Appetite controlled by a cholecystokinin nucleus of the solitary tract to hypothalamus neurocircuit
- University of Aberdeen, United Kingdom
- University of Cambridge, United Kingdom
- Harvard Medical School, United States
- Yale University School of Medicine, United States
Abstract
The nucleus of the solitary tract (NTS) is a key gateway for meal-related signals entering the brain from the periphery. However, the chemical mediators crucial to this process have not been fully elucidated. We reveal that a subset of NTS neurons containing cholecystokinin (CCKNTS) is responsive to nutritional state and that their activation reduces appetite and body weight in mice. Cell-specific anterograde tracing revealed that CCKNTS neurons provide a distinctive innervation of the paraventricular nucleus of the hypothalamus (PVH), with fibers and varicosities in close apposition to a subset of melanocortin-4 receptor (MC4RPVH) cells, which are also responsive to CCK. Optogenetic activation of CCKNTS axon terminals within the PVH reveal the satiating function of CCKNTS neurons to be mediated by a CCKNTS→PVH pathway that also encodes positive valence. These data identify the functional significance of CCKNTS neurons and reveal a sufficient and discrete NTS to hypothalamus circuit controlling appetite.
https://doi.org/10.7554/eLife.12225.001eLife digest
Obesity primarily results from eating more food than the body requires, the energy from which is then stored as fat. In recent years obesity has become increasingly common, with the resulting health problems presenting one of the major healthcare challenges of the twenty-first century. New ways to tackle the obesity epidemic are therefore required to improve human health on a global scale.
To regulate how much food is eaten, the gut sends chemical messengers to the brain about how much food has been consumed. These messengers activate particular cells in the brain that signal to other brain regions to trigger a decision about whether we’ve had enough food to eat. This raises a question: if we can artificially activate these cells, can we ‘trick’ the brain into thinking that food has been consumed?
A brain region called the nucleus of the solitary tract (NTS) is known to play a key role in receiving signals from the gut about meals. By studying mice, D’Agostino et al. found that cells in the NTS that make a brain hormone called cholecystokinin (CCK) are particularly activated by food.
Further experiments then used a technique called optogenetics to activate these cells in mice that had free access to different types of food. This activation significantly reduced how hungry the mice were, causing them to eat less food and lose weight. D’Agostino et al. also showed that CCK cells relay the signal about food intake to a brain region called the hypothalamus.
Overall, D’Agostino et al. have found a way to trick the brain into thinking that food has been eaten when it actually hasn’t, and for this reason mice eat less without feeling hungry and lose weight. The next step is to try and find a way to activate the CCK cells in obese humans who have health complications associated with excess body weight.
https://doi.org/10.7554/eLife.12225.002Introduction
Obesity has emerged as one of the global healthcare challenges of the 21st century. Common obesity is primarily a consequence of food intake beyond the body’s energetic requirements, with the excess energy consequently stored as fat. Mechanistically, the brain integrates and responds to multiple homeostatic hormones, neurotransmitters, nutrients and peripherally generated neural signals to maintain energetic balance (Dietrich and Horvath, 2013; Morton et al., 2014). One of the primary integration nodes within the brain for meal-related and metabolic signals from the periphery is the nucleus of the solitary tract (NTS) (Grill and Hayes, 2012; Rinaman, 2010; Schwartz, 2010; Wu et al., 2012). The NTS hosts a variety of factors to control homeostatic functions, including a small subset of neurons that express cholecystokinin (CCKNTS) (Garfield et al., 2012; Herbert and Saper, 1990; Vitale et al., 1991).
CCK consists of a family of peptides, the best characterized of which is a 33 amino acid peptide secreted from endocrine cells in the jejunum in response to nutrients in the intestinal lumen. This gut-derived peptide has a number of gastrointestinal (GI) functions including the promotion of satiation/satiety (Gibbs et al., 1973; Saito et al., 1981; Smith and Gibbs, 1994). Peripherally derived CCK does not readily penetrate the brain (Fan et al., 1997; Passaro et al., 1982). However, its short-term effect on appetite has been in part attributed to stimulation of vagal sensory neurons influencing the brainstem (Fan et al., 1997; Gibbs et al., 1973; Saito et al., 1981; Smith and Gibbs, 1994).
CCK is also synthesized within the brain, where it is post-translationally processed into an 8 amino acid peptide (CCK-8) that reduces food intake when centrally infused (Blevins et al., 2000; Hirosue et al., 1993). However, the source of brain CCK that controls appetite has not been established. Recently developed chemogenetic and optogenetic approaches now provide a means to decipher the real-time contribution of discrete neuronal populations and networks to behavior (Sternson and Roth, 2014; Tye and Deisseroth, 2012), although, no direct functional assessment of CCKNTS neurons has yet been undertaken.
Results and discussion
CCKNTS neurons are responsive to nutritional state
Although CCK is produced in the NTS (Garfield et al., 2012; Herbert and Saper, 1990), little is known about the function of this discrete source of brain CCK. To facilitate visualization and characterization of CCKNTS, we employed a knock-in mouse line expressing Cre recombinase at the Cck locus (Cck-iCre) crossed with a Cre-dependent enhanced yellow fluorescent protein reporter (Ai3) line (CckYFP; Figure 1A). CCK-eYFP cells were most abundantly expressed within the caudal aspect of the NTS (Figure 1B,C), a brain region innervated by gastrointestinal vagal afferents and involved in nutrient sensing (Appleyard et al., 2005; Blouet and Schwartz, 2012; Ritter, 2011; Schwarz et al., 2010).
Figure 1
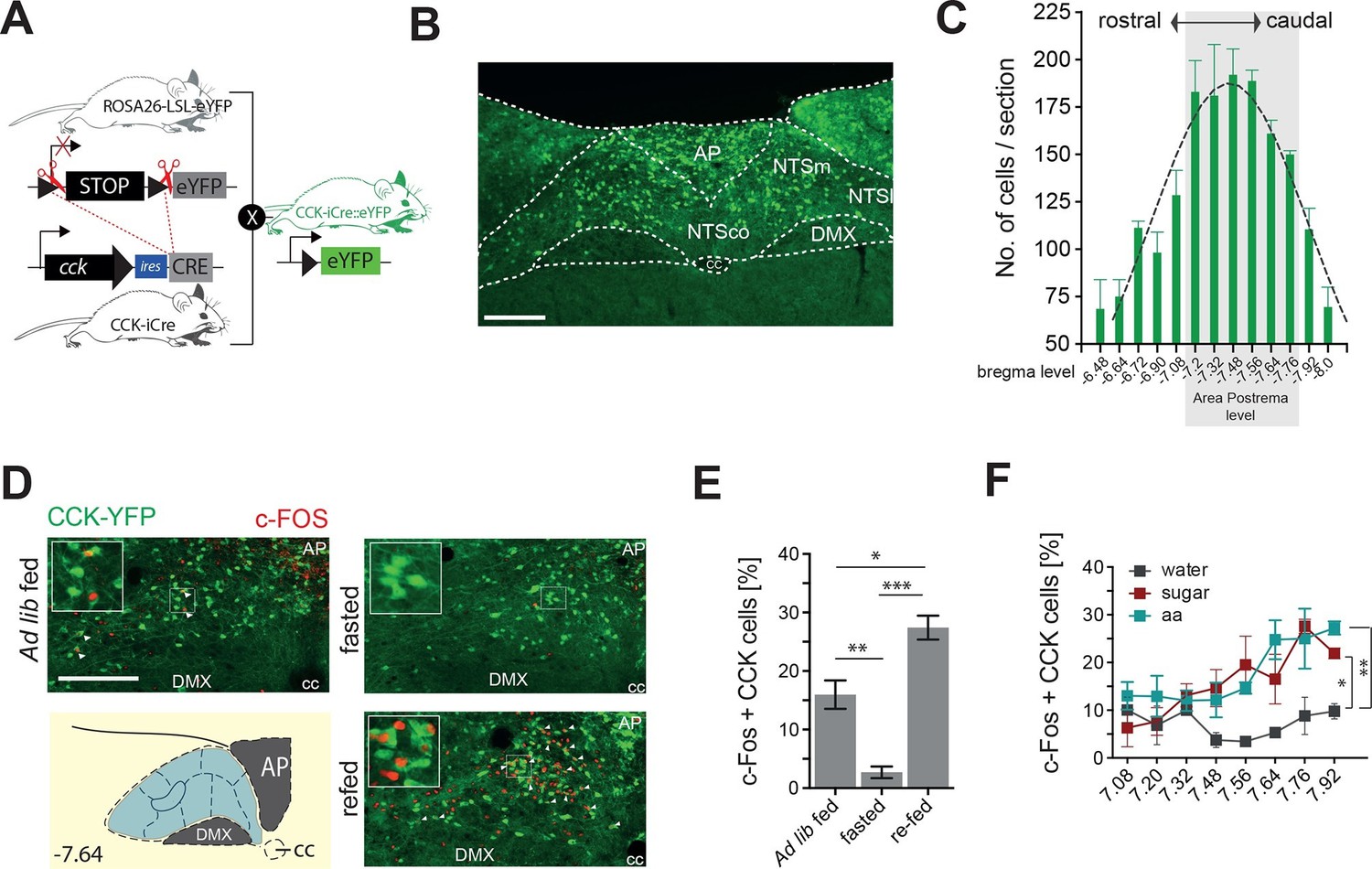
CCKNTSneurons are activated by feeding.
