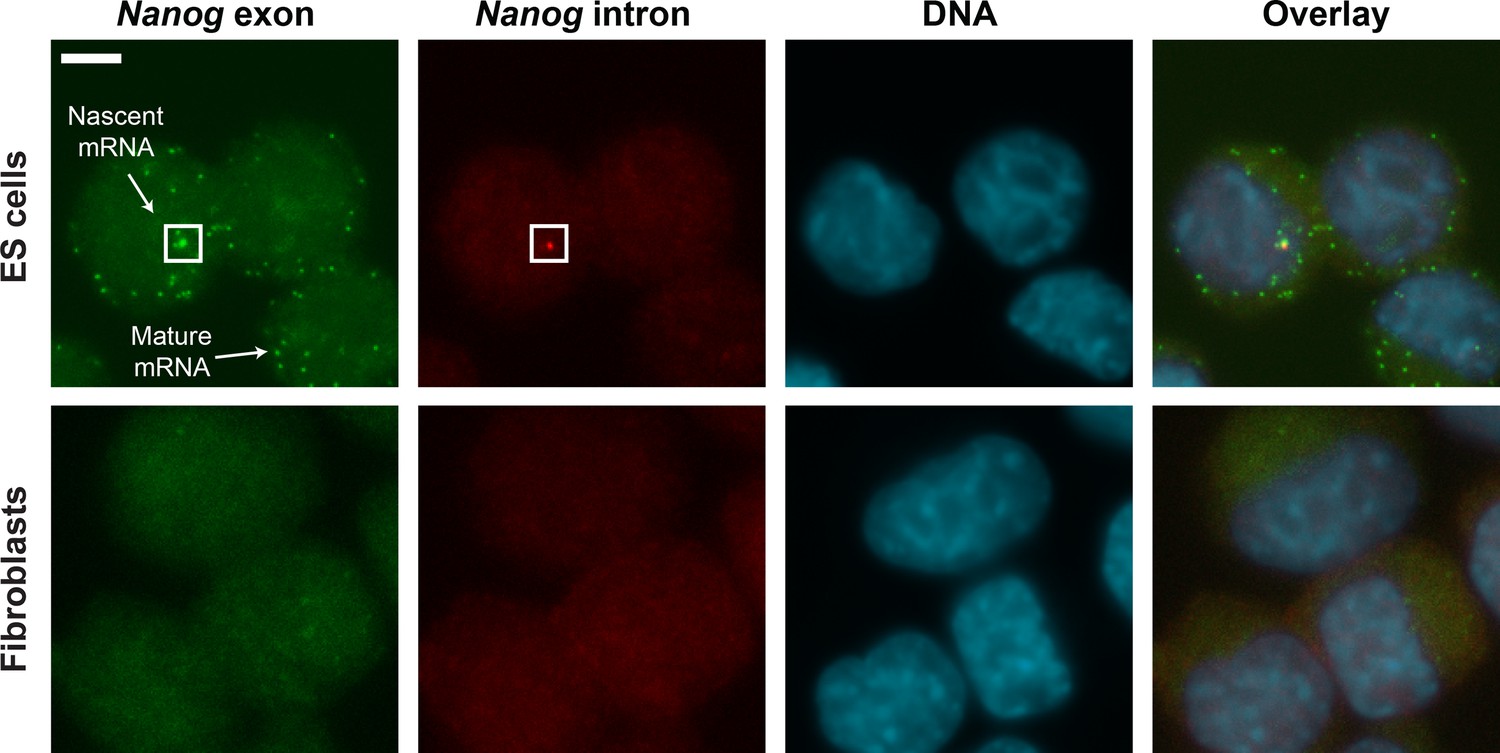
smFISH images of Nanog mRNA in ES cells and Fibroblasts.
Mouse ES cells (top row) labeled for Nanog exons (first column, green), Nanog introns (second column, red) and DNA (DAPI, third column, blue). Fibroblasts (bottom row) were used as negative control. Spots in the exon and intron channels were seen in ES cells but not in Fibroblasts. Co-localized exon and intron spots were identified as nascent mRNA (square), whereas spots found only in the exon channel were identified as mature mRNA. Scale bar, 5 µm.
