Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation
- Charité University Medicine, Germany
- Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases, Germany
- Max Planck Institute for Molecular Genetics, Germany
- Guangzhou Women and Children's Medical Center, China
- University of Potsdam, Germany
- Radboud University Medical Center, Netherlands
Figures
Figure 1 with 1 supplement
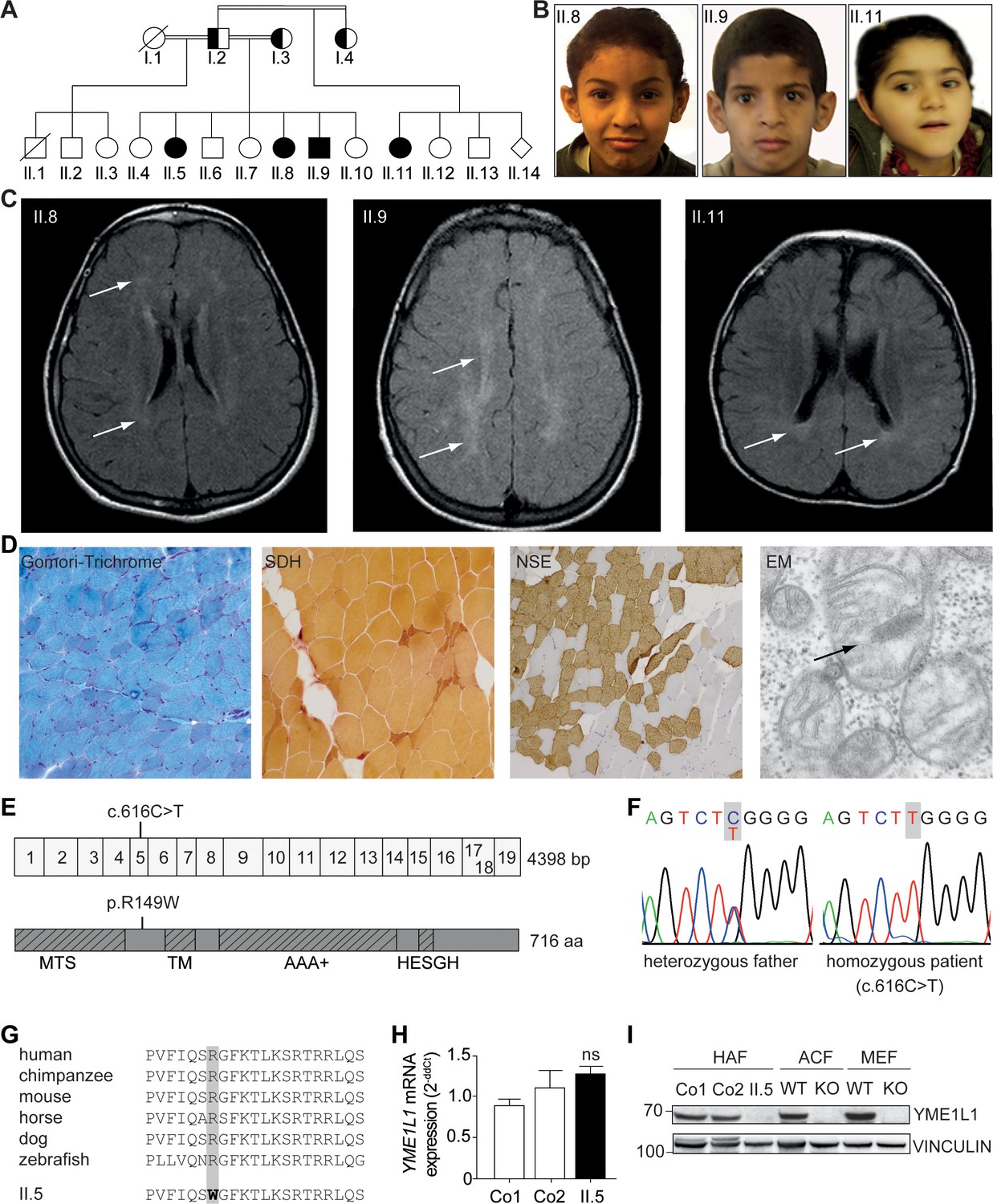
Phenotype and genotype of patients with YME1L1 mitochondriopathy.