(A) Schematic of mouse crossing to generate a CCK-iCre::R26-loxSTOPlox-eYFP mouse line (CckYFP). (B) Representative expression of CckYPF in a NTS coronal section. (C) Quantification of CckYPF-expressing cells across the rostral-to-caudal extent of the NTS. (D) Representative c-Fos-IR in CckYPF NTS cells in ad libitum fed, fasted or fasted then re-fed mice (white arrows denote colocalized neurons) and (E) quantification of c-FOS-positive CckYPF NTS cells (n = 3–4; one-way ANOVA F(2,7) = 39.82, p = 0.0001; Sidak’s post hoc comparison). (F) Quantification of c-FOS-IR across the rostral-to-caudal extent of the NTS in CckYPF cells by bregma level following intragastric delivery of water, amino acids (aa) or sucrose (n = 3–5 per group; one-way ANOVA, F(2,21) = 7.280, p = 0.0040; Tukey’s post hoc comparison). *p<0.05, **p< 0.01, ***p< 0.001. Scale bar in B and D represents 200 μm. AP, area postrema; DMX, dorsal motor nucleus; NTSco, nucleus of the solitary tract, commissural part; NTSm,nucleus of the solitary tract, medial part; NTSl, nucleus of the solitary tract, lateral part.
To determine whether CCKNTS cells are responsive to food intake, CckYFP mice were exposed to either ad libitum fed, dark cycle fasted or dark cycle fasted followed by 2 hr re-feeding conditions, and a surrogate marker of neuronal activation, c-Fos immunoreactivity (IR), was assessed. In contrast to light cycle ad libitum fed and the dark cycle fasted conditions, ingestion of food on an empty stomach significantly increased c-Fos-IR within CCKNTS cells, indicating responsiveness to food consumption (Figure 1D,E). To clarify whether this response is related to the nutrients, as opposed to stomach stretching or orosensorial aspects of feeding, we next investigated whether CCKNTS cells are responsive to nutrients if directly delivered to the stomach. Dark cycle fasted CckYFP mice were intragrastrically delivered isovolumetric (0.5 ml) non-nutritive water or isocaloric (1 kcal) sucrose or amino acids. As observed with chow intake, gavage of sucrose or amino acids significantly increased c-Fos-IR within CCKNTS cells compared with water (Figure 1F). These results suggest that CCKNTS cells are activated by nutrient intake.
CCKNTS neuron activation reduces food intake and body weight
We next considered whether activation of CCKNTS neurons could promote satiety by communicating a nutrient consumption signal. Cck-iCre mice were bilaterally injected into the NTS with AAVs that mediate the Cre-dependent expression of designer receptors exclusively activated by designer drugs (DREADDs; expressed as DREADD-mCherry fusion proteins, hM3Dq; Cck-iCre-hM3Dq-mCherryNTS) (Figure 2A). DREADDs are designer muscarinic receptor variants that can only be activated by an otherwise biologically inert designer drug, clozapine-N-oxide (CNO) (Alexander et al., 2009; Krashes et al., 2011). Using this approach, we achieved expression of the DREADD-fused mCherry reporter protein in the caudal aspect of the NTS (Figure 2B). CNO activated Cck-iCre-hM3Dq-mCherryNTS cells in vivo as shown by increased c-Fos-IR (Figure 2C) and ex vivo using electrophysiology in NTS slices (Figure 2D). Activation of CCKNTS neurons in ad libitum fed Cck-iCre-hM3Dq-mCherryNTS mice injected with CNO (0.3 mg kg-1, IP) suppressed food intake for 24 hr (Figure 2E). Likewise, CCKNTS neuron activation suppressed re-feeding in food deprived mice (Figure 2F).
Figure 2
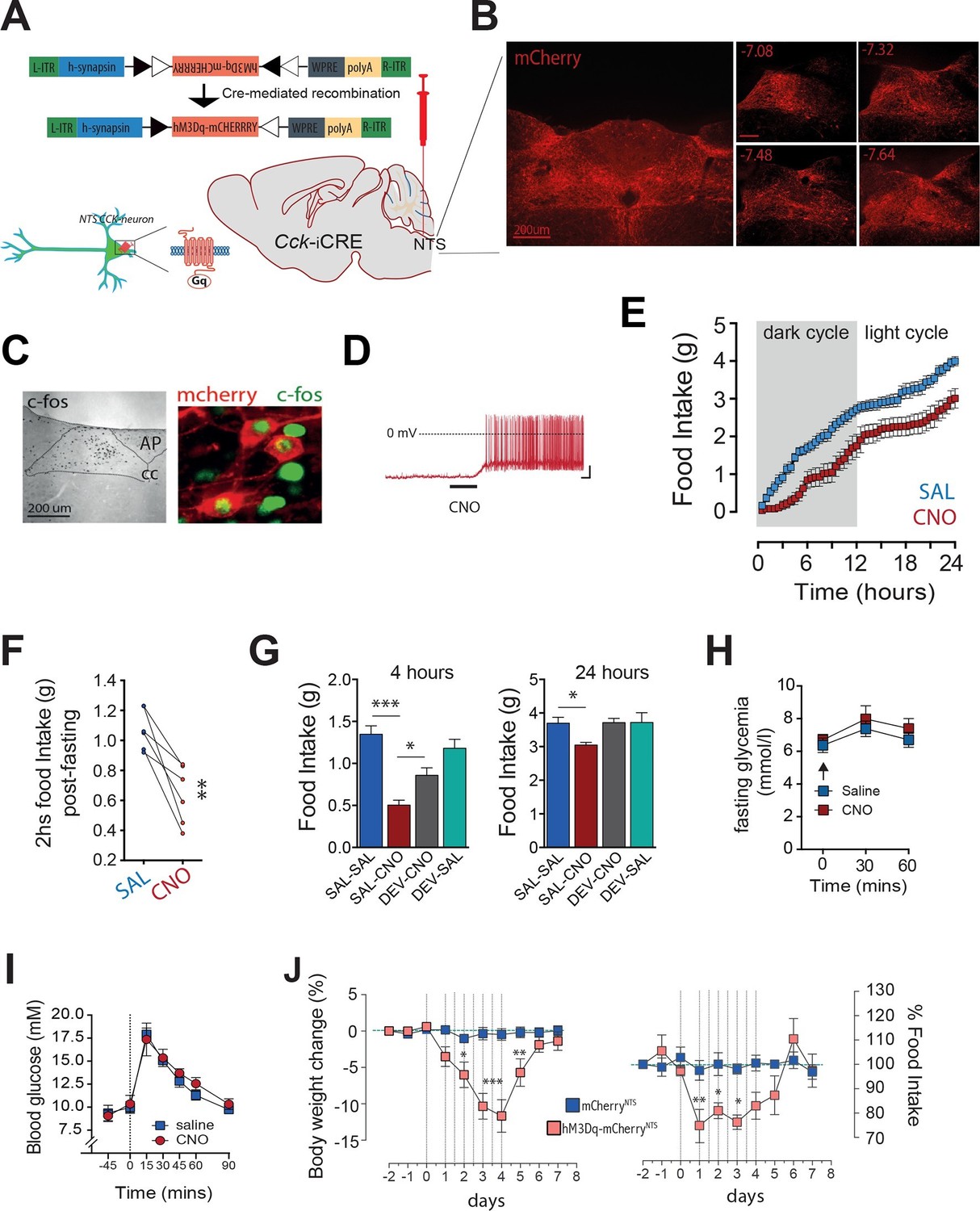
Activation of CCKNTS neurons reduces feeding and body weight.