Figure 1—figure supplement 3
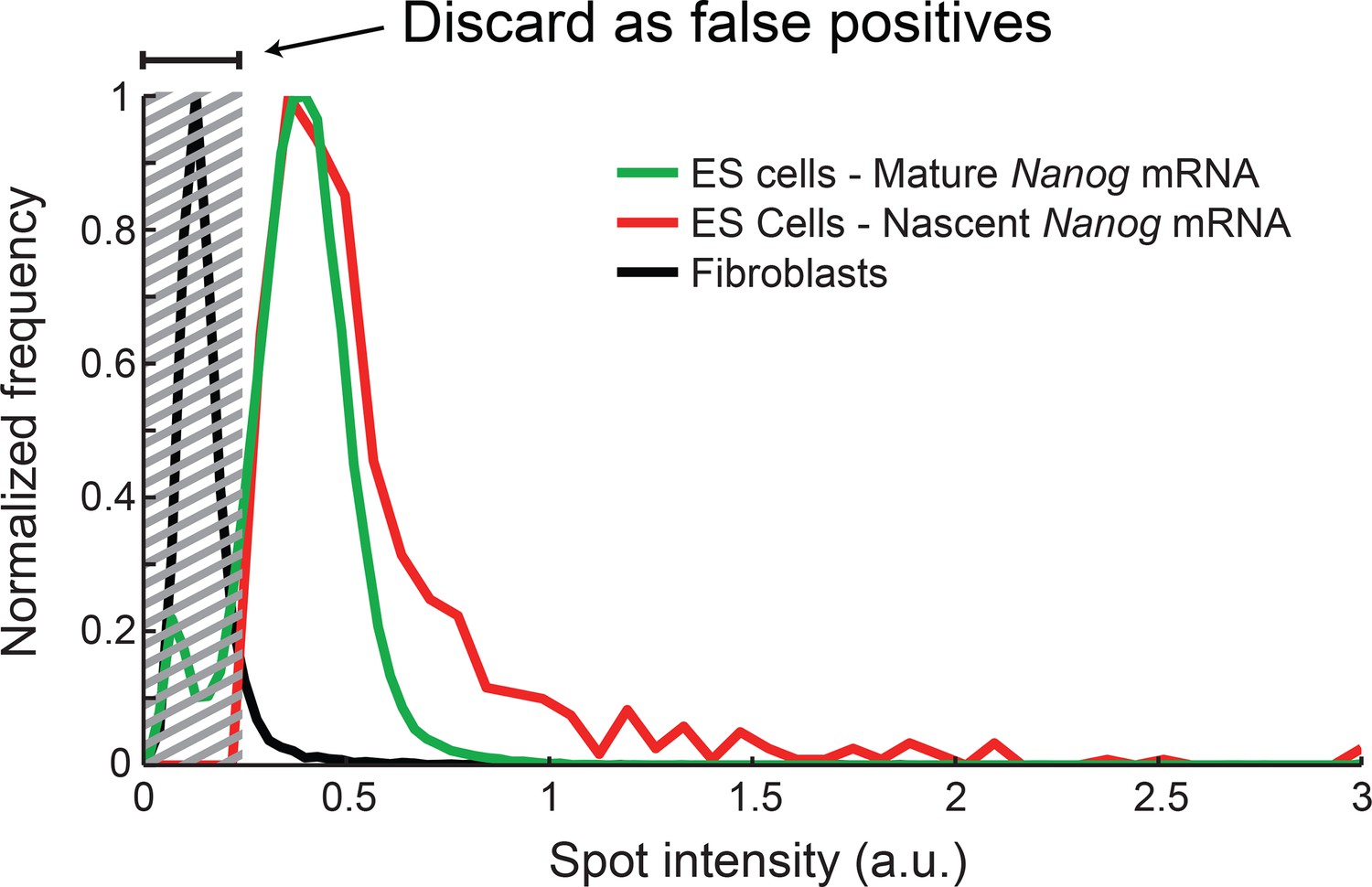
Distribution of Nanog mRNA spot intensities.
The distribution of exon-channel spot intensities for Nanog mature mRNA (green, >10000 spots), nascent mRNA (red, >1000 spots), and spots found in Fibroblasts (black, >1000 spots). The histograms were used to discard false positive spots (gray region) and to identify the signal intensity corresponding to a single mRNA.
Figure 1—figure supplement 4
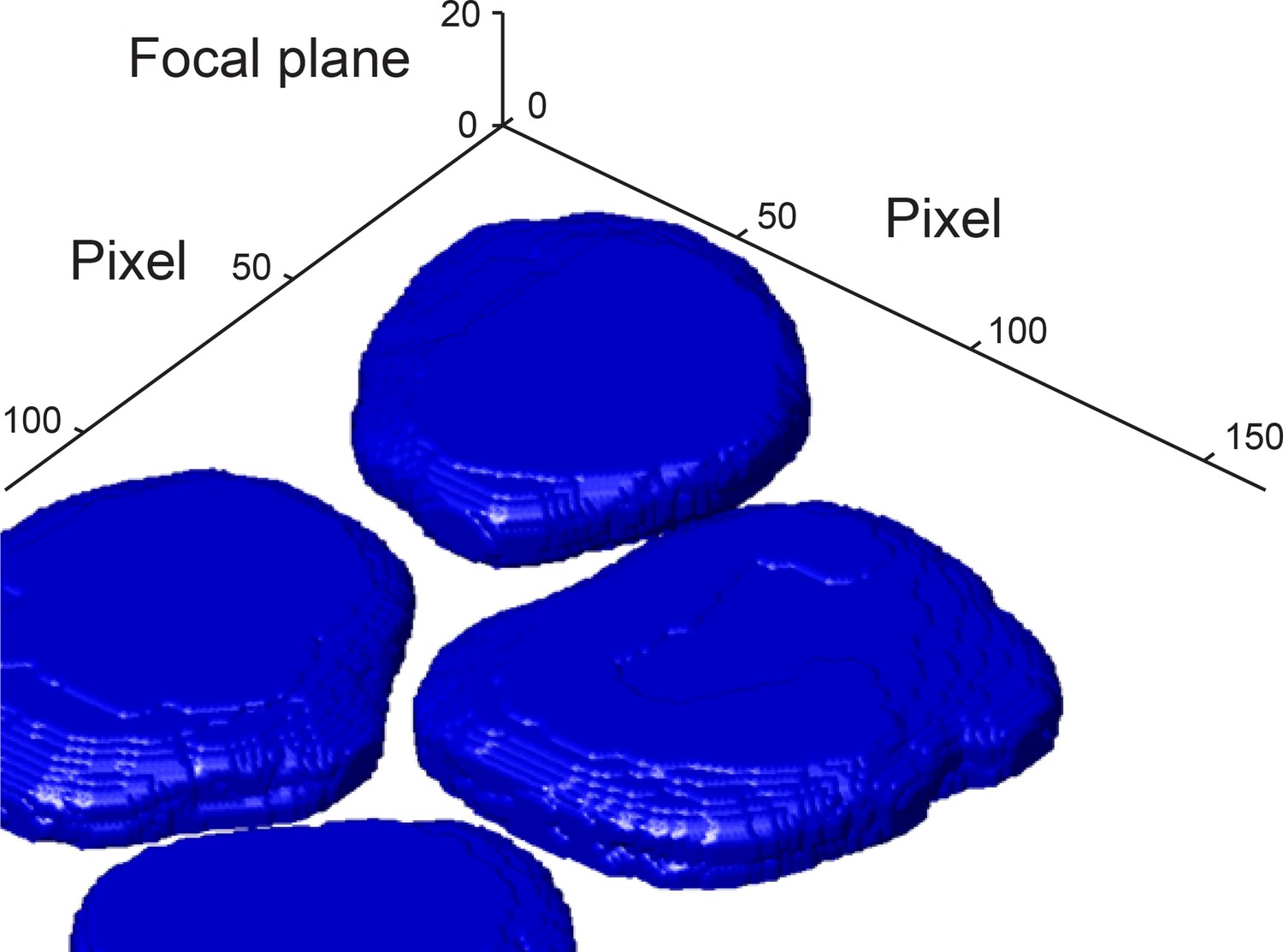
3D reconstruction of nuclei from the DAPI channel.
