The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition
- University of Georgia, United States
- Carver College of Medicine, University of Iowa, United States
- Howard Hughes Medical Institute, University of Iowa, United States
- Mount Sinai Hospital, Canada
- The Hospital for Sick Children, University of Toronto, Canada
- University of Toronto, Canada
- Blizard Institute, United Kingdom
- Queen Mary University of London, United Kingdom
- Wellcome Trust Sanger Institute, United Kingdom
- University of California, Los Angeles, United States
Figures
Figure 1
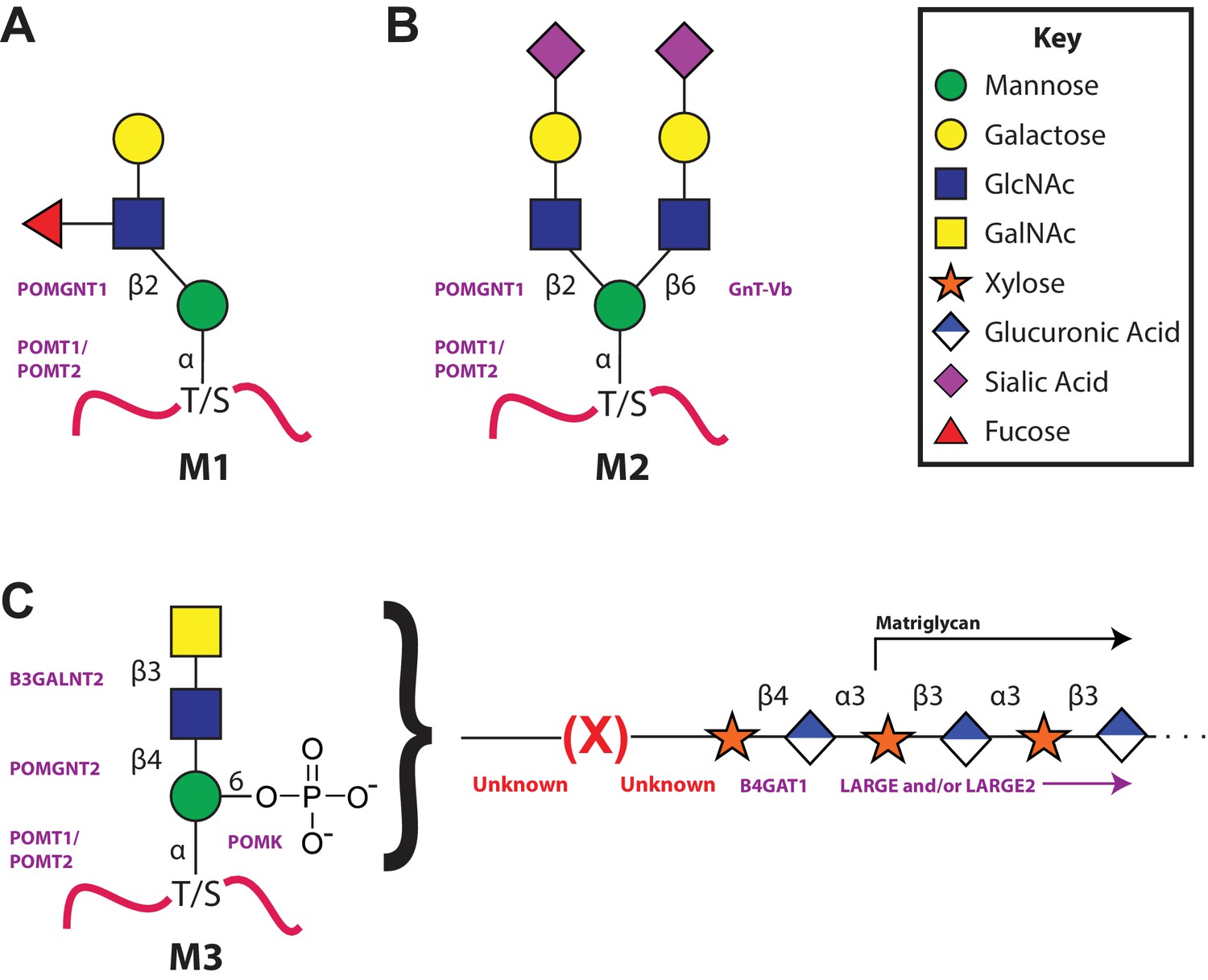
The 3 subclasses of the O-Mannosylation pathway.
Following addition of Man to serine/threonine residues on protein substrates by POMT1/2 there is a divergence in elaboration. Known enzymes are displayed in purple and unknown enzymes prior to this manuscript are displayed in red. (A) POMGnT1 adds a beta-2 linked GlcNAc onto the underlying Man. Shown is just one potential extended M1 glycan structure. (B) Following the action of POMGnT1, GnT-Vb can add a beta-6 linked GlcNAc onto a M1 glycan structure to convert it to a M2 glycan structure. Shown is just one potential extended M2 glycan structure. (C) Instead of extension in a beta-2 linkage, M3 glycan structures get the core Man extended by POMGNT2 with a beta-4 linked GlcNAc. This disaccharide is further elaborated to contain a phosphodiester linkage to an unknown moiety (shown as (X)) that becomes modified with repeats of (Xyl-GlcA).
Figure 2 with 2 supplements
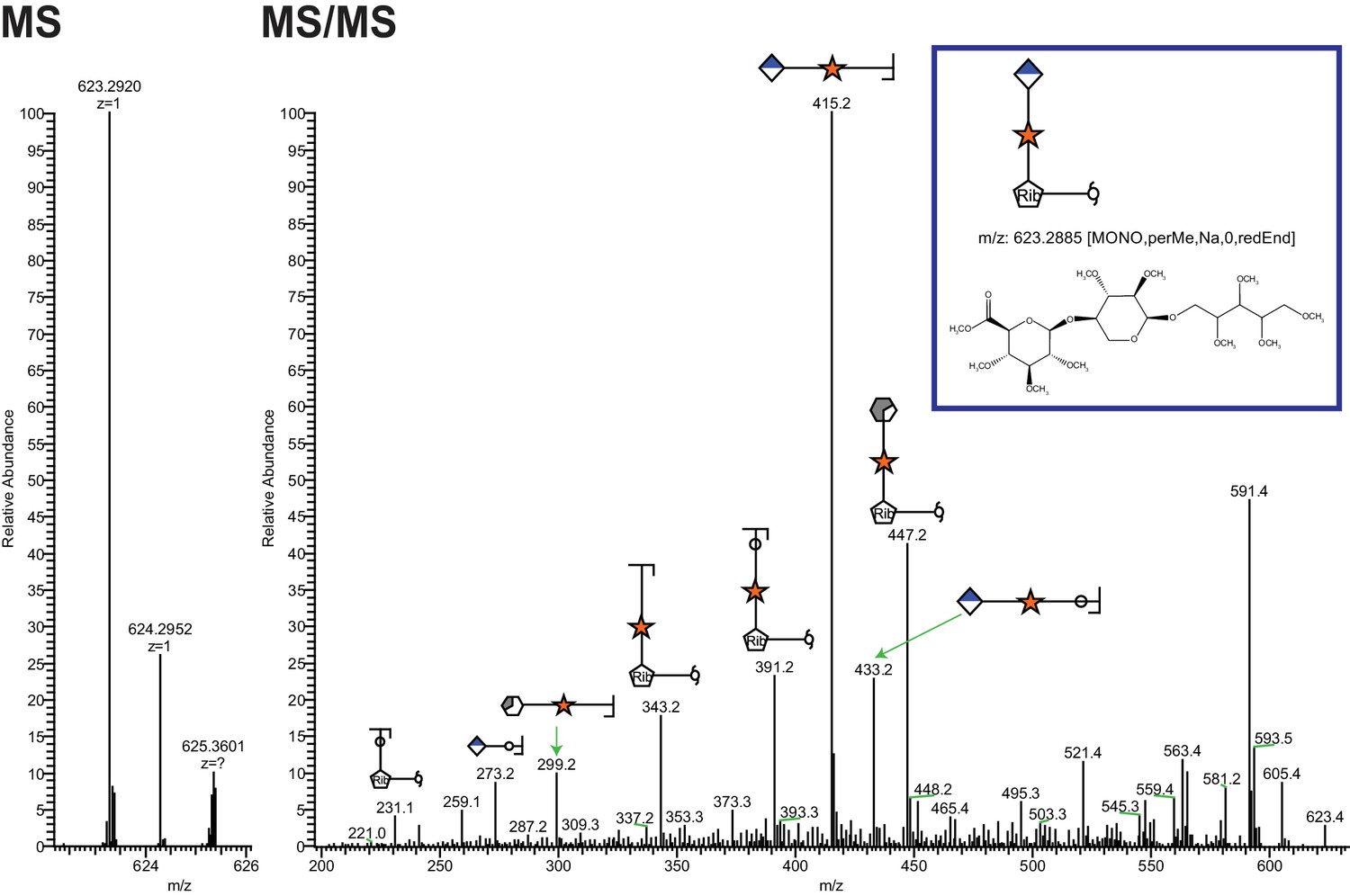
Ribitol-Xyl-GlcA is released from α-DG upon cleavage of phosphodiester linkage.
