Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation
- Charité University Medicine, Germany
- Cologne Excellence Cluster on Cellular Stress Responses in Aging-Associated Diseases, Germany
- Max Planck Institute for Molecular Genetics, Germany
- Guangzhou Women and Children's Medical Center, China
- University of Potsdam, Germany
- Radboud University Medical Center, Netherlands
Abstract
Mitochondriopathies often present clinically as multisystemic disorders of primarily high-energy consuming organs. Assembly, turnover, and surveillance of mitochondrial proteins are essential for mitochondrial function and a key task of AAA family members of metalloproteases. We identified a homozygous mutation in the nuclear encoded mitochondrial escape 1-like 1 gene YME1L1, member of the AAA protease family, as a cause of a novel mitochondriopathy in a consanguineous pedigree of Saudi Arabian descent. The homozygous missense mutation, located in a highly conserved region in the mitochondrial pre-sequence, inhibits cleavage of YME1L1 by the mitochondrial processing peptidase, which culminates in the rapid degradation of YME1L1 precursor protein. Impaired YME1L1 function causes a proliferation defect and mitochondrial network fragmentation due to abnormal processing of OPA1. Our results identify mutations in YME1L1 as a cause of a mitochondriopathy with optic nerve atrophy highlighting the importance of YME1L1 for mitochondrial functionality in humans.
https://doi.org/10.7554/eLife.16078.001Introduction
Mitochondriopathies often present as multisystemic diseases and can be caused by mutations in nuclear or mitochondrial DNA-encoded genes (Nunnari, 2012). Here, we report a novel form of mitochondriopathy with infantile-onset developmental delay, muscle weakness, ataxia, and optic nerve atrophy caused by a homozygous mutation in the yeast mitochondrial escape 1-like 1 gene YME1L1.
YME1L1 was first identified in yeast, in a screen for gene products that elevate the rate of mitochondrial DNA migration to the nucleus (Campbell et al., 1994; Thorsness et al., 1993). The lack of yme1 impairs respiratory growth of yeast highlighting its important function in mitochondrial maintenance (Nakai et al., 1995; Pearce and Sherman, 1995). YME1L1, a member of the AAA family of ATPases (ATPases associated with a variety of cell activities), is a nuclear genome-encoded ATP-dependent metalloprotease embedded in the inner mitochondrial membrane (IM), with its protease domain facing the intermembrane space (IMS) (also termed i-AAA protease) (Leonhard et al., 1996; Shah et al., 2000; Weber et al., 1996). Its import into mitochondria is accompanied by proteolytic processing via the mitochondrial processing peptidase (MPP), which cleaves off the mitochondrial targeting sequence (MTS) (Rainey et al., 2006). The mature protein assembles into a homo-oligomeric complex within the IM (Leonhard et al., 1996; Van Dyck and Langer, 1999). YME1L1 degrades both IMS and IM proteins such as lipid transfer proteins (Potting et al., 2013), components of protein translocases of the IM (Baker et al., 2012; Rainbolt et al., 2015), and the dynamin-like GTPase optic atrophy 1 (OPA1) (Anand et al., 2014; Rainbolt et al., 2015; van der Bliek and Koehler, 2003).
The OPA1 gene (MIM*605290), mutated in dominant optic atrophy (Delettre et al., 2000), encodes a mediator of mitochondrial fusion that also orchestrates mitochondrial cristae morphogenesis (Cipolat et al., 2004; Frezza et al., 2006; Olichon et al., 2003). OPA1 is processed by two peptidases in the IM, YME1L1 and OMA1 (overlapping with the m-AAA protease 1 homolog), which thereby balance fusion and fission of mitochondria (MacVicar and Langer, 2016). Long OPA1 forms (L-OPA1) carry out mitochondrial fusion, while short forms (S-OPA1) are dispensable for fusion but contribute to mitochondrial fission when accumulated (Anand et al., 2014; Ehses et al., 2009; Griparic et al., 2007; Head et al., 2009; Song et al., 2007). Loss of YME1L1 accelerates OMA1-dependent L-OPA1 cleavage, resulting in S-OPA1 accumulation, increased mitochondrial fission, and mitochondrial network fragmentation (Anand et al., 2014; Wai et al., 2015). Despite previous attempts to link YME1L1 to human disease (Coenen et al., 2005), its physiological role in humans remains to be elucidated.
Results and discussion
Homozygous YME1L1 missense mutation causes mitochondriopathy
We report for the first time that patients with a homozygous mutation in the YME1L1 gene develop an infantile-onset mitochondriopathy. Four affected children of healthy, consanguineous parents of Saudi Arabian descent were born at term without complications and with normal anthropometric data (Figure 1A). Medical history gave no indication for perinatal brain damage as a cause for the reported patient phenotypes. All four patients presented with intellectual disability, motor developmental delay, expressive speech delay, optic nerve atrophy associated with visual impairment, hearing impairment, but no facial dysmorphism (Figure 1B, Table 1). Inconsistent features were microcephaly (II.9, II.11) or macrocephaly (II.5), ataxia (II.5, II.8, II.9), hyperkinesia (II.9), and athetotic and stereotypic movements (II.11). Cranial MRI revealed leukoencephalopathy in all patients and signs of brain atrophy in patients II.9 and II.11 (Figure 1C). Lactate levels and lactate/pyruvate ratios were elevated in blood and/or cerebrospinal fluid (CSF) of three patients as indicators of a mitochondriopathy, while alanine levels were normal in blood and cerebrospinal fluid samples. Analysis of a muscle biopsy specimen from patient II.5 revealed mitochondria with altered cristae morphology (so-called ‘parking lots’) and paracristalline inclusions as well as a neurogenic pattern with grouped fibers indicating denervation, but no conspicuous ragged red fibers in the Gomori trichrome staining (Figure 1D). Results of chromosome analysis, mitochondrial DNA sequencing and genetic testing for fragile X syndrome, Nijmegen breakage syndrome, and ataxia teleangiectasia were normal. Similarly, biochemical analyses gave no indication for adrenoleukodystrophy, GM1/GM2 gangliosidoses, Sandhoff disease, Tay-Sachs disease, Gaucher disease, Fabry disease, Krabbe disease, Mucopolysaccharidosis type IVB, neuronal ceroid-lipofuscinoses 1 and 2, and congenital disorder of glycosylation (CDG) syndrome (data not shown).
Figure 1 with 1 supplement see all
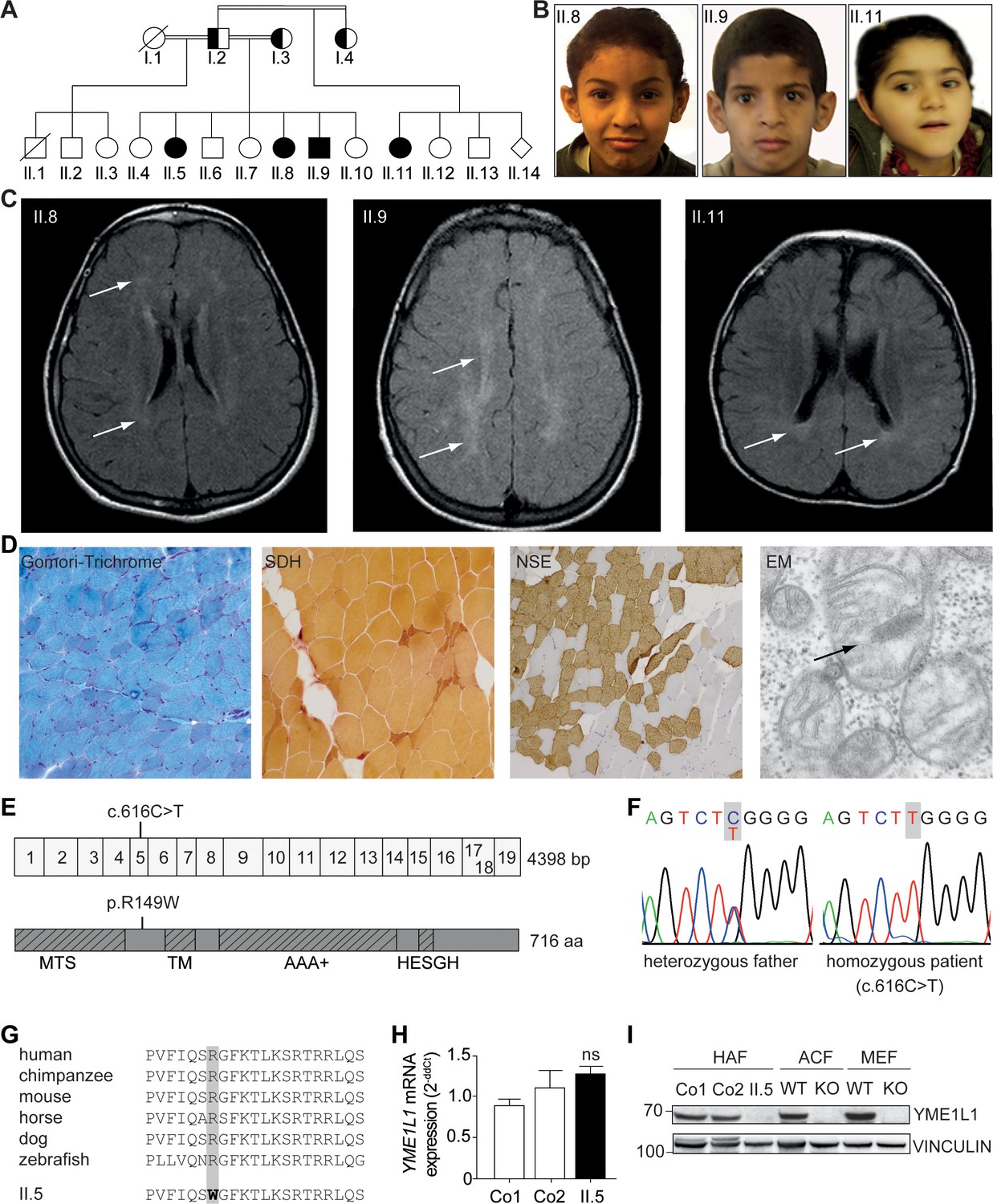
Phenotype and genotype of patients with YME1L1 mitochondriopathy.