(A–D) Bilateral stereotaxic injection of Cre-dependent excitatory hM3Dq-mCherry virus into the NTS of male Cck-iCre mice facilitated activation of CCKNTS neurons. (A) Schematic and Cre-mediated recombination of hM3Dq-mCherry allele. (B) Representative image of Cre-dependent expression of hM3Dq-mCherry within the caudal aspect of the NTS of a Cck-iCre mouse (coronal sections; numbers indicate bregma levels, scale bar represents 200 μm). (C) c-Fos-IR in the NTS and co-expression (green) in hM3Dq-mCherry-transduced CCKNTS neurons (red) (scale bar represents 200 μm). (D) Membrane potential and firing rate of Cck-iCre-hM3Dq-mCherryNTS neurons increased upon 5 μM CNO application. (E) Cck-iCre-hM3Dq-mCherryNTS mice exhibited a significant reduction of spontaneous feeding following CNO, compared to saline, administration (n = 6; RM ANOVA, main effect of treatment [F(1,5) = 22.41, p = 0.0052], main effect of time [F(47,235) = 101.6, p<0.0001], and interaction [F(47,235) = 1.807, p = 0.0023]); tick marks on x-axis represent 3 hr, measurements collected with 30-min intervals) and (F) a reduction of post-fast re-feeding following CNO compared to saline administration (n = 6; paired t test, t(5) = 4.769, p = 0.005). (G) CNO-induced reduction in spontaneous feeding was attenuated by pre-treatment with the CCK-A-receptor antagonist, devazepide (DEV; 1 mg/kg), (n = 6; ANOVA, F(3,20) = 16.81, p<0.0001; Sidak’s post hoc comparison ***p<0.001, *p< 0.05). (H) CNO did not change fasting glucose level or (I) glucose disposal rate following a systemic glucose load (1 g/kg, IP). (J) Repeated CNO administrations over 4 days reduced body weight (n = 6; RM ANOVA, main effect of treatment [F(1,100) = 60.78, p<0.0001], main effect of time [F(9,100) = 8.877, p<0.0001], and interaction [F(9,100) = 7.483, p<0.0001]) and decreased food intake (main effect of treatment [F(1,100) = 16.13, p = 0.0001], main effect of time [F(9,100) = 4.106, p = 0.0002], and interaction [F(9,100) = 3.307, p = 0.0014]; Sidak’s post hoc comparisons, *p<0.05, **p<0.01, ***p<0.001) in Cck-iCre-hM3Dq-mCherryNTS as compared to Cck-iCre-mCherryNTS mice.
We next tested whether CCK itself mediates these food supressing effects of CCKNTS neuron activation. Cck-iCre-hM3Dq-mCherryNTS mice were pre-treated with the CCK-A receptor antagonist devazepide which blunted CNO-induced appetite suppression both 4 hr and 24 hr after treatment (Figure 2G), with no effects in control mice. These results demonstrate CCK-A receptors as downstream effectors of CCKNTS neuron-mediated reduction in feeding.
Given our recent findings that parabrachial CCKergic (CCKPBN) transmission regulates hepatic glucose production (Flak et al., 2014; Garfield et al., 2014), we next investigated whether CCKNTS neurons impact glucose homeostasis. Unlike CCKPBN neurons, chemogenetic activation of CCKNTS neurons in Cck-iCre-hM3Dq-mCherryNTS mice altered neither circulating blood glucose levels (Figure 2H) nor glucose disposal following a systemic glucose load (Figure 2I). These results reveal a neuroanatomical functional divergence in the energy-intake- versus energy-metabolic features of CCKergic neurotransmission within the brain, with CCKNTS neurons modulating food intake, but not glucose homeostasis.
We next tested whether long-term stimulation of CCKNTS neurons promotes a sustained reduction in food intake and body weight loss. We treated Cck-iCre-hM3Dq-mCherryNTS and control Cck-iCre-mCherryNTS mice with CNO for 4 days (two injections per day). Repeated activation of CCKNTS neurons resulted in a pronounced reduction in food intake with a concomitant reduction in body weight (Figure 2J). Following withdrawal of CNO treatment, mice returned to the original body weight within approximately 48 hr and to baseline food intake after a transient phase of rebound feeding (Figure 2J). These data demonstrate that sustained activation of CCKNTS neurons is sufficient to promote pronounced and reversible anorexia and body weight loss in mice.
Together, these data reveal that activation of CCKNTS neurons promotes anorexia and weight loss, attenuating the homeostatic drive to eat even in face of marked negative energy balance (as occurs in food deprivation).
A CCKNTS→PVH circuit controls appetite
As a first step to delineate the circuits through which CCKNTS neurons modulate food intake, we surveyed the neuronal activation-like profile of Cck-iCre-hM3Dq-mCherryNTS mice treated with CNO or saline. Chemogenetic activation of CCKNTS neurons elicited striking c-Fos-IR in the paraventricular nucleus of the hypothalamus (PVH) in food-deprived mice compared with saline-treated mice (Figure 3—figure supplement 1). The PVH represents a crucial hub for the brain’s regulation of energy balance and integrates a diverse range of nutritionally related hormonal and synaptic inputs, including inputs from the NTS (Grill and Hayes, 2012; Riche et al., 1990). To evaluate whether CCKNTS neurons directly innervate the PVH, a Cre-inducible AAV vector expressing Channelrhodopsin-2-eYFP (ChR2-eYFP) was stereotaxically injected into the NTS of Cck-iCre mice (Figure 3A,B). Cre-mediated recombination of the vector led to expression of eYFP in CCKNTS cells and their projection fields (due to axonal localization of the ChR2-eYFP fusion protein), revealing robust CCKNTS → PVH innervation (Figure 3C), with no substantial projections at other anterior hypothalamic regions (Figure 3—figure supplement 2A–C). CCKNTS efferent projections were also observed to innervate the ventral and more caudal aspect of the dorsomedial nucleus of the hypothalamus and, to a lesser extent, the arcuate nucleus (Figure 3—figure supplement 2D–F).
