Phylogenomic analysis supports the ancestral presence of LPS-outer membranes in the Firmicutes
- Institut Pasteur, France
- Biocenter LMU, Germany
- Institut Pasteur
- University Lyon I, France
Figures
Figure 1
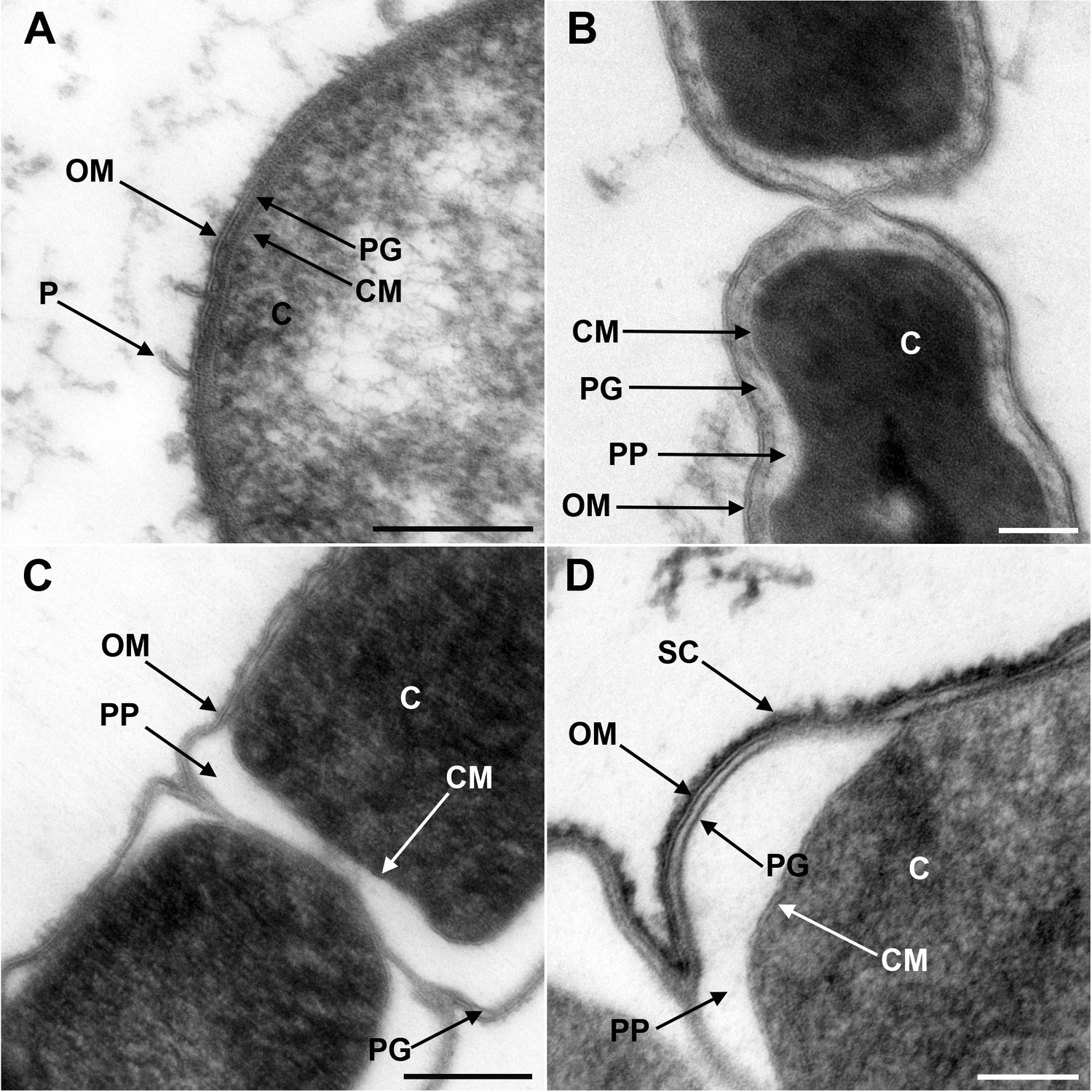
Transmission electron microscopy of a member of Negativicutes and a member of Halanaerobiales.
Ultrathin sections of high-pressure frozen cells of the Negativicutes member Megamonas rupellensis (A,C), and the Halanerobiales member Halanaerobium saccharolyticum (B,D). A Gram-negative like cell wall architecture is visible for both taxa (A,B): a cytoplasmic membrane (CM) surrounding the cytoplasm (C), a thin peptidoglycan layer (PG), and an outer membrane (OM). Pili-like structures (P) are also visible in M. rupelllensis. In some cases and due to a preparation artifact caused by swelling of the cells, the OM detaches from the IM creating an enlarged periplasmic space (PP) between two dividing cells (C,D). In these cases, the peptidoglycan becomes more apparent as it is also the case for an electron dense surface coat (SC), which might represent lipopolysaccharide (LPS) or a potential S-layer. Scale bars: 200 nm (A,C) and 100 nm (B,D).