(A, B) The four affected patients are children of healthy, consanguineous parents of Saudi Arabian decent (□ male; ○ female; ◊ unknown gender;  deceased;
deceased;  heterozygous, clinically not affected; ⚫/■ homozygous, affected;
heterozygous, clinically not affected; ⚫/■ homozygous, affected;  , consanguineous marriage). (C) Cranial MRIs reveal hyperintense changes (arrows) as a sign for leucencephalopathy, and cerebral atrophy (FLAIR, axial images). (D) Histological analysis of patient II.5 muscle biopsy specimen, from left to right: Gomori trichrome stain for muscle fibers revealed no conspicuous ragged red fibers indicative of a mitochondriopathy. Succinate dehydrogenase (SDH) and neuron specific enolase (NSE) staining revealed a neurogenic pattern with grouped fibers indicating denervation (magnification 200x). Electron microscopy (EM) revealed paracristalline inclusions (arrow) and altered cristae structure (“parking lots”, magnification 15,000x). (E) Whole exome sequencing discovered the homozygous mutation c.616C>T in exon 5 of the YME1L1 gene (NM_014263), localized at position 149 of the YME1L1 protein, and leading to an amino acid exchange of arginine to tryptophan in the mitochondrial targeting site (MTS; p.R149W, NP_055078). YME1L1 contains highly conserved domains: MTS, transmembrane domain (TM), ATPase domain (AAA+), motif of metalloprotease (Zinc) activity (HESGH). (F) Electropherogram depicting homozygous missense mutation in patient II.5, which is heterozygous in the father. (G) The mutation lies within a protein region, highly conserved throughout different species. (H) YME1L1 mRNA levels do not differ between patient and control primary human adult fibroblasts (Co, Control; II.5, patient II.5; ns, not significant; one-way ANOVA; p=0.2314; n=6 ) (I) Steady state levels of YME1L1 are below detection levels or profoundly reduced in cell lysates of patient II.5 YME1L1R149W; Yme1l1 knockout mouse fibroblasts serve as negative controls. YME1L1 protein levels in mitchondrial compartment fractions can be found Figure 1—figure supplement 1 (HAF, primary human adult fibroblasts; ACF, immortalized murine adult cardiac fibroblasts; MEF, immortalized murine embryonic fibroblast; Co, Control; II.5, patient II.5; WT, wild type; KO, Yme1l1 knockout, n=5).
, consanguineous marriage). (C) Cranial MRIs reveal hyperintense changes (arrows) as a sign for leucencephalopathy, and cerebral atrophy (FLAIR, axial images). (D) Histological analysis of patient II.5 muscle biopsy specimen, from left to right: Gomori trichrome stain for muscle fibers revealed no conspicuous ragged red fibers indicative of a mitochondriopathy. Succinate dehydrogenase (SDH) and neuron specific enolase (NSE) staining revealed a neurogenic pattern with grouped fibers indicating denervation (magnification 200x). Electron microscopy (EM) revealed paracristalline inclusions (arrow) and altered cristae structure (“parking lots”, magnification 15,000x). (E) Whole exome sequencing discovered the homozygous mutation c.616C>T in exon 5 of the YME1L1 gene (NM_014263), localized at position 149 of the YME1L1 protein, and leading to an amino acid exchange of arginine to tryptophan in the mitochondrial targeting site (MTS; p.R149W, NP_055078). YME1L1 contains highly conserved domains: MTS, transmembrane domain (TM), ATPase domain (AAA+), motif of metalloprotease (Zinc) activity (HESGH). (F) Electropherogram depicting homozygous missense mutation in patient II.5, which is heterozygous in the father. (G) The mutation lies within a protein region, highly conserved throughout different species. (H) YME1L1 mRNA levels do not differ between patient and control primary human adult fibroblasts (Co, Control; II.5, patient II.5; ns, not significant; one-way ANOVA; p=0.2314; n=6 ) (I) Steady state levels of YME1L1 are below detection levels or profoundly reduced in cell lysates of patient II.5 YME1L1R149W; Yme1l1 knockout mouse fibroblasts serve as negative controls. YME1L1 protein levels in mitchondrial compartment fractions can be found Figure 1—figure supplement 1 (HAF, primary human adult fibroblasts; ACF, immortalized murine adult cardiac fibroblasts; MEF, immortalized murine embryonic fibroblast; Co, Control; II.5, patient II.5; WT, wild type; KO, Yme1l1 knockout, n=5).
-
Figure 1—source data 1
Raw data Graph 1H.
Raw values of YME1L1 mRNA expression in human adult fibroblasts of controls and patient measured via TaqMan qPCR (n=6).
- https://doi.org/10.7554/eLife.16078.003
Table 1
YME1L1 mitochondriopathy phenotype.