Following treatment of purified α-DG-340 with aqueous HF, released glycans were captured, permethylated and analyzed by tandem mass spectrometry. On the left is a zoom view of the full MS scan demonstrating the intact mass and to the right the fragmentation pattern leading to the identification of GlcA-Xyl-pentitol. Experiments were performed in triplicate with identical findings each time.
Figure 2—figure supplement 1
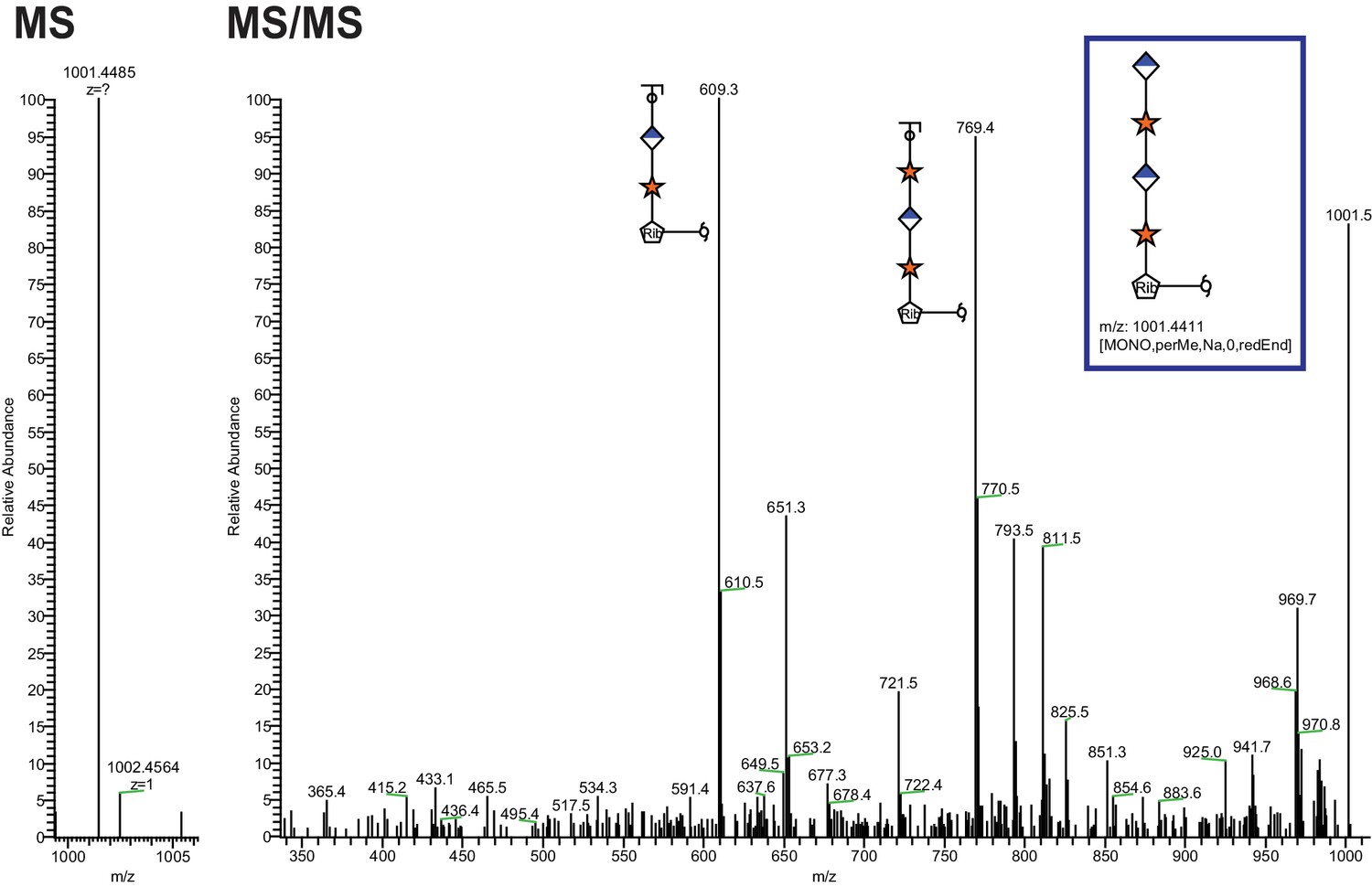
Extended Ribitol-(Xyl-GlcA)n Structures are also observed.
Identical analysis as in Figure 1 of a more extended structure (GlcA-Xyl)2-pentitol.
Figure 2—figure supplement 2
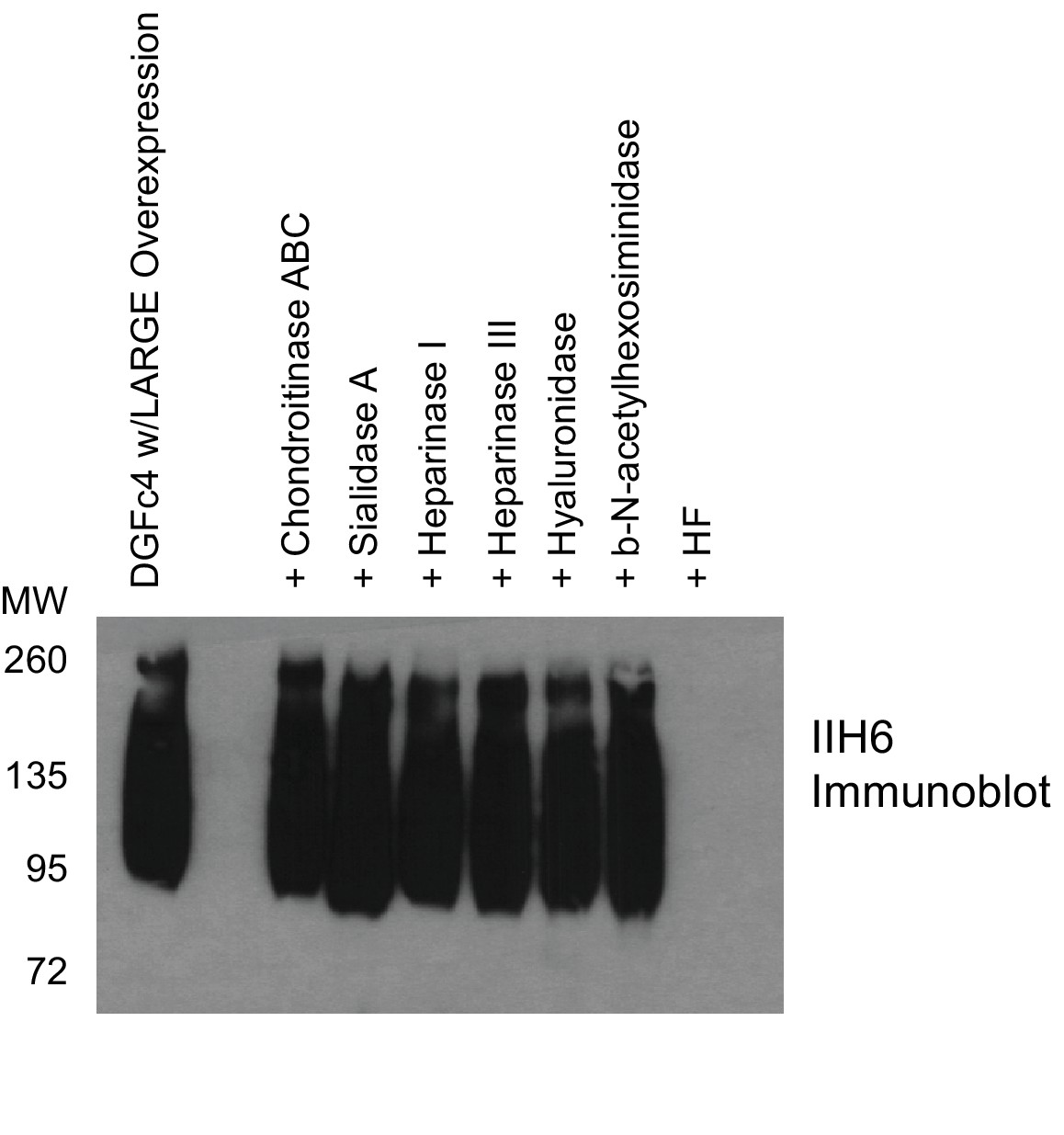
Phosphodiester cleavage but not glycosidase treatment abolishes IIH6 reactivity on α-DG.
A purified C-terminal truncated version of α-DG (expressed from DGFc4) from HEK293H cells also overexpressing LARGE was treated with a variety of glycosidases or treated with aqueous HF overnight and then separated by SDS-PAGE and immunoblotted with IIH6 to detect functionally glycosylated α-DG. Only HF treatment abolished IIH6 reactivity. One representative result of three is shown.
