NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies
- Institut Curie, PSL Research University, France
- CNRS UMR144, France
- Institut Curie, France
- Inserm, UMR 1037-CRCT, France
- Faculté des Sciences Pharmaceutiques, Université Toulouse III-Paul Sabatier, France
- Institut Claudius Regaud, France
- Le Pôle Technologique du Centre de Recherches en Cancérologie de Toulouse, plateau de protéomique, France
- Hybrigenics Service, France
Figures
Figure 1 with 4 supplements
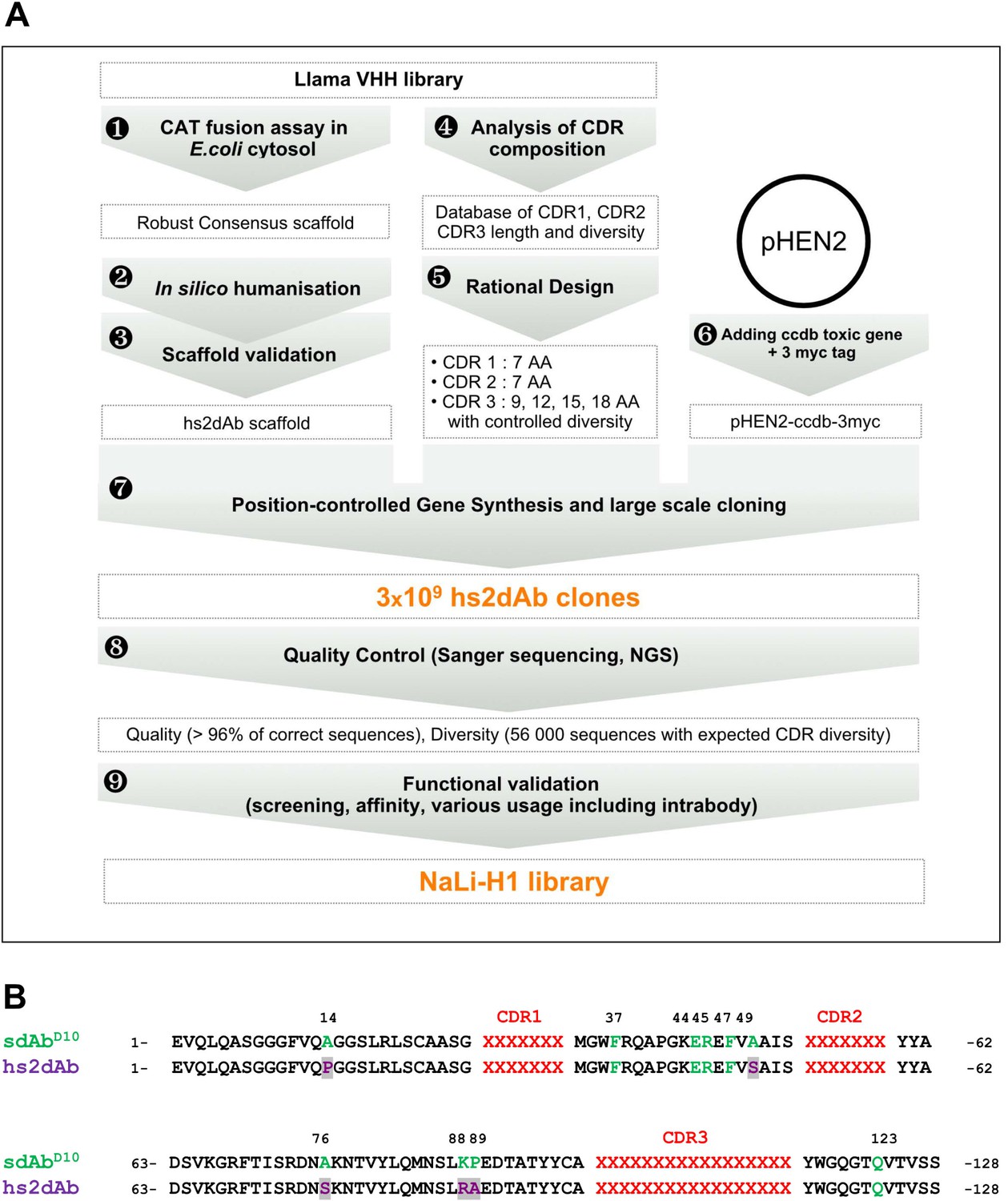
Overview of scaffold selection, diversity design, and synthetic production of the NaLi-H1 library.
The development of the NaLi-H1 library followed three lines of optimization. (i) A novel scaffold was defined by selecting a set of robust nanobodies using a CAT fusion assay (1). A consensus was derived and mutations were introduced to humanize the scaffold (2). Usability and efficacy of the novel scaffolds (VHH and humanized) were then confirmed evaluating their display on phage, expression in CHO cells and use as intrabodies (3).In silico design was completed analyzing natural CDR diversity (4) and using this information to design synthetic CDRs. A fixed size of 7 aa was chosen for the CDR1 and CDR2. 4 sizes (9, 12, 15 and 18 amino acids) were chosen for CDR3. Finally, the pHEN2 vector was improved by implementing a triple myc tag and inserting a toxic gene (ccdb) to ensure low frequency of empty clones during library construction (6). Gene synthesis (using a tri-nucleotide approach) was ordered, synthetic sequences cloned into the novel pHEN2+ vector, transformed into bacteria and plated on 430 15 cm plates. 3 × 109 clones were obtained. Quality control was carried out using Sanger sequencing of 315 randomly picked clones and large scale sequencing of 56 000 clones. No redundant clone was found. The NaLi-H1 was then screened in various conditions and diversity, efficacy, versatility and affinity evaluated.
Figure 1—figure supplement 1
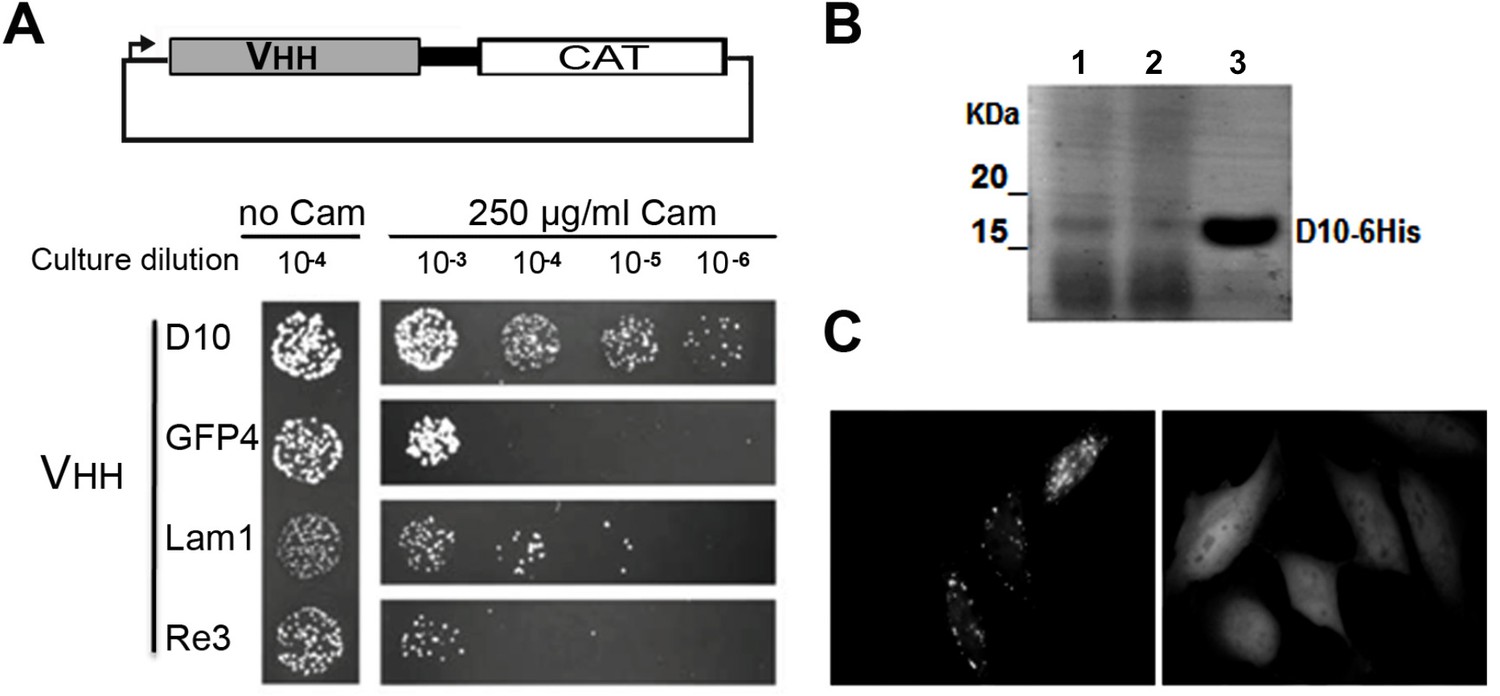
Robust scaffold identification.
(A) Chloramphenicol acetyl transferase carboxy terminal fusion is a folding reporter allowing for the selection of soluble amino terminal VHH. Top: scheme of the construct expressed from pAO-VHH-CAT-HA vector. Bottom: Relative colony growth of selected VHH (GFP4 and Lam1 chromobodies, or thermostable Re3) on chloramphenicol selection medium (Cam). Serial dilution of E.coli culture expressing VHH. (B) Analysis of heat purified sdAbD10 by SDS-PAGE. Clone D10 was expressed in E.coli and protein secreted in the periplasm were extracted (lane 1). Periplasmic extract was subjected to heat treatment at 70°C and insoluble proteins were pelleted by centrifugation (lane 2). The soluble supernatant containing the VHH was then concentrated using Amicon filters (lane 3). (C) HeLa cells expressing a GFP fusion of sdAbD10 showing homo-dispersed fluorescence (right) compared to typical randomly chosen aggregating llama VHH considered as non intrabody (left).
