Neural Circuits: Decoding obesity in the brainstem
- Northwestern University Feinberg School of Medicine, United States
When an obese person loses weight they will often continue to experience hunger after they stop dieting and merely try to maintain a lower body weight (Sumithran et al., 2011). This situation exemplifies the complexity of regulating appetite and energy balance. Feelings of hunger or satiety result from integrating a wide range of signals from our environment with a similarly large spectrum of signals from inside the body. These internal signals might be nerve impulses from the gut, nutrient levels in the blood, or protein-based hormones. For example, fat tissue makes and releases a hormone called leptin that inhibits hunger to regulate the body’s energy balance.
The brainstem is heavily involved in these processes, integrating signals from the gut and relaying this information to a part of the brain called the hypothalamus. However, while many researchers have focused on the neurons within the feeding centers in the hypothalamus, the neurons within the brainstem have received less attention. Specifically, we don't know how the parts of the hypothalamus that respond to leptin communicate with brainstem regions that respond to more short-term signals of satiety. The specific pathways of neurons that carry and process signals from the gut have also remained less characterized.
Now, in eLife, Giuseppe D’Agostino, Lora Heisler and colleagues – who are based at the Universities of Aberdeen and Cambridge, Harvard Medical School and Yale University School of Medicine – advance this area of study by genetically dissecting a circuit of neurons that originates from the brainstem (Figure 1). D’Agostino et al. report how this circuit contributes to satiety and how it integrates with long-term control of appetite and energy balance via direct connections with neurons in the hypothalamus (D'Agostino et al., 2016).
Figure 1
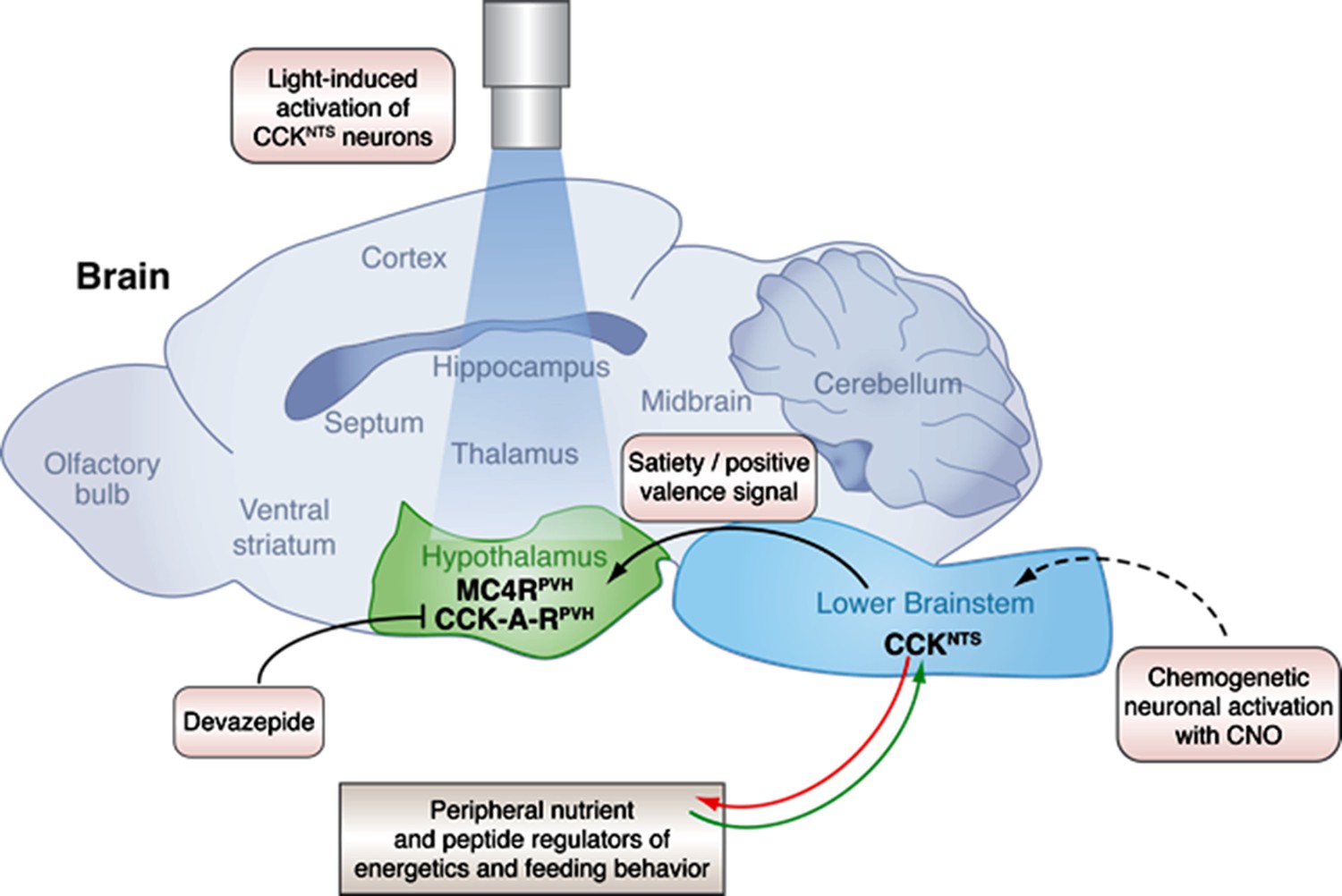
A neural circuit in the brainstem integrates signals from peripheral tissues to control feeding behavior and energy balance.