The boundary of each nucleus was detected in each focal plane. The nuclei boundaries were used to reconstruct the 3D shape of each nucleus. For more information see Materials and methods 4. Pixel size is 130 nm × 130 nm. Focal planes have 500 nm spacing.
Figure 1—figure supplement 5
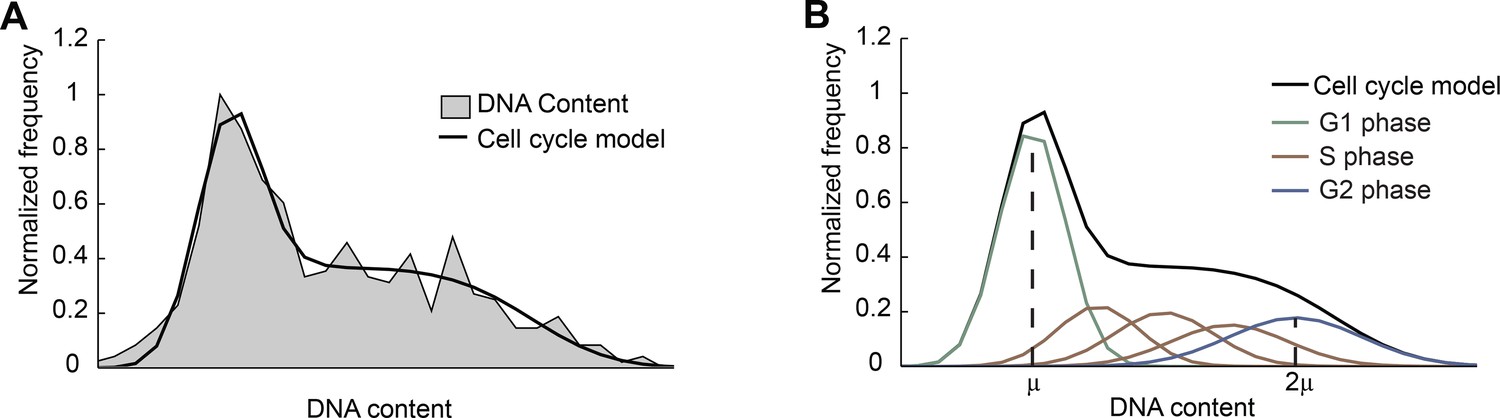
Fitting the DNA-content histogram to a cell-cycle model.
(A) The DNA-content histogram from mouse ES cells (gray, >700 cells) was fitted to the Fried/Baisch cell cycle model (Johnston et al., 1978) (black). (B) In the model (black), the distribution of DNA contents per cell is the sum of multiple Gaussians (colored lines) with equal coefficients of variation (CV = σ/μ, the ratio of the standard deviation to the mean): The DNA content of cells in G1 phase is represented by a single Gaussian distribution (green) with mean μ and standard deviation σ. The DNA of cells in G2/M phase is represented by a Gaussian distribution (blue) with mean 2μ and standard deviation 2σ. The DNA content of cells in S phase is approximated by a sum of 3 Gaussians (brown). For more information see Materials and methods 6.2.
Figure 2 with 1 supplement
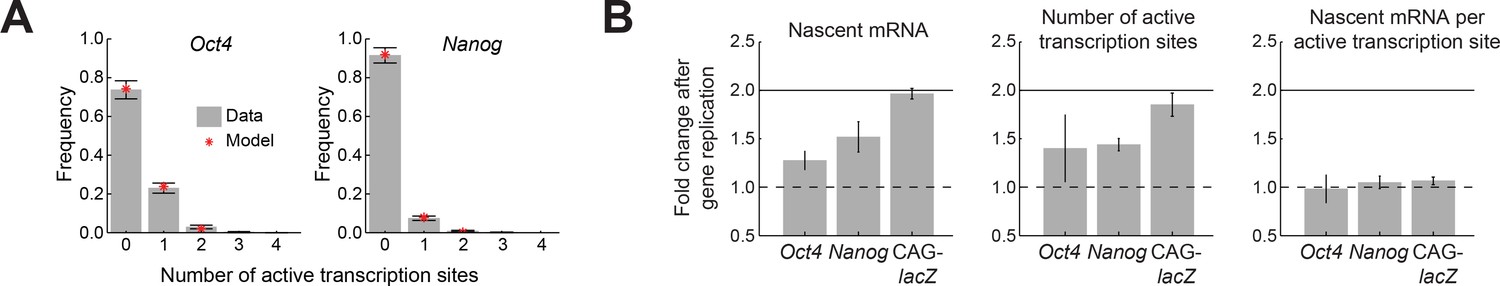
Oct4 and Nanog exhibit independent allele activity and dosage compensation.