Figure 1—figure supplement 2
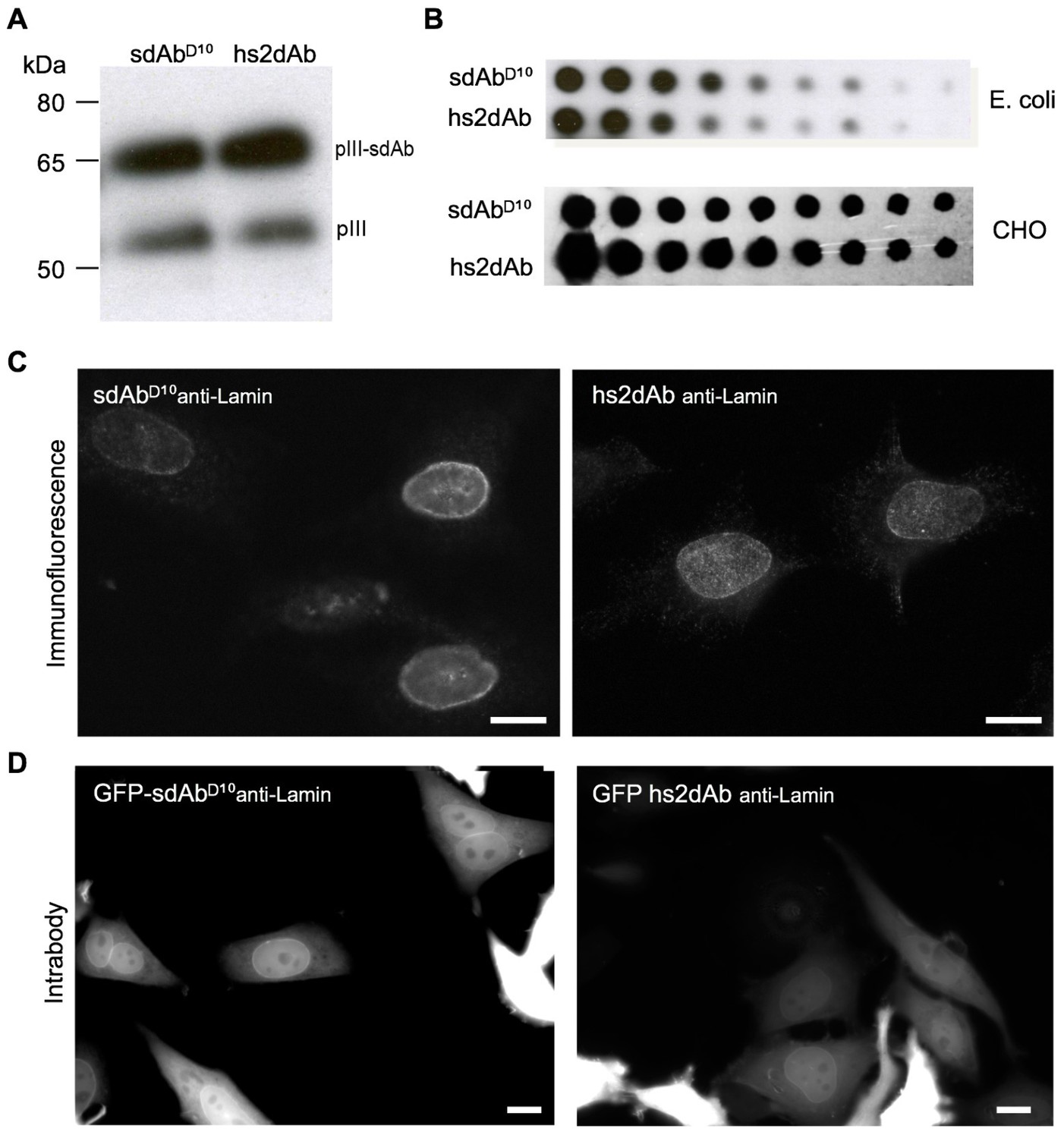
CDR3 loop grafting and synthetic scaffold validation.
(A) Phages presenting each scaffolds (sdAbD10 and hs2dAb) bearing anti-lamin CDRs were produced in E.coli and supernatant were detected in Western Blot with an anti-pIII antibody (NEB). Two bands are visible, one for wild-type pIII and one for the pIII fusion with single domain antibodies. (B) Dot blot analysis of the production of both scaffolds either as single domain antibodies in E. coli supernatant or as fusions with a human Fc domain and secreted by CHO cells (Moutel et al., 2009). Serial dilutions of supernatant were revealed with an anti-His tag antibody or an anti-human Fc antibody. (C) Immunofluorescence of HeLa cells with recombinant antibody in both scaffolds labeling the nuclear rim structure characteristic of the nuclear lamina. (D) The anti-lamin based on the two scaffolds were transiently expressed in HeLa cells as GFP fusion. Living cells were imaged after 24 hr and showed that the hs2dAb recognized its intracellular target lamin. Bar = 10 µm.
Figure 1—figure supplement 3
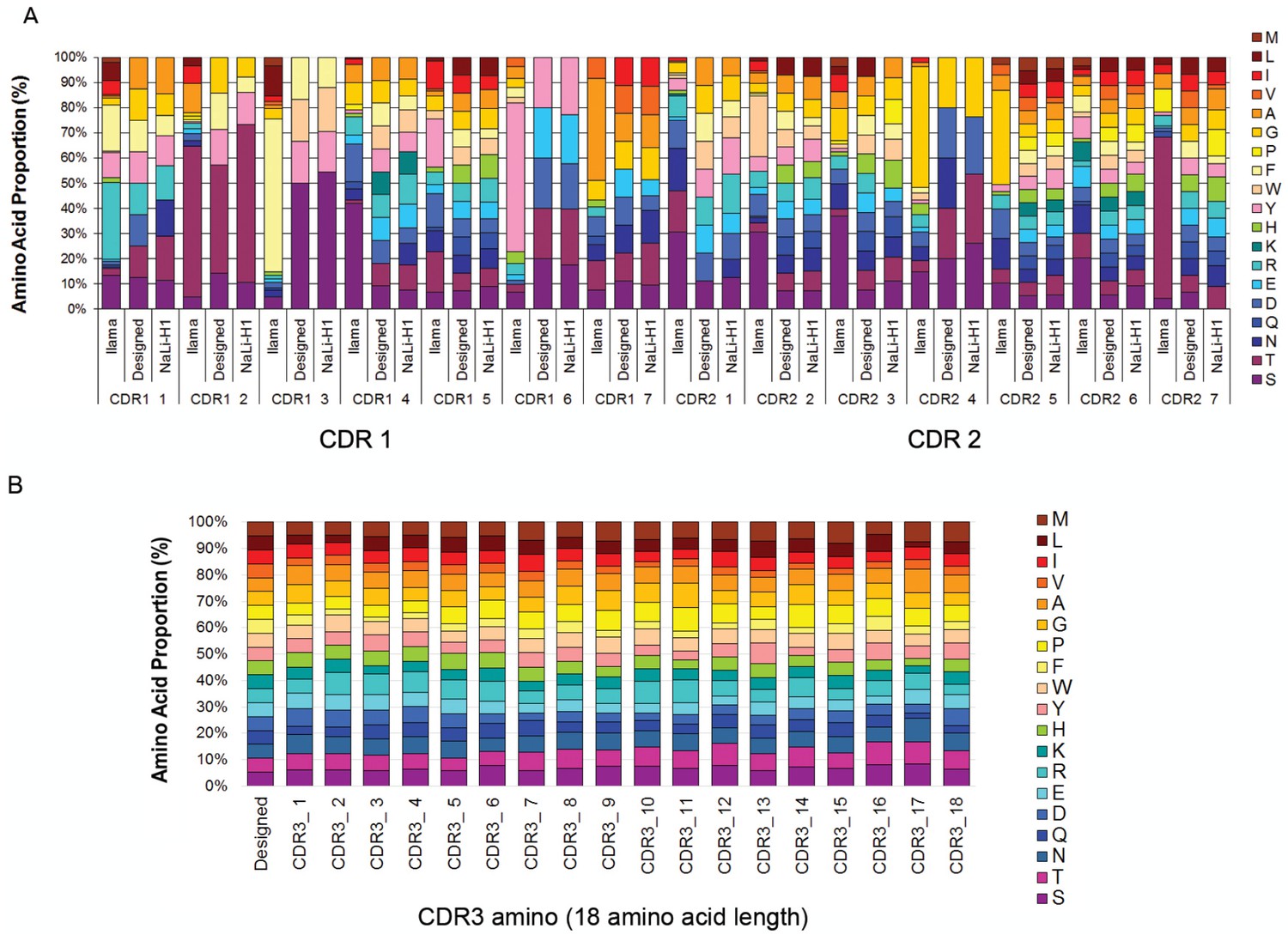
Nali-H1 library diversity.
(A) Comparison between relative amino acid frequencies in CRD1 and CDR2 observed from 250 llama VHH isolated from a naïve library (llama), the rationally designed (Designed) and the effective diversity in the synthetic library (Nali-H1) as computed after sequencing of 2500 clones using NGS. The position of each amino acid is indicated (CDR1 1; CDR1 2; etc). Amino Acid are indicated in single letter code. (B) Comparison between the designed diversity set for every amino acid position in the CDR3 region with the effective diversity observed after sequencing 2500 clones using NGS. Note that various CDR3 amino acid lengths are present in the library. They were almost evenly distributed with a little bias for shorter CDR3s (9 aa: 26%; 12 aa: 27.2%; 15 aa: 23.7%; 18 aa: 23.1%). For simplicity, we report in the figure the diversity observed for up to 18 aa. Note also that although cysteine occurs in natural llama CDRs, they were avoided by design and, accordingly, not found in Nali-H1 CDRs.