Figure 2 with 3 supplements
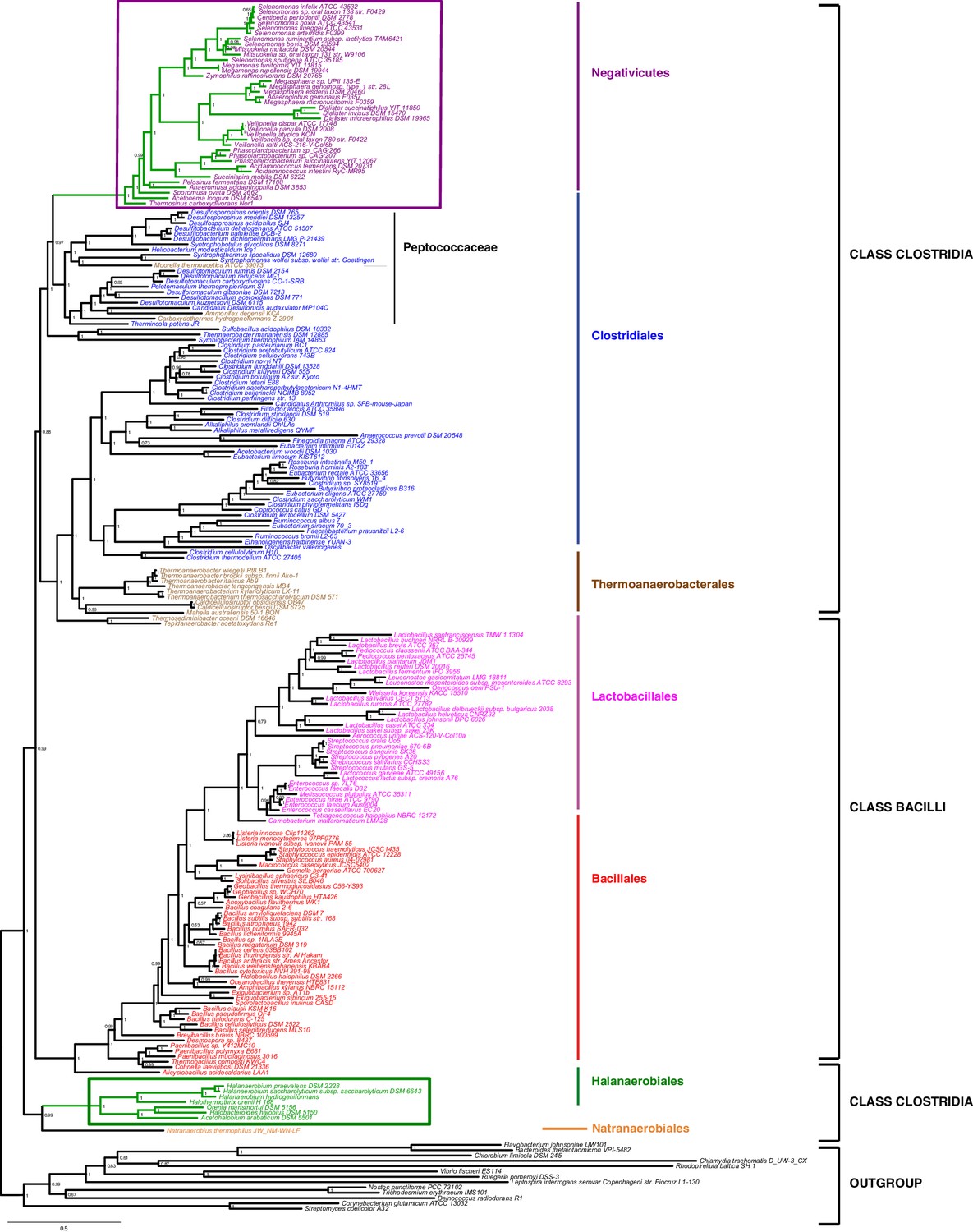
Phylum-level phylogeny of the Firmicutes.
Bayesian phylogeny of the Firmicutes based on a concatenation of 47 orthologous ribosomal proteins comprising 5551 amino acid positions and the CAT+GTR+Γ4 model. Values at nodes represent Bayesian posterior probabilities. The scale bar represents the average number of substitutions per site. For details on analyses, see Materials and methods.
Figure 2—figure supplement 1