Figure 3 with 3 supplements see all
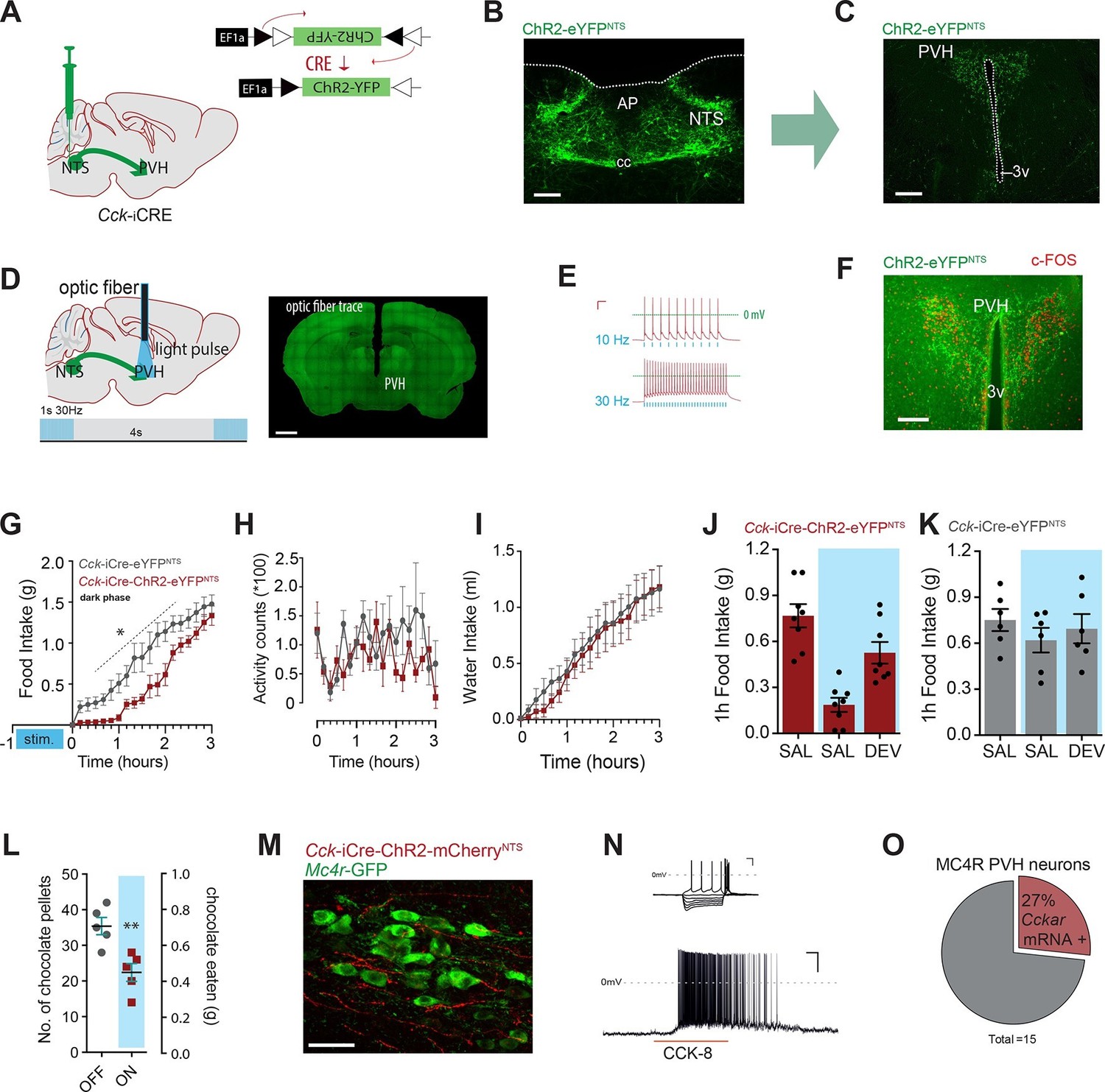
Activation of CCKNTS neurons efferent to the PVH suppresses appetite.
(A) Schematic of CCKNTS→ PVH targeting strategy using bilateral NTS delivery of Cre-dependent ChR2-eYFP expressing vector in Cck-iCre mice. (B) Selective eYFP expression following Cre-mediated recombination in the caudal aspect of the NTS (scale bar represents 200 μm). (C) CCKNTS efferents (green) innervate the PVH (scale bar represents 400 μm). (D) CCKNTS axon targeting for photostimulation, positioning of the optic fiber and photostimulation parameters (scale bar represents 400 μm). (E) Current clamp recording of a CCKNTS neuron expressing ChR2 (scale bar 20 mV/100 ms). (F) Bilaterally transduced CCKNTS axons in the PVH and c-Fos-IR following PVH photostimulation (scale bar represents 200 μm). (G) In vivo optogenetic photostimulation of NTSCCK→PVH terminals in Cck-iCre-ChR2-eYFPNTS significantly reduced nocturnal feeding (n = 6, RM ANOVA: main effect of treatment (F(1,10) = 8.663, p = 0.0147), main effect of time (F(18,180) = 97.25, p<0.0001) and interaction (F(18,180) = 2.788, p = 0.0003) Sidak’s post hoc comparisons, *at least p<0.05) (H) without altering locomotor activity (RM ANOVA: main effect of treatment (F(1, 10) = 1.510, p = 0.0.2472), main effect of time (F(18,180) = 1.797, p = 0.0285) and interaction (F(18,180) = 1.198, p = 0.2671) or (I) water consumption (RM ANOVA: main effect of treatment (F(1,10) = 0.0924, p = 0.7673), main effect of time (F(18,180) = 50.68, p<0.0001) and interaction (F(18,180) = 0.2666, p = 0.9989) as compared to control Cck-iCre-eYFPNTS. Tick marks on x-axis represent 10-min intervals. (J) Real time food intake reduction following optogenetic activation of NTSCCK→PVH terminals in fasted Cck-iCre-ChR2-eYFPNTS and reversion following injection of CCK-A receptor antagonist (devazipide; DEV) (n = 8; RM one-way ANOVA, treatment F(1.809,12.66) = 16.15, p = 0.0004; individual F(7,14) = 0.4241, p = 0.8714, Sidak’s post hoc comparison **p<0.005, *p<0.05). (K) Neither photostimulation nor DEV treatment alter food intake in fasted Cck-iCre-eYFPNTS control mice (n = 6; RM one-way ANOVA, treatment F(1.294,6.469) = 1.486, p = 0. 2780; individual F(5,10) = 5.089, p = 0.014). (L) Photostimulation of NTSCCK→PVH terminals reduces total intake of chocolate pellets over 30 min following 18–20 hr of food deprivation (n = 5, paired two-tailed t-test, t(4) = 6.949, p = 0.0023), as compared to no photostimulation. (M) Representative confocal image of NTSCCK→PVH mCherry fibers and varicosities in close apposition to putative PVH MC4R-GFP neurons (scale bar represents 20 μm; 10 μm stack, maximum intensity projection). (N) Top, IV relationship of a PVH MC4R-GFP neuron produced by the superimposition of membrane potential deflection in response to a series of current injections of constant increment (scale bar 20 mV/200 ms); bottom, current clamp recording of the above neuron following bath application of CCK-8 (500 nM; scale bar 20 mV/30 s). (O) Percentage of PVH MC4R-neurons expressing CCK-A receptor mRNA as assessed by single-cell qPCR. NTS, nucleus of the solitary tract; PVH, paraventricular nucleus of the hypothalamus; AP, area postrema; cc, central canal; 3v, third ventricle.
To probe for the physiological significance of this CCKNTS → PVH circuit, Cck-iCre mice injected into the NTS with AAVs encoding for ChR2-eYFP (a blue-light-sensitive cation channel) were implanted with an optic fiber above the PVH to stimulate these CCKNTS efferents (Figure 3A–D). Ex vivo application of blue light (473 nm) to CCKNTS neurons expressing ChR2 using slice electrophysiology resulted in faithful action potential discharge with stimulation/discharge fidelity preserved at both 10 Hz and 30 Hz (but not 50 Hz, data not shown) in Cck-iCre-ChR2-eYFPNTS cells (Figure 3E). In vivo photostimulation of CCKNTS axon terminals in the PVH elicited c-Fos-IR in PVH neurons in close proximity of transduced CCKNTS axons (Figure 3F).