Figure 1—figure supplement 4
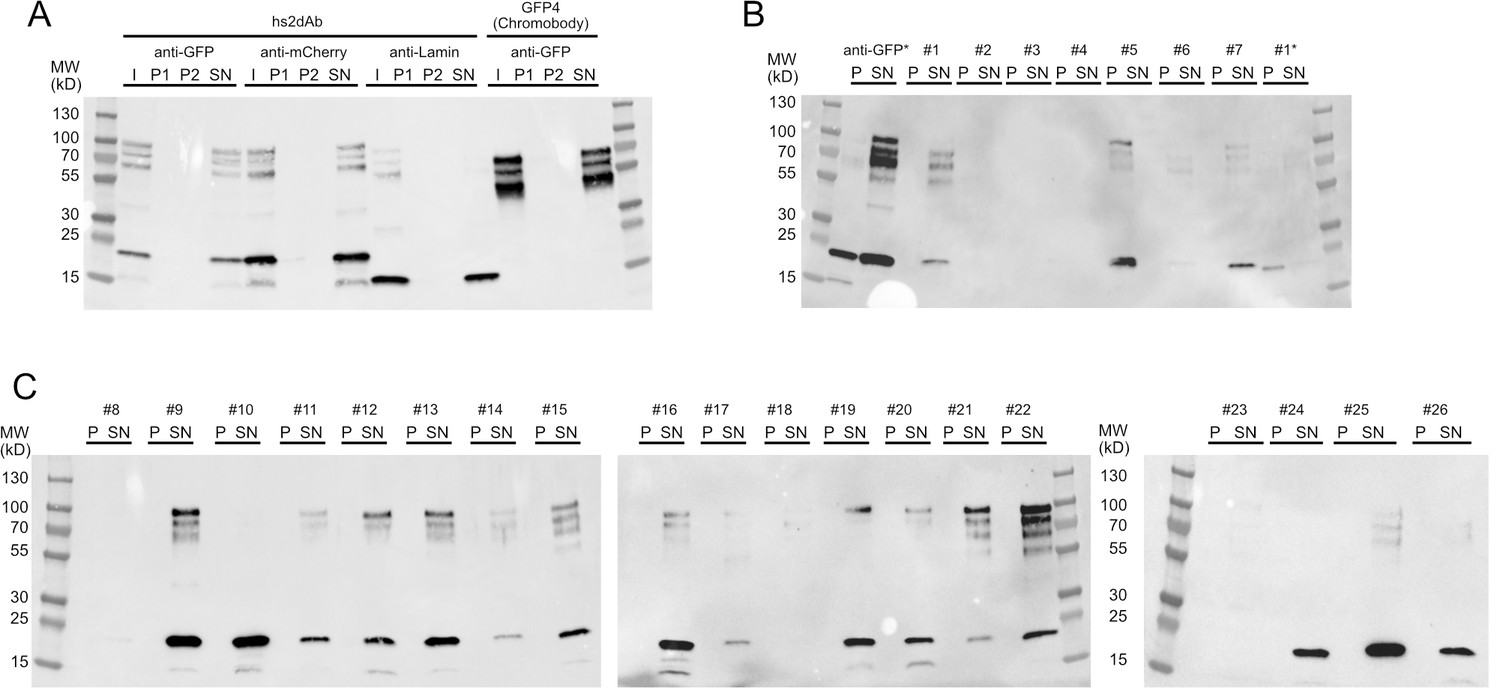
Solubility of secreted hs2dAb antibodies.
(A) Sequential centrifugation carried out using clones selected from the NaLi-H1 library (anti-GFP, anti-mCherry) or obtained after CDR grafting into the hs2dAb scaffold (anti-lamin). As a reference, the behaviour of a widely used natural anti-GFP antibody (GFP4 or Chromobody) was analysed. I: Input; P1: pellet after the 20 000 g centrifugation, P2: pellet obtained after the 250 000 g centrifugation, SN: supernatant of the 250 000 g centrifugation. Samples were analysed after western blotting using an anti-HIS antibody. Note that the apparent size of the lamin antibody and the Chromobody is lower because these antibodies are fused to only one myc tag (expressed from the parental pHEN2-His-Myc plasmid). (B), (C): a set of clones randomly chosen in the NaLi-H1 library was expressed in a multiwell format and analysed as in A. Only-the pellet (P) and the supernatant (SN) of the 250 000 g centrifugation are shown. Two samples (an anti-GFP hs2dAb and a randomly picked clone #1) were heated at 90°C for 10 min before being analysed by sequential centrifugation (anti-GFP* and #1*). Note that in these conditions, a fraction of the hs2dAb is unfolded and found in the pellet. A significant resistance to temperature is however observed. Note that high molecular weight bands were systematically seen which may suggest that a fraction of the hs2dAb is in a multimeric form. The same behaviour was observed for the natural Chromobody.
Figure 2 with 3 supplements
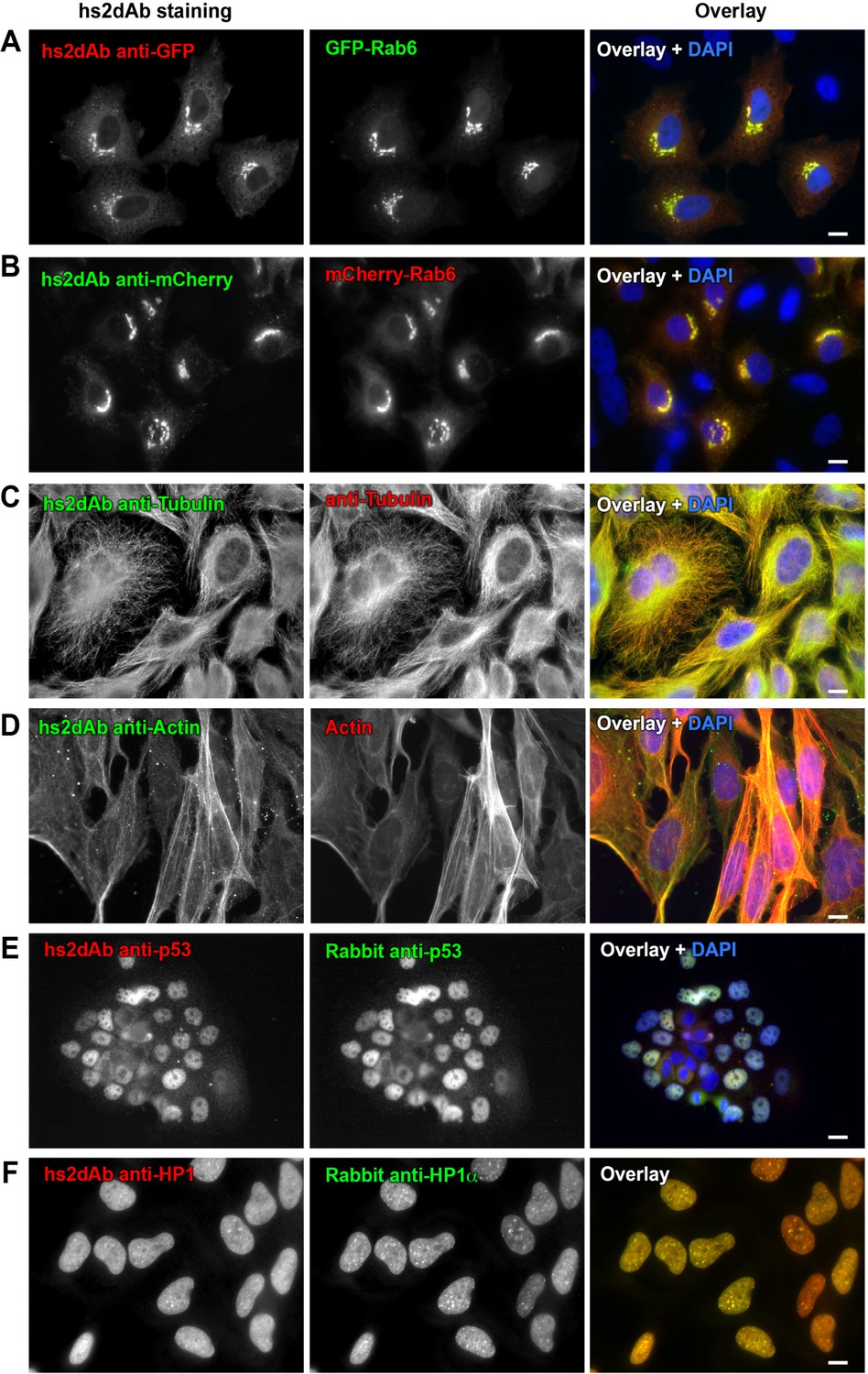
Selection of functional hs2dAb against various antigens.