(A) The distribution of number of active transcription sites for Oct4 (left; >700 cells) and Nanog (right; >1,000 cells), in cells having two copies of each gene. In both cases, the measured distribution (gray) is described well by a theoretical model assuming independent activity of the two alleles (binomial distribution, red). Error bars represent the estimated SEM due to finite sampling. (B) The fold change in transcriptional activity following gene replication for Oct4, Nanog, and a control reporter gene (CAG-lacZ). For Oct4 and Nanog, the average number of nascent mRNA (left) increases less than two-fold following gene replication, while a two-fold increase is observed in the control reporter gene. The change in number of nascent mRNA reflects an increase in the number of active transcription sites (middle), with no change in the number of nascent mRNA at each transcription site (right). Error bars represent SEM from 3 experiments with >200 cells per cell-cycle phase in each experiment.
Figure 2—figure supplement 1

Nascent mRNA correlation between two gene copies.
(A) Heat maps of the number of nascent mRNA at the two gene copies within the same cell. Left, Oct4 (1 experiment with >200 cells). Right, Nanog (1 experiment with >200 cells). The Pearson’s correlation coefficient (r; mean ± SEM from 3 experiments with >200 cells per experiment) between gene copies is indicated on each plot, as well as the p-value (mean ± SEM from 3 experiments with >200 cells per experiment) obtained using a Student’s t-distribution (calculated using the MATLAB function corr). (B) The data in Panel A were reshuffled by pairing the nascent mRNA at one gene copy from a given cell with the nascent mRNA from a gene copy at another, randomly selected cell.
Figure 3 with 5 supplements
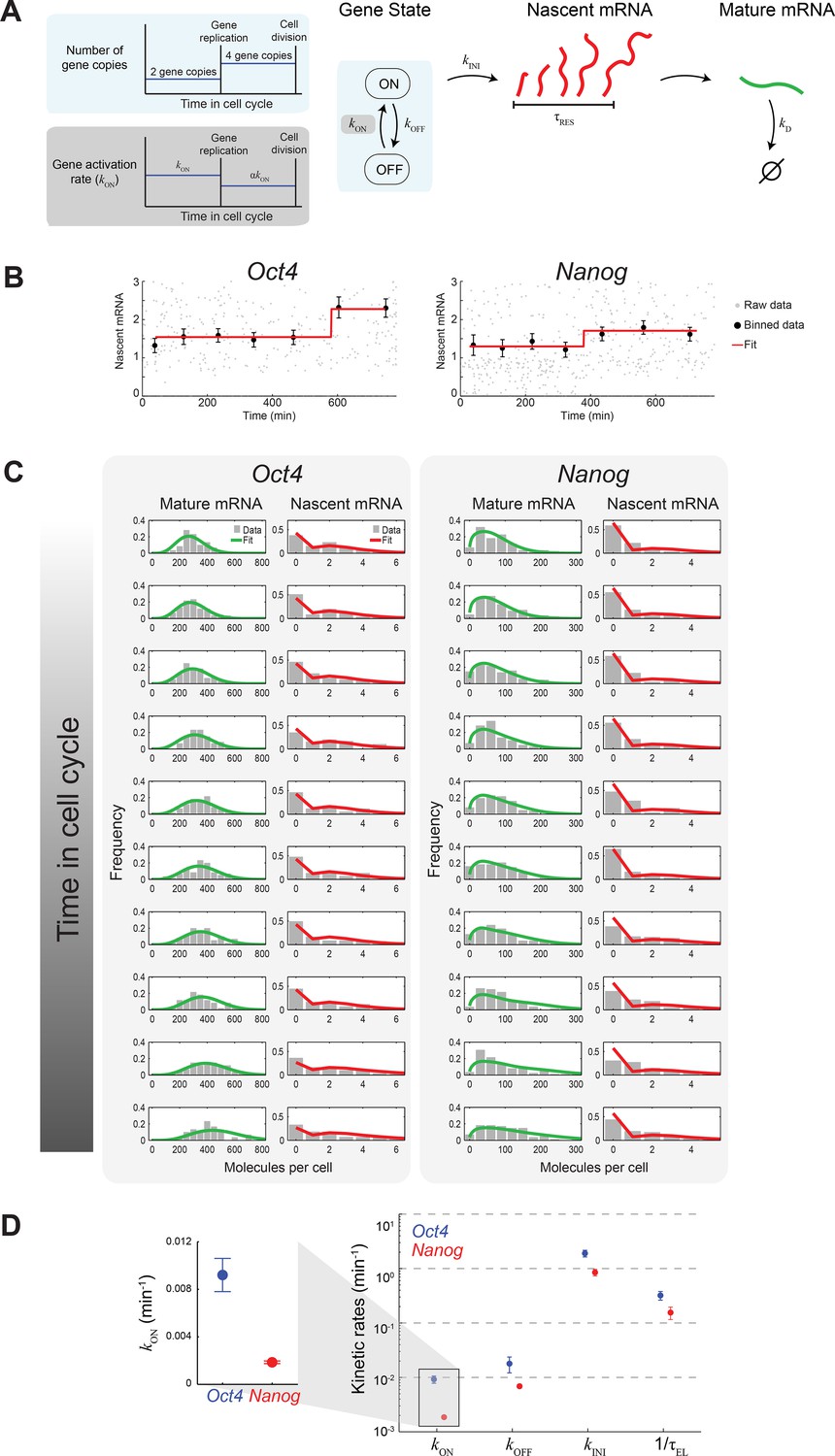
Extracting the stochastic kinetics of Oct4 and Nanog.