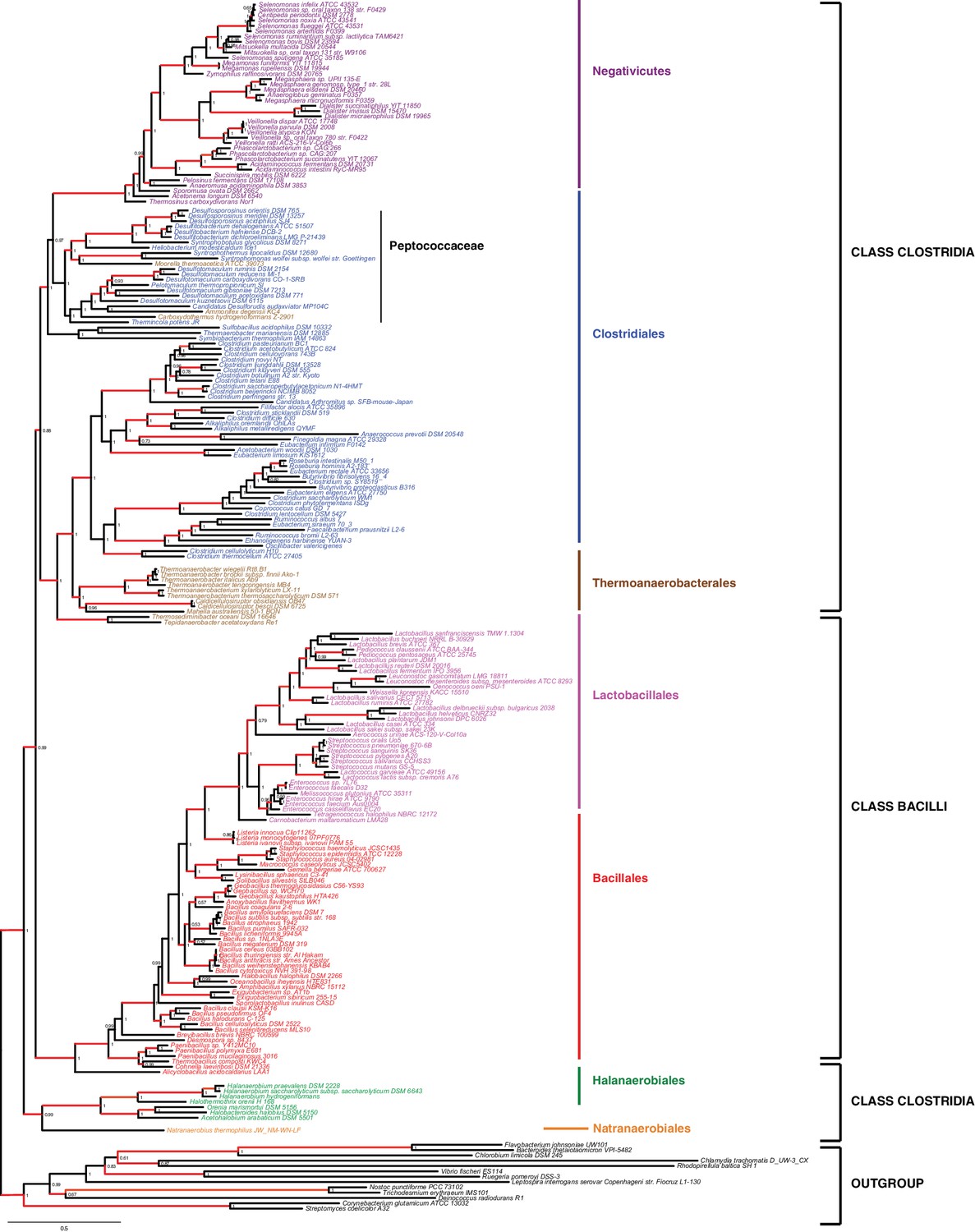
Results of IC congruence test for the 47 ribosomal proteins.
IC values were mapped onto the ribosomal protein concatenation phylogeny shown in Figure 2. Branches in red indicate congruence among markers according to IC tests. Raw results of the test are provided as Additional Data in Dryad (Antunes et al., 2016).
Figure 2—figure supplement 2
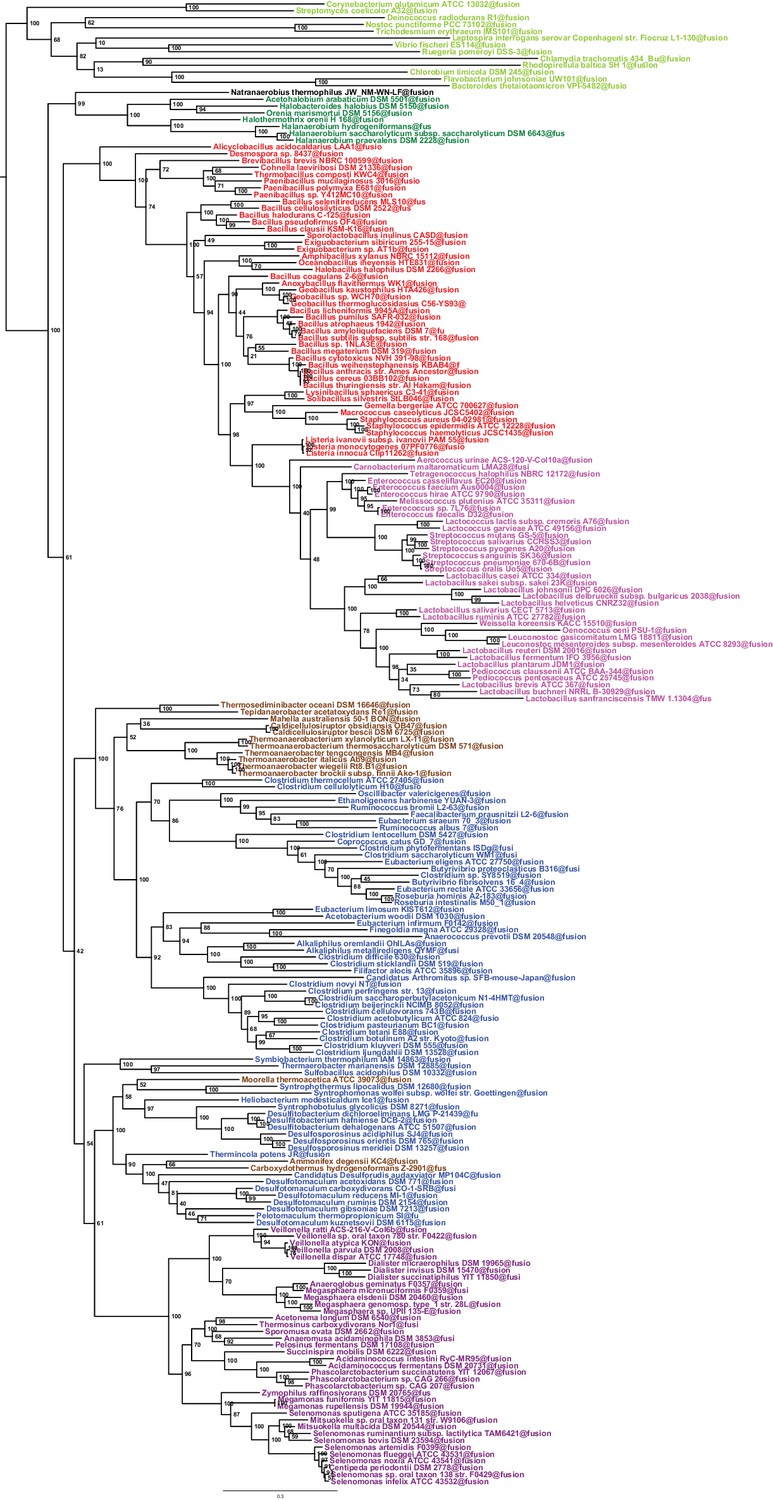
Maximum likelihood phylogeny of the Firmicutes.