(A) HeLa cells were transfected with GFP-Rab6, fixed using paraformaldehyde, permeabilized using saponin and stained with non-purified myc-tagged hs2dAb (R3TF3) directed against EGFP and revealed with anti-Myc-Tag (9E10) and Cy3-labeled secondary antibodies. (B) HeLa cells transfected with mCherry-Rab6, fixed and permeabilized as in A and stained using a myc-tagged non purified hs2dAb against mCherry (C11). The hs2dAb was then revealed using 9E10 and A488-labeled secondary antibodies. (C) Cells were fixed in methanol and co-stained with a non-purified anti-tubulin hs2dAb (D5) fused to a human Fc domain and a mouse monoclonal anti-tubulin antibody (DM1A), and revealed using an anti-Human Fc-A488 and an anti-Mouse-Cy3 secondary antibody, respectively. (D) hs2dAb F4 anti-beta-actin was used to stain MRC5 cells fixed with paraformaldehyde and post fixed with methanol. The hs2dAb was then revealed using 9E10 and A488-labeled secondary antibodies. Cells were co-stained by red fluorescent phalloidin to detect actin stress fibers. (E) A431 cells were fixed with 3% paraformaldehyde, permeabilized with 0.1% Triton and stained with the anti-p53 hs2dAb (B7) fused to a human Fc domain together with a rabbit polyclonal antibody directed against p53. Immuno-labeling was revealed using anti-Human Fc-Cy3 and anti-Rabbit-A488 secondary antibodies. (F) The hs2dAb antibody directed against HP1α (A5) fused to a human Fc domain was used to stain HeLa cells fixed with paraformaldehyde and permeabilized with 0.1% TritonX100. Cells were co-stained using a polyclonal rabbit antibody directed against HP1α and immuno-labeling was revealed using anti-Human Fc-Cy3 and anti-Rabbit-A488 secondary antibodies. (scale bar = 10 µm).
Figure 2—figure supplement 1
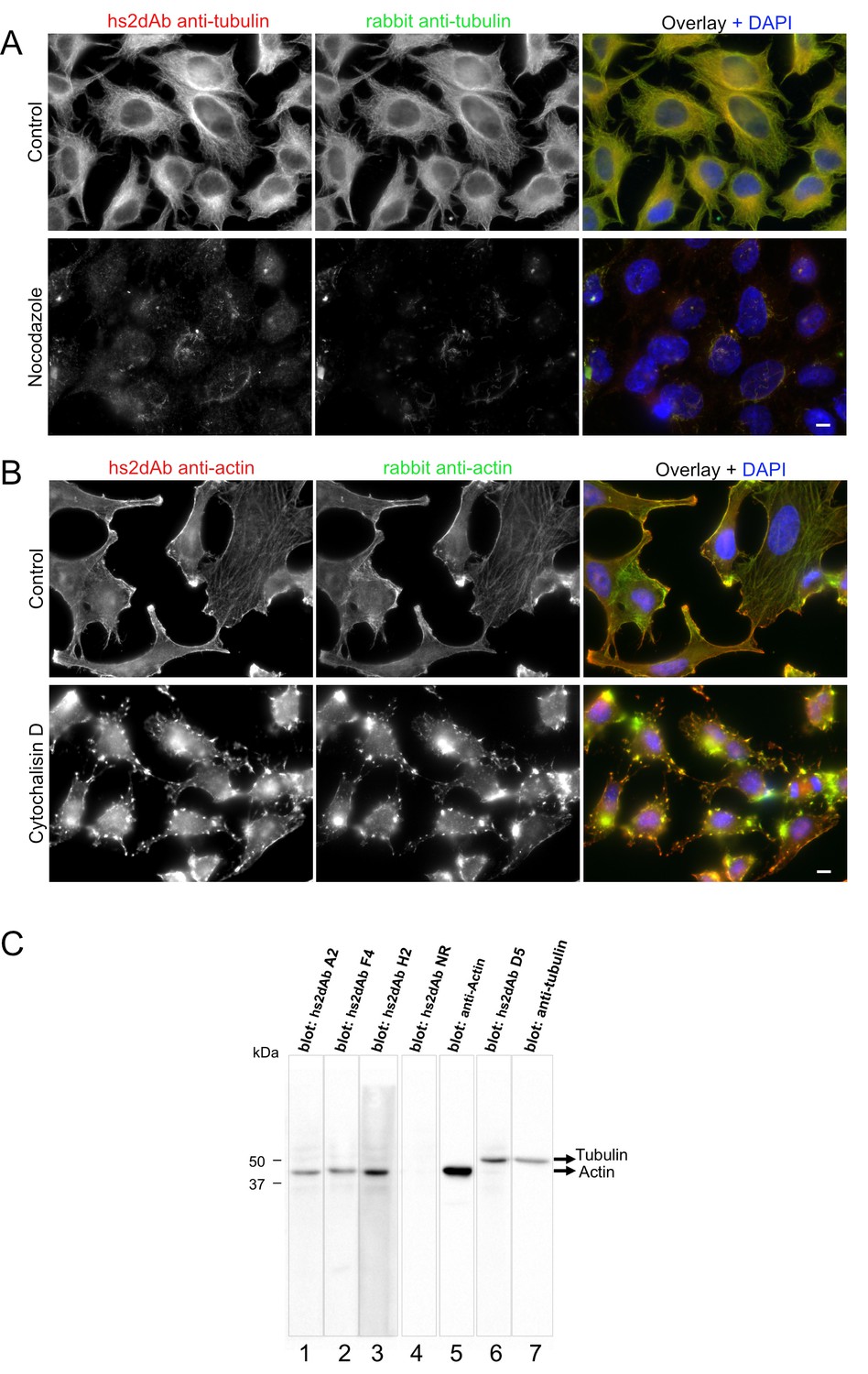
Specificity of hs2dAb directed against tubulin and actin.
(A) HeLa cells were left untreated (top) or incubated with 10 µM nocodazole for 90 min at 37°C (bottom). Cells were then permeabilized using 0.2% tritonX100, fixed using methanol (−20°C, 4 min) and immunolabelled using an anti-tubulin hs2dAb (green) and a polyclonal anti-tubulin antibody (Red). Staining was essentially lost upon nocodazole treatment with only few nocodazole-stable microtubules being labelled. Bar = 10 µm (B) HeLa cells were left untreated (top) or incubated with 5 µM cytochalasin D for 60 min at 37°C (bottom). Cells were then fixed using paraformaldehyde, permeabilized with saponin and immunolabelled using the anti-actin hs2dAb H2 (green) and a polyclonal anti-actin antibody (Red). Staining was strongly reorganized upon cytochalasin D treatment. Bar = 10 µm (C) SDS-PAGE of 40 µg sample per well of WI38 whole cell extract was blotted and separated in stripes for each lane. Stripes were then incubated for immuno-detection with various hs2dAb (lane 1–3: anti-actin hs2dAb; lane 4: non relevant [NR] control; lane 6: anti-tubulin hs2dAb) directly used from E.coli culture supernatant and further revealed using myc-HRP antibodies. Control monoclonal anti-actin or anti-tubulin were used on stripes 5 and 7, respectively.
Figure 2—figure supplement 2
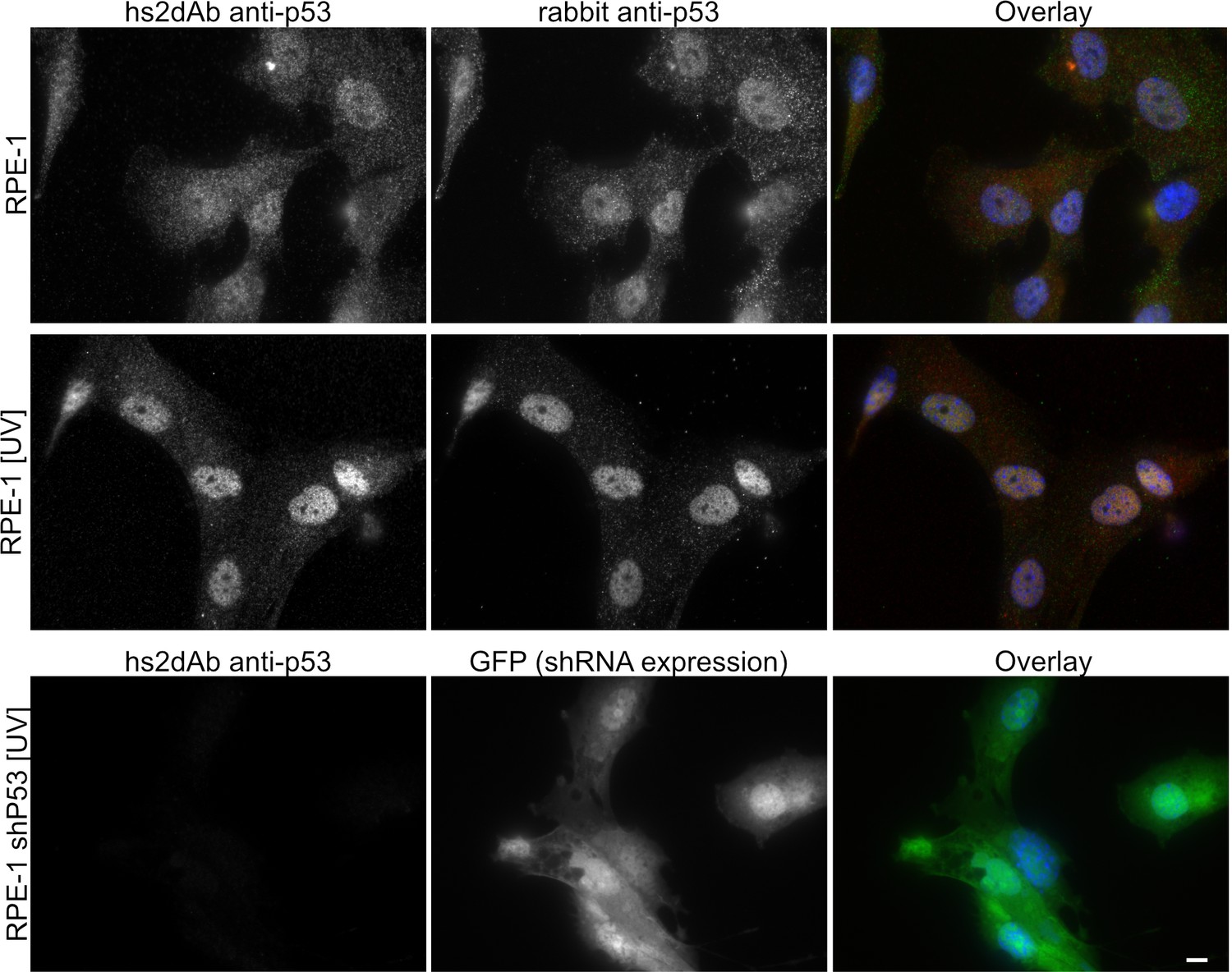
Specificity of the anti-p53 hs2dAb.