(A, B) The four affected patients are children of healthy, consanguineous parents of Saudi Arabian decent (□ male; ○ female; ◊ unknown gender;  deceased;
deceased;  heterozygous, clinically not affected; ⚫/■ homozygous, affected;
heterozygous, clinically not affected; ⚫/■ homozygous, affected;  , consanguineous marriage). (C) Cranial MRIs reveal hyperintense changes (arrows) as a sign for leucencephalopathy, and cerebral atrophy (FLAIR, axial images). (D) Histological analysis of patient II.5 muscle biopsy specimen, from left to right: Gomori trichrome stain for muscle fibers revealed no conspicuous ragged red fibers indicative of a mitochondriopathy. Succinate dehydrogenase (SDH) and neuron specific enolase (NSE) staining revealed a neurogenic pattern with grouped fibers indicating denervation (magnification 200x). Electron microscopy (EM) revealed paracristalline inclusions (arrow) and altered cristae structure (“parking lots”, magnification 15,000x). (E) Whole exome sequencing discovered the homozygous mutation c.616C>T in exon 5 of the YME1L1 gene (NM_014263), localized at position 149 of the YME1L1 protein, and leading to an amino acid exchange of arginine to tryptophan in the mitochondrial targeting site (MTS; p.R149W, NP_055078). YME1L1 contains highly conserved domains: MTS, transmembrane domain (TM), ATPase domain (AAA+), motif of metalloprotease (Zinc) activity (HESGH). (F) Electropherogram depicting homozygous missense mutation in patient II.5, which is heterozygous in the father. (G) The mutation lies within a protein region, highly conserved throughout different species. (H) YME1L1 mRNA levels do not differ between patient and control primary human adult fibroblasts (Co, Control; II.5, patient II.5; ns, not significant; one-way ANOVA; p=0.2314; n=6 ) (I) Steady state levels of YME1L1 are below detection levels or profoundly reduced in cell lysates of patient II.5 YME1L1R149W; Yme1l1 knockout mouse fibroblasts serve as negative controls. YME1L1 protein levels in mitchondrial compartment fractions can be found Figure 1—figure supplement 1 (HAF, primary human adult fibroblasts; ACF, immortalized murine adult cardiac fibroblasts; MEF, immortalized murine embryonic fibroblast; Co, Control; II.5, patient II.5; WT, wild type; KO, Yme1l1 knockout, n=5).
, consanguineous marriage). (C) Cranial MRIs reveal hyperintense changes (arrows) as a sign for leucencephalopathy, and cerebral atrophy (FLAIR, axial images). (D) Histological analysis of patient II.5 muscle biopsy specimen, from left to right: Gomori trichrome stain for muscle fibers revealed no conspicuous ragged red fibers indicative of a mitochondriopathy. Succinate dehydrogenase (SDH) and neuron specific enolase (NSE) staining revealed a neurogenic pattern with grouped fibers indicating denervation (magnification 200x). Electron microscopy (EM) revealed paracristalline inclusions (arrow) and altered cristae structure (“parking lots”, magnification 15,000x). (E) Whole exome sequencing discovered the homozygous mutation c.616C>T in exon 5 of the YME1L1 gene (NM_014263), localized at position 149 of the YME1L1 protein, and leading to an amino acid exchange of arginine to tryptophan in the mitochondrial targeting site (MTS; p.R149W, NP_055078). YME1L1 contains highly conserved domains: MTS, transmembrane domain (TM), ATPase domain (AAA+), motif of metalloprotease (Zinc) activity (HESGH). (F) Electropherogram depicting homozygous missense mutation in patient II.5, which is heterozygous in the father. (G) The mutation lies within a protein region, highly conserved throughout different species. (H) YME1L1 mRNA levels do not differ between patient and control primary human adult fibroblasts (Co, Control; II.5, patient II.5; ns, not significant; one-way ANOVA; p=0.2314; n=6 ) (I) Steady state levels of YME1L1 are below detection levels or profoundly reduced in cell lysates of patient II.5 YME1L1R149W; Yme1l1 knockout mouse fibroblasts serve as negative controls. YME1L1 protein levels in mitchondrial compartment fractions can be found Figure 1—figure supplement 1 (HAF, primary human adult fibroblasts; ACF, immortalized murine adult cardiac fibroblasts; MEF, immortalized murine embryonic fibroblast; Co, Control; II.5, patient II.5; WT, wild type; KO, Yme1l1 knockout, n=5).
-
Figure 1—source data 1
Raw data Graph 1H.
Raw values of YME1L1 mRNA expression in human adult fibroblasts of controls and patient measured via TaqMan qPCR (n=6).