We investigated whether specifically activating CCKNTS axon terminals within the PVH (10 ms, 30 Hz, 1 s on, every 4 s) prior the initiation of mouse nocturnal feeding could communicate a post-prandial-like signal, and thereby reduce subsequent spontaneous feeding. Monitoring home-cage food intake revealed that optogenetic activation of the CCKNTS→PVH circuit produced an almost complete, and reversible, suppression of spontaneous feeding (Figure 3G). Importantly, optogenetic activation of the CCKNTS→PVH pathway neither suppresses locomotor activity (Figure 3H) nor water consumption (Figure 3I). Furthermore, real-time optogenetic stimulation of this circuit (10 ms, 30 Hz, 1 s on, every 4 s) significantly suppressed re-feeding in food deprived Cck-iCre-ChR2-eYFPNTS mice (Figure 3J), but not control Cck-iCre-eYFPNTS mice (Figure 3K). The reduction in food intake in Cck-iCre-ChR2-eYFPNTS stimulated mice was blunted by pre-treatment with the CCK-A receptor antagonist devazepide (Figure 3J), which did not affect food intake in control mice (Figure 3K). Next, we tested the role of the CCKNTS→PVH circuitry to supress feeding in response to palatable food (chocolate pellets). Food deprived Cck-iCre-ChR2-eYFPNTS mice were offered chocolate pellets via a computer-controlled delivery system using a within-subjects design (optogenetic activation or no activation). Optogenetic activation of CCKNTS→PVH efferent reduced the total number of chocolate pellets consumed (Figure 3L), indicating that the appetite-suppressing properties of the CCKNTS→PVH circuit maintains salience even when the homeostatic drive to eat following food deprivation is boosted with a hedonic component.
Converging pharmacological and genetic data has established the importance of melanocortin-4 receptor (MC4R)-expressing neurons in the regulation of energy balance (Fan et al., 1997; Huszar et al., 1997; Krashes et al., 2016), with the PVH having been identified as a principal site of their satiety-promoting action (Balthasar et al., 2005; Garfield et al., 2015; Shah et al., 2014). To determine whether MC4R-expressing neurons are positioned to be a functional exponent of CCKNTS→PVH efferents, we first employed a modified bacterial artificial chromosome (BAC) transgenic MC4R-GFP reporter mouse line (Ghamari-Langroudi et al., 2015; Liu et al., 2003) and crossed it with the Cck-iCre line. CCKNTS neurons were transduced with a mCherry-conjugated ChR2 AAV vector in the Cck-iCre::MC4R-GFP double transgenic line. We observed CCKNTS→PVH mCherry-positive fibers and varicosities in close apposition to MC4R-expressing neurons (Figure 3M and Figure 3—figure supplement 3). Moreover, using patch clamp electrophysiology, we observed that approximately 23% (5/23 cells) of PVH MC4R-expressing neurons were excited by CCK-8 application (Figure 3N).
To increase the stringency of genetic identification of MC4R-expressing neurons, we also used a second and recently described knock-in mouse line (Mc4r-t2a-Cre) that express Cre recombinase from the endogenous and transcriptionally active Mc4r locus (Garfield et al., 2015) and in which MC4R-expressing neurons are fluorescently labeled by means of Cre-enabled tdTomato expression (Mc4r-t2a-Cre::tdTomato; Figure 3—figure supplement 3A). Single-cell qPCR of manually sorted PVH Mc4r-t2a-Cre::tdTomato-expressing neurons revealed Cckar mRNA to be expressed in 27% (4/15) of MC4R PVH cells analyzed (Figure 3O and Figure 3—figure supplement 3C). Likewise, electrophysiological recordings revealed that approximately 30% (6/20 cells) of PVH Mc4r-t2a-Cre::tdTomato neurons were excited by CCK-8 application, in a CCK-A receptor-dependent manner (0/11 cells; Figure 3—figure supplement 3D). Thereby, these data identify CCK as a novel peptide neurotransmitter activating the appetite-controlling PVH MC4R neurons.
A CCKNTS→PVH circuit encodes positive valence
In addition to the homeostatic regulation of energy balance, the NTS is also associated with negative valance and anorexia related to nausea (Lachey et al., 2005; Rinaman, 2004; Swank and Bernstein, 1994). We next considered whether the CCKNTS→PVH circuit is associated with the induction of an aversive physiological state. The motivational valence of this circuit was assessed using a real-time place preference test (Stamatakis and Stuber, 2012). We observed no place preference when Cck-iCre-ChR2-eYFPNTS or Cck-iCre-eYFPNTS controls were tested under normal energy balance conditions, an important indication that activation NTSCCK→PVH efferents is not aversive (Figure 4A–C). However, while Cck-iCre-eYFPNTS controls maintained no chamber preference when food deprived, calorie depleted Cck-iCre-ChR2-eYFPNTS mice exhibited a significant preference for the photostimulation-paired chamber (Figure 4B,C). This behavioral response is reminiscent of that observed following direct optogenetic activation of PVH MC4R-expressing neurons (Garfield et al., 2015). We further examined this observation using a T-shaped maze. In this test, exploration of one of the three maze’s arms was paired with the photostimulation of the NTSCCK→PVH efferent (Figure 4D). Over the three testing trials Cck-iCre-ChR2-eYFPNTS mice, but not Cck-iCre-eYFPNTS controls, developed a clear preference for the photostimulation-paired arm (Figure 4E,F). Thus, when hungry and in the absence of food, mice sought out activation of the NTSCCK efferent to PVH, revealing the CCKNTS→PVH circuit encodes positive valence. These data suggest that the activation of this circuit provides relief from the unpleasantness of energy deficit by mimicking a post-prandial phase.
Figure 4
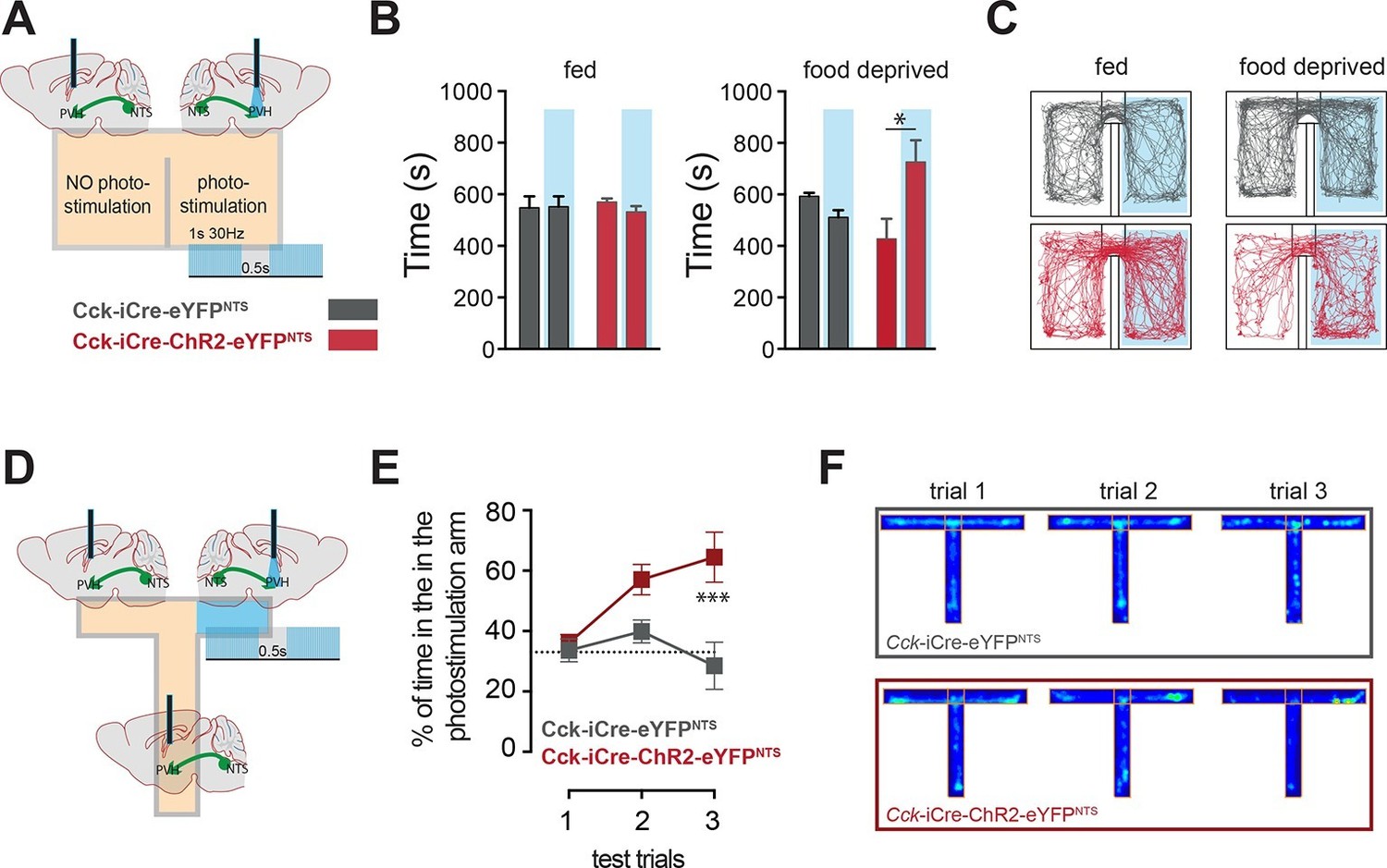
CCKNTS neurons signal positive valence via the PVH.