(A) RPE-1 were left untreated or irradiated with UV light (100 J/m2). Cells were then fixed and stained using an anti-p53 hs2dAb together with an anti-p53 rabbit polyclonal antibody. Both the polyclonal and the hs2dAb antibodies detected the strong increase of p53 localization in the nuclei. (B) RPE-1 cells stably expressing an shRNA directed against p53 together with GFP were irradiated as in A and stained using the hs2dAb. No labeling was obtained in p53 KD cells. Bar = 10 µm.
Figure 2—figure supplement 3
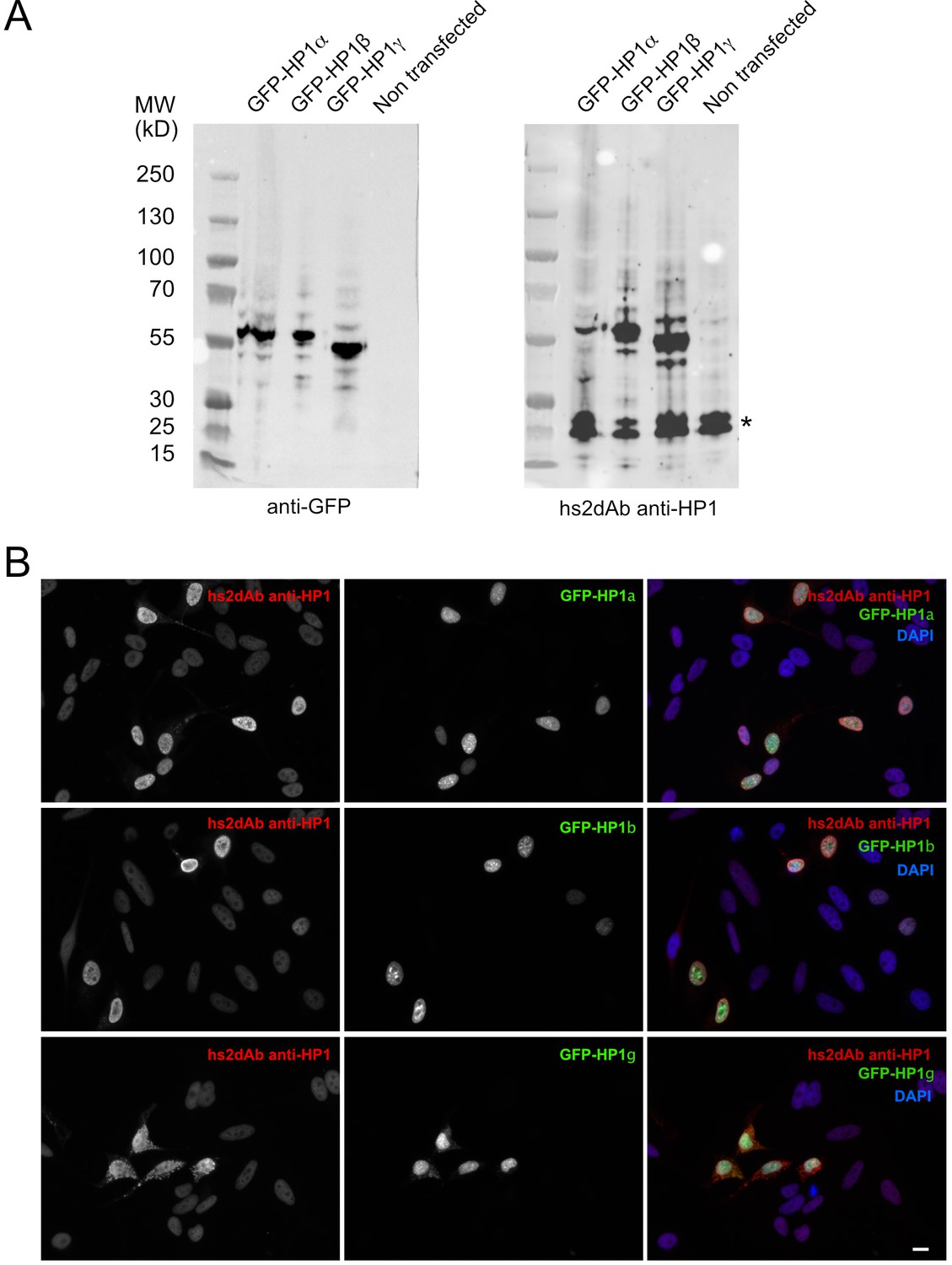
Specificity of the anti-HP1 hs2dAb.
(B). (A) GFP tagged HP1α, HP1b and HP1γ were expressed in Hela cells. Cells were then lysed and extract separated by SDS-PAGE, transferred on nitrocellulose filters. Immunodetection was carried out using an anti-GFP antibody (left) or an anti-HP1 hs2dAb. Although the selection was carried out against was HP1α, the hs2dAb efficiently detected GFP tagged HP1α, HP1β and HP1γ. The band indicated by a star likely represent the endogenous HP1 proteins. (B) Cells were transfected as in (A), fixed and analyzed by immunofluorescence using the anti-HP1 hs2dAb. Nuclei were stained using DAPI. GFP tagged HP1α, HP1b and HP1γ were all efficiently detected by the recombinant antibody. Bar = 10 µm.
Figure 3 with 1 supplement
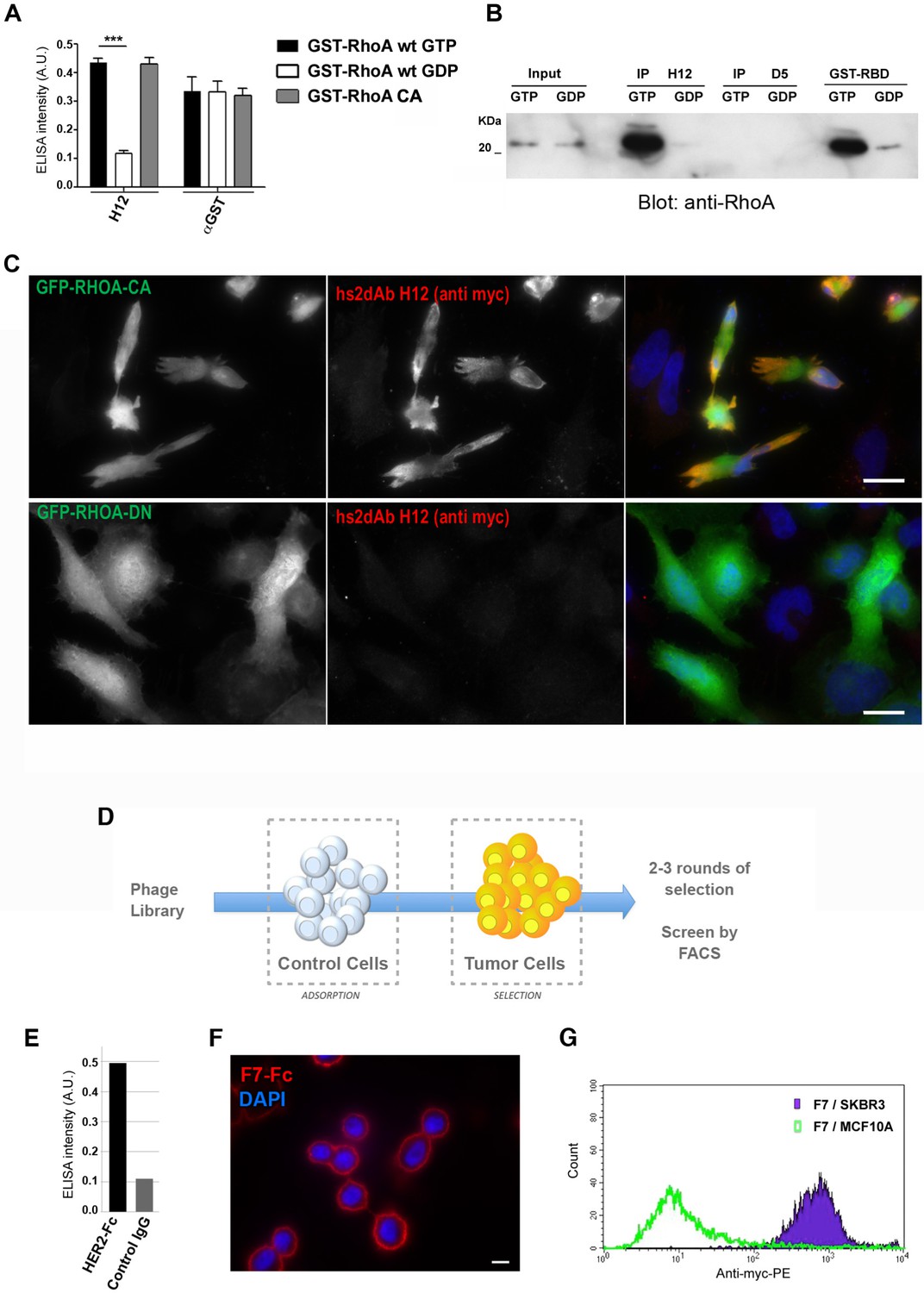
Subtractive selection led to conformational or cell type specific hs2dAbs.