(A) A stochastic 2-state model for gene activity, which incorporates cell cycle and gene copy-number effects. Each gene copy stochastically switches between ‘ON’ and ‘OFF’ states. Transcription is stochastically initiated only in the ‘ON’ state. After initiation, the nascent transcript (red) elongates with constant speed, and is then converted into a mature mRNA molecule (green). Mature mRNA are degraded stochastically. Gene copies are independent, and their number changes from 2 to 4 following gene replication (left, cyan box). At the end of the cell cycle, mRNA molecules are binomially partitioned between the two daughter cells. Dosage compensation is included though a decrease in the rate of activation following gene replication (left, grey box). (B) Estimating the gene replication time and the fold-change in transcriptional activity for Oct4 (left; >700 cells) and Nanog (right; >1000 cells). The number of nascent mRNA was plotted against the time within the cell cycle for each cell (grey points), and the data were binned into populations of equal cell number (black markers). The binned data were fit to a step function (red), used to estimate the gene replication time and the fold-change in number of nascent mRNA before/after gene replication. Error bars represent SEM. (C) The distribution of mature and nascent mRNA copy number over time, for Oct4 (left; >700 cells) and Nanog (right; >1000 cells). The cell population was partitioned into 12 time windows, equally-spaced within the cell cycle (rows; we discarded the first and last windows, where the low cell numbers lead to a large error in the ERA calculation [Kafri et al., 2013]). The measured distributions (gray) are overlaid with the model predictions for mature (green) and nascent (red) mRNA. (D) The probabilistic rates of the transcription process and the gene elongation rate, for Oct4 (blue) and Nanog (red). The rates were estimated from the best theoretical fit of the mature and nascent mRNA distributions (panel C). The rate that varies most between Oct4 and Nanog is the probability of switching to an active transcription state, kON, which is ~5-fold higher for Oct4 (inset). Error bars represent SEM from 3 experiments with >600 cells per experiment.
Figure 3—figure supplement 1
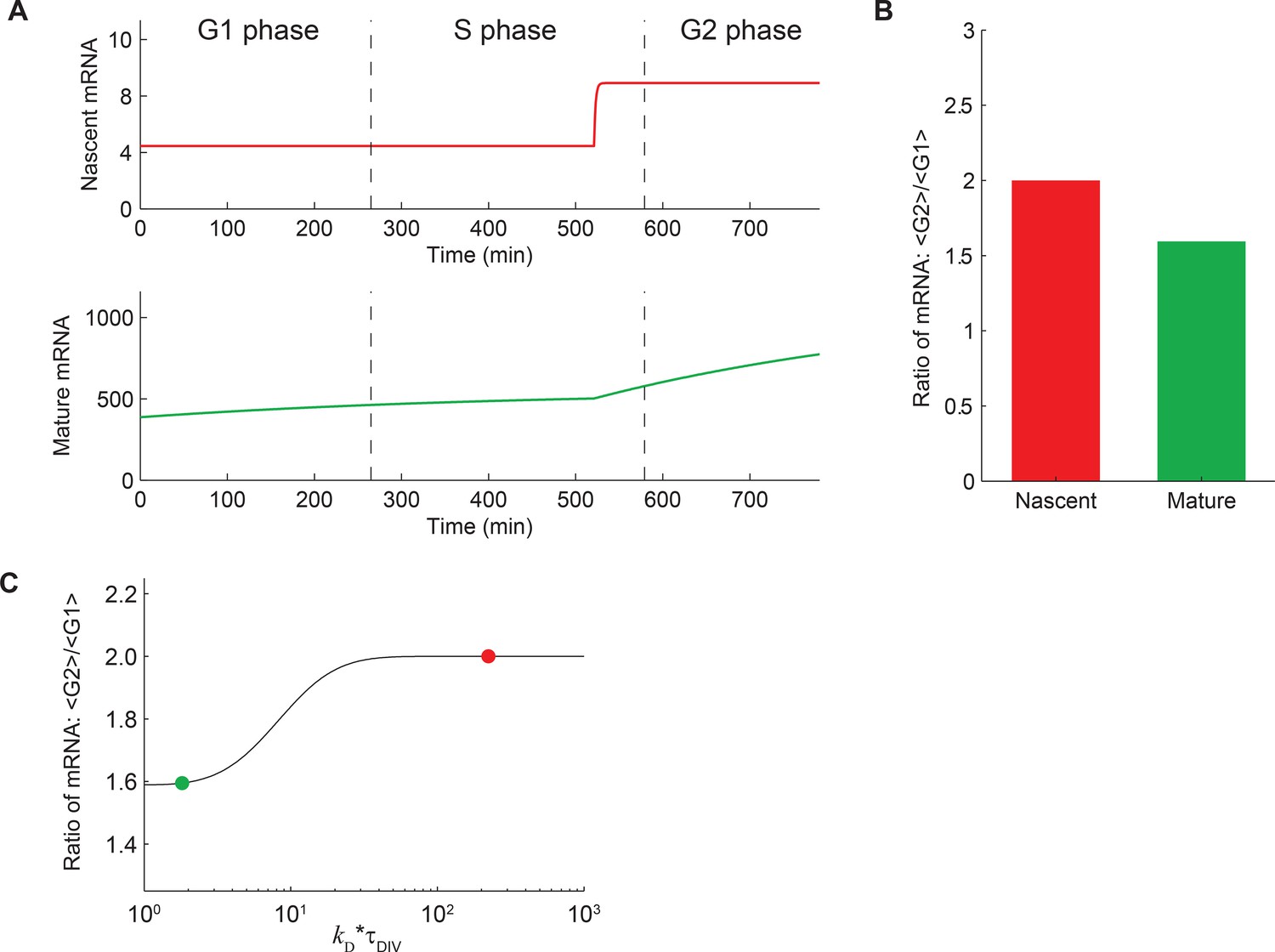
Expected behavior of mature and nascent mRNA numbers over time.