Figure 3 with 1 supplement
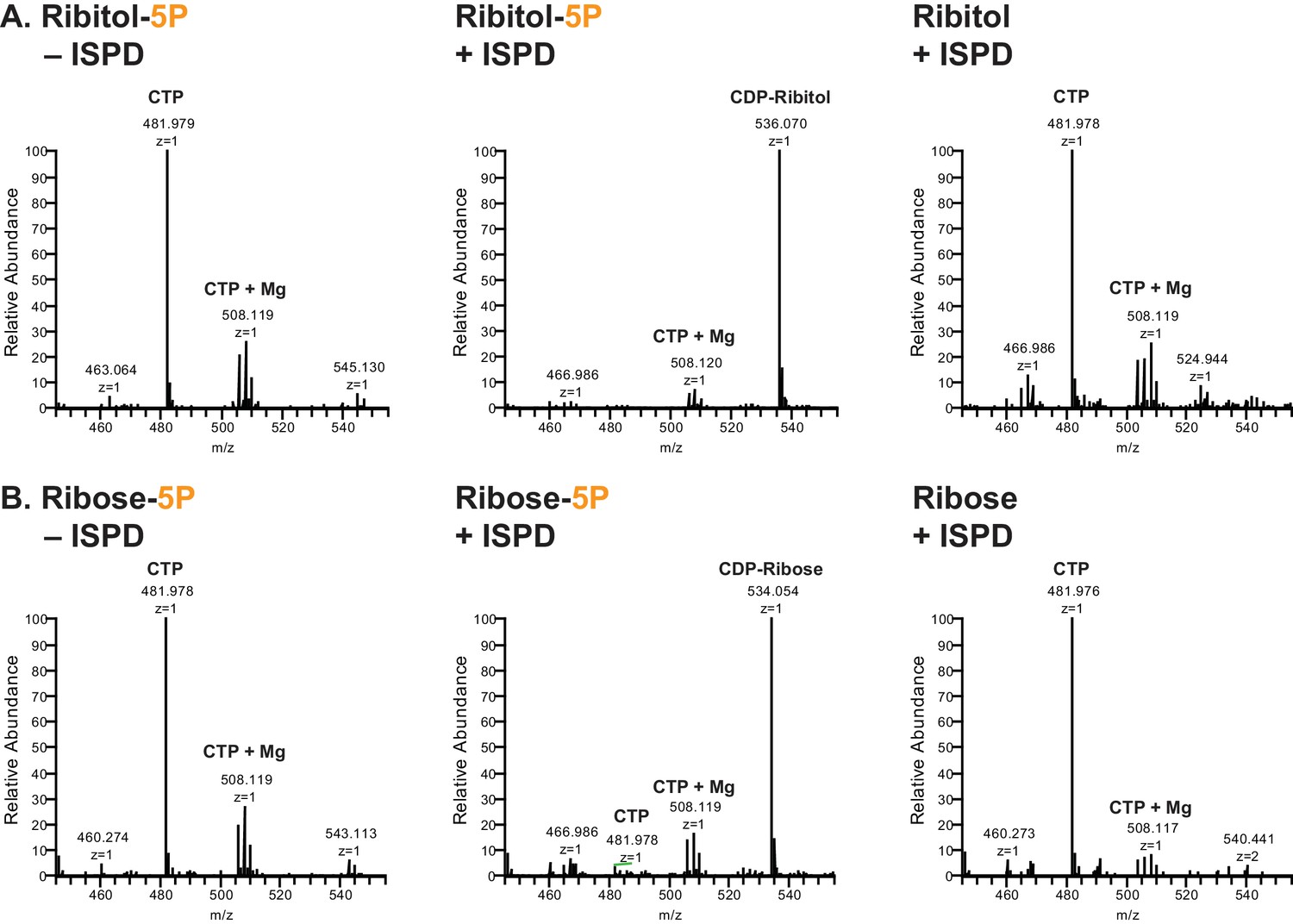
ISPD synthesizes CDP-Ribitol from CTP and Ribitol-phosphate.
(A) Purified recombinant ISPD (+ISPD) or buffer control (-ISPD) was incubated with CTP and either ribitol-5-phosphate (Ribitol-5P) or ribitol (Ribitol) and the donor substrate (CTP, MW 481.98) and/or product (CDP-ribitol, MW 536.07) was detected by mass spectrometry. CDP-ribitol was only observed in the presence of ISPD, CTP, and Ribitol-5P. (B) Purified recombinant ISPD (+ISPD) or buffer control (-ISPD) was incubated with CTP and either ribose-5-phosphate (Ribose-5P) or ribose (Ribose) and the donor substrate (CTP, MW 481.98) and/or product (CDP-ribose, MW 534.05) detected by mass spectrometry. CDP-ribose was only observed in the presence of ISPD, CTP, and Ribose-5P. Note that CDP-ribose has a molecular weight that is 2 daltons less than CDP-ribitol. Shown are representative images of an N = 3.
Figure 3—figure supplement 1
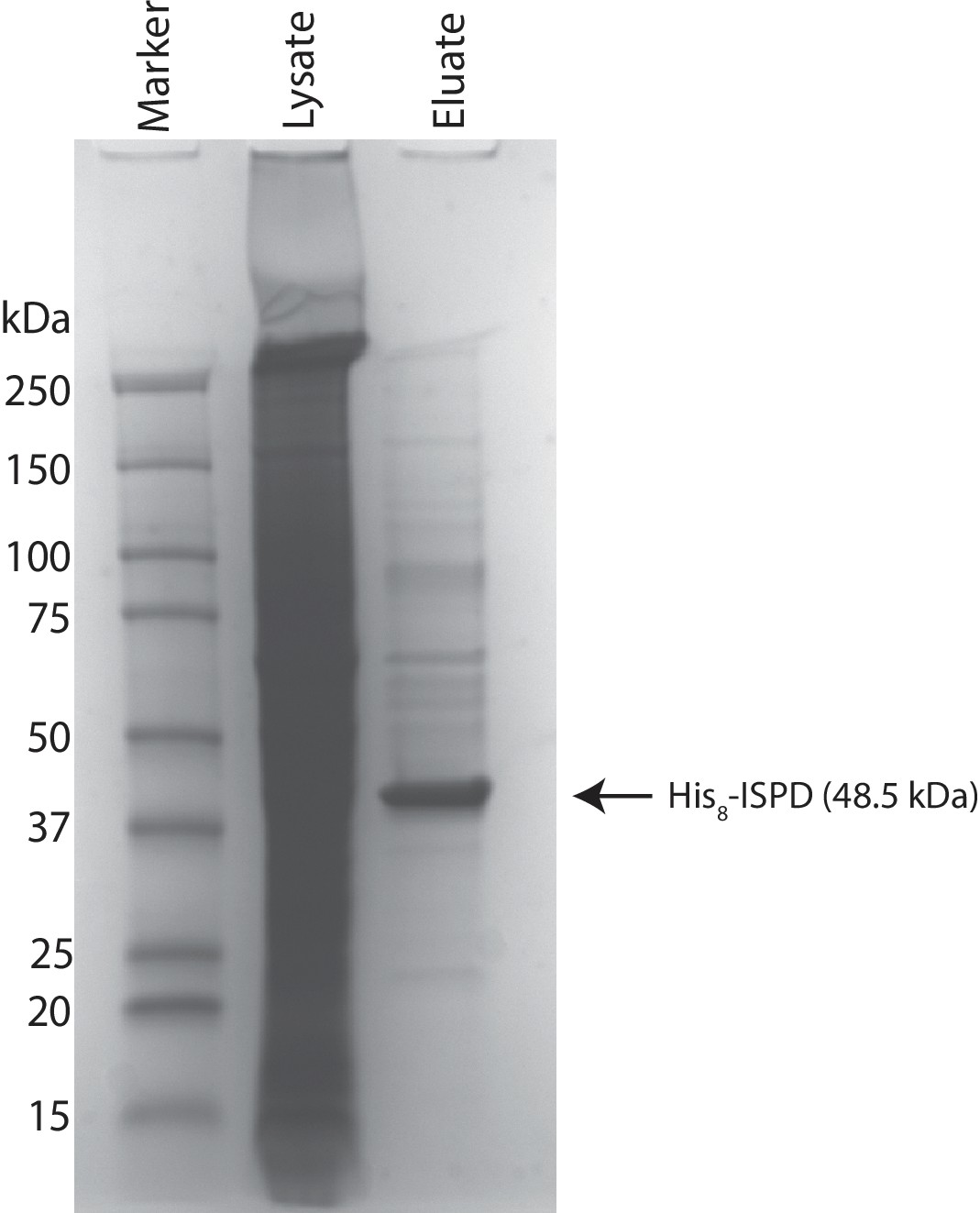
Coomassie stained SDS-PAGE gel of purified ISPD.
His8-tagged ISPD was expressed in HEK293F cells and purified from whole cell lysates via immobilized metal affinity chromatography and the resulting purified protein was separated via SDS-PAGE and visualized with Coomassie-G250 staining.