The tree was obtained by PhyML 3.0 based from the same concatenation of 47 orthologous ribosomal proteins as the Bayesian tree in Figure 2 and the LG+Γ4 model. Values at nodes represent non-parametric bootstrap values calculated on 100 replicates of the original dataset. The scale bar represents the average number of substitutions per site.
Figure 2—figure supplement 3

Results of AU test for 12 alternative topologies.
Full results of the test are provided as Additional Data in Dryad (Antunes et al., 2016).
Figure 3
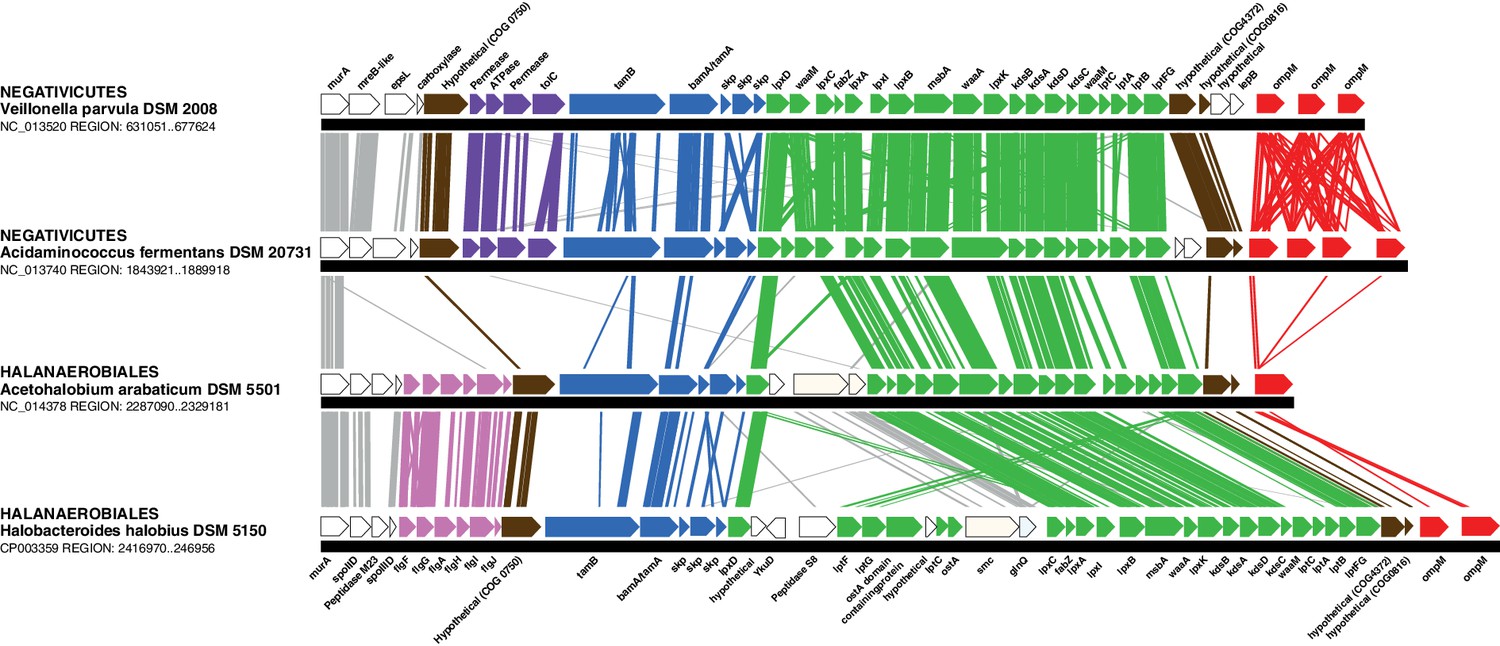
Conserved genomic locus for cell envelope components.
Co-localization of the genes coding for LPS synthesis and transport, OMP assembly and structural OMPs in the Negativicutes and the Halanaerobiales. Representatives of the 2 families of Negativicutes and the 2 families of Halanaerobiales are shown (for full distribution and accession numbers see Supplementary file 1). Genes are colored according to their functional class: LPS synthesis and transport (green), OMP assembly (blue), flagellum (light pink), OM-PG attachment (red), hypothetical (brown), efflux (purple) (see text for discussion). White boxes indicate proteins not known to being related to the OM or non-conserved proteins whose connection with the OM is unclear. The figure was obtained by EasyFig (Sullivan et al., 2011), where vertical lines represent BLAST hits with a cutoff of 0.0001.