(A and D) Experimental strategies for the interrogation of the valence encoded by NTSCCK→PVH terminals. (B) Food-deprived Cck-iCre-ChR2-eYFPNTS mice exhibited a significant place preference for the photostimulation-paired chamber during a real-time place preference assay, as compared to control Cck-iCre-eYFPNTS mice (n = 5–6; Two-way ANOVA – fed: no effect of photostimulation (F(1,18) = 0.3244, p = 0.5760), no effect of ChR2 (F(1,18) = 0.001145, p = 0.9734) or interaction (F(1,18) = 0.5007, p = 0.4883); food deprived: main effect of photostimulation (F(1,18) = 5.289, p = 0.0336), no effect of ChR2 (F(1,18) = 0.08811, p = 0.7700) and interaction (F(1,18) = 12.63, p = 0.0023); Sidak’s post hoc comparisons,*p = 0.05). (C) Representative real-time place preference location plots one representative mouse per condition. (E) Cck-iCre-ChR2-eYFPNTS developed preference for the photostimulation-paired arm in a three-trial T-maze test (n = 5–6, main effect of treatment (F(1,27) = 15.93, p = 0.0005) main effect of trials (F(2,27) = 3.36, p = 0.0498) and interaction (F(2,27) = 4.36, p = 0.0228); Sidak’s post hoc comparisons, ***p<0.001), as compared to Cck-iCre::eYFPNTS. (F) Representative T-maze location plots from one representative mouse per condition. NTS, nucleus of the solitary tract; PVH, paraventricular nucleus of the hypothalamus.
While further studies attempting site-specific Cck loss of function are needed to fully clarify the physiological necessity of NTS CCKergic transmission in eating behavior and body weight regulation, here we reveal that activation of CCKNTS produces a prolonged effect on appetite and a rapid reduction in body weight. We did not find evidence that this reduction in energy intake is due to an aversive state, but rather, reveal that the CCKNTS→PVH circuit transmits positive valence in an energy-state-dependent manner; a particularly attractive prospect considering that in patient populations the negative valence of energy deficit is a major factor contributing to low compliance on weight-loss diets.
Materials and methods
Animals
CCK-ires-Cre (Cck-iCre; Ccktm1.1(cre)Zjh/J; Stock No. 012706), R26-loxSTOPlox-eYFP (Ai3; B6.Cg-Gt(ROSA)26Sortm3(CAG-EYFP)Hze/J; Stock No. 007903), MC4R-GFP (B6.Cg-Tg(Mc4r-MAPT/Sapphire)21Rck/J; Stock No. 008323) or R26-loxSTOPlox-tdTomato (Ai9; B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J; Stock No.007909) were obtained from Jackson Laboratories (Bar Harbor, ME). Mc4r-t2a-Cre mice were previously described (Garfield et al., 2015). Mice were provided with standard mouse chow and water ad libitum, unless otherwise noted and housed at 22–24°C with a 12-hr light/12-hr dark cycle. All experimental procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986 or the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Drugs
Drugs for in vivo use were prepared in sterile saline and administered intraperitoneally (IP). Clozapine-N-oxide (CNO; Tocris Bioscience, Bristol, UK; Cat. No. 4936) was administered at 0.3 mg/kg. Devazepide (Tocris Bioscience, Cat. No. 2304) was dissolved in DMSO, further diluted with sterile saline and administered at 1 mg/kg, IP, 40 min before CNO injection or optogenetic photostimulation. CCK-8 (Tocris, Cat. No. 1166) and SR 27,897 (CCK-A antagonist; Tocris Bioscience, Cat. No. 27897) were dissolved in artificial cerebrospinal fluid (aCSF).
Viral vectors
Request a detailed protocolDREADD and Optogenetics viruses employed were obtained from University North Carolina Vector Core (Chapel Hill, NC). DREADD constructs were packaged in AAV serotype-8 and injected at a titer of 1.3 × 1012 vg/ml. AAV-EF1a-DIO-EYFP, AAV/EF1a-DIO-ChR2(E123T/T159C)-eYFP, AAV/EF1a-DIO-ChR2(E123T/T159C)-mCherry were packaged in AAV serotype-2 and injected at a titer of about 1 × 1012 vg/ml. Nucleus-specific delivery of AAVs was achieved through stereotaxic injections.
Stereotaxic surgery
Request a detailed protocolNTS delivery of AAVs was achieved through a modified stereotaxic procedure. Briefly, 5–8 week-old male mice were anesthetized with a mixture of ketamine and xylazine dissolved in saline (100 and 10 mg/kg, respectively; 10 ml/kg). Mice were placed in a stereotaxic frame (Kopf Instruments) using ear bar with head angled at about 45°. Under magnification, an incision was made at the level of the cisterna magna and neck muscles carefully retracted. A 33G needle was used for dura incision. The obex served as reference point for injection. Injections were performed using a glass micropipette (diameter 20–40 μm). NTS coordinates were A/P, -0.2; M/L, ±0.2; D/V, -0.2 from the obex. Virus was delivered under air pressure using a PLI-100A Pico-Injector (Harvard Apparatus, Cambridge, UK). About 150 nl of virus/side were delivered with multiple microinjections over 4–5 min. The pipette remained in place for a minimum of 3 min after injection. Animals were administered an analgesic (5 mg/kg Carprofen, subcoutaneously) for 2 days post-operatively and given a minimum of 14 days recovery before being used in any DREADD experiments. During this time, mice were acclimated to handling and IP injections. For optogenetic experiments, optic fibers were placed above the PVH at least 4 weeks after the NTS viral delivery to ensure labelling of distal projection sites. Based on the Franklin and Paxinos Mouse Brain Atlas (Franklin and Paxinos, 2008), the following coordinates were used for targeting optic fibers (200 μm diameter core; CFMLC22U-20; Thorlabs, Newton, NJ) at the PVH (mm from Bregma): A/P, −0.65; M/L, ± 0; D/V, −4.20 mm. After placement of the optic fiber, mice were allowed additional 14 days post-surgical recovery, as described above. Mice were excluded from behavioral analysis if post hoc histological validation showed either no viral transduction or misplaced optical fibers.
Energy balance and body weight studies
Request a detailed protocolDark-cycle food intake. Mice were injected with saline or CNO (0.3 mg/kg; IP Tocris Bioscience) 30 min prior to the onset of the dark. At onset of dark, food was returned and intake recorded automatically via the TSE Phenomaster system (TSE, Bad Homburg, Germany) or manually (Figure 2g). Post-fast re-feed. Mice were fasted overnight. The following morning, mice were treated IP with saline or CNO and food returned 30 min later and intake recorded manually. Daily treatment. Mice were treated IP twice daily for 4 days with 0.3 mg/kg of CNO or saline near the onset of the dark and light cycles. Food intake and body weight were recorded daily prior to the dark cycle. Nutrient gavage. Mice were fasted overnight and the following morning, received intragastric 0.5 ml volume gavage of water, amino acids (Peptone enzymatic digest from casein, Sigma-Aldrich, Dorset, UK; Cat. No. 70172) or sucrose (Sigma-Aldrich, Cat. No. 84100). Amino acids and sucrose were diluted in drinking water and delivered in an amount of 1kcal. Blood glucose studies. Mice were food deprived overnight and blood glucose concentration was determined from tail bleeds using OneTouch Ultra glucometer and test strips (LifeScan, Johnson and Johnson Medical Limited, Livingstone, UK). Basal blood glucose concentration was determined prior to injection of any substances.