(A) A deterministic theoretical model of transcription, which includes the effects of gene replication and cell division, was used to predict the numbers of nascent (red, top) and mature (green, bottom) mRNA at different times in the cell cycle. For demonstration, the model parameters were given values measured for Oct4. For more details see Materials and methods 9. It can be seen that, even though the mRNA levels are cyclostationary (i.e. the number of mRNA at the end of the cell cycle is twice that at the beginning), the level of mature mRNA does not reach steady state during the cell cycle. This is because the lifetime of mature mRNA (7.1 hr; Supplementary file 1) is comparable to the duration of individual cell cycle phases. In contrast, the number of nascent mRNA reaches steady state soon after gene replication because of its short lifetime (residence time 3.5 min; Figure 3D). (B) The ratio of mean mRNA level in G2 phase to that in G1 is predicted to be 2 for nascent mRNA, but <2 for mature mRNA. (C) The predicted ratio of mean mRNA level in G2 phase to that in G1 as a function of the ratio of cell cycle duration to mRNA lifetime (kD*τDIV; black line). The values for nascent (red) and mature (green) mRNA are indicated on the plot.
Figure 3—figure supplement 2
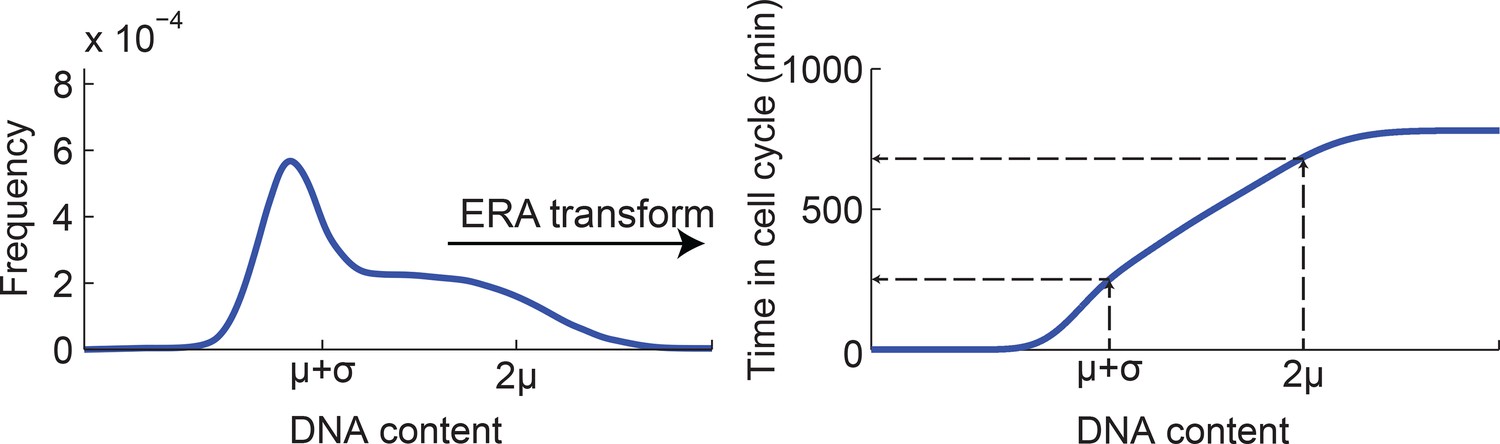
Mapping DNA content to time in the cell cycle using ergodic rate analysis.
Ergodic rate analysis (see Materials and methods 7) was used to transform the DNA content distribution (left, model fit of the experimental data, see Figure 1—figure supplement 5) to a mapping between DNA content and time within the cell-cycle (right). For example, the DNA contents values μ+σ and 2μ (extracted from the cell cycle model) are mapped to distinct times within the cell cycle.
Figure 3—figure supplement 3
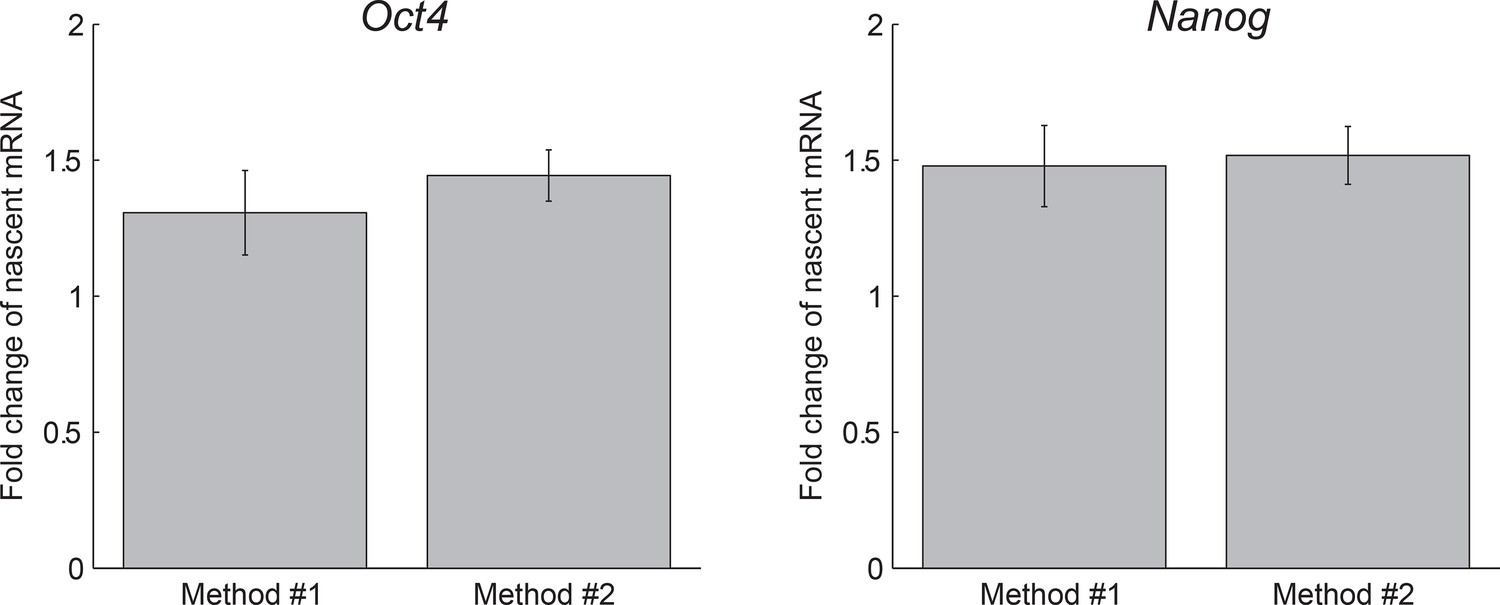
Agreement between methods of measuring dosage compensation.