Figure 4 with 2 supplements
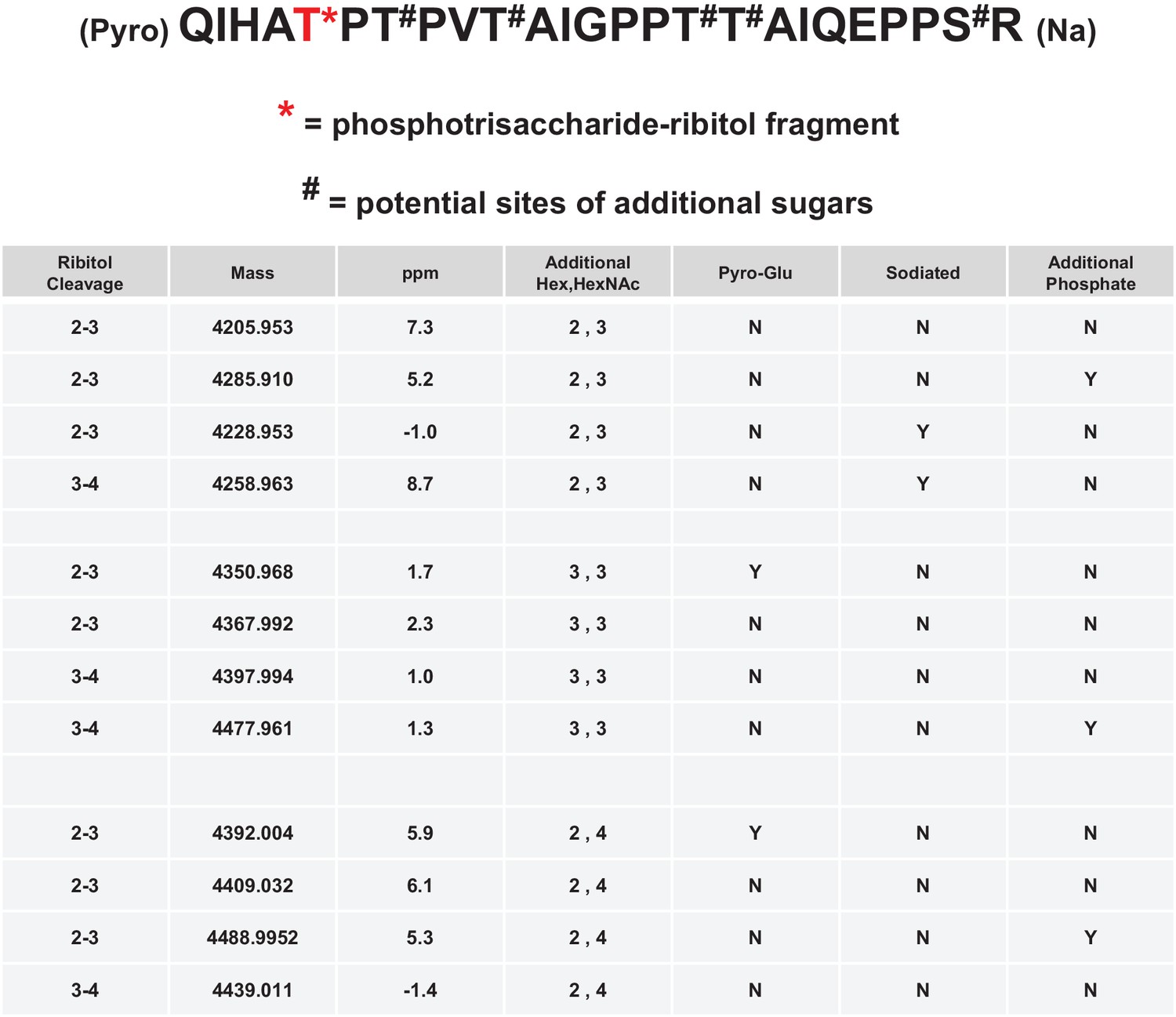
Mild periodate cleavage reveals that ribitol is connected to the M3 glycan.
Following treatment of purified α-DG-340 under mild periodate conditions, the protein was reduced, alkylated, and digested with trypsin. The resulting peptides were analyzed by tandem mass spectrometry. The α-DG-340 tryptic peptide was detected with multiple glycoforms containing a phosphotrisaccharide glycopeptide. Specifically, at least 12 tryptic peptides were identified at less than 9 ppm mass accuracy that contained the phosphotrisaccharide with a ribitol fragment at threonine 317 (the red T*) in two separate analyses. These peptides differed in additional glycosylation by either hexose (Hex) or HexNAc sugars at additional hydroxyl amino acids (indicated by #). We also observed that the N-terminal glutamine was cyclized to pyroglutamic acid on some peptides (Pyro) and that some glycopeptides were sodiated (Na). Furthermore, we noticed the presence of a second phosphate on some phosphoglycopeptides. The 12 peptides observed are grouped by having the same number of additional Hex and HexNAc glycans (2,3 or 3,3 or 2,4). For all 3 sets of glycopeptides we observed glycopeptides that had ribitol fragments between C2 and C3 as well as C3 and C4 indicating that C2, C3, and C4 are not involved in linkages to other moieties. We also observed for each set of glycopeptides the presence of an additional phosphate.
Figure 4—figure supplement 1

Tandem MS/MS spectra of glycopeptides.
Shown are MS/MS spectra for 2 of the 12 glycopeptides described in Figure 3. Glycan oxonium ions as well as b- and y- fragments are labeled on the spectra. The amino acid sequence displays the observed b- (blue) and y-(red) ions observed as well as highlights the glycan modified amino acids in red. In all cases the phosphotrisaccharide-ribitol fragment (2–3 or 3–4) is assigned to Thr-317 (the first red labeled T in the sequence).
Figure 4—figure supplement 2

Dot blot analysis reveals loss of IIH6 signal for α-DG treated with mild periodate.
Following treatment (or mock treatment with the omission of NaIO4) of purified α-DG-340 under mild periodate conditions, the protein was spotted at 3 ug or 0.3 ug and immunoblotted with the IIH6 antibody. Mild periodate treatment almost completely abolishes IIH6 reactivity even at 3 ug spotting. Shown is one of the two blots performed.
Figure 5 with 2 supplements
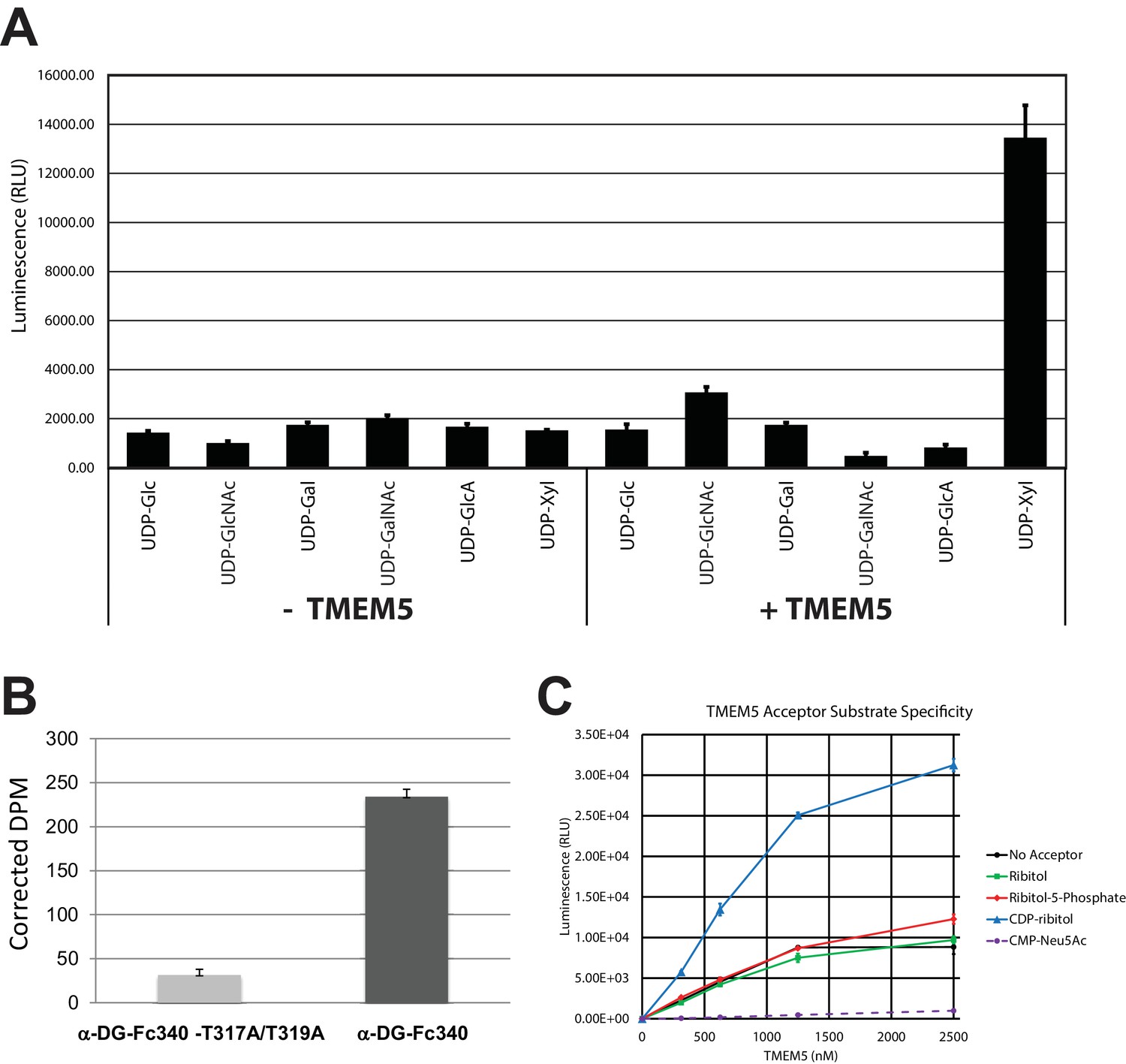
TMEM5 is a UDP-Xyl transferase that modifies phosphodiester linked ribitol.