- https://doi.org/10.7554/eLife.16078.003
Figure 1—figure supplement 1
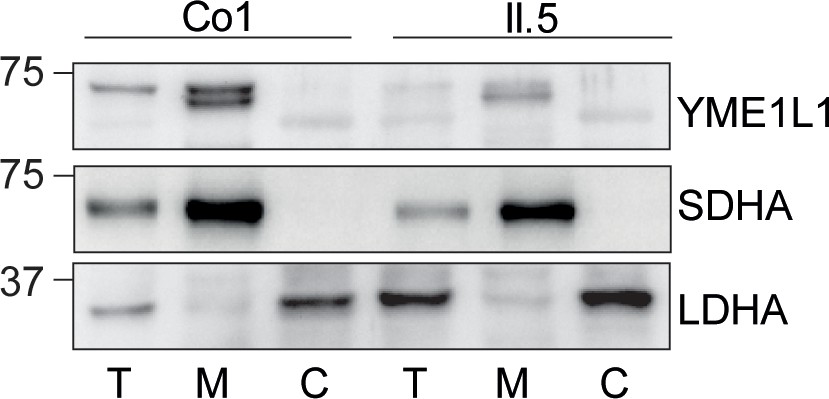
(I) YME1L1R149W signal is barely detectable in total cell lysates (T), significantly reduced in the mitochondrial (M) but not present in the cytosolic (C) fraction of patient primary human adult fibroblasts (Co, Control; II.5, patient II.5; SDHA, succinate dehydrogenase; LDHA, lactate dehydrogenase; n=3).
https://doi.org/10.7554/eLife.16078.004
Figure 2
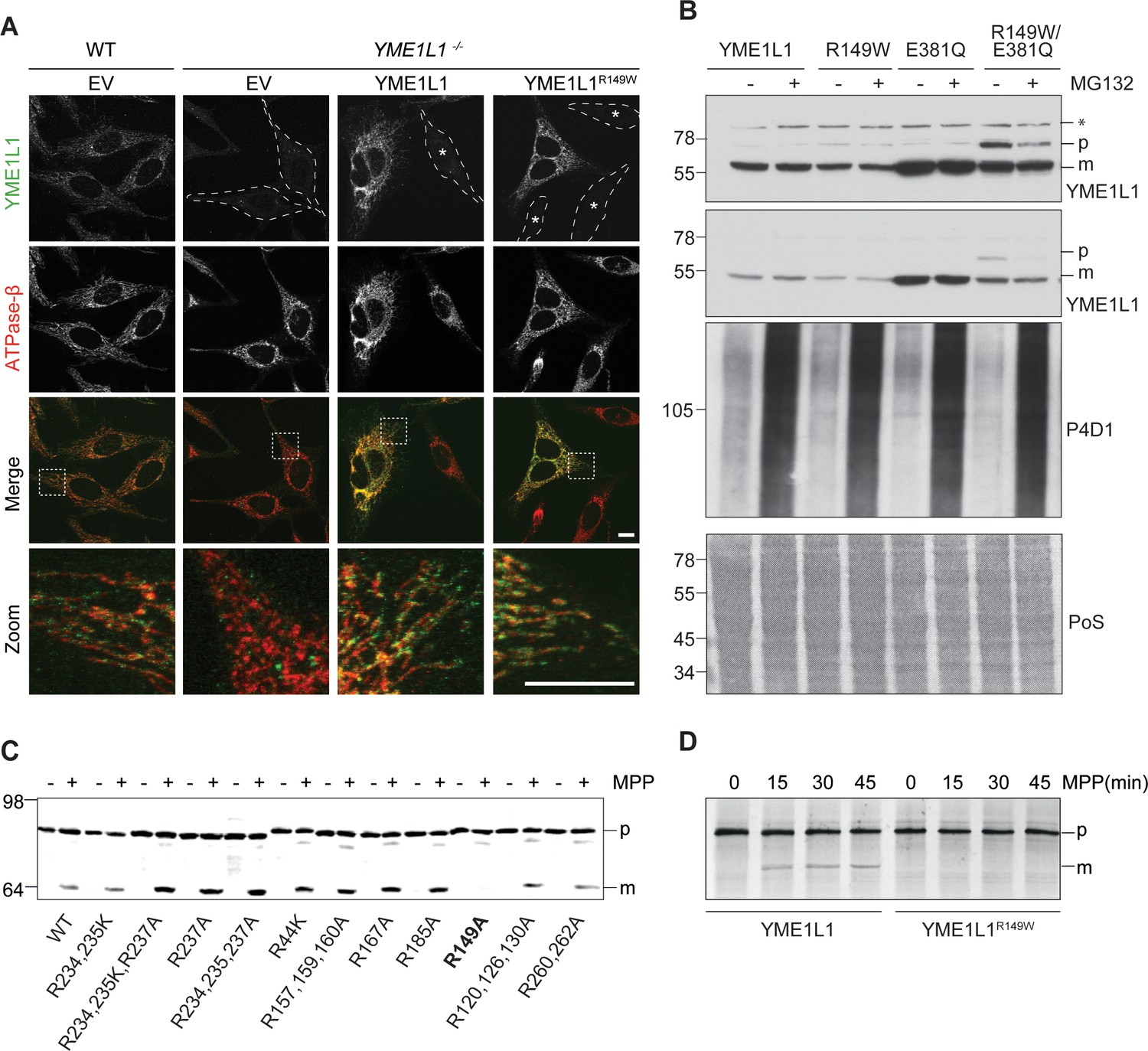
Mutation of arginine 149 impairs maturation of YME1L1 by MPP upon import into mitochondria.