(A–C) H12 is a conformational hs2dAb binding only to the GTP bound, activated state, of the RHOA GTPase: (A) ELISA using the H12 or anti-GST antibodies to reveal recombinant GST-RHOA wild type proteins loaded with either 100 µM GTP gamma S (Black) or 1 mM GDP (White), or constitutively active mutant proteins GST-RHOA Q63L (Grey). Means ± SEM. (B) A CBD tagged H12 pull down from HeLa cell extract loaded with100 µM GTP gamma S (GTP) or with 1 mM GDP as inputs. Western blot reveals RHOA at a similar level in 5% of both input but only on the GTP loaded extract in the CBD-H12 pull down. D5 anti tubulin was used as a negative control and the standard GST-RBD (RHO binding domain of Rhotekin) as a positive control of active RHO pull down. (C) Immunofluorescence on HeLa cells overexpressing GFP-RHOA CA (constitutively active) mutant or GFP-RHOA DN dominant negative mutant. H12 staining detected using a myc tag antibody revealed only cells overexpressing the constitutively active mutant with a pattern stronger at the cell periphery were RHOA activation is high. (D) Tumor cell surface subtractive selection scheme. (E) ELISA of hs2dAb F7 anti-HER2 on HER2 fused with a rabbit Fc versus binding on rabbit Fc at equimolar concentration. (F) hs2dAb F7 anti-HER2 decorated the SKBR3 membrane in immunofluorescence. SKBR3 cells were fixed with 3% paraformaldehyde and stained with F7 revealed by an anti-HisTag (Sigma) and an anti-MouseCy3 secondary antibody (Jackson). (G) FACS analysis of F7 anti-HER2 on SKBR3 HER2 positive cells versus MCF10A HER2 negative cells. (Scale bar = 10 µm).
Figure 3—figure supplement 1
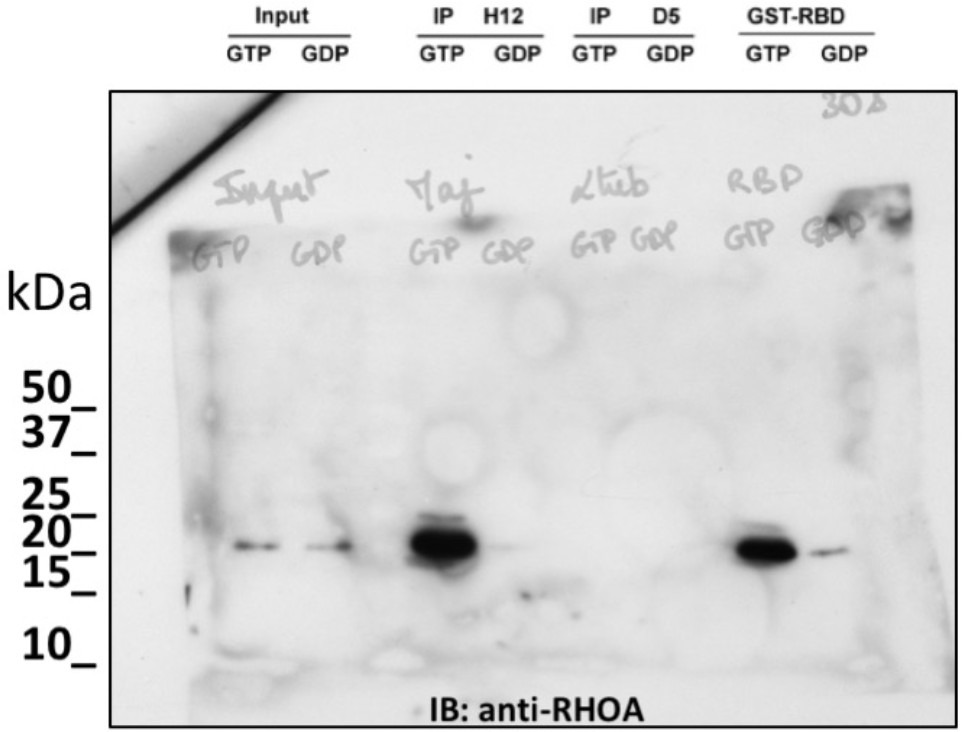
non cropped western blot corresponding to Figure 3B detection RHOA.
https://doi.org/10.7554/eLife.16228.013
Figure 4

Fluorescent intrabodies tracking endogenous proteins.
Intracellular expression of hs2dAb. (A) (top panel) HeLa cells were co-transfected with GFP-Rab6 and a hs2dAb-mCherry anti-EGFP plasmids. The hs2dAb mCherry anti-EGFP colocalized perfectly with the Rab6 Golgi staining. (bottom panel) HeLa cells were co-transfected with Myr-palm-mCherry and a VHH-EGFP anti-mCherry plasmids. The VHH-EGFP anti-mCherry interacted with its target in vivo and colocalized perfectly with the mCherry staining at the plasma membrane. (B) SKBR3 cells were transfected with an anti-p53 hs2dAb-mCherry alone (top panel), or together with full length p53-EGFP which concentrated the hs2dAb into the nucleus (bottom panel). (C) GFP, used as a control (top panel) or a GFP-tagged anti-HP1 hs2dAb (bottom panel) were transiently expressed in HeLa cells (green). In contrast to the GFP control, the GFP-tagged anti-HP1 strongly accumulated in the nucleus where it labeled nuclear condensations. (Scale bar = 10 µm)
Figure 5 with 4 supplements
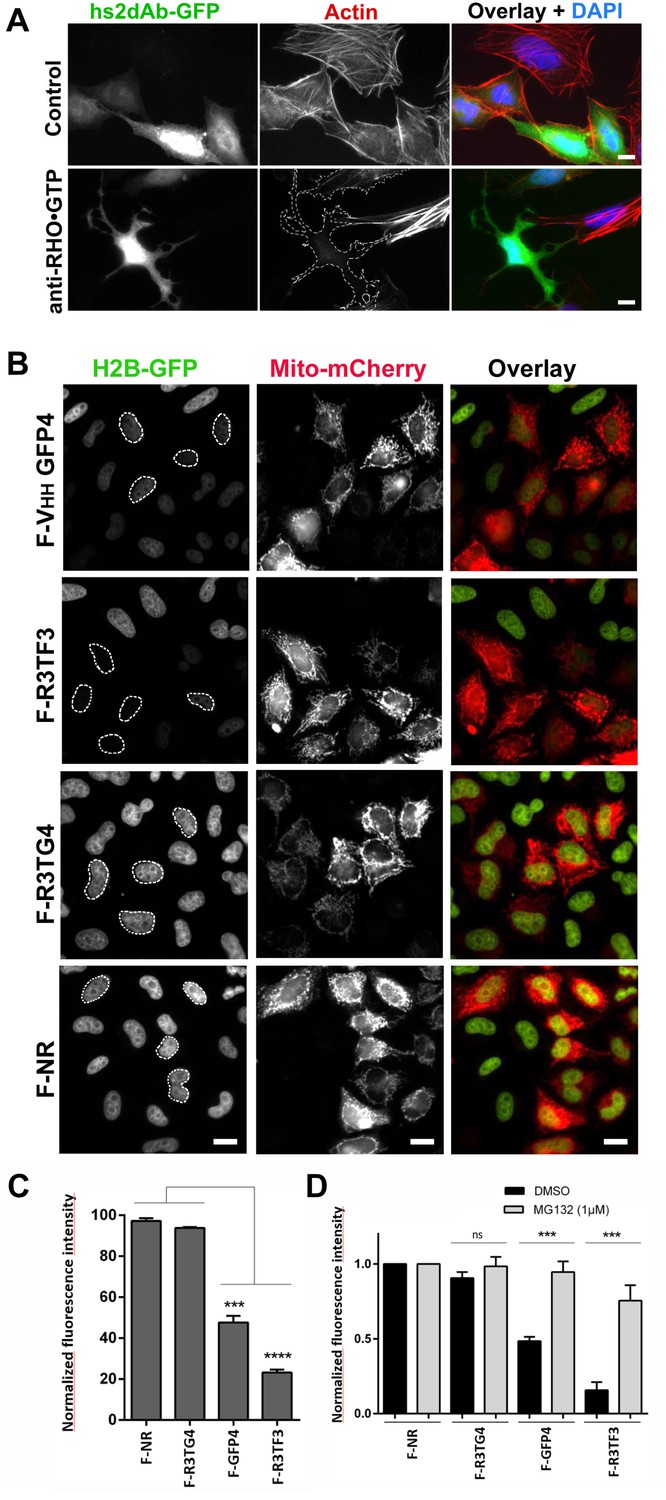
Targeting cellular proteins using inhibitory antibodies or by functionalizing antibodies to induce protein knockdown.