The extracted fold change in nascent Oct4 (left) and Nanog (right) mRNA following gene replication, as measured using two methods: Method #1, comparing the mean number of nascent mRNA of cells in G1 phase to that of cells in G2 phase (see Figure 1F). Error bars represent SE.M. from 3 experiments with >200 cells per phase in each experiment. Method #2, extracting the fold change from a step-function fit to the nascent mRNA over time (see Figure 3B). Error bars represent SEM from 3 experiments with >600 cells per experiment.
Figure 3—figure supplement 4
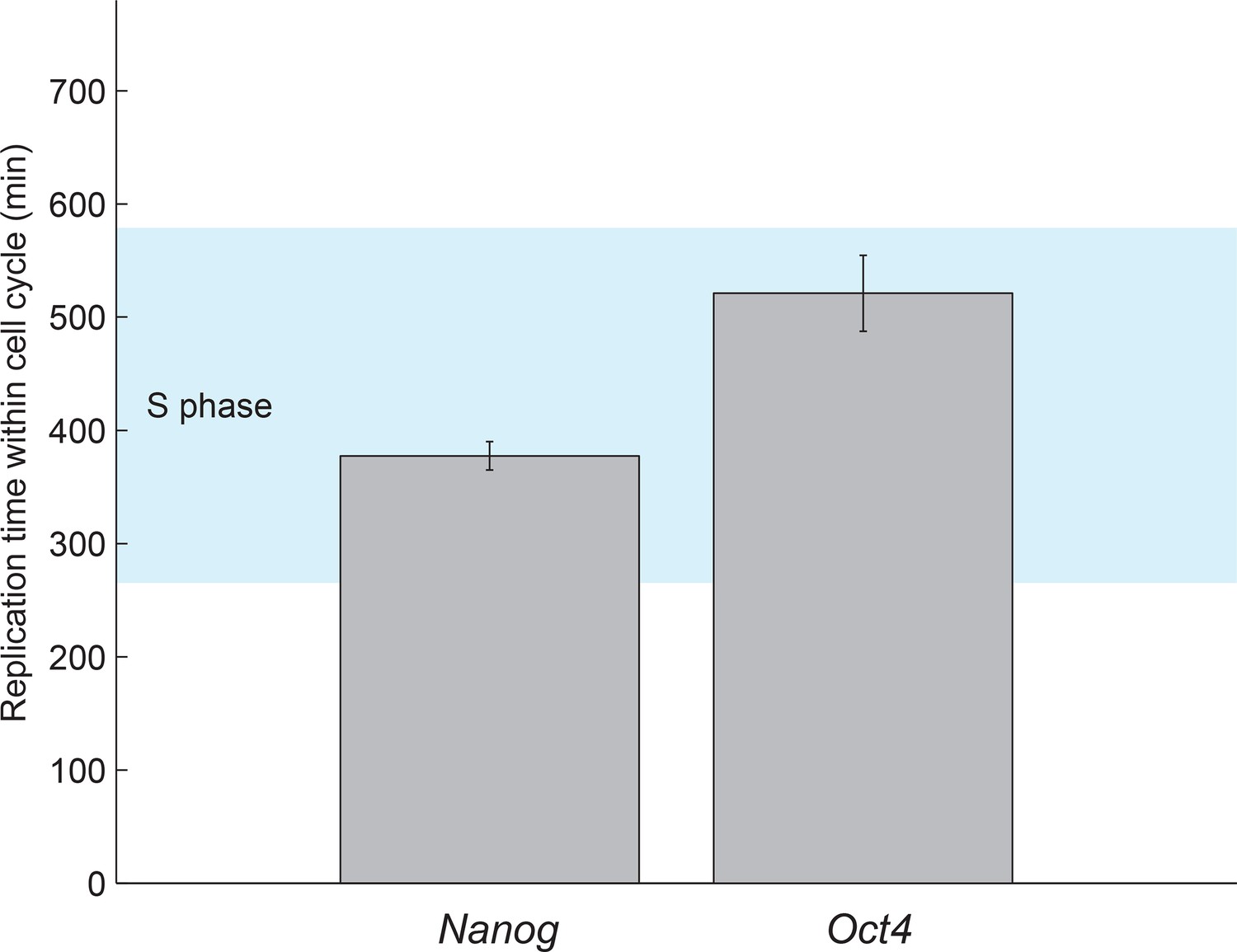
Estimated gene replication times fall within S phase.