(A) Various UDP-sugar nucleotides were incubated with (+) or without (-) purified transmembrane deleted and tagged TMEM5 and then analyzed for hydrolysis using the UDP-Glo assay (N = 3). The formation of UDP was only observed when dTM-TMEM5 was incubated with UDP-Xyl. (B) Recombinant purified full length TMEM5 and radiolabeled UDP-Xyl [Xyl-14C] was incubated with either purified α-DG-Fc340 or mutant α-DGFc340 where the sites for M3 addition had been mutated to alanines (α-DG-Fc340-T317A/T319A) and incorporation of radioactivity into the proteins were determined by scintillation chromatography (N = 3). α-DGFc340 that contains the sites for M3 addition is the preferred substrate for addition of radiolabeled Xyl by TMEM5. (C) dTM-TMEM5 (2500, 1250, 625, or 312.5 nM) and 50 µM UDP-Xyl donor substrate were incubated without or with 1 mM of the indicated acceptor molecules in 0.1 M MES pH 6.0 and 10 mM MgCl2 for 18 hr at 37°C, and the release of UDP was detected by the UDP-Glo assay. RLU: Relative Luminescence Units. Data are represented as the mean of three trials and error bars represent standard deviation.
Figure 5—figure supplement 1
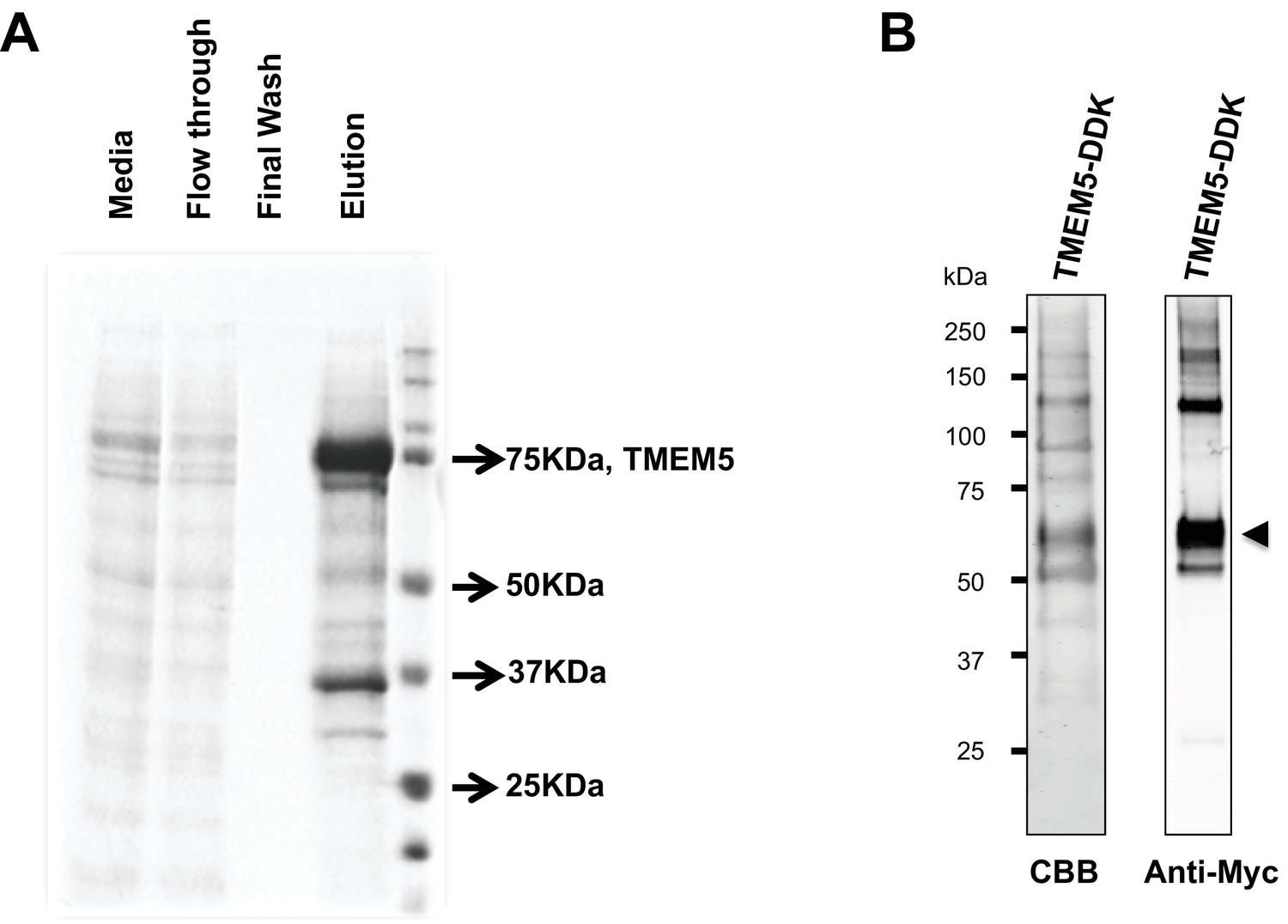
Analysis of purified TMEM5 constructs.
(A) GFP/HIS-tagged dTM-TMEM5 was expressed in HEK293F cells and purified from the media via immobilized metal affinity chromatography and the resulting purified protein was separated via SDS-PAGE and visualized with Coomassie Brilliant blue and used in experiments shown in Figure 5A. (B) Purified full length TMEM5 C-terminally tagged with Myc/DDK-epitope tag was purchased from Origene and analyzed by Coomassie Brilliant blue and immunoblotting with anti-C-Myc following separation via SDS-PAGE and used in experiments shown in Figure 5B.
Figure 5—figure supplement 2
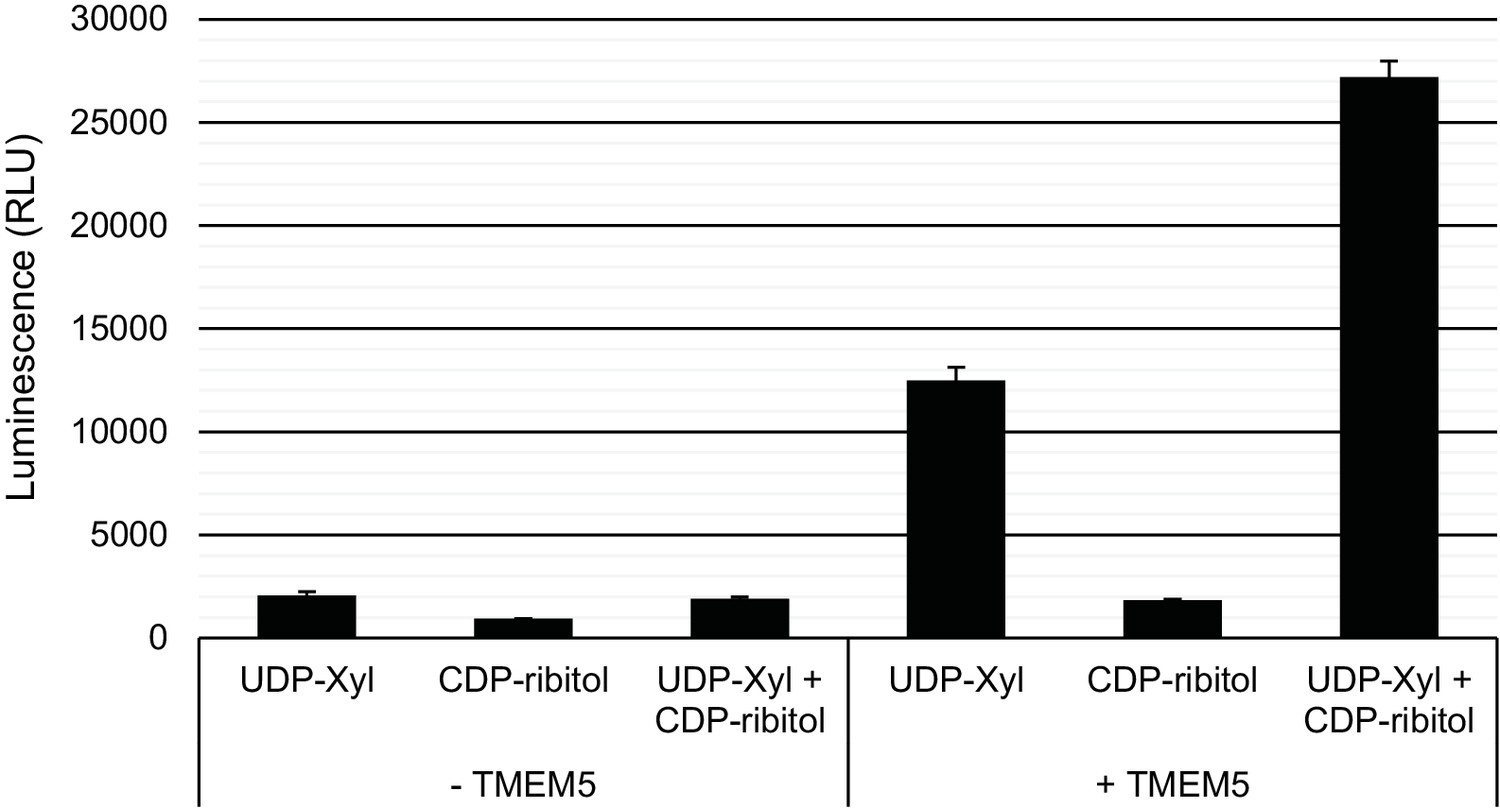
Specificity of TMEM5 for CDP-ribitol as an acceptor.