The lower brainstem (light blue) receives input (green arrow) from peripheral tissues such as the gut, and also sends output (red arrow) to these tissues. Now, in experiments on mice, D’Agostino et al. have characterized a neuronal pathway that starts with neurons that express CCK in the nucleus of the solitary tract (NTS), which is part of the lower brainstem. Genetically engineering the CCKNTS neurons so that a chemical called CNO can activate them showed that activating the neurons signals satiety and strongly decreases eating in mice. This leads to significant weight loss in the mice. The CCKNTS neurons send signals to a part of the hypothalamus (green) called the paraventricular nucleus (PVH). Specifically, the ends of the CCKNTS neurons form connections with neurons expressing melanocortin 4 receptors (MC4RPVH) in the PVH; the MC4RPVH neurons are thought to regulate appetite. Using light to activate the endings of the CCKNTS neurons that contact the PVH (via an optogenetics approach) also promoted satiety and a positive emotional state (or positive valence) in behavior tests. This strongly suggests that the connections between the CCKNTS neurons and the PVH are important. Inhibiting the CCK-A receptors (CCK-A-RPVH) with a chemical called devazepide meant that the chemogenetic and optogenetic activation of CCKNTS neurons no longer inhibited food intake. This figure was created with help from Billie Marcheva.
Over 40 years of research has implicated a molecule called cholecystokinin (or CCK) as a key regulator of satiety (Gibbs et al., 1973; Cai et al., 2014). CCK produced by the small intestine activates a part of the brainstem called the nucleus of the solitary tract (or NTS), but only if so-called melanocortin 4 receptors are expressed in this region (Fan et al., 2004). The NTS sends this nutritional information onto parts of the hypothalamus and other nearby regions of the brain (Cai et al., 2014). These brain regions then integrate signals about the animal’s nutrient state and appetite (Grill and Hayes, 2012)
D’Agostino et al. have now characterized a neural circuit in the NTS that expresses its own CCK instead. First, they showed that these CCKNTS neurons became active in mice that were fed nutrients (e.g. amino acids or sucrose) but not in mice fed only water. This shows that the activity of these neurons depends on the nutrient state of the animal.
D’Agostino et al. then engineered these neurons so that they respond to a specific chemical. Activating the neurons with this chemical stopped the mice from eating even after fasting overnight. The mice lost a lot of weight if their CCKNTSneurons were activated for prolonged periods, but rapidly regained weight when the chemical activation was stopped. However, this chemical activator had no effect on the mice’s feeding behavior when D’Agostino et al. prevented CCK from binding to its receptors on its target neurons. This suggests that the CCK receptor is directly responsible for the signals sent from the NTS
D’Agostino et al. found that the CCKNTS neurons form direct connections with the paraventricular nucleus in the hypothalamus. Stimulating this connection via an optogenetic technique stopped the mice from eating, even after they had been deprived of food. Further analysis showed the nerve endings of the CCKNTS neurons at the paraventricular nucleus were also very close to a subset of paraventricular nucleus cells that express melanocortin 4 receptors.
Leptin signals how much fat is stored in the body and it was already known that this hormone regulates the activity of neurons in the brainstem that respond to CCK (Flak et al., 2014). D’Agostino et al. now give new insight into how leptin and CCK signals are integrated in the brainstem and the paraventricular nucleus, and provide further support for the idea that the connection between these signals can act in both directions (Wu et al., 2012; Morton et al., 2005; Hayes et al., 2010). Collectively, D’Agostino et al.’s findings broaden our understanding of the neuronal circuitry that processes an animal’s appetite and behavioral responses to its own energy state. The findings also describe the underlying communication, and important “gateways”, between the peripheral tissues, brainstem and hypothalamus.
At present, it is still not known what signal first activates the CCKNTS neurons. This means that it remains unknown how the CCKNTS neurocircuit integrates information from the gut. Also, while activating the CCKNTS circuit did not promote satiety if the CCK receptors were blocked, it is not clear whether gut-derived CCK or other gut hormones can activate the CCKNTS neurons to promote satiety.
The pathway characterized by D’Agostino et al. clearly has a strong effect on regulating body weight. As such, it will be important to ask whether this specific neurocircuit can be exploited to treat obesity. If this is the case, it’s worth noting that such a treatment is unlikely to use CCK itself. This is partly because this molecule is rapidly degraded in the body, and partly because people may become tolerant to CCK’s appetite-suppressing effects if it is given for prolonged periods. Importantly, other promising molecules that can activate CCK receptors have already been investigated in mice as potential weight loss therapies (Irwin et al., 2012). Perhaps these potential drugs could be improved and made more specific such that they only target a subset of CCK-expressing neurons within the gut and the brain. Thus, characterizing the CCKNTS neurons further, and uncovering how they are activated by the gut, may reveal specific molecules that could be targeted by drugs to suppress appetite for long enough to combat obesity.
References
-
Central amygdala PKC-δ+ neurons mediate the influence of multiple anorexigenic signalsNature Neuroscience 17:1240–1248.https://doi.org/10.1038/nn.3767
-
Leptin-inhibited PBN neurons enhance responses to hypoglycemia in negative energy balanceNature Neuroscience 17:1744–1750.https://doi.org/10.1038/nn.3861
-
Leptin action in the forebrain regulates the hindbrain response to satiety signalsJournal of Clinical Investigation 115:703–710.https://doi.org/10.1172/JCI200522081
-
Long-term persistence of hormonal adaptations to weight lossNew England Journal of Medicine 365:1597–1604.https://doi.org/10.1056/NEJMoa1105816
Article and author information
Author details
Jonathan Cedernaes

Publication history
Copyright
© 2016, Cedernaes et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,312
- views
-
- 291
- downloads
-
- 5
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 5
- citations for umbrella DOI https://doi.org/10.7554/eLife.16393
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Neural Circuits: Decoding obesity in the brainstem
eLife 5:e16393.
https://doi.org/10.7554/eLife.16393




{kind=link}