| Pedigree ID (gender) | II.5 (f) | II.8 (f) | II.9 (m) | II.11 (f) | ||
|---|---|---|---|---|---|---|
| Age at last assessment (y) | 15.8 | 12.3 | 10.3 | 5.2 | ||
| Category | Feature | HPO | ||||
| Inheritance | ||||||
| AR | AR | AR | AR | |||
| Growth | ||||||
| Height | Short stature SD;% | 0004322 | + -2.19; 1 | + -2.09; 2 | + -1.89; 3 | - 0.38; 35 |
| Weight | Low weight SD;% | 0004325 | + -2.8; <1 | - -1.18; 12 | - 0.07; 53 | - -1.35; 12 |
| Neonatal period | ||||||
| Neonatal asphyxia | 0012768 | - | - | - | + | |
| Head and Neck | ||||||
| Head | Microcephaly | 0000252 | - | - | + | + |
| Macrocephaly | 0000256 | + | - | - | - | |
| Face | Midface retrusion | 0011800 | - | - | + | + |
| Congenital facial diplegia | 0007188 | + | - | - | + | |
| Ears | Hearing impairment | 0000365 | - | + | + | + |
| Sensorineural hearing impairment | 0000407 | n.a. | n.a. | + | + | |
| Macrotia | 0000400 | - | + | + | - | |
| Eyes | Pigmentary retinopathy | 0000580 | + | - | - | - |
| Optic nerve hypoplasia | 0000609 | + | + | + | + | |
| Cherry red spot of the macula | 0010729 | - | - | - | + | |
| Strabismus | 0000486 | - | - | + | + | |
| Hypermetropia | 0000540 | - | - | - | + | |
| Myopia | 0000545 | + | + | + | - | |
| Amblyopia | 0000646 | - | + | + | + | |
| Abnormality of visual evoked potentials | 0000649 | n.a. | - | + | + | |
| Abdomen | ||||||
| Gastro-intestinal | Constipation (in infancy) | 0002019 | + | n.a. | + | + |
| Spleen | Splenomegaly | 0001744 | + | - | + | - |
| Skeletal | ||||||
| Feet | Bilateral talipes equinovarus | 0001776 | - | - | - | + |
| Muscle, soft tissue | ||||||
| Increased variability in muscle fiber diameter | 0003557 | + | n.a. | n.a. | n.a. | |
| Neurologic | ||||||
| CNS | Hypotonia, neonatal, generalized | 0008935 | - | - | - | + |
| Infantile muscular hypotonia | 0008947 | - | - | - | + | |
| Global developmental delay (onset, months) | 0001263 | + | + | + | + (6) | |
| Motor delay | 0001270 | + | + | + | + | |
| Gait apraxia | 0010521 | - | - | - | + | |
| Athetosis | 0002305 | - | - | - | + | |
| Intellectual disability, moderate; (IQ) | 0002342 | + (48) | n.a. | n.a. | + (39) | |
| Incomprehensible speech | 0002546 | n.a. | - | - | + | |
| Poor speech | 0002465 | n.a. | - | + | - | |
| Absent speech | 0001344 | - | + | - | - | |
| Seizures (onset, months) | 0001250 | - | - | - | + (6) | |
| Dysmetria | 0001310 | + | + | + | - | |
| Ataxia | 0001251 | + | + | + | - | |
| Brain atrophy | 0012444 | - | - | - | + | |
| Ventriculomegaly | 0002119 | - | - | - | + | |
| Delayed CNS myelination | 0002188 | + | - | - | - | |
| Abnormality of the cerebral white matter | 0002500 | + | + | + | + | |
| Abnormality of the basal ganglia | 0002134 | - | - | + | - | |
| Cerebellar hypoplasia (progressive) | 0001321 | + | - | - | + | |
| EEG with focal sharp waves | 0011196 | n.a. | - | + | - | |
| Abnormal auditory evoked potentials | 0006958 | n.a. | n.a. | + | + | |
| PNS | Decreased sensory nerve conduction velocity | 0003448 | - | + | n.a. | - |
| Behavioural Psychiatric | Hyperactivity | 0000752 | - | + | - | - |
| Attention deficit hyperactivity disorder | 0007018 | - | - | - | + | |
| Stereotypical body rocking | 0012172 | - | - | - | + | |
| Laboratory anomalies | ||||||
| Mildly elevated creatine phosphokinase | 0008180 | - | - | - | + | |
To identify the genetic basis of the disease, we performed whole-exome sequencing (WES) followed by Sanger sequencing and bioinformatic analysis. We thereby identified a homozygous missense mutation in a highly conserved region of the YME1L1 gene in the affected children (c.616C>T, NM_014263; chr10:27425300; GRCh 38.5, raw data available: sequence reads archive (SDA) accession no. SRP073309) that segregates with the phenotype in the index family. We did not detect mutations in other genes linked previously to neurologic diseases. (De Michele et al., 1998; Hazan et al., 1994; Warnecke et al., 2007). The YME1L1 gene mutation c.616C>T causes the exchange of highly conserved hydrophilic arginine to hydrophobic tryptophan (p.R149W, NP_055078) within the N-terminal MTS (Figure 1E–G). YME1L1R149W was below detection levels or profoundly reduced in patient whole cell lysates and in mitochondrial enriched fractions of patient fibroblasts, while mRNA levels remain unaltered (Figure 1H,I, Figure 1–source data 1, Figure 1–figure supplement 1). These results identify a disease-causing mutation in arginine 149 of YME1L1, which impairs protein accumulation.
YME1L1R149W abrogates maturation of YME1L1 upon import into mitochondria
The affected amino acid residue arginine 149 is present within a predicted mitochondrial targeting sequence of YME1L1 and may affect targeting of the mutant protein to mitochondria. We therefore transiently expressed YME1L1 and YME1L1R149W in YME1L1-/- HeLa cells generated by CRISPR/Cas9-mediated genome editing (Figure 2A). Both YME1L1 and YME1L1R149W co-localized with the mitochondrial marker protein ATPase subunit ß, demonstrating mitochondrial targeting of the mutant protein. Consistently, inhibition of the proteasome did not stabilize mutant YME1L1 (Figure 2B), suggesting that reduced levels of YME1L1R149W are not caused by proteasomal degradation of non-imported YME1L1 precursor proteins. We therefore excluded a degradation of mutant YME1L1R149W via the proteasome.
Figure 2
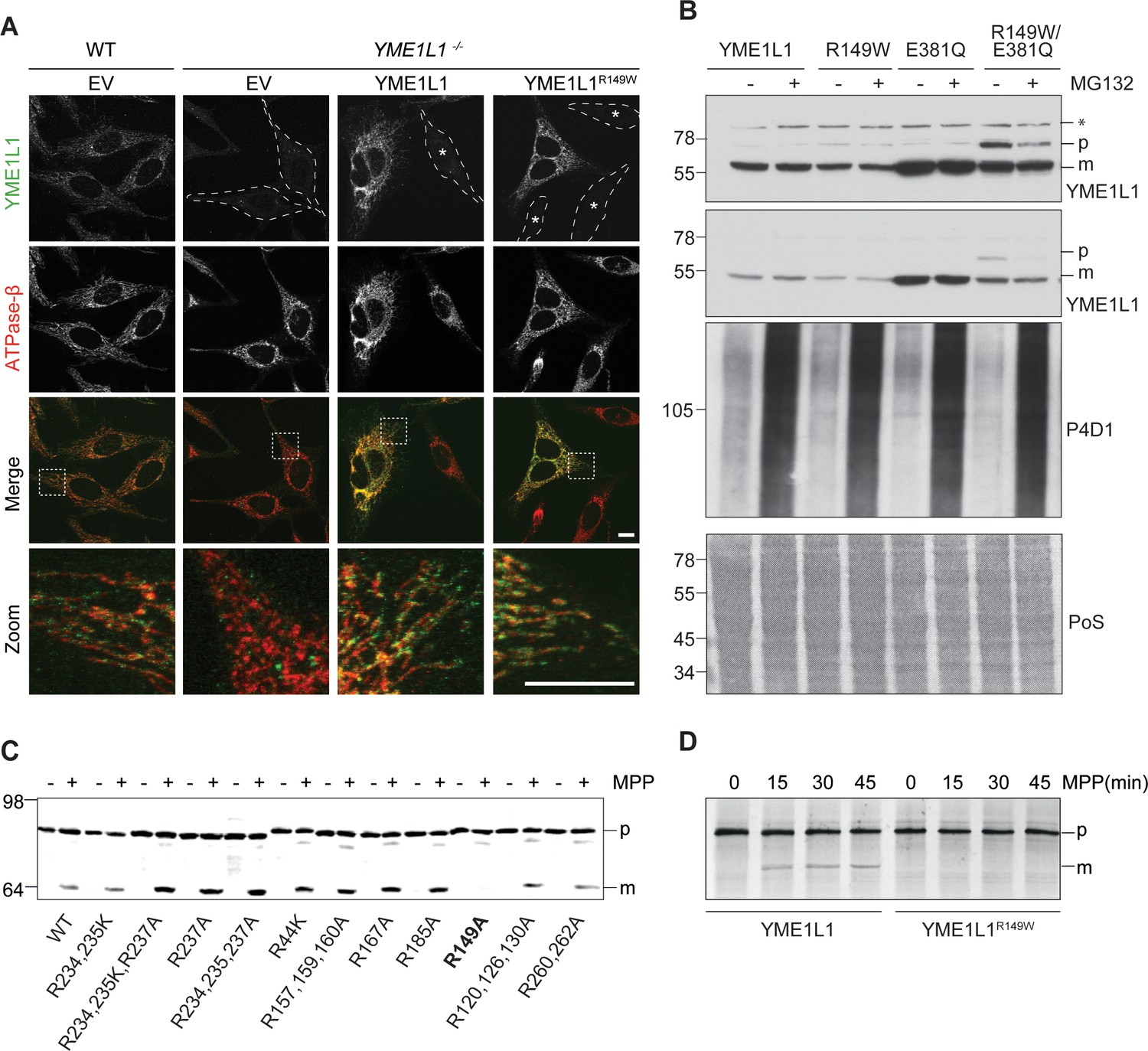
Mutation of arginine 149 impairs maturation of YME1L1 by MPP upon import into mitochondria.