Figure 4 with 1 supplement
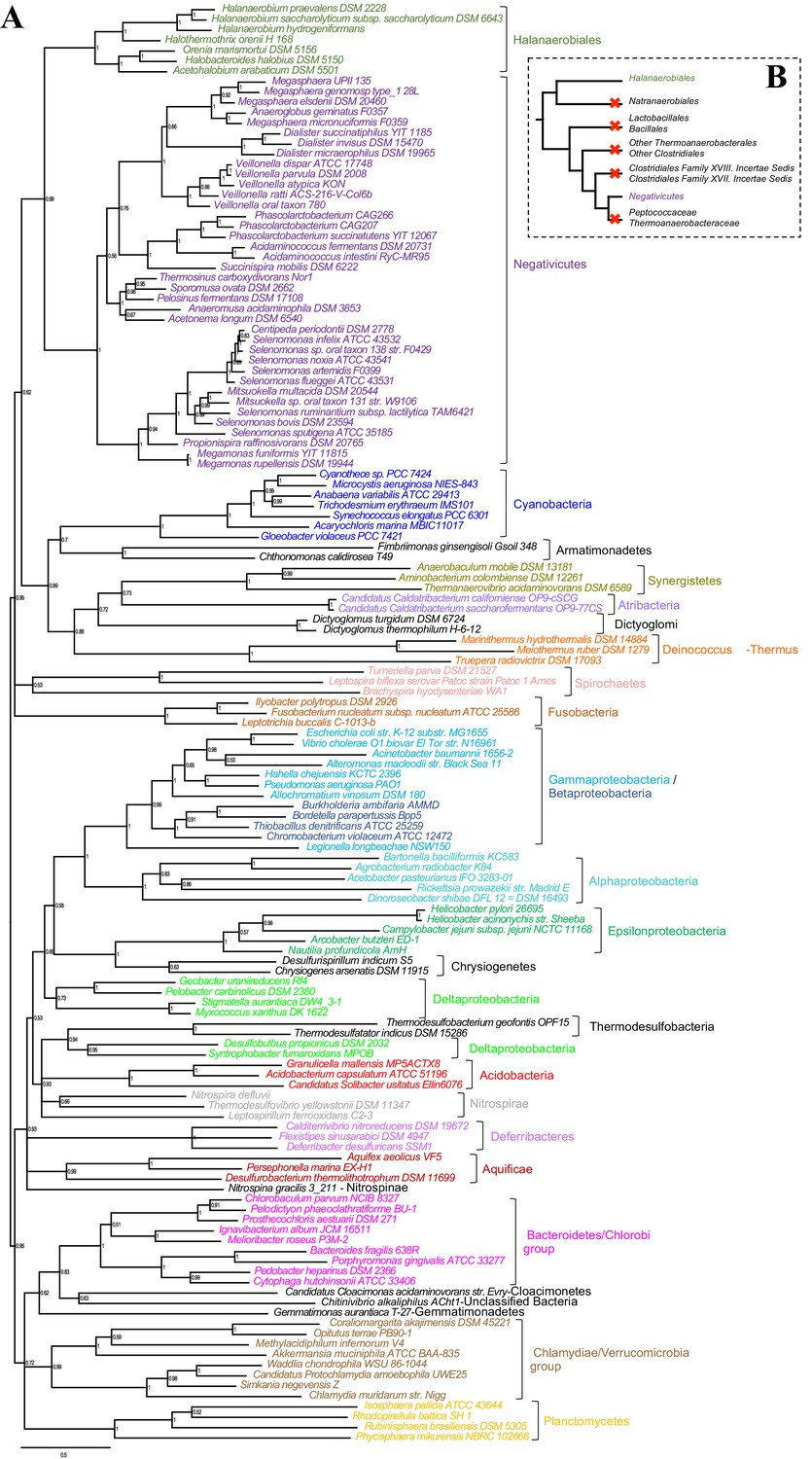
Phylogenetic tree of core LPS components.
(A) Bayesian phylogeny based on a concatenation of orthologs of the four core components of the LPS biosynthesis pathway (lpxABCD), comprising 898 amino acid positions and the CAT+GTR+Γ4 model. Values at nodes represent Bayesian posterior probabilities. The scale bar represents the average number of substitutions per site. For details on analyses, see Materials and methods. (B) Schematic representation of the Firmicutes phylum-level phylogeny from Figure 2, onto which putative losses of the OM are mapped (red crosses). See text for discussion.