The boundaries of S phase were estimated from the fit of the cell cycle model (see Figure 1F and Figure 1—figure supplement 5). The gene replication times estimated from a step-function fit to nascent mRNA over time (Figure 3B) fall within S phase for both Oct4 and Nanog. Error bars represent SEM from 3 experiments with >600 cells per experiment.
Figure 3—figure supplement 5
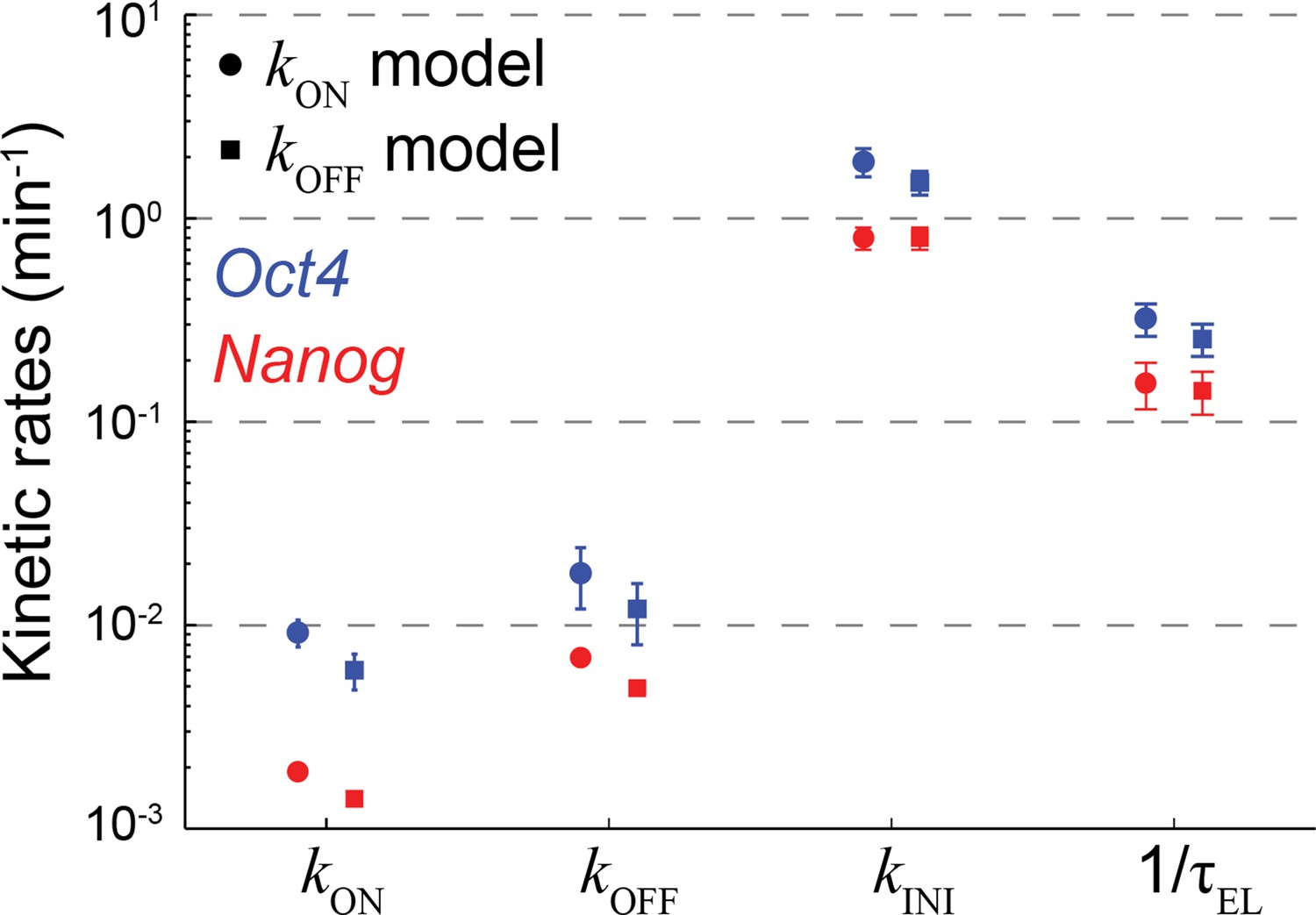
The effect of model representation of dosage compensation on the estimated rates of transcription.
The probabilistic rates of the transcription process and the gene elongation rate for Oct4 (blue) and Nanog (red). The rates were estimated from the best theoretical fit of the mature and nascent mRNA distributions (Figure 3C), using two versions of the stochastic 2-state model for gene activity. The models differ in their representation of dosage compensation: The kON model (circles) includes a decrease in the rate of gene activation following gene replication (Figure 3A), whereas the kOFF model (squares) includes instead an increase in the rate of gene inactivation. For each model, the amount of dosage compensation was calculated to reflect the measured increase in the number of nascent mRNA over time (Figure 3B). Error bars represent SEM from 3 experiments with >600 cells per experiment.
Additional files
-
Supplementary file 1
Literature estimates of transcription parameters used in this study.
- https://doi.org/10.7554/eLife.12175.017
-
Supplementary file 2
Estimated parameters of transcription for Oct4 and Nanog.
- https://doi.org/10.7554/eLife.12175.018
-
Supplementary file 3
Sequences of smFISH probes.
- https://doi.org/10.7554/eLife.12175.019
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Single-cell analysis of transcription kinetics across the cell cycle
eLife 5:e12175.
https://doi.org/10.7554/eLife.12175




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}