(A) YME1L1R149W is targeted to mitochondria. HeLa cells were transiently transfected with wild-type YME1L1, YME1L1R149W or the empty vector (EV) as control. Cells were analyzed by indirect immunofluorescence and co-localization of the immunofluorescent signals of antibodies against YME1L1 and ATP Synthase subunit beta (ATPase-ß) (WT, Wildtype; YME1L1-/-, Knockout; EV, empty vector; YME1L1R149W, patient mutation; Scale bar 10 µM). (B) Inhibition of the proteasome with MG132 (20 mM, 18 h) does not stabilize precursor or mature YME1L1 in Flp-In T-Rex HEK293T cells expressing YME1L1 or YME1L1 mutant variants: R149W, E381Q or R149W/E381Q. P4D1 antibodies were used as a control for ubiquitin accumulation and proteasome inhibition. (R149W, patient mutation; E381Q, dominant negative mutation of ATPase domain; R149W/E381Q, double mutant; *, unspecific signal; p, premature; m, mature; PoS, Ponceau staining; n=2). (C) Maturation of YME1L1 is mediated by mitochondrial processing peptidase (MPP) and impaired upon mutation of arginine 149. After site-directed mutagenesis of N-terminal arginine residues in YME1L1, the mutant proteins were expressed in a cell-free system and processing by recombinant MPP was examined (WT, wild type YME1L1; p, premature m, mature, n=2–3). (D) Cell-free MPP cleavage-assay: MPP can cleave YME1L1 but not YME1L1R149W from the premature to its mature form (min, minutes; p, premature; m, mature; YME1L1R149W, patient mutation; n=3).
Nuclear-encoded YME1L1 requires cleavage of the MTS from premature YME1L1 (~80 kDa) upon import into mitochondria to give rise to the mature protein (~63 kDa), which is able to assemble into a homooligomeric proteolytic complex in the IM (Graef et al., 2007; Stiburek et al., 2012; Van Dyck and Langer, 1999). MTS cleavage of YME1L1 precursor is bioinformatically predicted to be mediated by MPP (NM_014263; Mitoprot) (Claros and Vincens, 1996). Since the identified human mutation affects an arginine and such residues are known to function as MPP recognition and cleavage sites (Niidome et al., 1994), we asked whether arginine 149 is part of the MPP cleavage site in YME1L1. We mutated various arginine residues in the N-terminal region of YME1L1 and analyzed maturation of corresponding YME1L1 variants synthesized in a cell-free expression system using recombinant MPP (Figure 2C). YME1L1 was converted by MPP in its mature form (Figure 2C). Mutation of arginine at position 149 abrogated MPP-mediated processing of YME1L1, suggesting that amino acid 151 forms the N-terminal amino acid of mature YME1L1. Replacement of arginine 149 by tryptophan similarly abolished maturation of YME1L1 by MPP in vitro (Figure 2D).
YME1L1R149W undergoes rapid proteolysis
Grossly reduced YME1L1 protein levels coinciding with unaltered mRNA levels indicate a post-transcriptional mechanism responsible for the reduction of YME1L1R149W. Stabilization of the precursor form of YME1L1 by abolishing the MPP recognition site may allow another peptidase to cleave YME1L1. To identify the latter mechanism, we screened candidate mitochondrial proteases for their effect on YME1L1 and YME1L1R149W stably expressed in HEK293T cells, but could not detect a difference in YME1L1/YME1L1R149W level upon depletion of individual proteases (data not shown). However, by preventing the proteolytic activity of YME1L1 upon ectopic expression of the dominant-negative YME1L1E381Q, harboring a point mutation in the ATPase domain, we were able to stabilize not only the known YME1L1 substrate PRELID1 (Potting et al., 2013) but also observed accumulation of both the precursor and mature forms of YME1L1 (Figure 3A). The mature form of YME1L1 remained stable in cells harboring YME1L1E381Q upon further incubation in the presence of cycloheximide, while ectopic expression of the wild type variant resulted in significantly lower levels of YME1L1 and reduced stability (Figure 3A). These observations point to an auto-catalytic degradation of YME1L1R149W. We therefore combined the dominant negative mutation E381Q with the pathogenic mutation R149W in YME1L1 in cis and examined the stability of this variant (Figure 3A). We observed an accumulation of mature YME1L1R149W/E381Q upon ectopic expression in HEK293T cells (Figure 3A). Both mature YME1L1R149W/E381Q and larger forms with a similar size to the precursor form of YME1L1 accumulated in these cells. Stabilization of the precursor form of YME1L1 in these cells may allow another peptidase to cleave YME1L1 resulting in the accumulation of slightly smaller forms of YME1L1. These results suggest that YME1L1 mediates degradation of mutant YME1L1R149W, which accumulates in the precursor form due to the mutation of the MPP cleavage site and thus is likely recognized as a misfolded protein in the IM. As the mutant YME1L1 variants were expressed in wild type HEK293T cells, we cannot distinguish whether degradation of YME1L1R149W occurs intramolecularly and/or by endogenous YME1L1 under these experimental conditions. However, the lack of wild-type YME1L1 in patient cells with homozygous YME1L1 mutations together with our ectopic expression studies argue for auto-catalytic degradation of YME1L1R149W.
Figure 3
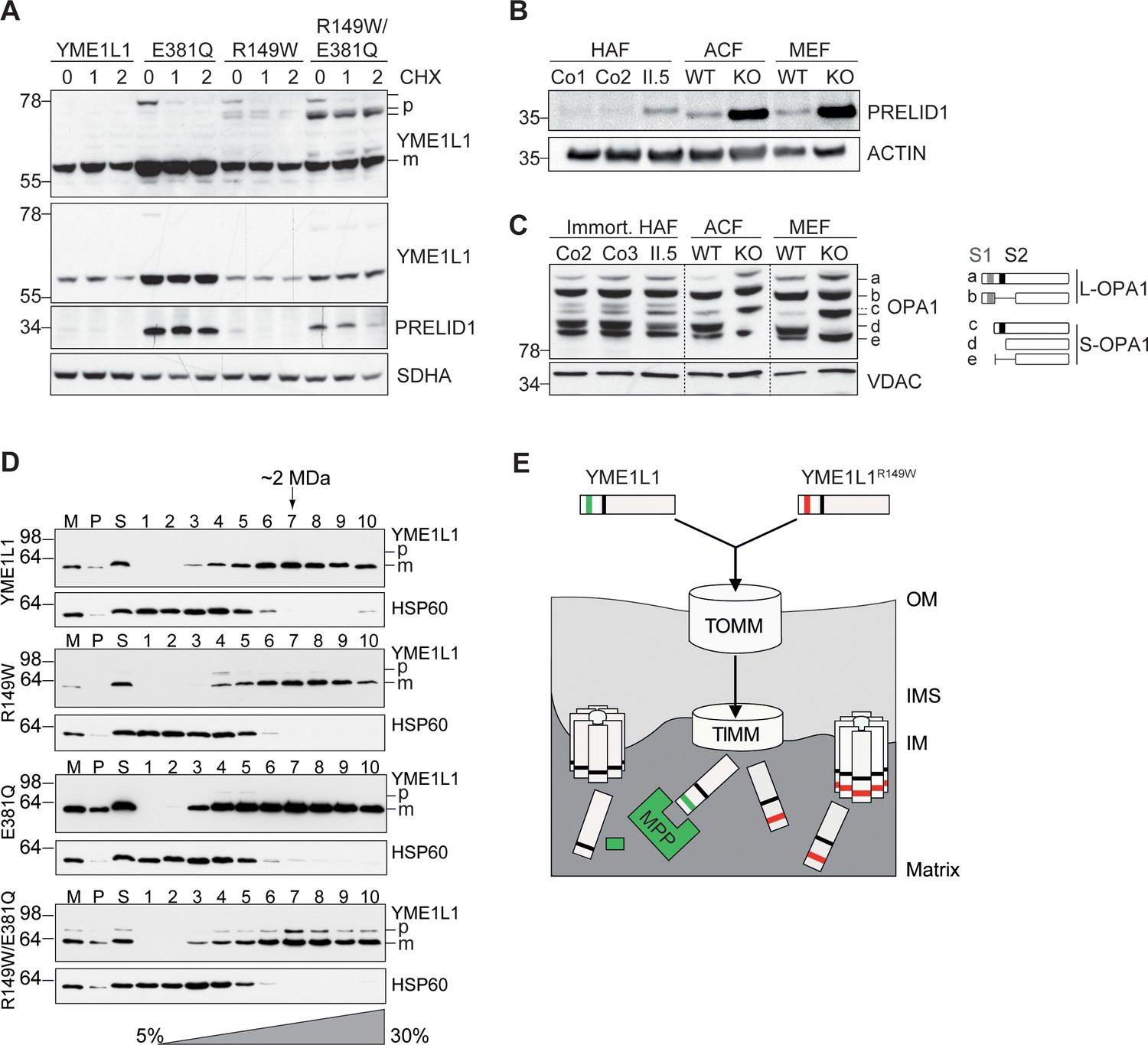
Mutation of arginine 149 destabilizes YME1L1 but retains residual YME1L activity.