Figure 4—figure supplement 1
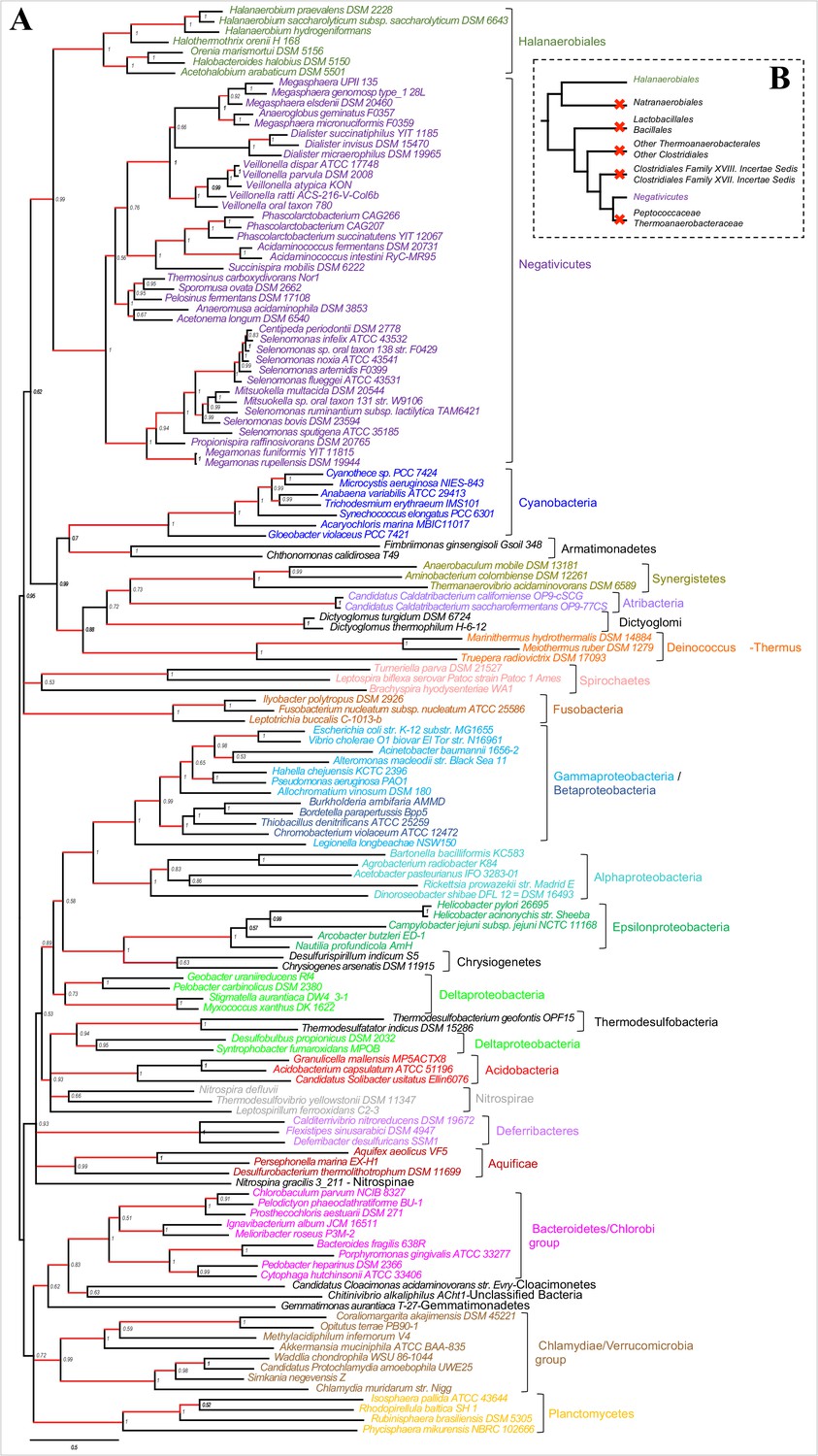
Results of IC congruence test for the 4 LPS core proteins.
IC values were mapped onto the LPS core proteins concatenation phylogeny shown in Figure 3. Branches in red indicate congruence among markers (IC values>1). Full results of the test are provided as Additional Data in Dryad (Antunes et al., 2016).
Figure 5 with 3 supplements
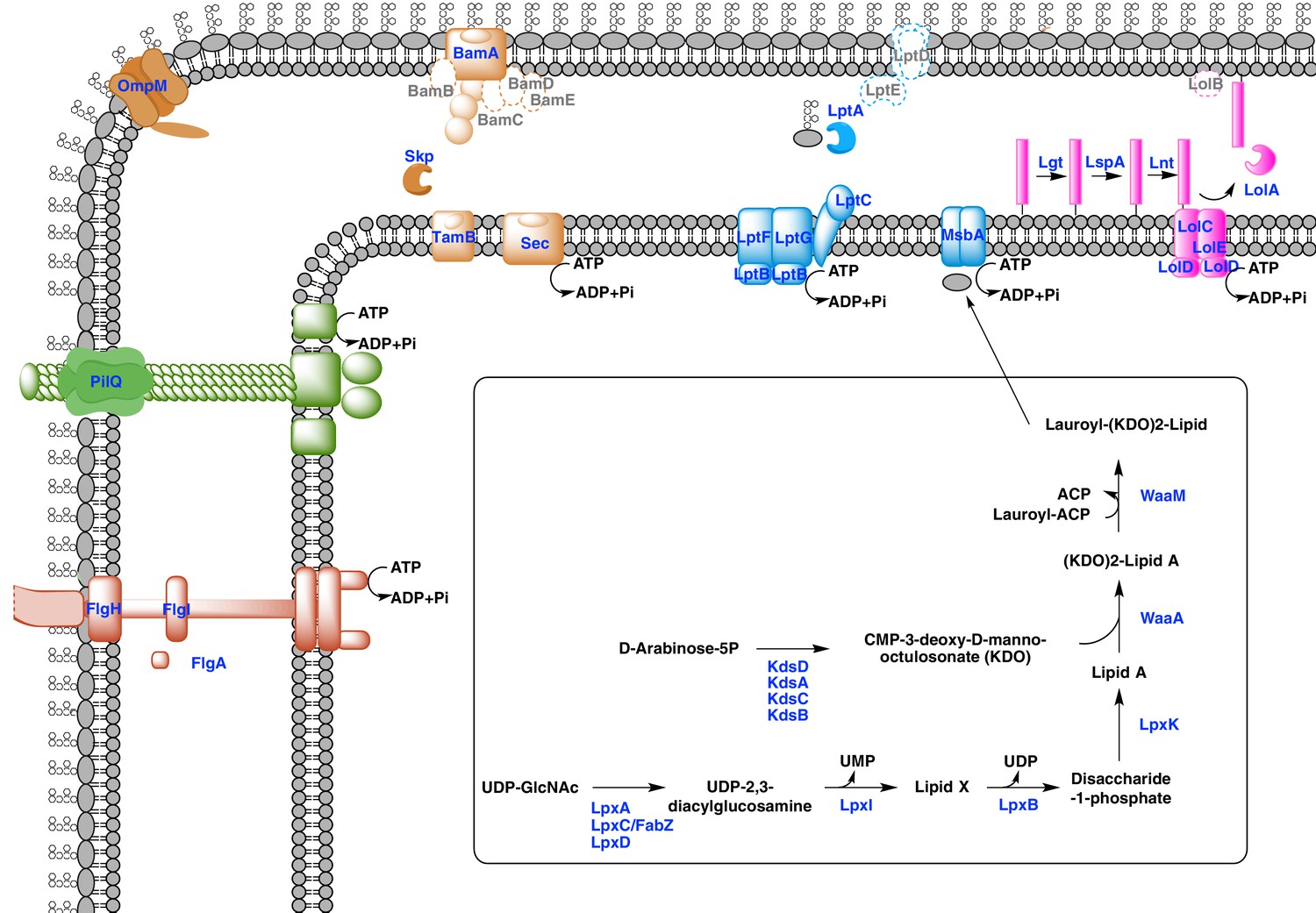
Sketched diagram of inferred characteristics of the diderm Firmicutes cell envelope.