(A) YME1L1R149W is targeted to mitochondria. HeLa cells were transiently transfected with wild-type YME1L1, YME1L1R149W or the empty vector (EV) as control. Cells were analyzed by indirect immunofluorescence and co-localization of the immunofluorescent signals of antibodies against YME1L1 and ATP Synthase subunit beta (ATPase-ß) (WT, Wildtype; YME1L1-/-, Knockout; EV, empty vector; YME1L1R149W, patient mutation; Scale bar 10 µM). (B) Inhibition of the proteasome with MG132 (20 mM, 18 h) does not stabilize precursor or mature YME1L1 in Flp-In T-Rex HEK293T cells expressing YME1L1 or YME1L1 mutant variants: R149W, E381Q or R149W/E381Q. P4D1 antibodies were used as a control for ubiquitin accumulation and proteasome inhibition. (R149W, patient mutation; E381Q, dominant negative mutation of ATPase domain; R149W/E381Q, double mutant; *, unspecific signal; p, premature; m, mature; PoS, Ponceau staining; n=2). (C) Maturation of YME1L1 is mediated by mitochondrial processing peptidase (MPP) and impaired upon mutation of arginine 149. After site-directed mutagenesis of N-terminal arginine residues in YME1L1, the mutant proteins were expressed in a cell-free system and processing by recombinant MPP was examined (WT, wild type YME1L1; p, premature m, mature, n=2–3). (D) Cell-free MPP cleavage-assay: MPP can cleave YME1L1 but not YME1L1R149W from the premature to its mature form (min, minutes; p, premature; m, mature; YME1L1R149W, patient mutation; n=3).
Figure 3
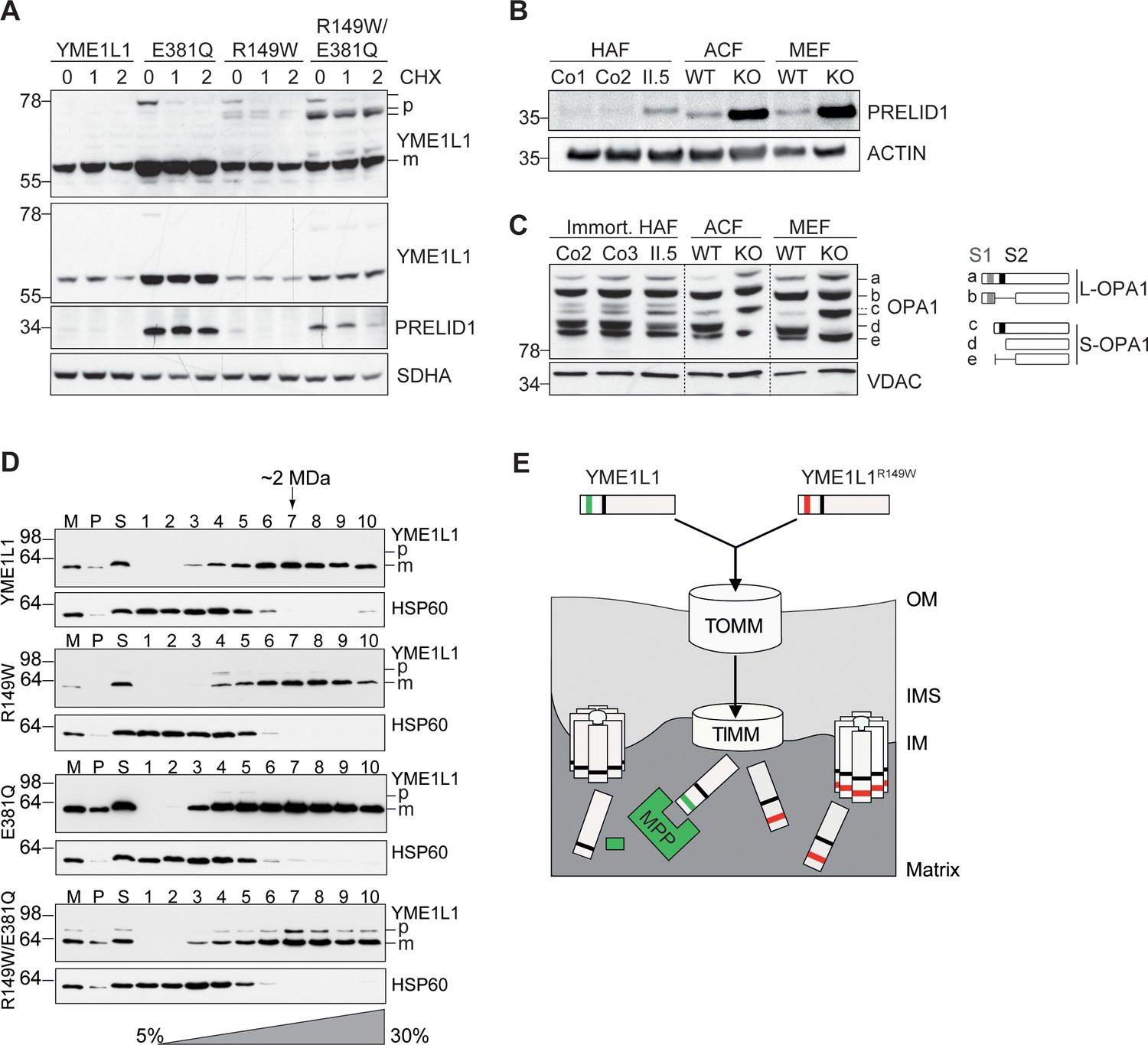
Mutation of arginine 149 destabilizes YME1L1 but retains residual YME1L activity.
(A) Stability of YME1L1 or YME1L1 mutant variants (R149W, E381Q and R149W/E381Q) expressed in Flp-In T-Rex HEK293T cells. The dominant negative E381Q mutation in the ATPase domain of YME1L1 prevents degradation of YME1L1R149W (E381Q, dominant negative mutation of ATPase domain; R149W, patient mutation; R149W/E381Q, double mutant; CHX, cycloheximide; h, hours; p, premature; m, mature; SDHA, succinate dehydrogenase; n=2). (B) Homozygous mutation in YME1L1 results in an accumulation of PRELID1 