(A) Stability of YME1L1 or YME1L1 mutant variants (R149W, E381Q and R149W/E381Q) expressed in Flp-In T-Rex HEK293T cells. The dominant negative E381Q mutation in the ATPase domain of YME1L1 prevents degradation of YME1L1R149W (E381Q, dominant negative mutation of ATPase domain; R149W, patient mutation; R149W/E381Q, double mutant; CHX, cycloheximide; h, hours; p, premature; m, mature; SDHA, succinate dehydrogenase; n=2). (B) Homozygous mutation in YME1L1 results in an accumulation of PRELID1 in the human patient. Yme1l1 knockout mouse fibroblasts serve as positive controls to demonstrate impaired proteolysis of PRELID1 (HAF, human adult primary fibroblasts; ACF, immortalized murine adult cardiac fibroblasts; MEF, immortalized murine embryonic fibroblast; Co, Control; II.5, patient; WT, wild type; KO, knockout; n=5). (C) Mutation of arginine 149 of YME1L1 impairs processing of OPA1 with a decrease of short OPA1 form d levels. The formation of OPA1 form d indicates residual YME1L1 activity in human patient fibroblasts (immort. HAF, immortalized human adult fibroblasts; ACF, immortalized murine adult cardiac fibroblasts; MEF, immortalized murine embryonic fibroblast; Co, Control; II.5, patient; WT, wild type; KO, knockout; n=6). The schematic diagram illustrates the proteolytic processing of OPA1 by YME1L1 on processing site 2 (S2) and OMA1 on processing site 1 (S1). The presence of long OPA1 forms (L-OPA1) is required for the maintenance of mitochondrial inner membrane fusion, whereas accumulation of short OPA1 forms (S-OPA1) is associated with accelerated fission. (D) Mitochondria-enriched membrane fractions from Flp-In T-Rex HEK293T cells expressing YME1L1 or YME1L1 mutant variants (R149W, E381Q and R149W/E381Q) were solubilized in digitonin and analyzed by sucrose gradient centrifugation. Fractions were collected and separated on SDS-PAGE for immunoblotting to detect high MW complexes of YME1L1. HSP60 complexes were used as a control (M, mitochondrial input, P, S, pellet and supernatant fraction after solubilization; HSP60, heat shock protein 60; E381Q, dominant negative mutation of ATPase domain; R149W, patient mutation; R149W/E381Q, double mutant). (E) Premature YME1L1/ YME1L1R149W is imported into the mitochondrial matrix via translocons of the outer mitochondrial membrane (TOMM) and inner mitochondrial membrane (TIMM). Here, MPP binds and cleaves the N-terminal mitochondrial targeting site (MTS) from premature YME1L1 but not YME1L1R149W, which then allows the mature and premature YME1L1R149W protein to assemble as proteolytic complex.
Impaired proteolysis by YME1L1R149W
To confirm a functional impairment of YME1L1R149W, we analyzed the effect of YME1L1R149W on two substrate proteins, the protein of relevant evolutionary and lymphoid interest (PRELID1) and OPA1, in patient-derived skin fibroblasts. PRELID1, a lipid transfer protein for phosphatidic acid in the IMS, is constitutively degraded by YME1L1 (Potting et al., 2013). PRELID1 accumulated in primary human fibroblasts (HAF) of patient II.5 when compared to controls (Figure 3B), consistent with the low levels of YME1L1R149W present in mitochondria (Figure 1I, Figure 1–figure supplement 1). Deletion of murine Yme1l1 leads to accelerated processing of L-OPA1 by OMA1 (Anand et al., 2014; Rainbolt et al., 2016) and drives mitochondrial fragmentation in vitro (Baker et al., 2014) and in vivo (Korwitz et al., 2016; Wai et al., 2015). We similarly identified an altered cleavage pattern of OPA1 in patient cells, albeit more modestly than in knockout mouse cells (Figure 3C).
This suggests that YME1L1R149W retains proteolytic activity. As proteolysis by YME1L1 depends on its oligomerization, we examined in further experiments the assembly of YME1L1R149W expressed in HEK293 cells by sucrose gradient centrifugation (Figure 3D). To allow detection of YME1L1R149W precursor forms and impair its degradation, we also examined the assembly of YME1L1R149W/E381Q expressed in HEK293 cells. Similar to YME1L1, the mutant variants of YME1L1 were recovered in protein complexes of ~2 MDa (Figure 3D). Notably, we also observed assembly of the precursor form of YME1L1R149W/E381Q, demonstrating that the disease-causing mutation R149W does not impair the formation of the i-AAA protease complex. Thus, assembled YME1L1R149W precursor forms may explain the residual activity of YME1L1 in patient fibroblasts (Figure 3E). Alternatively, and not mutually exclusive, low levels of mature YME1L1R149W may maintain residual proteolytic activity in the patient system.
Together, we conclude that YME1L1R149W is a hypomorphic allele that retains partial YME1L1 activity, which reconciles the embryonic lethality (Wai et al., 2015) of Yme1l1 knockout mice with the milder phenotype in our patients with a homozygous YME1L1 missense mutation.
YME1L1R149W causes mitochondrial fragmentation
Given the role of unbalanced mitochondrial dynamics in neurodegenerative diseases (Burté et al., 2015) and the observed, impaired OPA1 processing, we focused our attention on mitochondrial morphology in patient fibroblasts. We detected an increase in shortened and fragmented mitochondrial networks in HAF of patient II.5 relative to a control (Figure 4A,B, Videos 1–4, Figure 4–source data 1), consistent with the more severe fragmentation phenotype in Yme1l1 KO mouse fibroblasts (Anand et al., 2014). We therefore transiently expressed human YME1L1R149W and YME1L1 in murine adult cardiac Yme1l1-/-fibroblasts (Wai et al., 2015) and observed that human YME1L1 but not YME1L1R149W was able to complement mitochondrial fragmentation. Of note, the human mutant variant was still able to partially tubulate mitochondria (Figure 4C,D, Figure 4–source data 1), consistent with the notion that YME1L1R149W retains residual function.
Figure 4
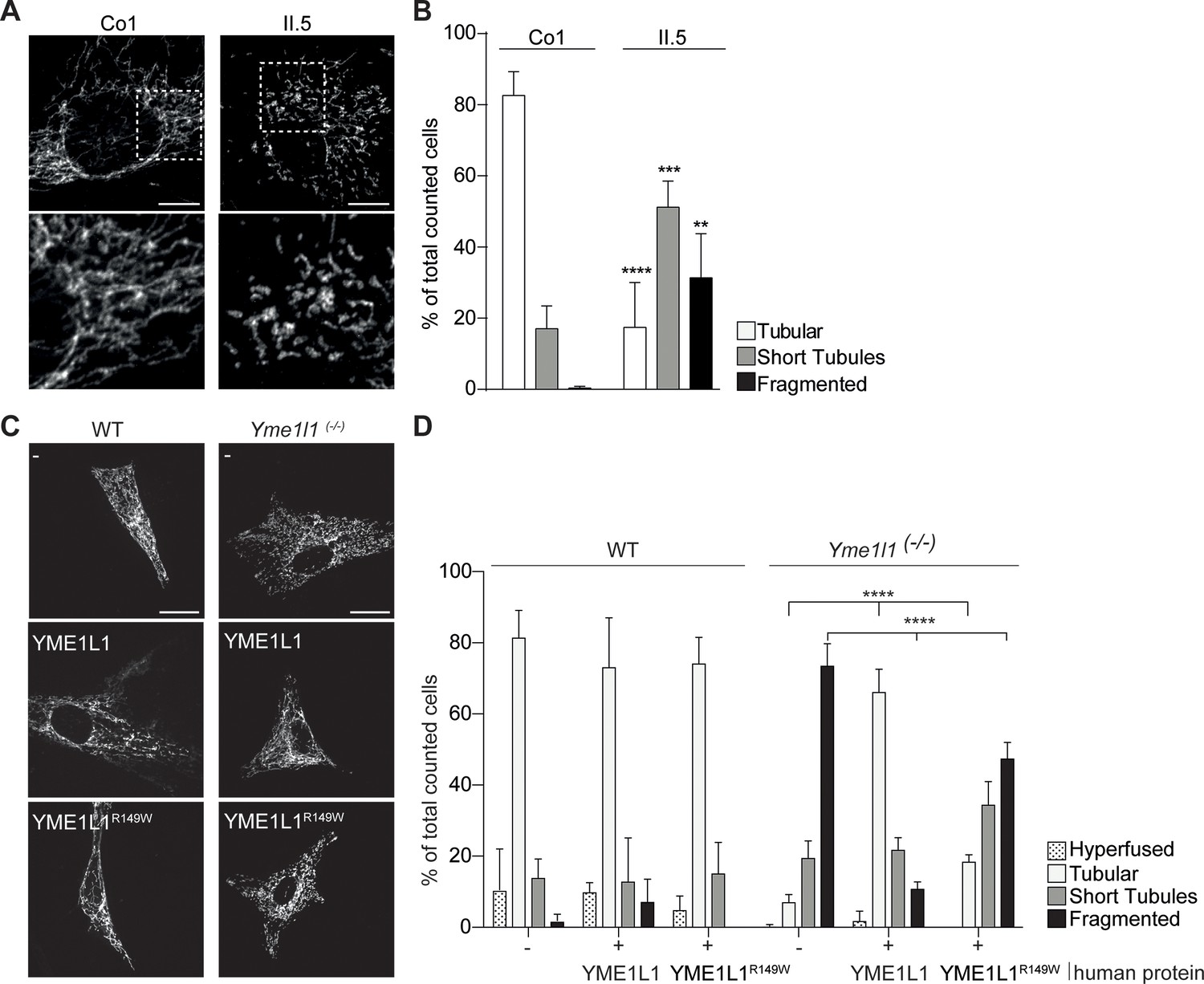
YME1L1R149W causes mitochondrial fragmentation.