(A) HeLa cells expressing transiently a EGFP-tagged non relevant hs2dAB (top panel) or EGFP-H12 anti RHO-GTP (bottom panel) were fixed 20 hr post transfection and stained using DAPI and Alexa 594 phalloidin to detect actin stress fibers. The H12 hs2dAb induced actin stress fibers disappearance and major cell shape change (see cells outlined with a dotted line) (B) Protein knockdown of H2B-EGFP mediated by functionalized inhibitory antibodies. HeLa S3 cell stably expressing histone H2B-EGFP were transfected with vectors expressing antibodies fused to an F-box (F-Ib) to induce degradation of the targeted cellular antigen. The F-GFP4 VHH (DegradFP) was used as a positive control (top panel) and a non-relevant hs2dAb as a negative control (bottom panel). F-Ib were expressed using a bi-cistronic vector driving the co-expression of mitochondrial targeted mCherry. Protein interference is analyzed in cells displaying mCherry positive mitochondria (mitoCherry channel). Efficient protein knockdown is obtained using the R3TF3 anti-EGFP intrabody. Note that not all nanobodies can be used as F-Ib because R3TG4 does not induce protein degradation. (Scale bars = 20 µm) (C) Fluorescence decay measurement of the protein interference assay was quantified by flow cytometry (10000 cells analyzed, from 3 independent replicates). GFP fluorescence intensity was quantified in the transfected and the untransfected subpopulations for each F-Ib. The ratio of each median of fluorescence (transfected versus untransfected population) was calculated as a percentage of GFP fluorescence intensity for one F-Ib. A strong decrease in fluorescence corresponding to protein knockdown was observed with F-GFP4 VHH and F-R3TF3 hs2dAb intrabodies while the non-relevant negative control and R3TG4 did not induce a decrease of fluorescence. (D) Cells were analyzed as in C but the cells were incubated in 1 µM MG132 or DMSO for 44 hr after transfection by the different F-Ib and fluorescence intensity was normalized using the non-relevant control. Protein knockdown was inhibited by MG132.
Figure 5—figure supplement 1
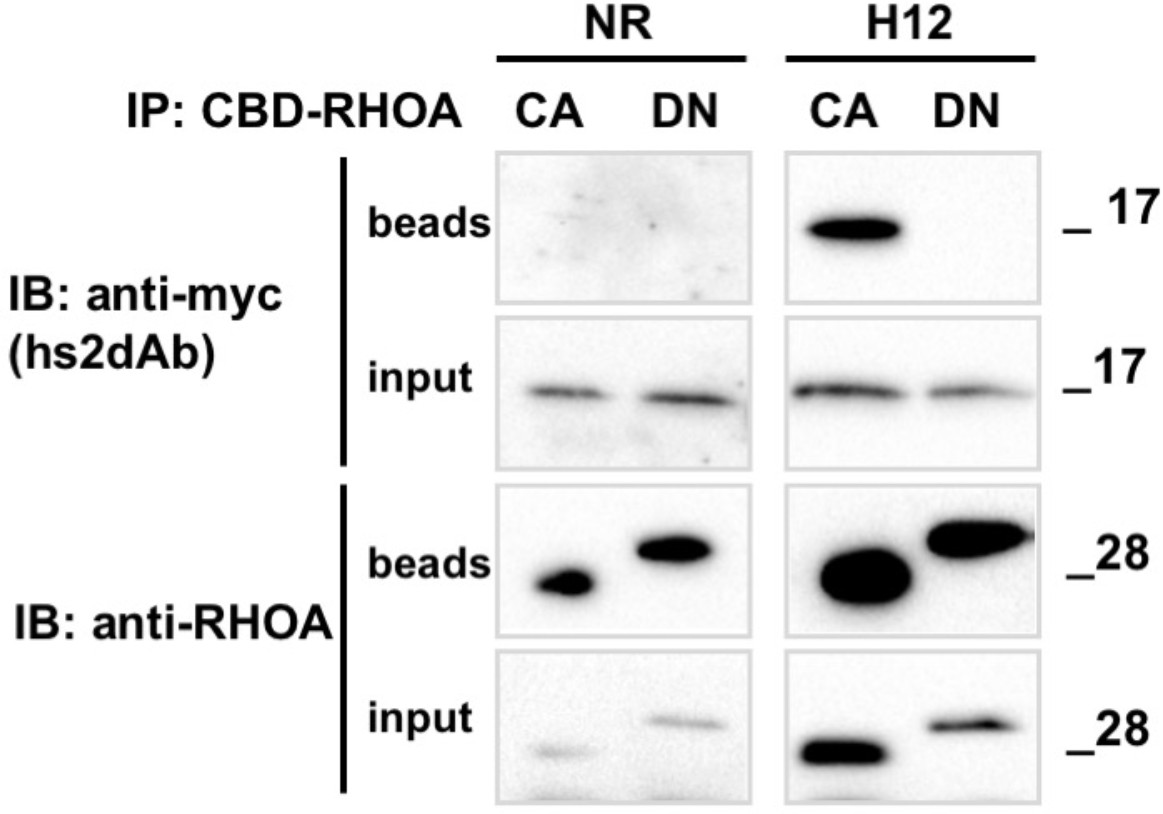
Conformational selectivity of the H12 intrabody towards RHOA.GTP.
HeLa cells were co-transfected for 24 hr with the anti RHO hs2dAb H12 fused to carboxy-terminal myc tag (H12-myc) or a non relevant control (NR-myc) that was a negative clone in a panning against FITC together with chitin binding domain (CBD) fusion of either the dominant negative mutant RHOA-N19 (DN) or the constitutively active mutant RHOA-L63 locked in the GTP bound state (CA). Chitin beads pull down of CBD-RHOA-DN or CBD-RHOA-CA revealed the selective co-precipitation of H12-myc together with the RHO active mutant. The total level of CBD-RHOA or hs2dAb-myc proteins was revealed by loading 5% of the respective input. CBD-RHOA proteins were detected with anti CBD tag and the hs2dAb antibodies with an anti-myc tag antibody.
Figure 5—figure supplement 2
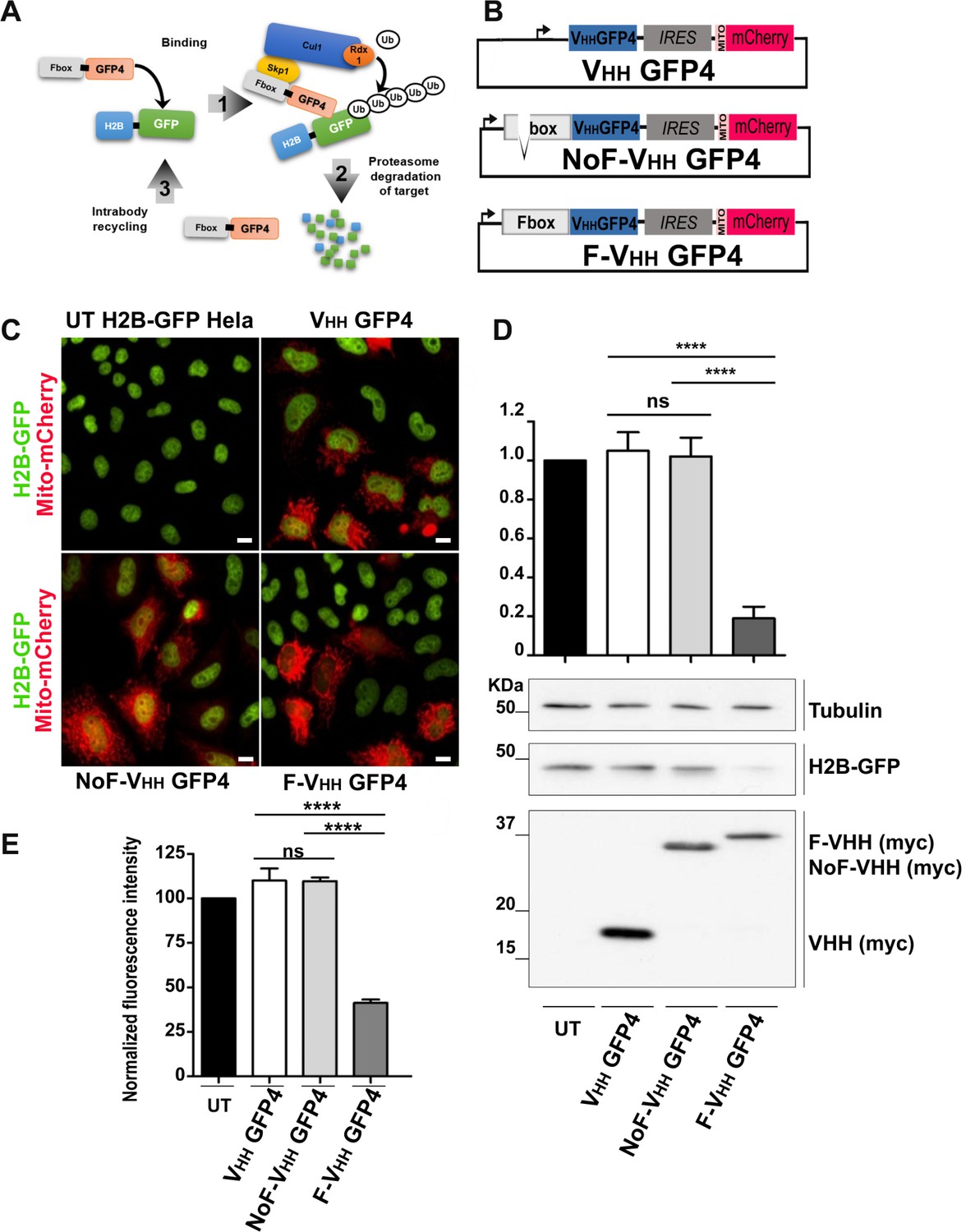
Protein knockdown set up using F-Ib degradation with anti-GFP intrabodies.
(A) Model for F-Ib degradation by fusing Fbox domain to the VHH GFP4 inducing GFP target protein ubiquitiation followed by proteasome-dependent degradation of the target protein. (B) Schematic illustration of the bicistronic vectors: an Fbox or NoFbox domain, respectively, is fused to the N terminal part of hs2dAb and a transfection marker MTS-mCherry, labeling mitochondria in red, is co-expressed using an Internal Ribosome Entry Site (IRES). (C) Fluorescence visualization HeLa S3 cells stably expressing H2B-GFP and transfected with NoF-VHH GFP4 or F-VHH GFP4. Degradation by the degradFP was observed in cells expressing F-VHH GFP4. Scale bar = 10 µm (D) Western blot quantification of protein knockdown mediated by F-GFP4. (E) Quantification of GFP fluorescence by flow cytometry in MTS Cherry positive cells.