in the human patient. Yme1l1 knockout mouse fibroblasts serve as positive controls to demonstrate impaired proteolysis of PRELID1 (HAF, human adult primary fibroblasts; ACF, immortalized murine adult cardiac fibroblasts; MEF, immortalized murine embryonic fibroblast; Co, Control; II.5, patient; WT, wild type; KO, knockout; n=5). (C) Mutation of arginine 149 of YME1L1 impairs processing of OPA1 with a decrease of short OPA1 form d levels. The formation of OPA1 form d indicates residual YME1L1 activity in human patient fibroblasts (immort. HAF, immortalized human adult fibroblasts; ACF, immortalized murine adult cardiac fibroblasts; MEF, immortalized murine embryonic fibroblast; Co, Control; II.5, patient; WT, wild type; KO, knockout; n=6). The schematic diagram illustrates the proteolytic processing of OPA1 by YME1L1 on processing site 2 (S2) and OMA1 on processing site 1 (S1). The presence of long OPA1 forms (L-OPA1) is required for the maintenance of mitochondrial inner membrane fusion, whereas accumulation of short OPA1 forms (S-OPA1) is associated with accelerated fission. (D) Mitochondria-enriched membrane fractions from Flp-In T-Rex HEK293T cells expressing YME1L1 or YME1L1 mutant variants (R149W, E381Q and R149W/E381Q) were solubilized in digitonin and analyzed by sucrose gradient centrifugation. Fractions were collected and separated on SDS-PAGE for immunoblotting to detect high MW complexes of YME1L1. HSP60 complexes were used as a control (M, mitochondrial input, P, S, pellet and supernatant fraction after solubilization; HSP60, heat shock protein 60; E381Q, dominant negative mutation of ATPase domain; R149W, patient mutation; R149W/E381Q, double mutant). (E) Premature YME1L1/ YME1L1R149W is imported into the mitochondrial matrix via translocons of the outer mitochondrial membrane (TOMM) and inner mitochondrial membrane (TIMM). Here, MPP binds and cleaves the N-terminal mitochondrial targeting site (MTS) from premature YME1L1 but not YME1L1R149W, which then allows the mature and premature YME1L1R149W protein to assemble as proteolytic complex.