Photostimulation protocol
Request a detailed protocolFiber optic cables (1.5 m long, 200 μm diameter, 0.22 NA; Doric Lenses, Franquet, Quebec, Canada) were firmly attached to the implanted fiber optic cannulae with zirconia sleeves (Doric Lenses). Photostimulation was programmed using a pulse generator software (Prizmatix, Givat-Shmuel, Israel) that controlled a blue light laser (473 nm; Laserglow, Toronto, Canada) via a USB-TTL interface (Prizmatix).
Photostimulation for feeding experiments: light pulse trains 10-ms pulse width, 30 Hz, 1 s on, 4 s off. Photostimulation for place preference and T-maze tests: light pulse trains 10-ms, 30 Hz, 1 s on, 0.5 s off. Light power was adjusted such that the light power exiting the fiber optic cable was at least 10 mW using a digital optical power meter (PM100D, Thorlabs) and an online light transmission calculator for brain tissue (http://web.stanford.edu/group/dlab/cgi-bin/graph/chart.php). After the completion of photostimulation experiments, mice were perfused and the approximate locations of fiber tips were identified according to the atlas coordinates (Franklin and Paxinos, 2008). After removal, optic fiber were connected to optic fiber cable and tested for light transmission.
Behavioral testing
Request a detailed protocolMice were tested for real-time place preference in a model in which one chamber was paired with 30-Hz photostimulation and the other, identical chamber resulted in no photostimulation. Total test duration was 20 min. A similar protocol was used for the T-maze experiments. For the T-maze test mice were tested in three 10-min trials with an inter-trail interval of 15–20 min, during which mice were returned to the home cage. Time spent in the photostimulation versus non-photostimulation zones was recorded via a CCD camera interfaced with Any-maze software (Stoelting, Wood Dale, IL).
Brain tissue preparation and immunohistochemistry
Request a detailed protocolFollowing deep terminal anesthesia with pentobarbital, mice were transcardially perfused with phosphate-buffered saline (PBS) followed by 10% neutral buffered formalin (Fisher Scientific, Loughborough, UK). Brains were extracted, cryoprotected in 30% sucrose in PBS, sectioned coronally on a freezing sliding microtome (Bright solid state freezer series 8000, Bright Instruments, Luton, UK) at 30 μm and collected in four equal series. IHC was performed using standard methods and as previously described (Garfield et al., 2012). Briefly, sections were washed in PBS before blocking in 0.5% BSA/0.25% Triton X-100 in PBS for 1 hr at room temperature. Tissue was incubated overnight at room temperature in blocking buffer containing the primary antibodies: rabbit anti-c-FOS (EMD Millipore, Livingston, UK; Cat. No. PC05; diluted 1/5000), or chicken anti-GFP (Abcam, Cambridge, UK; Cat. No. ab13970; diluted 1/1000). The next day, sections were washed in PBS then incubated in blocking buffer containing appropriate secondary antibody (Alexa Fluor, Life Technologies; diluted 1/500) for 1 hr. 3,3′-Diaminobenzidine (DAB) staining was used for used for detection of c-Fos-IR using: a rabbit anti-c-FOS (EMD Millipore Cat. No. PC05; diluted 1/5000), secondary biotin-SP donkey anti-rabbit (Jackson Immunoresearch, West Grove, PA; Cat. No. 711-065-152; diluted 1/250), an avidin/biotin-based peroxidase system (Vectastain Elite ABC Kit, Vector Laboratories, Burlingame, CA; Cat. No. PK-6100) and a developing kit (DAB Peroxidase Substrate Kit, Vector Laboratories, Cat. No. SK-4100). c-Fos-IR was pseudocolored using Photoshop software to appear colored in images. Sections were mounted onto microscope slides and coverslipped in an aqueous mounting medium (Vectashield Antifade Mounting Medium, Vector Laboratories, Cat. No. H-1000). Slides were imaged on a VS120 slide scanner (Olympus, Essex, UK) or AXIOSKOP2 (Zeiss, Oberkochen Germany). For counting of c-FOS-IR nuclei, the boundaries of the nucleus were defined using neuroanatomical landmarks and the Franklin and Paxinos Mouse Brain Atlas (Franklin and Paxinos, 2008).
Electrophysiological studies
Request a detailed protocolFor electrophysiological validation mice were anesthetized with sodium pentobarbital (Euthatal) and decapitated. The brain was rapidly removed and placed in cold (0–4°C), oxygenated (95%O2/5%CO2) ‘slicing’ solution containing (in mM) sucrose (214), KCl (2.5), NaH2PO4 (1.2), NaHCO3 (26), MgSO4 (4), CaCl2 (0.1), D-glucose (10). The brain was glued to a vibrating microtome (Campden Instruments, Loughborough, UK) and 200-μm thick coronal sections of the brainstem containing the NTS were prepared. Slices were immediately transferred to a’ recording’ solution containing (in mM) NaCl (127), KCl (2.5), NaH2PO4 (1.2), NaHCO3 (26), MgCl2 (1.3), CaCl2 (2.4), D-glucose (10), in a continuously oxygenated holding chamber at 35°C for a period of 25 min. Subsequently, slices were allowed to recover in ‘recording’ solution at room temperature for a minimum of 1 hr before recording. For whole-cell recordings, slices were transferred to a submerged chamber and a Slicescope upright microscope (Scientifica, Uckfield, UK) was used for infrared - differential interference contrast and fluorescence visualization of cells. During recording slices were continuously perfused at a rate of ca. 2 ml/min with oxygenated ‘recording’ solution (as above) at room temperature. All pharmacological compounds were bath applied. Whole cell current-clamp recordings were performed with pipettes (3–7 MΩ when filled with intracellular solution) made from borosilicate glass capillaries (World Precision Instruments, Aston, UK) pulled on a Zeitz DMZ micropipette puller (Zeitz Instruments GmBH, Martinsried, Germany). The intracellular recording solution contained (in mM) K-gluconate (140), KCl (10), HEPES (10), EGTA (1), Na2ATP (2), pH 7.3 (with KOH). Recordings were performed using a Multiclamp 700B amplifier and pClamp10 software (Molecular Devices, Sunnyvale, CA). Liquid junction potential was 16.4mV and not compensated. The recorded current was sampled at 10 kHz and filtered at 2 kHz unless otherwise stated. Photostimulation of channelrhodopsin2 was achieved by 470 nm blue light delivered via the microscope objective. Light was generated by a pE-4000 LED illumination system (CoolLED, Andover, UK) driven via clampex 10.4 software (Molecular Devices, Sunnyvale, CA). For CCK-8 application on MC4R PVH neurons, brain coronal slices from 6–8 week-old Mc4r-GFP or Mc4r-t2a-Cre::tdTomato mice were prepared. Extracted brains were immediately submerged in ice-cold, carbogen-saturated (95% O2, 5% CO2) high-sucrose solution (238 mM sucrose, 26 mM NaHCO3, 2.5 mM KCl, 1.0 mM NaH2PO4, 5.0 mM MgCl2, 10.0 mM CaCl2, 11 mM glucose). Then, 300-µm thick coronal sections were cut with a Leica VT1000S vibratome and incubated in oxygenated aCSF (126 mM NaCl, 21.4 mM NaHCo3, 2.5 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 10 mM glucose) at 34˚C for 30 min. Then, slices were maintained and recorded at room temperature (20–24˚C). The intracellular solution for current clamp recordings contained the following (in mM): 128 K gluconate, 10 KCl, 10 HEPES, 1 EGTA, 1 MgCl2, 0.3 CaCl2, 5 Na2ATP, 0.3 NaGTP, adjusted to pH 7.3 with KOH. CCK-8 (100 nM) and SR 27,897 (CCK-A antagonist; 250 nM) were applied to the bath through perfusion. Synaptic blockers (1 mM kynuerenate and 100 µM picrotoxin) were added to the aCSF to synaptically isolate MC4R PVH neurons.