Figure 5—figure supplement 3

non cropped western blot corresponding to Figure 5—figure supplement 1 detection of RHOA, Myc tagged hs2dAb intrabodies, and GAPDH which is not in the main figure.
https://doi.org/10.7554/eLife.16228.019
Figure 5—figure supplement 4

non cropped western blot corresponding to Figure 5—figure supplement 1D detection of tubulin, GFP and myc tag.
https://doi.org/10.7554/eLife.16228.020
Figure 6
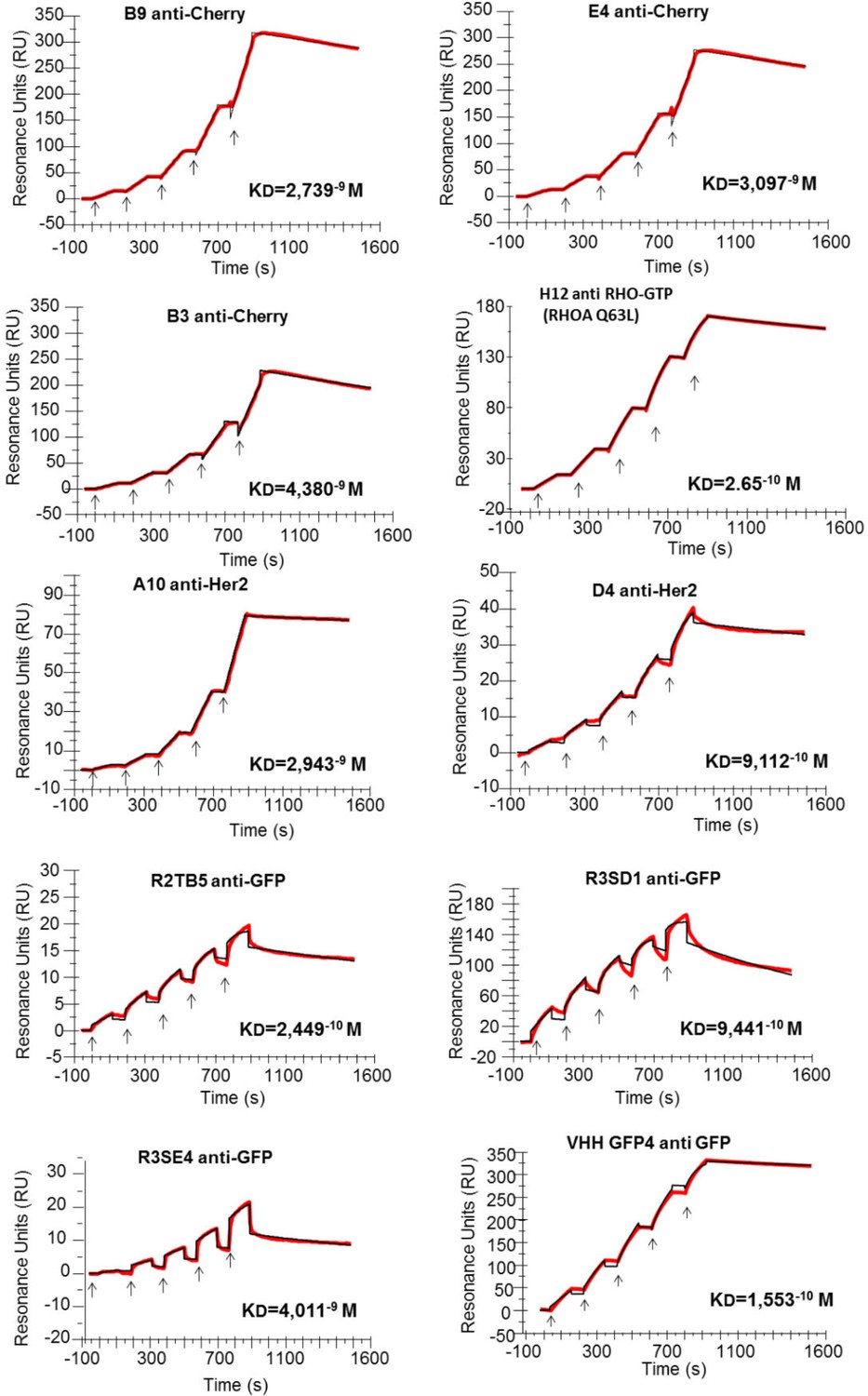
Affinity determination.
Single cycle kinetics analysis was simultaneously performed on immobilized His fusion VHH antibodies (250–300 RU), with five injections of analytes (EGFP, HER2, RHOAQ63L and mCherry) at 3.125 nM, 6.25 nM, 12.5 nM, 25 nM, and 50 nM. Analytes injections lasted for 120 s each and were separated by 10 s dissociation phases. At this time of buffer exchange, a slight refraction index discrepancy between the sample and the flow buffer can induce a drop in resonance unit. This common bulk effect, which is clearly visible on sensorgrams with a smaller scale range on the RU axis (ie: R3SE4, R2TB5), does not affect the measurement of off-rate constant. Off-rate constant was calculated from an extended dissociation period of 10 min following the last injection according to the single cycle kinetics method. Each sensorgram (expressed in RUs as a function of time in seconds) represents a differential response where the response on an empty reference channel (Fc1) was subtracted. The red curves correspond to the data and the black curves represent the fit done by the BIAevaluation software. Note that the fitted curve is almost identical to the data curve in some cases like for example the RHOA Q63L or the HER2 binding measurement.
Videos
Video 1
mcherry-Rab6 was transiently expressed in HeLa cells together with an anti-mCherry hs2dAb fused to GFP. 24 hr after transfection, cells were imaged using a spinning disk confocal microscope.
https://doi.org/10.7554/eLife.16228.015Tables
Table 1
Summary of screenings showing the number of unique clones giving positive signal. (ND means non determined)
Positive clones | ||||
|---|---|---|---|---|
| Antigen | Phage ELISA | IF/FACS | Intrabody | Rounds of panning |
| GFP | 37 | 10/ND | 4/10 | 2 |
| mCherry | ND | 6/ND | 2/6 | 3 |
| Tubulin | ND | 3/ND | 0/3 | 2 |
| Actin | 16 | 7/ND | 1/7 | 3 |
| p53 | 12 | 6/ND | 2/6 | 2 |
| RHOA-GTP | 24 | 8/ND | 3/8 | 4 |
| Her2 | 6 | 5/10 | ND | 3 |
Table 2
Binding affinities of 9 selected hs2dAb fused to a 6HIS tag measured by surface plasmon resonance single cycle kinetics method. Dissociation equilibrium constant KD corresponds to the ratio between off-rate and on-rate kinetic constant Koff/Kon. Non relevant hs2dAb were used as negative controls and gave no detectable binding signal. A positive control endowed with subnanomolar affinity, the GFP binder VHH-GFP4, was analyzed in parallel to the GFP hs2dAbs. A KD of 1.55–10 M was measured for VHH-GFP4 which is similar to published values. The binding properties of the conformational H12 hs2dAb to the GTP loaded RHOA subfamily were measured using the L63 or L61 constitutively active mutants of RHO, RHOB, RHOC, RAC1 and CDC42 related small GTPases, as well as the negative mutant T19N of RHOA. ('no' means no detectable binding).
| hs2dAb-6xHis | Antigen | kon (M−1 s−1) | koff (s−1) | KD(M) |
|---|---|---|---|---|
| R2TB5 anti-GFP | GFP | 1.24 10+6 | 3.05 10−4 | 2.45 10−10 |
| R3SD1 anti-GFP | GFP | 7.07 10+5 | 6.68 10−4 | 9.44 10−10 |
| R3SE4 anti-GFP | GFP | 1.45 10+5 | 5.83 10−4 | 4.01 10−9 |
| Llama VHH GFP4 | GFP | 2.99 10+5 | 4.65 10−5 | 1.55 10−10 |
| D4 anti-Her2 | Her2 | 1.79 10+5 | 1.63 10−4 | 9.11 10−10 |
| A10 anti-Her2 | Her2 | 1.66 10+4 | 4.88 10−5 | 2.94 10−9 |
| B9 anti-Cherry | mCherry | 6.14 10+4 | 1.68 10−4 | 2.74 10−9 |
| E4 anti-Cherry | mCherry | 6.57 10+4 | 2.03 10−4 | 3.10 10−9 |
| B3 anti-Cherry | mCherry | 6.19 10+4 | 2.71 10−4 | 4.38 10−9 |
| H12 anti-RHO.GTP | RHOA Q63L | 4.81 10+5 | 1.28−4 | 2.65 10−10 |
| H12 anti-RHO.GTP | RHOB Q63L | 2.24 10+5 | 3.59−4 | 1.57 10−9 |
| H12 anti-RHO.GTP | RHOC Q63L | 1.12 10+6 | 5.41−5 | 4.79 10−11 |
| H12 anti-RHO.GTP | RHOA T19N | no | no | no |
| H12 anti-RHO.GTP | RAC1 Q61L | 7.53 10+5 | 2.55−4 | 3.3 10−10 |
| H12 anti-RHO.GTP | CDC42 Q61L | no | no | no |
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
NaLi-H1: A universal synthetic library of humanized nanobodies providing highly functional antibodies and intrabodies
eLife 5:e16228.
https://doi.org/10.7554/eLife.16228




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}