GFP-dTMTMEM5 was incubated with either 50 µM UDP-Xyl, 1 mM CDP-ribitol, or both in 0.1 M MES pH 6.0 and 10 mM MgCl2 for 18 hr at 37°C, and the release of UDP (or CDP) was detected by the UDP-Glo assay. RLU: Relative Luminescence Units. Data are represented as the mean of three trials and error bars represent standard deviation.
Figure 6 with 1 supplement

Knockdown of zebrafish tmem5 recapitulates characteristic defects in WWS.
(A) Compared with uninjected control and p53 MO injected embryos, embryos injected with tmem5 MO alone or together with p53 MO developed mild to severe hydrocephalus (asterisk) from 32 hr post fertilization (h.p.f.) (95% and 88%, respectively). Each bar represents a combination of two independent blindly-scored experiments. n = 114–150 embryos. Scale bar, 100 µm. (B) One-way ANOVA analysis revealed that tmem5 MO injected embryos have significantly reduced eye size (bracket in panel a) at 48 -h.p.f. Inhibition of MO off-target effects by co-injection of p53 MO does not rescue the reduced eye size. n = 15 embryos in each group. Error bars, s.d. **p< 0.01. NS, not significant. (C) Knockdown of tmem5 compromised the sarcolemma integrity as indicated by EBD infiltrated muscle fibers (arrows) at 48 h.p.f. Muscle fibers undergoing degeneration pull away from chevron-shaped myosepta. Scale bar, 50 µm. (D) Compared with uninjected control, western blotting with IIH6 antibody showed a 44% reduction in glycosylated α-dystroglycan in tmem5 MO-injected embryos at 48 h.p.f. Equal loading was demonstrated by Ponceau S and unknown glycoproteins (~35 kDa) detected by IIH6 in all lanes.
Figure 6—figure supplement 1
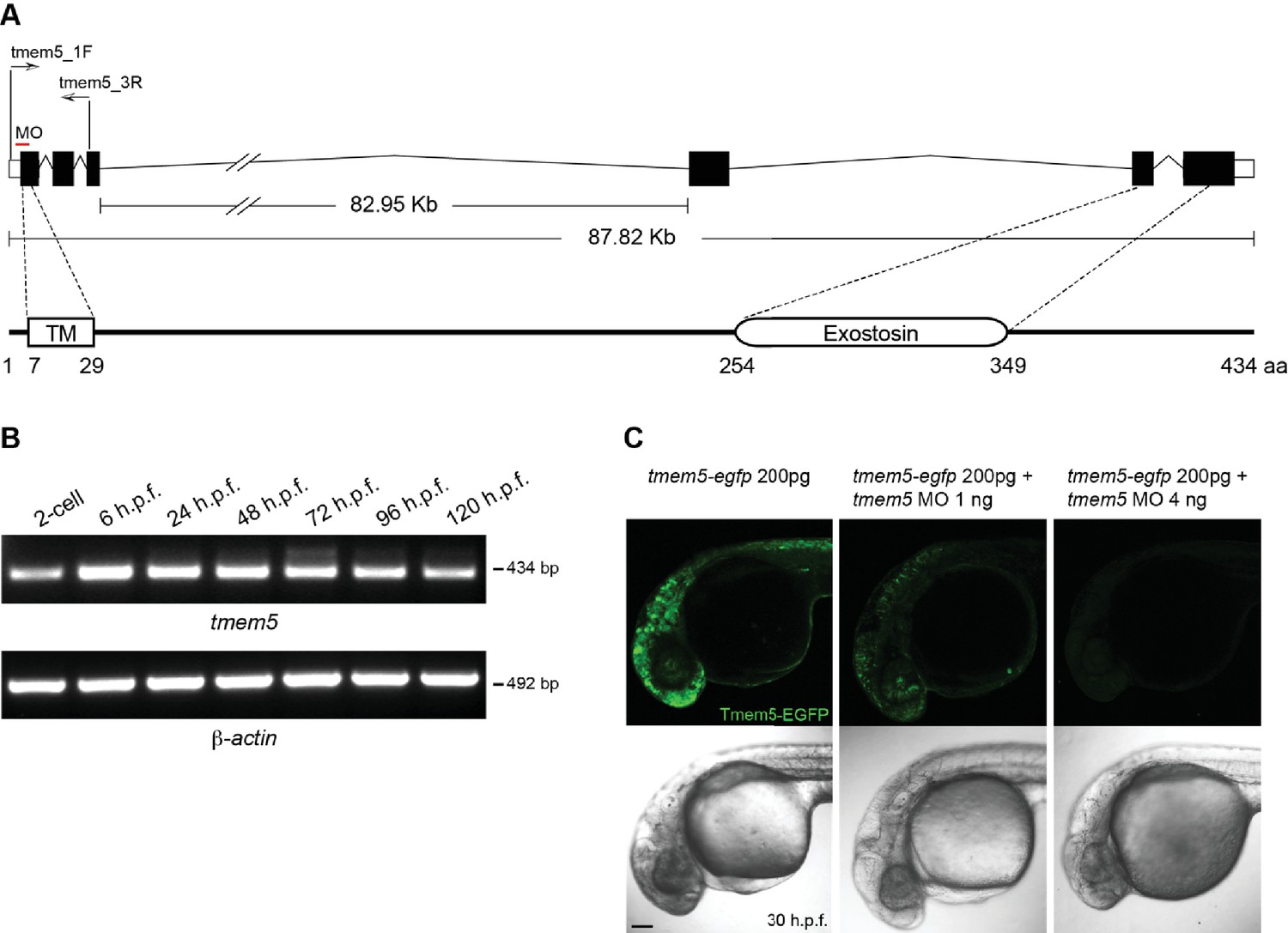
Zebrafish tmem5 temporal gene expression and MO specificity.
(A) A schematic representation of the zebrafish tmem5 transcript and protein domains. White boxes indicate the untranslated region (UTR). Black boxes indicate coding exons. The translational blocking MO target site and PCR primer binding sites are indicated. The amino acid sequence of zebrafish Tmem5 is ~73% similar and ~55% identical to that of human orthologue. Conserved protein domains are boxed: TM, transmembrane domain (7–29 aa); Exostosin (GT-47 homology) domain (254–349 aa). (B) The expression of tmem5 transcript was detected by RT-PCR using primer tmem5_1F and tmem5_3R. Zebrafish tmem5 is expressed maternally at the 2-cell stage, during the maternal-zygotic transition (6 h.p.f) and zygotically at later stages (24, 48, 72, 96 and 120 h.p.f.). (C) To assess tmem5 MO specificity, tmem5-egfp mRNA (200 pg) was injected into single cell, followed by injection of tmem5 MO (1 ng or 4 ng) into the yolk flow. Embryos that received only tmem5-egfp mRNA showed a strong Tmem5-EGFP expression at 30 h.p.f., whereas embryos received a second injection of 1 ng or 4 ng tmem5 MO showed a moderate and strong reduction in Tmem5-EGFP expression, respectively. These results confirm the specificity of tmem5 MO. n = 20 embryos in each group. Scale bar, 100 µm.
Figure 7 with 2 supplements
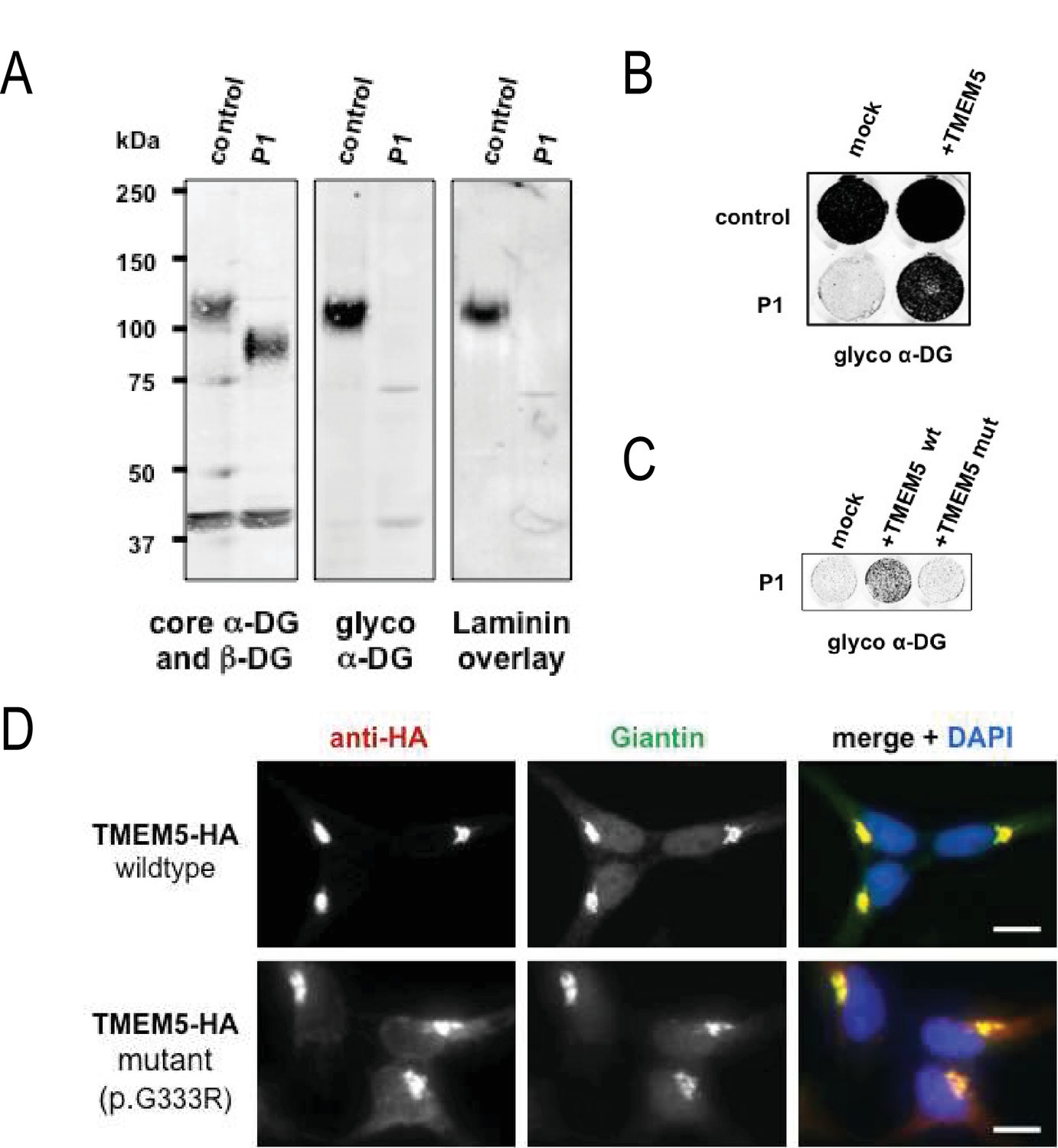
A missense mutation of TMEM5 in a WWS family leads to a loss of functional α-DG.