Figure 4
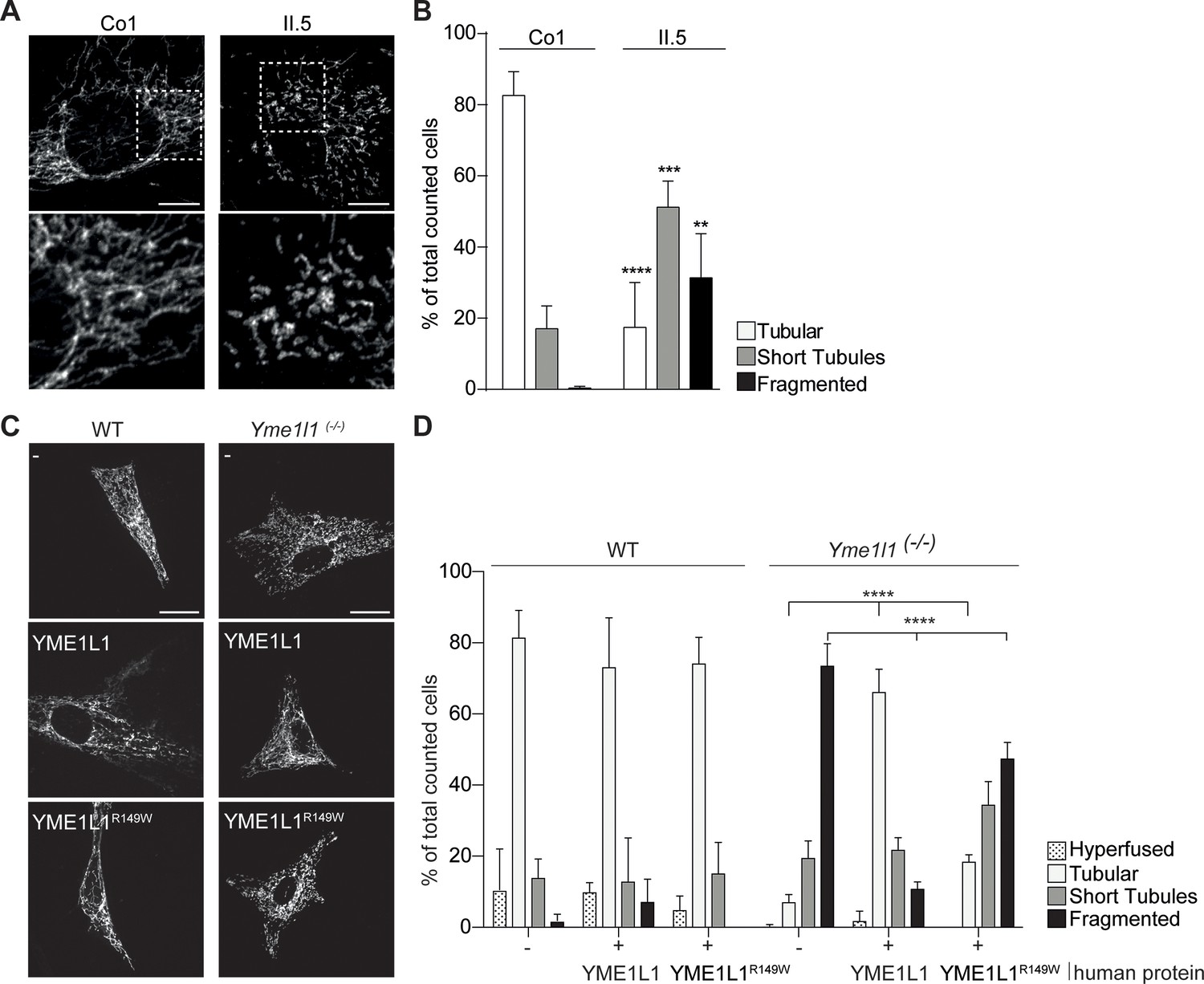
YME1L1R149W causes mitochondrial fragmentation.