The main processes discussed in the text are shown schematically. Components that were not detected in the genomes of Negativicutes and Halanaerobiales are indicated with a dashed outline and grey font.
Figure 5—figure supplement 1

Flagellar gene cluster of Negativicutes and Halanaerobiales.
Structure of the region coding for flagellar components in representative members of Negativicutes and Halanaerobiales, and its conservation with respect to their closely related monoderm relatives Therminicola potens, and Natranaerobius thermophiles, respectively. By comparison is shown the structure of the operon in Escherichia coli as representative of a classical diderm. Colors are only meant to highlight synteny.
Figure 5—figure supplement 2
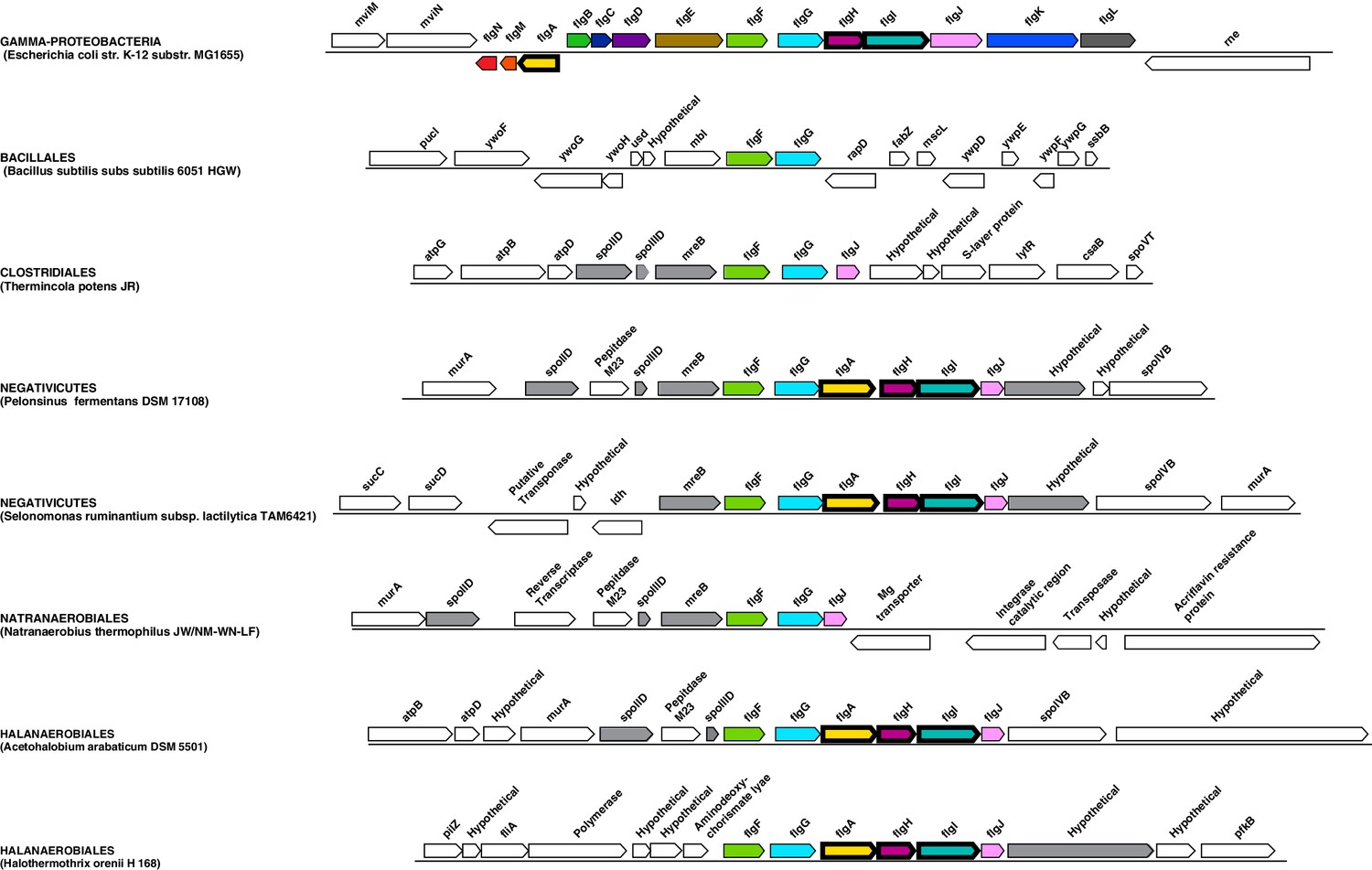
Genomic context of the genes coding for flagellar rings in Halanaerobiales and Negativicutes.