Single-cell qPCR
Request a detailed protocolAdult (4–5 week old) Mc4r-t2a-Cre::tdTomato male mice (n=2) were anesthetized with isoflurane. Brains were extracted and immediately chilled in ice-cold, carbogen-saturated (95% O2, 5% CO2) high-sucrose solution (238 mM sucrose, 26 mM NaHCO3, 2.5 mM KCl, 1.0 mM NaH2PO4, 5.0 mM MgCl2, 10.0 mM CaCl2, 11 mM glucose). Next, 300-µm thick coronal sections were cut with a Leica VT1000S Vibratome and incubated in oxygenated aCSF (126 mM NaCl, 21.4 mM NaHCO3, 2.5 mM KCl, 1.2 mM NaH2PO4, 1.2 mM MgCl2, 2.4 mM CaCl2, 10 mM glucose) at 34°C for 30 min. The PVH was visualized by fluorescence stereoscope then micro-dissected and enzymatically dissociated according to a published protocol (Saxena et al., 2012), except that trituration was performed with fire-polished Paster pipettes. From the resulting cell suspension, tdTomato+ cells were individually washed and collected (Hempel et al., 2007), frozen at -80°C and then processed for cDNA synthesis and amplification (Picelli et al., 2014). To control for mRNA contamination during cell collection, an equivalent volume of cell-picking buffer was sampled and processed along with each batch of cell samples. After 20 cycles of amplification by polymerase chain reaction (PCR), cDNA was purified (Picelli et al., 2014) and eluted in 30 μl PCR-grade water, and then analyzed by quantitative PCR (qPCR) for Actb, tdTomato, and Cckar in duplicate reactions. Actb and Cckar qPCR assays were obtained from Integrated DNA Technologies (IDT, Coralville, IA; Cat. No. Mm.PT.58.28904620.g and Mm.PT.58.12665706IDT). The tdTomato assay was custom synthesized by IDT from the following sequences: left, ACCCAGACCGCCAAGCTGAA; right primer, AGTTCATCACGCGCTCCCACT; internal probe, GCCCCCTGCCCTTCGCCTGG.
Statistics
Statistical analyses were performed using Prism 6 (Graphpad Software, La Jolla, CA). Data were analyzed using t-test, one-way ANOVA, two-way or repeated measures (RM) ANOVA with post hoc comparisons, where appropriate. N represents independent biological replicates. No statistical methods were used to predetermine sample sizes. Sample size was computed based on pilot data and published literature. Data are presented as mean ± SEM and statistical significance was set at p<0.05.
References
-
Hypothalamic control of energy balance: insights into the role of synaptic plasticityTrends in Neurosciences 36:65–73.https://doi.org/10.1016/j.tins.2012.12.005
-
Leptin-inhibited PBN neurons enhance responses to hypoglycemia in negative energy balanceNature Neuroscience 17:1744–1750.https://doi.org/10.1038/nn.3861
-
BookThe Mouse Brain in Stereotaxic Coordinates (3rd Edition)Amsterdam London: Elsevier Academic Press.
-
A neural basis for melanocortin-4 receptor–regulated appetiteNature Neuroscience 18:863–871.https://doi.org/10.1038/nn.4011
-
Cholecystokinin decreases food intake in ratsJournal of Comparative and Physiological Psychology 84:488–495.https://doi.org/10.1037/h0034870
-
Cholecystokinin octapeptide analogues suppress food intake via central CCK-a receptors in miceThe American Journal of Physiology 265:R481–486.
-
Rapid, reversible activation of AgRP neurons drives feeding behavior in miceJournal of Clinical Investigation 121:1424–1428.https://doi.org/10.1172/JCI46229
-
Melanocortin-4 receptor–regulated energy homeostasisNature Neuroscience 19:206–219.https://doi.org/10.1038/nn.4202
-
Transgenic mice expressing green fluorescent protein under the control of the melanocortin-4 receptor promoterJournal of Neuroscience 23:7143–7154.
-
Neurobiology of food intake in health and diseaseNature Reviews Neuroscience 15:367–378.https://doi.org/10.1038/nrn3745
-
Full-length RNA-seq from single cells using Smart-seq2Nature Protocols 9:171–181.https://doi.org/10.1038/nprot.2014.006
-
Neuropeptides and catecholamines in efferent projections of the nuclei of the solitary tract in the ratThe Journal of Comparative Neurology 293:399–424.https://doi.org/10.1002/cne.902930306
-
Hindbrain contributions to anorexiaAJP: Regulatory, Integrative and Comparative Physiology 287:R1035–R1036.https://doi.org/10.1152/ajpregu.00437.2004
-
Brainstem integrative function in the central nervous system control of food intakeForum of Nutrition 63:141–151.https://doi.org/10.1159/000264402
-
MC4R-expressing glutamatergic neurons in the paraventricular hypothalamus regulate feeding and are synaptically connected to the parabrachial nucleusProceedings of the National Academy of Sciences of the United States of America 111:13193–13198.https://doi.org/10.1073/pnas.1407843111
-
Satiating effect of cholecystokininAnnals of the New York Academy of Sciences 713:236–241.https://doi.org/10.1111/j.1749-6632.1994.tb44071.x
-
Activation of lateral habenula inputs to the ventral midbrain promotes behavioral avoidanceNature Neuroscience 15:1105–1107.https://doi.org/10.1038/nn.3145
-
Chemogenetic tools to interrogate brain functionsAnnual Review of Neuroscience 37:387–407.https://doi.org/10.1146/annurev-neuro-071013-014048
-
Optogenetic investigation of neural circuits underlying brain disease in animal modelsNature Reviews Neuroscience 13:251–266.https://doi.org/10.1038/nrn3171
Article and author information
Author details
Funding
Wellcome Trust (WT098012)
- Lora K Heisler
Biotechnology and Biological Sciences Research Council (BB/K001418/1)
- Giuseppe D'Agostino
- Lora K Heisler
Wellcome Trust (105625/Z/14/Z)
- Giuseppe D'Agostino
National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK075632)
- Bradford B Lowell
National Institute of Diabetes and Digestive and Kidney Diseases (P30 DK046200)
- Bradford B Lowell
National Institute of Diabetes and Digestive and Kidney Diseases (P30 DK 057521)
- Bradford B Lowell
American Heart Association (14POST20100011)
- John N Campbell
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
Authors wish to thank members of staff of the Medical Research Facility (University of Aberdeen) for assisting with mouse care and husbandry and Raffaella Chianese for assisting with mouse genotyping. This work was supported by the Wellcome Trust (LKH; WT098012), Biotechnology and Biological Sciences Research Council (LKH and GD’A; BB/K001418/1), Wellcome Trust and the University of Aberdeen (GD’A; 105625/Z/14/Z), NIDDK (BBLo; R01 DK075632, P30 DK046200, P30 DK 057521) and American Heart Association (JNC; 14POST20100011). Single-cell qPCR experiment was supported in part by the Molecular Medicine Core facility at Beth Israel Deaconess Medical Center.
Ethics
Animal experimentation: All experimental procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986 (Project License No. 60/4565).
Copyright
© 2016, D'Agostino et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 8,787
- views
-
- 1,582
- downloads
-
- 165
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 165
- citations for umbrella DOI https://doi.org/10.7554/eLife.12225
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Appetite controlled by a cholecystokinin nucleus of the solitary tract to hypothalamus neurocircuit
eLife 5:e12225.
https://doi.org/10.7554/eLife.12225




{kind=link}
{kind=link}
{kind=link}
{kind=link}