(A, B) YME1L1R149W causes fragmentation (fragmentation or short tubules) of mitochondrial networks in patient fibroblasts (Co, Control, II.5, patient II.5; one-way ANOVA; n=150–200 cells; 4 replicates; scale bar 10 µm). (C) Co-transfection of mitochondrial GFP and human YME1L1/ YME1L1R149W protein in WT and Yme1l1-/- MEFs revealed that YME1L1 but not YME1L1R149Wcan rescue mitochondrial fragmentation in Yme1l1-/- MEFs and (D) increases the number of cells with tubular networks. Expression of YME1L1R149W partially rescues the fragmentation phenotype. In WT MEFs, YME1L1R149W expression results in only a mild, but not significant decline of cells with a tubular mitochondrial network (WT, wildtype; Yme1l1-/-, Yme1l1 Knockout; two-way ANOVA; n=3; scale bar 10 µm; **p<0.01; ***p<0.001; ****p<0.0001).
-
Figure 4—source data 1
Raw data Graph 4B.
Relative mitochondrial morphology quantification after immunocytochemistry with TOMM20 antibody and DAPI in human adult fibroblasts of control and patient (n=150–200 cells, 4 replicates).
- https://doi.org/10.7554/eLife.16078.009
-
Figure 4—source data 2
Raw data Graph 4D.
Relative mitochondrial morphology quantification after immunocytochemistry with TOMM20 antibody and DAPI in WT and Yme1l1-/- adult cardiac murine fibroblasts overexpressing YME1L1 or YME1L1R149W (n=3).
- https://doi.org/10.7554/eLife.16078.010
Figure 5

YME1L1 mutation impairs fibroblast cell growth and proliferation.
(A) Significant reduction of cell culture growth (y-axis scale *104) and (B) proliferation of patient primary human adult fibroblasts after 120h under culture conditions (Co, Control; II.5, patient II.5; one-way ANOVA; ***p<0.001; ****p<0.0001; n=5 ). (C) No increase in apoptosis-sensitivity upon 2 µM staurosporine (STS) treatment in patient HAF after 6h (SDS-PAGE for cleaved PARP level (cPARP) (Co, Control; II.5, patient II.5; one-way ANOVA; p6h = 0.4283; n=3; ). (D) Normal respiratory chain subunit activity in patient HAF (U, units; mU, milli U; CS, Citrate synthase). (E) Cytochrome c oxidase subunit 4 (COX4) and NADH ubiquinone oxidoreductase 1 beta subcomplex 6 (NDUFB6) protein levels were not elevated in patient fibroblasts (Co, Control; II.5, patient II.5; n=3). (F) Decreased levels of mitochondrial compartment markers TOMM20 (translocon of outer mitochondrial membrane 20) for the outer membrane (OM), Cytochrome c for the intermembrane space (IMS), SDHA (succinate dehydrogenase) for the inner membrane (IM), and Cyclophilin D for the matrix in whole cell lysates of patient II.5 (Co, Control; II.5, patient II.5; n=3).
-
Figure 5—source data 1
Raw data Graph 5A.
Raw values of fluorometric measurement with Cell-titer Blue Cell Viability Assay application in human adult fibroblasts of controls and patient (n=5).
- https://doi.org/10.7554/eLife.16078.016
-
Figure 5—source data 2
Raw data Graph 5B.
Raw values of colorimetric measurement with BrdU Cell Proliferation ELISA Assay application in human adult fibroblasts of controls and patient (n=5).
- https://doi.org/10.7554/eLife.16078.017
-
Figure 5—source data 3
Raw data Graph 5C.
Raw values of cleaved-PARP antibody signal intensity after staurosporine treatment in human adult fibroblasts of controls and patient and SDS-Page (n=3).
- https://doi.org/10.7554/eLife.16078.018
Videos
Video 1
Three dimensional rendering of mitochondrial structure of a control fibroblast.
MitoTracker Deep Red- mitochondria; DAPI-nucleus.
Video 2
Three dimensional rendering of mitochondrial structure of a patient II.5 fibroblast.
MitoTracker Deep Red- mitochondria; DAPI-nucleus.
Video 3
Mitochondrial dynamics in a control fibroblast.
MitoTracker Green-mitochondria, live cell imaging.
Video 4
Mitochondrial dynamics in a patient II.5 fibroblast.
MitoTracker Green-mitochondria, live cell imaging.