(A, B) YME1L1R149W causes fragmentation (fragmentation or short tubules) of mitochondrial networks in patient fibroblasts (Co, Control, II.5, patient II.5; one-way ANOVA; n=150–200 cells; 4 replicates; scale bar 10 µm). (C) Co-transfection of mitochondrial GFP and human YME1L1/ YME1L1R149W protein in WT and Yme1l1-/- MEFs revealed that YME1L1 but not YME1L1R149Wcan rescue mitochondrial fragmentation in Yme1l1-/- MEFs and (D) increases the number of cells with tubular networks. Expression of YME1L1R149W partially rescues the fragmentation phenotype. In WT MEFs, YME1L1R149W expression results in only a mild, but not significant decline of cells with a tubular mitochondrial network (WT, wildtype; Yme1l1-/-, Yme1l1 Knockout; two-way ANOVA; n=3; scale bar 10 µm; **p<0.01; ***p<0.001; ****p<0.0001).
-
Figure 4—source data 1
Raw data Graph 4B.
Relative mitochondrial morphology quantification after immunocytochemistry with TOMM20 antibody and DAPI in human adult fibroblasts of control and patient (n=150–200 cells, 4 replicates).
- https://doi.org/10.7554/eLife.16078.009
-
Figure 4—source data 2
Raw data Graph 4D.
Relative mitochondrial morphology quantification after immunocytochemistry with TOMM20 antibody and DAPI in WT and Yme1l1-/- adult cardiac murine fibroblasts overexpressing YME1L1 or YME1L1R149W (n=3).
- https://doi.org/10.7554/eLife.16078.010
Video 1
Three dimensional rendering of mitochondrial structure of a control fibroblast.
MitoTracker Deep Red- mitochondria; DAPI-nucleus.
Video 2
Three dimensional rendering of mitochondrial structure of a patient II.5 fibroblast.
MitoTracker Deep Red- mitochondria; DAPI-nucleus.
Video 3
Mitochondrial dynamics in a control fibroblast.
MitoTracker Green-mitochondria, live cell imaging.
Video 4
Mitochondrial dynamics in a patient II.5 fibroblast.
MitoTracker Green-mitochondria, live cell imaging.
YME1L1R149W impairs cell proliferation
To further address the pathophysiological mechanism underlying the neurological phenotype in humans, we analyzed proliferation and apoptosis of HAF. Proliferation of patient II.5 fibroblasts was significantly reduced compared to those of healthy controls, while the level of induced apoptotic cell death remained unaffected (Figure 5A–C, Figure 5–source data 1, Figure 5–source data 1, Figure 5–source data 1). Analysis of the respiratory chain in patient fibroblasts revealed the specific enzymatic activities to be within the normal physiological range and subunit levels to be unaltered (Figure 5D,E), consistent with the observation in Yme1l1–depleted hearts (Wai et al., 2015). However, the reduced steady state levels of various mitochondrial compartment markers in patient cells indicate a general reduction of mitochondrial mass in YME1L1R149W cells (Figure 5E, Figure 5F).
Figure 5

YME1L1 mutation impairs fibroblast cell growth and proliferation.
(A) Significant reduction of cell culture growth (y-axis scale *104) and (B) proliferation of patient primary human adult fibroblasts after 120h under culture conditions (Co, Control; II.5, patient II.5; one-way ANOVA; ***p<0.001; ****p<0.0001; n=5 ). (C) No increase in apoptosis-sensitivity upon 2 µM staurosporine (STS) treatment in patient HAF after 6h (SDS-PAGE for cleaved PARP level (cPARP) (Co, Control; II.5, patient II.5; one-way ANOVA; p6h = 0.4283; n=3; ). (D) Normal respiratory chain subunit activity in patient HAF (U, units; mU, milli U; CS, Citrate synthase). (E) Cytochrome c oxidase subunit 4 (COX4) and NADH ubiquinone oxidoreductase 1 beta subcomplex 6 (NDUFB6) protein levels were not elevated in patient fibroblasts (Co, Control; II.5, patient II.5; n=3). (F) Decreased levels of mitochondrial compartment markers TOMM20 (translocon of outer mitochondrial membrane 20) for the outer membrane (OM), Cytochrome c for the intermembrane space (IMS), SDHA (succinate dehydrogenase) for the inner membrane (IM), and Cyclophilin D for the matrix in whole cell lysates of patient II.5 (Co, Control; II.5, patient II.5; n=3).
-
Figure 5—source data 1
Raw data Graph 5A.
Raw values of fluorometric measurement with Cell-titer Blue Cell Viability Assay application in human adult fibroblasts of controls and patient (n=5).
- https://doi.org/10.7554/eLife.16078.016
-
Figure 5—source data 2
Raw data Graph 5B.
Raw values of colorimetric measurement with BrdU Cell Proliferation ELISA Assay application in human adult fibroblasts of controls and patient (n=5).
- https://doi.org/10.7554/eLife.16078.017
-
Figure 5—source data 3
Raw data Graph 5C.
Raw values of cleaved-PARP antibody signal intensity after staurosporine treatment in human adult fibroblasts of controls and patient and SDS-Page (n=3).
- https://doi.org/10.7554/eLife.16078.018
Here we report a new form of infantile-onset mitochondriopathy caused by a homozygous mutation in the YME1L1 gene. Patients suffer from severe intellectual disability, muscular impairments, and optic nerve atrophy. In contrast to complete gene deletion in the mouse (Wai et al., 2015), the homozygous c.616C<T missense mutation is compatible with life, likely owing to the residual function of YME1L1R149W. We demonstrate that the missense mutation affects the MPP processing site and impairs YME1L1 maturation, leading to its rapid degradation. We further show that the YME1L1R149W leads to a proliferation defect, abnormal OPA1 processing and mitochondrial fragmentation. Since fusion and fission of mitochondria are essential to preserve cellular functions, it is conceivable that defects in these processes, contribute to the patient phenotype in a cell-type specific manner. With our findings, we reveal an important role of YME1L1 for the maintenance of mitochondrial morphology in humans and link a loss of YME1L1 function to disease. YME1L1 thereby joins other members of the AAA family of ATPases that have already been linked to neurologic disease, such as spastin (SPG4, MIM*6042779 and paraplegin (SPG7, MIM*602783) gene, and in the gene encoding the YME1L1 substrate optic atrophy 1 (OPA1, MIM*605290) (De Michele et al., 1998; Hazan et al., 1994; Nielsen et al., 1997; Warnecke et al., 2007). Future studies on further patients are required to determine the full phenotype spectrum associated with YME1L1 gene mutations.
Material and methods
Subjects
Informed consent was obtained from the parents of the patients for the molecular genetic analysis, the publication of clinical data, photos, magnetic resonance images (MRI), and studies on fibroblasts. The human study was approved by the local ethics committee of the Charité (approval no. EA1/212/08).
Genetic analyses
Request a detailed protocolHomozygosity linkage intervals (chr10:24,333,063–49,978,774, LOD=2.533) with LOD>2 and length >1 Mb were identified using the Affymetrix SNP array 6.0. For whole exome sequencing, 1 µg genomic DNA extracted from peripheral blood was enriched by Agilent SureSelect Human All Exon enrichment kit version 3 and deep-sequenced by Illumina Hiseq2000 sequencer in a 101 bp single-end mode. The output sequences amounted to 12 Gb and >94% of the exon-coding regions were covered by at least 20 folds. The data analysis pipeline considered linkage interval, pathogenicity of variants, allele frequency in polymorphism databases (1000Genome, ESP, and an in-house database including 868 individual exomes of Middle East origin), functional involvement of specific genes, thus resulting in the identification of a homozygous YME1L1 gene mutation (chr10p12.1) (Najmabadi et al., 2011). We confirmed the absence of homozygous deleterious variants (missense, nonsense, frameshift, splice site change) in YME1L1 in the NHLBI Exome Sequencing Project (ESP, http://evs.gs.washington.edu/EVS/; ExAC 0.3 http://exac.broadinstitute.org/), the 1000 Genome database (http://www.1000genomes.org/), the dbSNP138 (http://www.ncbi.nlm.nih.gov/SNP/), and our in-house 898 Middle East origin exome database. The raw data is available under the sequence reads archive (SDA) accession no. SRP073309.