(A) Analysis of α-DG glycosylation status in TMEM5-deficient WWS patient (P1) dermal fibroblasts. WGA-enriched cell lysates from control and TMEM5 P1 fibroblasts were subjected to biochemical analysis. α-DG glycosylation status as assessed by western blotting using antibodies against the glycosylated form of α-DG (IIH6), core α-DG (G6317), and by laminin overlay assay (N = 3). An antibody against β-DG (AP83) was used to assess loading. Apparent molecular masses are indicated. (B) On-Cell western-based complementation assay (N = 3) of TMEM5-WWS patient (P1) fibroblasts after adenoviral gene transfer with wildtype TMEM5 expression construct. Rescue of α-DG functional glycosylation was monitored with α-DG glyco (IIH6) antibodies. (C) On-Cell western-based complementation assay (N = 3) of P1 fibroblasts after nucleofection with a wild-type or mutant (p.G333R) human TMEM5 expression construct. Rescue of α-dystroglycan functional glycosylation was detected with antibody to glycosylated α-dystroglycan (glyco α-DG, IIH6). (D) Subcellular localization of HA-tagged wildtype and WWS mutant TMEM5 protein, as assessed by immunofluorescence. HEK293T cells stably expressing HA-tagged proteins were stained with anti-HA (red), anti-Giantin (Golgi marker, green) and 4 ́,6-diamidino-2-phenylindole (DAPI, nuclei, blue). Individual stainings for HA and Giantin are shown in greyscale and a merged image is shown in color. Scale bars indicate 10 µm.
Figure 7—figure supplement 1
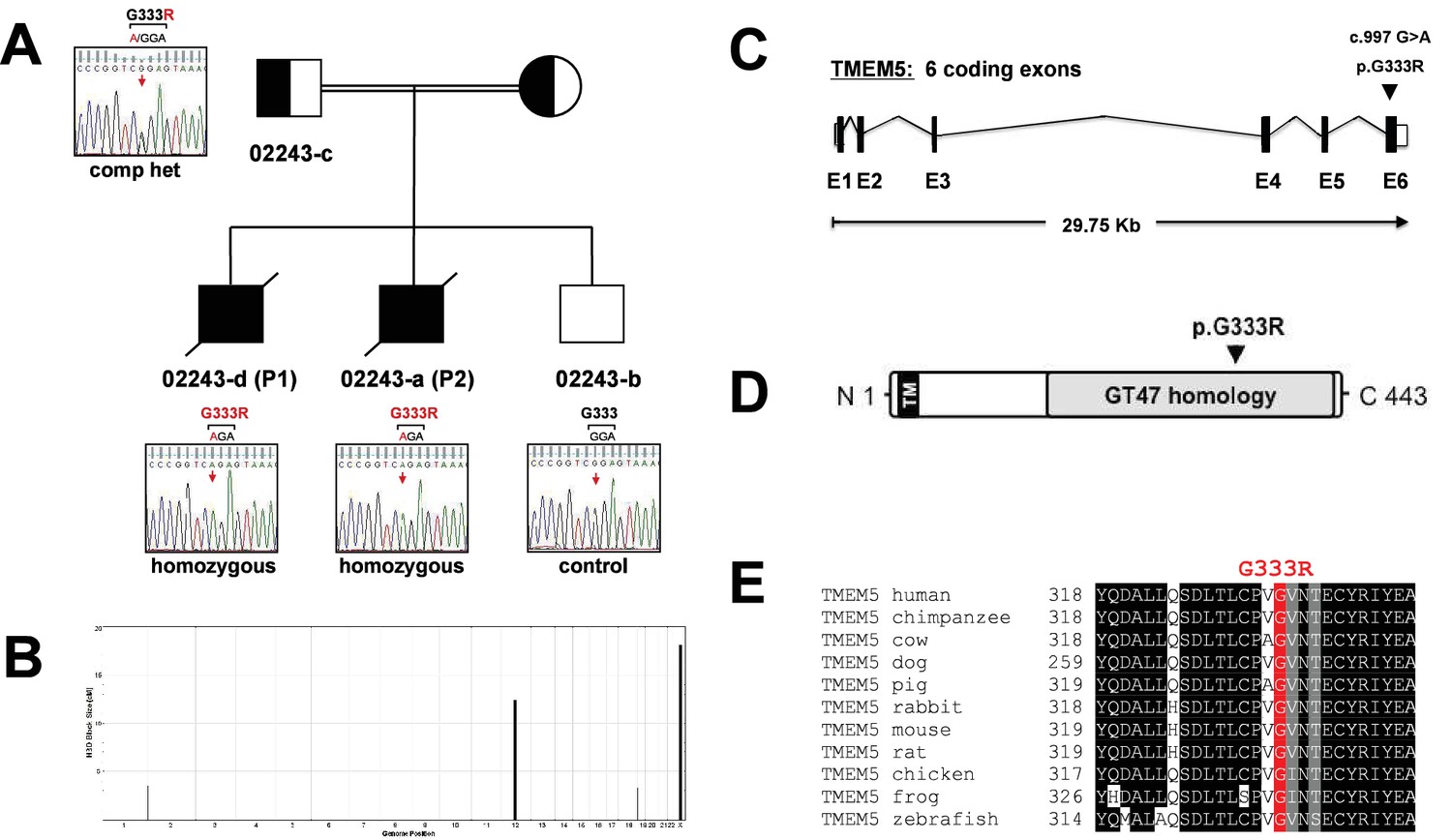
Identification and validation of TMEM5 as the disease gene in a WWS family.
(A) Pedigree structure of consanguineous family 02243. Sanger sequencing confirmed the sequence variants in 022443 family members identified by whole exome sequencing in P1 (02243-d). (B) Homozygosity-by-descent (HBD) shared between the affected siblings (P1 NS P2) but not with the unaffected sibling. HBD block size is plotted along the y-axis in cM and chromosomal position along the x-axis. (C) Schematic representation (not drawn to scale) of the TMEM5 exon-intron gene structure. Human TMEM5 cDNA (NM_014254.2, 5,524 bp) contains 6 coding exons spread across 29,756 bp genomic DNA (NG_033244.1). The identified pathogenic TMEM5 protein change is indicated. Coding exons are indicated by black boxes, and untranslated regions (UTR) by open boxes. (D) Schematic representation of predicted functional domains of TMEM5 protein (443aa). The transmembrane domain (TM) is indicated in black and predicted catalytic domain with homology to CAZy glycosyltransferase (GT47) family is highlighted in grey. The location of the identified amino acid substitution in the TMEM5-WWS family (p.G333R) is indicated. (E) Partial alignment of TMEM5 protein sequences indicates high conservation of the mutated residue p.G333 across various species.