Tables
Table 1
YME1L1 mitochondriopathy phenotype.
| Pedigree ID (gender) | II.5 (f) | II.8 (f) | II.9 (m) | II.11 (f) | ||
|---|---|---|---|---|---|---|
| Age at last assessment (y) | 15.8 | 12.3 | 10.3 | 5.2 | ||
| Category | Feature | HPO | ||||
| Inheritance | ||||||
| AR | AR | AR | AR | |||
| Growth | ||||||
| Height | Short stature SD;% | 0004322 | + -2.19; 1 | + -2.09; 2 | + -1.89; 3 | - 0.38; 35 |
| Weight | Low weight SD;% | 0004325 | + -2.8; <1 | - -1.18; 12 | - 0.07; 53 | - -1.35; 12 |
| Neonatal period | ||||||
| Neonatal asphyxia | 0012768 | - | - | - | + | |
| Head and Neck | ||||||
| Head | Microcephaly | 0000252 | - | - | + | + |
| Macrocephaly | 0000256 | + | - | - | - | |
| Face | Midface retrusion | 0011800 | - | - | + | + |
| Congenital facial diplegia | 0007188 | + | - | - | + | |
| Ears | Hearing impairment | 0000365 | - | + | + | + |
| Sensorineural hearing impairment | 0000407 | n.a. | n.a. | + | + | |
| Macrotia | 0000400 | - | + | + | - | |
| Eyes | Pigmentary retinopathy | 0000580 | + | - | - | - |
| Optic nerve hypoplasia | 0000609 | + | + | + | + | |
| Cherry red spot of the macula | 0010729 | - | - | - | + | |
| Strabismus | 0000486 | - | - | + | + | |
| Hypermetropia | 0000540 | - | - | - | + | |
| Myopia | 0000545 | + | + | + | - | |
| Amblyopia | 0000646 | - | + | + | + | |
| Abnormality of visual evoked potentials | 0000649 | n.a. | - | + | + | |
| Abdomen | ||||||
| Gastro-intestinal | Constipation (in infancy) | 0002019 | + | n.a. | + | + |
| Spleen | Splenomegaly | 0001744 | + | - | + | - |
| Skeletal | ||||||
| Feet | Bilateral talipes equinovarus | 0001776 | - | - | - | + |
| Muscle, soft tissue | ||||||
| Increased variability in muscle fiber diameter | 0003557 | + | n.a. | n.a. | n.a. | |
| Neurologic | ||||||
| CNS | Hypotonia, neonatal, generalized | 0008935 | - | - | - | + |
| Infantile muscular hypotonia | 0008947 | - | - | - | + | |
| Global developmental delay (onset, months) | 0001263 | + | + | + | + (6) | |
| Motor delay | 0001270 | + | + | + | + | |
| Gait apraxia | 0010521 | - | - | - | + | |
| Athetosis | 0002305 | - | - | - | + | |
| Intellectual disability, moderate; (IQ) | 0002342 | + (48) | n.a. | n.a. | + (39) | |
| Incomprehensible speech | 0002546 | n.a. | - | - | + | |
| Poor speech | 0002465 | n.a. | - | + | - | |
| Absent speech | 0001344 | - | + | - | - | |
| Seizures (onset, months) | 0001250 | - | - | - | + (6) | |
| Dysmetria | 0001310 | + | + | + | - | |
| Ataxia | 0001251 | + | + | + | - | |
| Brain atrophy | 0012444 | - | - | - | + | |
| Ventriculomegaly | 0002119 | - | - | - | + | |
| Delayed CNS myelination | 0002188 | + | - | - | - | |
| Abnormality of the cerebral white matter | 0002500 | + | + | + | + | |
| Abnormality of the basal ganglia | 0002134 | - | - | + | - | |
| Cerebellar hypoplasia (progressive) | 0001321 | + | - | - | + | |
| EEG with focal sharp waves | 0011196 | n.a. | - | + | - | |
| Abnormal auditory evoked potentials | 0006958 | n.a. | n.a. | + | + | |
| PNS | Decreased sensory nerve conduction velocity | 0003448 | - | + | n.a. | - |
| Behavioural Psychiatric | Hyperactivity | 0000752 | - | + | - | - |
| Attention deficit hyperactivity disorder | 0007018 | - | - | - | + | |
| Stereotypical body rocking | 0012172 | - | - | - | + | |
| Laboratory anomalies | ||||||
| Mildly elevated creatine phosphokinase | 0008180 | - | - | - | + | |
Additional files
-
Supplementary file 1
List of primers and antibodies.
(A) List of primers for qPCR and genotyping. (B) List of primary and secondary antibodies.
- https://doi.org/10.7554/eLife.16078.019
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation
eLife 5:e16078.
https://doi.org/10.7554/eLife.16078




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}