Structure of the region coding for components of the flagellar rings (flgA, flgH, flgI) in representative members of Negativicutes and Halanaerobiales, in comparison with their closely related monoderm relatives Therminicola potens, and Natranaerobius thermophiles, respectively. The genomic structures in Bacillus subtilis and Escherichia coli are also shown as the most studied models for monoderm and diderm flagella. Colors are only meant to highlight synteny.
Figure 5—figure supplement 3
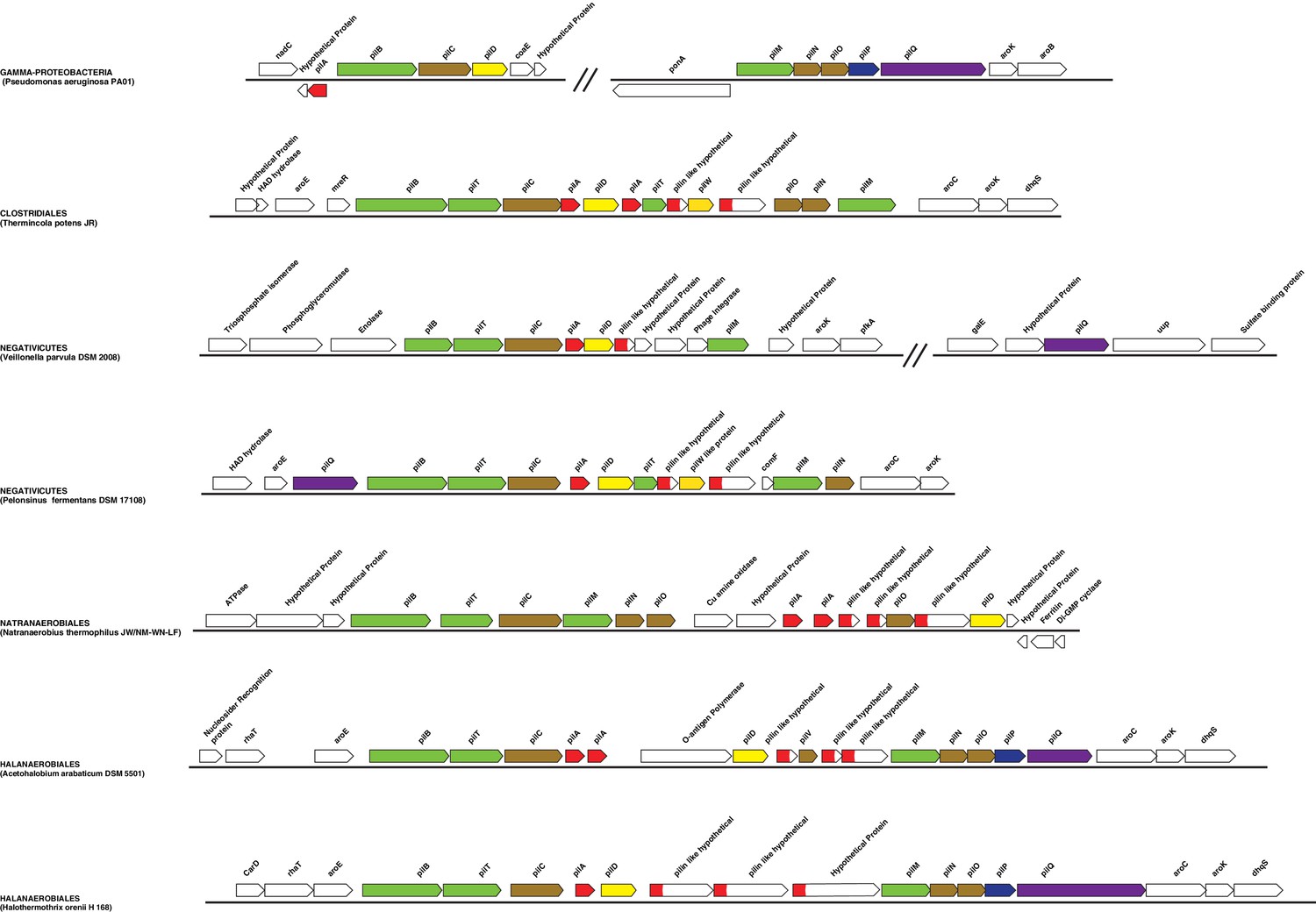
Structure of the main Type IV pilus cluster in Negativicutes and Halanaerobiales.
Structure of the region coding for components of the type IV pilus in representative members of Negativicutes and Halanaerobiales, in comparison with their closely related monoderm relatives Therminicola potens, and Natranaerobius thermophiles, respectively. The genomic structure in Pseudomonas aeruginosa is also shown as the most studied model for diderm type IV pili. Colors are only meant to highlight synteny.
Additional files
-
Supplementary file 1
Full distribution and accession numbers of the protein families discussed in the text.
- https://doi.org/10.7554/eLife.14589.015
-
Supplementary file 2
Full distribution and accession numbers of the four core LPS genes in a local databank of 121 complete genomes representatives of all major bacterial phyla as discussed in the text.
NA indicates that the taxon is unlisted in the NCBI Taxonomy database, while NH indicate that no homologues were found based on HMM search.
- https://doi.org/10.7554/eLife.14589.016
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Phylogenomic analysis supports the ancestral presence of LPS-outer membranes in the Firmicutes
eLife 5:e14589.
https://doi.org/10.7554/eLife.14589




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}