Figure 7—figure supplement 2
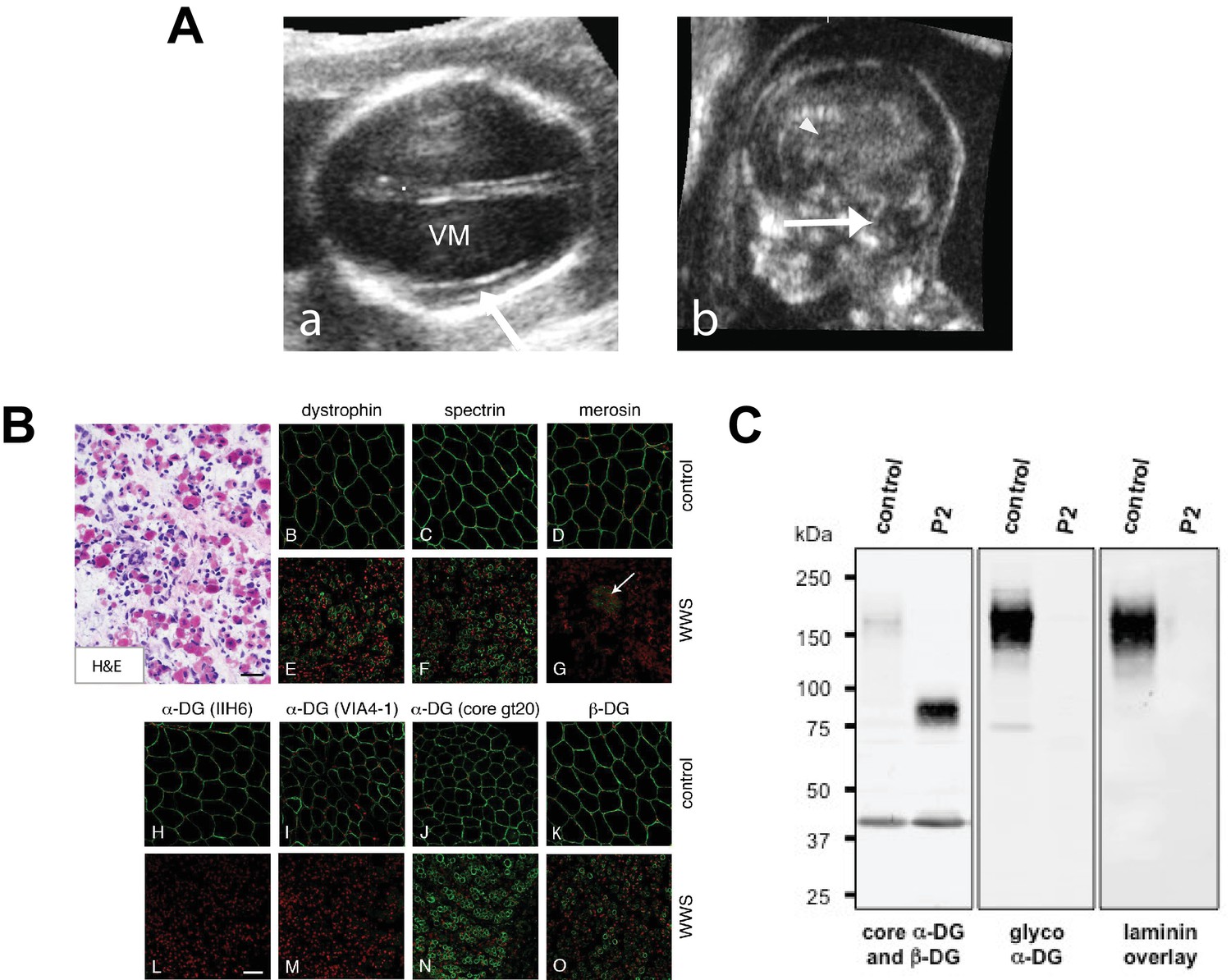
Clinical presentation and α-DG glycosylation defect in TMEM5-WWS patient P2.
(A) (a) Fetal ultrasound axial view at 17 weeks shows marked ventriculomegaly (VM) and smooth cortex without clear early Sylvian fissure formation (arrow), (b) Median view at 17 weeks shows kinked brainstem with prominent space under the cerebellar vermis (arrow) and small malformed corpus callosum (arrowhead). (B) Hematoxylin and eosin (H&E) staining of frozen cross-sections of fetal skeletal muscle from WWS patient (P2) show severe dystrophic histopathology with muscle fiber necrosis and regeneration, as well as endomysial fibrosis (panel A; scale bar = 50 μm). The immunofluorescence with two antibodies against glycosylated α-DG reveals a complete loss of functional α-DG glycosylation (panels L and M). Merosin (laminin α2; panel G) is greatly reduced in muscle fibers, but nearly normal in intramuscular nerve twigs (arrow). Antibodies against dystrophin, α-DG core protein (gt20), β-DG, and spectrin show mildly reduced to normal staining (scale bar 100 μm). The mild reduction in dystrophin, β-DG, and spectrin staining may be due to proteolysis following the death of the fetus prior to freezing the muscle sample. (C) Western blot of skeletal muscle from control and TMEM5-WWS P2. The patient samples reveal core α-DG hypoglycosylation and loss of α-DG functional glycosylation. Glycoproteins were WGA-enriched from muscle or cell lysates (250 μg protein/lane). The immunoblot was probed with antibodies against the glycosylated form of α-DG (IIH6) and core α-DG (G6317), as well as with β-DG (AP83) as a loading control. A laminin overlay experiment shows loss of α-DG ligand binding affinity in the patient sample.
Figure 8
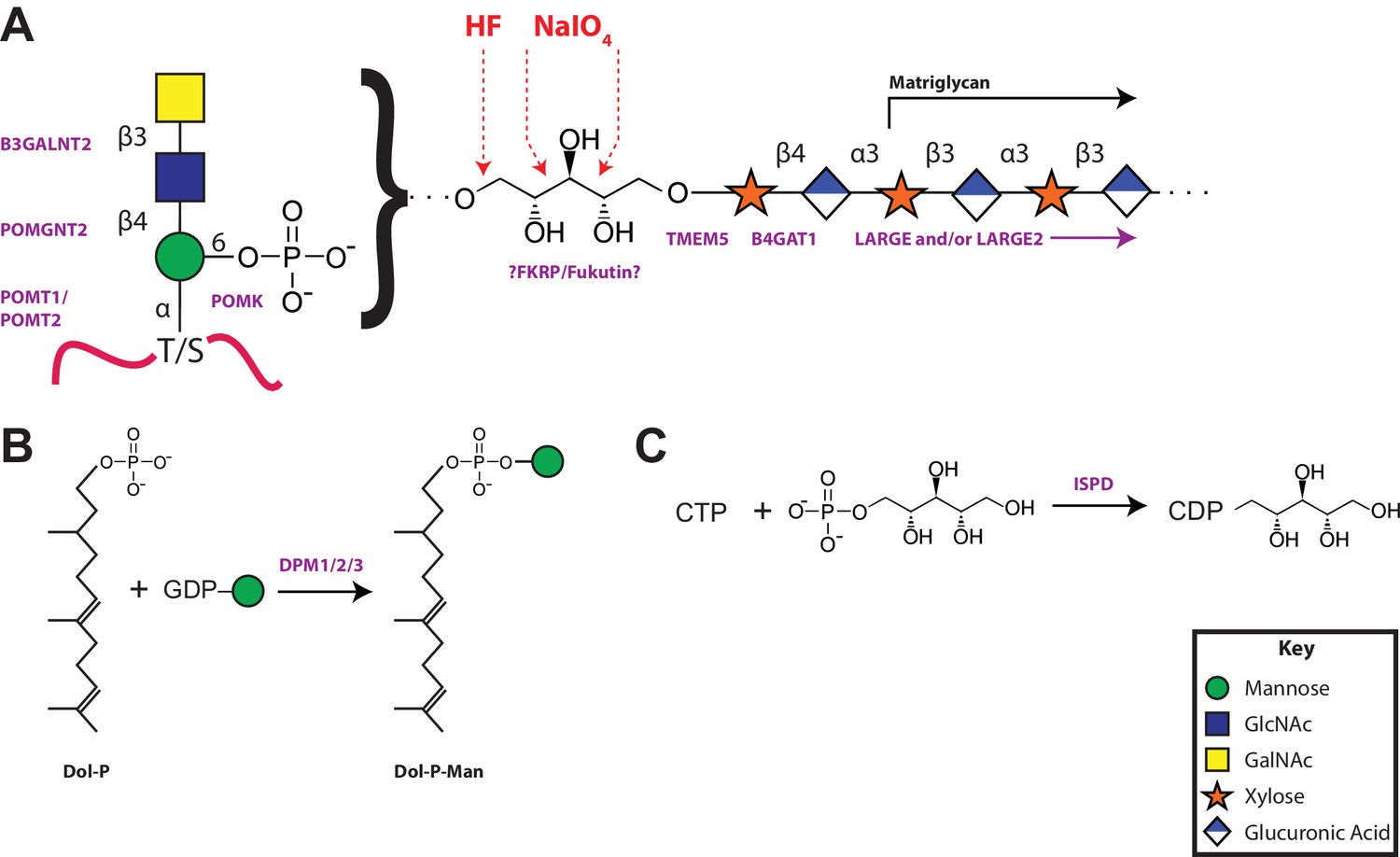
A model of the α-DG functional glycan structure and enzymes that contribute to its synthesis.
(A) The proposed functional M3 glycan structure is shown with known or putative (?FKRP/Fukutin?) enzymes involved labeled in purple that when defective lead to secondary dystroglycanopathies. Also detailed in red are the observed sites of ribitol cleavage by aqueous HF (HF) and mild periodate (NaIO4) treatment. (B) The DPM1/2/3 enzyme complex (labeled in purple) is required for the synthesis of Dolichol-phosphomannose (Dol-P-Man) from GDP-Man and Dolichol phosphate (Dol-P). (C) ISPD (labeled in purple) is required for the synthesis of CDP-ribitol from CTP and ribitol-phosphate. Defects in DPM1/2/3 and ISPD are considered tertiary dystroglycanopathies since they synthesize the sugar donors Dol-P-Man (POMT1/2) and CDP-ribitol (presumably Fukutin and/or FKRP) for subsequent glycosyltransferases.
Additional files
-
Source code 1
HBD analysis.
- https://doi.org/10.7554/eLife.14473.020
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition
eLife 5:e14473.
https://doi.org/10.7554/eLife.14473




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}