Sanger sequencing of the YME1L1 gene (NM_014263, GRCh 38.5) confirmed the mutation in the patients and established the genotype in the other family members and in additional families.
Mouse and human fibroblasts
Request a detailed protocolHAF were established from skin biopsies derived from patient II.5 and unrelated controls (Co1-3). MEF and adult cardiac fibroblasts (ACF) were derived from Yme1l1 mutant mice (Yme1l1loxP/loxP) as described previously (Anand et al., 2014; Wai et al., 2015; Yao and Shoubridge, 1999). HAF, MEF, and ACF were cultured in high glucose Dulbecco’s modified Eagle’s medium (Gibco, Darmstadt, Germany) supplemented with 15%, 10%, and 10% fetal bovine serum (FBS, Biochrome, Berlin, Germany; Life Technologies; Carlsbad, California), respectively, and 1% penicillin-streptomycin (Sigma Aldrich; St. Louis, Missouri). Fibroblasts were immortalized by stable expression of hTERT and E7 (Zhu et al., 1998).
Generation of YME1L1-/- HeLa cells
Request a detailed protocolYME1L1 KO cells were generated using the CRISPR/Cas9 system. Guide RNA (gRNA) and Cas9 were expressed using the px330 expression vector (Cong et al., 2013). The following gRNA target sequence was used: 5’-GGAACCGACCATATTACAACAGG-3´ (recommended by the lab of Alexander van der Bliek). Single clones were obtained by serial dilution after transfection and screened by SDS-PAGE and immunoblotting.
qPCR
Request a detailed protocolDNA extraction and cDNA synthesis were performed with established methods reported previously (Issa et al., 2013). To specifically amplify and detect murine Yme1l1, human YME1L1, Hprt (Hypoxanthine-guanine phosphoribosyl-transferase, reference gene), and RPII (RNA polymerase II, reference gene) cDNA, we designed sets of primers and TaqMan probes specified in Supplementary file 1A using the GenScript real-time PCR (TaqMan) Primer Design online software (www.genscript.com). qPCR and quantification was performed as described previously (Issa et al., 2013), and further statistics were performed using the GraphPad Prism 5 Software (GraphPad Software Inc., La Jolla, California).
Western blot
Request a detailed protocolProtein whole lysate extraction, fractionated mitochondrial and cytosolic protein extraction, and Western blots were performed with established methods reported previously (Issa et al., 2013; Potting et al., 2013). Antibodies are listed in Supplementary file 1B.
Isolation of a crude mitochondrial fraction
Request a detailed protocolThe protocol was adapted from (Vogel et al., 2005). Cells were harvested using trypsin and washed three times in PBS to remove the trypsin. Subsequently, the cells were resuspended in 3 ml isotonic Buffer (0,25 M Sucrose, 5 mM Tris-HCL pH 7,5, 1 mM EDTA and 0,1 mM PMSF). The cells were opened with eight strokes by 2500 rpm using a potter homogenisator. Unbroken cells and nuclei were removed by centrifugation at 600x g for 15 min at 4°C. The supernatant was applied to another centrifugation step at 10000x g for 25 min at 4°C. The supernatant contains the cytoplasmic fraction (C) whereas mitochondria are enriched in the pellet. The mitochondrial fraction (M) was washed two times in isotonic buffer and afterwards applied to SDS Page.
Cell viability, apoptosis, and proliferation
Request a detailed protocolViability and proliferation of HAF were quantified using the Fluorimetric CellTiter-Blue Cell Viability Assay (Promega, Madison, Wisconsin) and the colorimetric Cell Proliferation BrdU-ELISA (Roche Diagnostics, Rotkreuz, Switzerland), respectively, according to the manufacturer´s instructions. Apoptosis of HAF following treatment with staurosporine (Cell signaling, Cambridge, United Kingdom) was determined through quantification of cleaved PARP protein levels on Western blots.
Mitochondrial respiratory chain enzyme activity
Request a detailed protocolMitochondrial respiratory chain enzyme activities were measured in skin fibroblasts according to established procedures (Janssen et al., 2006; Smeitink et al., 2001). The values were expressed relative to the activity of Cytochrome C oxidase and/or Citrate synthase.
Overexpression of human YME1L1
Request a detailed protocolHuman wild type YME1L1 and mutant YME1L1R149W were stably expressed in Flp-In T-Rex HEK293T cells (Life Technologies) under the control of a tetracycline-regulated promotor according to manufacturer´s instructions. HEK293T cells were cultured as described previously (Potting et al., 2013). Transient transfection was performed using GeneJuice (EMD Millipore, Darmstadt, Germany) according to manufacturer´s instruction.
Sucrose gradient density centrifugation
Request a detailed protocolCells were harvested and washed three times in PBS. Subsequently, the cells were resuspended in isotonic buffer (0,25 M Sucrose, 5 mM Tris-HCL pH 7,5, 1 mM EDTA and 1X cOmplete, EDTA-free Protease Inhibitor Cocktail [Sigma]). The cells were opened with 15 strokes by 1000 rpm using a Potter S homogenisator (Sartorius, Göttingen, Germany). Unbroken cells and nuclei were removed by centrifugation at 600x g for 5 min at 4°C. The supernatant was applied to another centrifugation step at 10000x g for 10 min at 4°C. The mitochondrial-enriched membrane fraction (pellet) was re-suspended in lysis buffer (270 mM sucrose, 100 mM KCl, 20 mM 20 mM MgCl2, 10 mM Tris/HCl, pH 7.5, and 1X cOmplete, EDTA-free Protease Inhibitor Cocktail) containing 6 g of digitonin per g of protein at a concentration of 2.5 mg/ml for 20 min on ice. Supernatant from a clarifying centrifugation at 16,000 g for 10 min at 4°C was directly applied to a 5–25% sucrose gradient prepared using an automated gradient maker (Biocomp instruments, Fredericton, Canada) in 14x89mm Ultra-Clear centrifuge tubes (Beckman Instruments, Brea, California). Ultracentrifugation was performed at 71,000 x g for 16 hr at 4°C and 1.2 ml fractions (n=10) were collected by hand, precipitated by TCA and subjected to SDS-PAGE.
Cell-free synthesis of YME1L1 and processing by purified MPP in vitro
Request a detailed protocolThe cell-free synthesis of (Baker et al., 2014) S-labeled precursor proteins of human wild type and mutant YME1L1 was performed using the TNT Sp6 or Coupled Reticulocyte lysate system (Promega) according to the manufacturer's instructions. Radiolabeled wild type and mutant YME1L1 were incubated in cleavage buffer (20 mM HEPES, pH 7,4, 50 mM NaCl2, 1 mM Zn Cl2, 1 mM ATP, 5 mM Mg Cl2) at 30°C in the presence of absence of MPP as described previously (Nolden et al., 2005). Samples were analyzed by SDS-PAGE and autoradiography.
Immunocytochemistry
Request a detailed protocolImmunocytochemistry was performed as described previously (Anand et al., 2014; Issa et al., 2013). Antibodies are listed in Supplementary file 1B; DAPI was purchased from Sigma-Aldrich.
Mitochondrial network analysis
Request a detailed protocolHAF, MEF, and ACF were stained with anti-TOMM20 antibody and DAPI (4′,6-Diamidin-2-phenylindol). Images were taken as described below and quantified in double-blind fashion into four categories: hyperfused, tubular, short tubules and fragmented mitochondrial network as previously described (Anand et al., 2014). For the complementation experiments: co-transfection of 100 ng of mitochondrially-targeted GFP (mito-GFP) along with 1 µg of either YME1L1 or YME1L1R149W was performed, and GFP-positive fixed cells were scored for mitochondrial morphology.
Imaging
Request a detailed protocolFluorescently labeled fibroblasts were imaged with a fluorescent Olympus BX51 microscope using the software Magnafire 2.1B Version 2001 (Olympus, Tokyo, Japan) and a Spinning Disc Microscopy system (Carl Zeiss Microscopy, Oberkochen, Germany) with the ZEN 2012 Software, with an lsm5exciter Zeiss confocal microscope (Carl Zeiss Microscopy). Analysis of overexpression experiment of human wildtype and mutant YME1L1 was realized with an UltraVIEW VoX spinning disc Yokogawa CSU-X1 confocal microscope (PerkinElmer, Waltham, Massachusetts) on a Nikon Ti microscope equipped with a 60x (Apo TIRF 60x Oil N.A. 1.49) objective and camera (EMCCD C9100-50 CamLink). Imaging of HeLa cells was performed using a Meta 510 confocal laser scanning microscope (Carl Zeiss). All images were processed using Adobe Photoshop (Adobe Systems Inc., San José, California), Volocity (PerkinElmer), and ImageJ (National Institute of Health, USA).
Live-cell imaging
Request a detailed protocolHAF were cultured on 35 mm Fluorodish Cell Culture Dishes (World Precision Instruments, Sarasota, Florida) and incubated in pre-warmed medium containing 500 nM MitoTracker Green (Thermo Fisher Scientific, Waltham, Massachusetts) 30 min prior to imaging. Live-cell imaging was performed in fresh medium in the microscope incubation chamber (5% CO2, 37°C) of a Zeiss Axio Observer with Spinning Disc Technology Carl Zeiss Microscopy) for 7–10 min. Recordings were processed using Adobe Photoshop (Adobe Systems Inc.) and Magix Video Maker Deluxe (Magix Software GmbH, Berlin, Germany).
Electron microscopy (EM)
Request a detailed protocolFibroblasts and muscle biopsy specimen were prepared for TEM analysis and imaged as described previously (Anand et al., 2014; Stenzel et al., 2015; Wakabayashi et al., 2009).
Statistical methods
Request a detailed protocolAll results are presented as mean ± SD, GraphPad Prism 5 software was used for all statistical analyses, and the significance level was set at p<0.05. Differences between two groups were evaluated by un-paired or paired Student’s t-test, while for multi-group comparisons, one-way ANOVA with Tukey’s multiple-comparison test was used. For the mitochondrial fragmentation analysis (Figure 3D), two-way ANOVA followed by the Tukey’s multiple-comparisons test was used. Sample sizes, replicate numbers, and p-values are stated in the figure legends. Replicates are biological replicates except for experiments illustrated in Figures 1H and 2B.
Data availability
-
Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentationPublicly available at the NCBI Gene Expression Omnibus (accession no: SRP073309).
References
-
The i-AAA protease YME1L and OMA1 cleave OPA1 to balance mitochondrial fusion and fissionThe Journal of Cell Biology 204:919–929.https://doi.org/10.1083/jcb.201308006
-
Impaired folding of the mitochondrial small TIM chaperones induces clearance by the i-AAA proteaseJournal of Molecular Biology 424:227–239.https://doi.org/10.1016/j.jmb.2012.09.019
-
Disturbed mitochondrial dynamics and neurodegenerative disordersNature Reviews Neurology 11:11–24.https://doi.org/10.1038/nrneurol.2014.228
-
Computational method to predict mitochondrially imported proteins and their targeting sequencesEuropean Journal of Biochemistry 241:779–786.https://doi.org/10.1111/j.1432-1033.1996.00779.x
-
Mutation detection in four candidate genes (OXA1L, MRS2L, YME1L and MIPEP) for combined deficiencies in the oxidative phosphorylation systemJournal of Inherited Metabolic Disease 28:1091–1097.https://doi.org/10.1007/s10545-005-4483-y
-
A new locus for autosomal recessive hereditary spastic paraplegia maps to chromosome 16q24.3The American Journal of Human Genetics 63:135–139.https://doi.org/10.1086/301930
-
Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1The Journal of Cell Biology 187:1023–1036.https://doi.org/10.1083/jcb.200906084
-
Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavageThe Journal of Cell Biology 178:757–764.https://doi.org/10.1083/jcb.200704112
-
Linkage of a new locus for autosomal dominant familial spastic paraplegia to chromosome 2pHuman Molecular Genetics 3:1569–1573.https://doi.org/10.1093/hmg/3.9.1569
-
Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cellsThe Journal of Cell Biology 187:959–966.https://doi.org/10.1083/jcb.200906083
-
Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondriaThe Journal of Cell Biology 212:157–166.https://doi.org/10.1083/jcb.201507022
-
AAA proteases with catalytic sites on opposite membrane surfaces comprise a proteolytic system for the ATP-dependent degradation of inner membrane proteins in mitochondriaThe EMBO Journal 15:4218–4229.
-
OPA1 processing in cell death and disease - the long and short of itJournal of Cell Science 129:2297–2306.https://doi.org/10.1242/jcs.159186
-
CAG repeat expansion in autosomal dominant pure spastic paraplegia linked to chromosome 2p21-p24Human Molecular Genetics 6:1811–1816.https://doi.org/10.1093/hmg/6.11.1811
-
Arginine residues in the extension peptide are required for cleavage of a precursor by mitochondrial processing peptidase. Demonstration using synthetic peptide as a substrateThe Journal of Biological Chemistry 269:24719–24722.
-
Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosisJournal of Biological Chemistry 278:7743–7746.https://doi.org/10.1074/jbc.C200677200
-
Degradation of cytochrome oxidase subunits in mutants of yeast lacking cytochrome c and suppression of the degradation by mutation of yme1Journal of Biological Chemistry 270:20879–20882.https://doi.org/10.1074/jbc.270.36.20879
-
Human NADH:ubiquinone oxidoreductaseJournal of Bioenergetics and Biomembranes 33:259–266.https://doi.org/10.1023/A:1010743321800
-
OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1LThe Journal of Cell Biology 178:749–755.https://doi.org/10.1083/jcb.200704110
-
A mitochondrial rhomboid proteaseDevelopmental Cell 4:769–770.https://doi.org/10.1016/S1534-5807(03)00167-9
-
ATP-dependent proteases controlling mitochondrial function in the yeast Saccharomyces cerevisiaeCellular and Molecular Life Sciences 56:825–842.https://doi.org/10.1007/s000180050029
-
The dynamin-related GTPase Drp1 is required for embryonic and brain development in miceThe Journal of Cell Biology 186:805–816.https://doi.org/10.1083/jcb.200903065
-
Expression and functional analysis of SURF1 in Leigh syndrome patients with cytochrome c oxidase deficiencyHuman Molecular Genetics 8:2541–2549.https://doi.org/10.1093/hmg/8.13.2541
Article and author information
Author details
Funding
Sonnenfeld Stiftung
- Bianca Hartmann
Deutsche Gesellschaft für Muskelkranke e.V.
- Bianca Hartmann
Human Frontier Science Program
- Timothy Wai
Max-Planck-Gesellschaft
- Hao Hu
- Luciana Musante
- Hans-Hilger Ropers
- Thomas F Wienker
Deutsche Forschungsgemeinschaft (KO2891/1-1)
- Björn Fischer-Zirnsak
National Center for Research Resources (P20 RR016453)
- Hans-Hilger Ropers
EU FP 7 project GENCODYS (241995)
- Hans-Hilger Ropers
Deutsche Forschungsgemeinschaft (Reinhart Koselleck Grant)
- Thomas Langer
Deutsche Forschungsgemeinschaft (SFB665)
- Angela M Kaindl
Berlin Institute of Health (BIH, CRG1)
- Angela M Kaindl
Deutscher Akademischer Austauschdienst
- Angela M Kaindl
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors thank the family members who participated in this study. We acknowledge T. Burmester for sending us fibroblasts of the affected children, E. Morava-Kozicz, D. Emmerich for sending DNA samples of patients with YME1L1 mitochondriopathy-like phenotypes, as well as G. Stoltenburg-Didinger, V. Tarabykin, C. Birchmeier, N. Krämer, K. Voigt, and J. Fassbender for technical help and discussions. This work was supported by the German Research Foundation (SFB665, KO2891/1-1, Reinhart Koselleck grant), the Berlin Institute of Health (BIH, CRG1), the Sonnenfeld Stiftung, the German Society for Muscle diseases (DGM), the German Academic Exchange Service (DAAD), the NCRR P20-RR016453, the Robert C. Perry Fund (20061479), the Max-Planck Society, the EU FP 7 project GENCODYS (241995), and the Human Frontiers Science Program.
Ethics
Human subjects: Informed consent was obtained from the parents of the patients for the molecular genetic analysis, the publication of clinical data, photos, magnetic resonance images (MRI), and studies on fibroblasts. The human study was approved by the local ethics committee of the Charité (approval no. EA½12/08).
Animal experimentation: All animal experiments were carried out in accordance to the national ethic principles (registration no. T0344/12, Charité).
Copyright
© 2016, Hartmann et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,282
- views
-
- 666
- downloads
-
- 104
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 104
- citations for umbrella DOI https://doi.org/10.7554/eLife.16078
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation
eLife 5:e16078.
https://doi.org/10.7554/eLife.16078




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}