Origins of the 2009 H1N1 influenza pandemic in swine in Mexico
- Icahn School of Medicine at Mount Sinai, United States
- National Institutes of Health, United States
- Laboratorio Avi-Mex, Mexico
- Rega Institute, University of Leuven, Belgium
- University of Edinburgh, United Kingdom
Figures
Figure 1
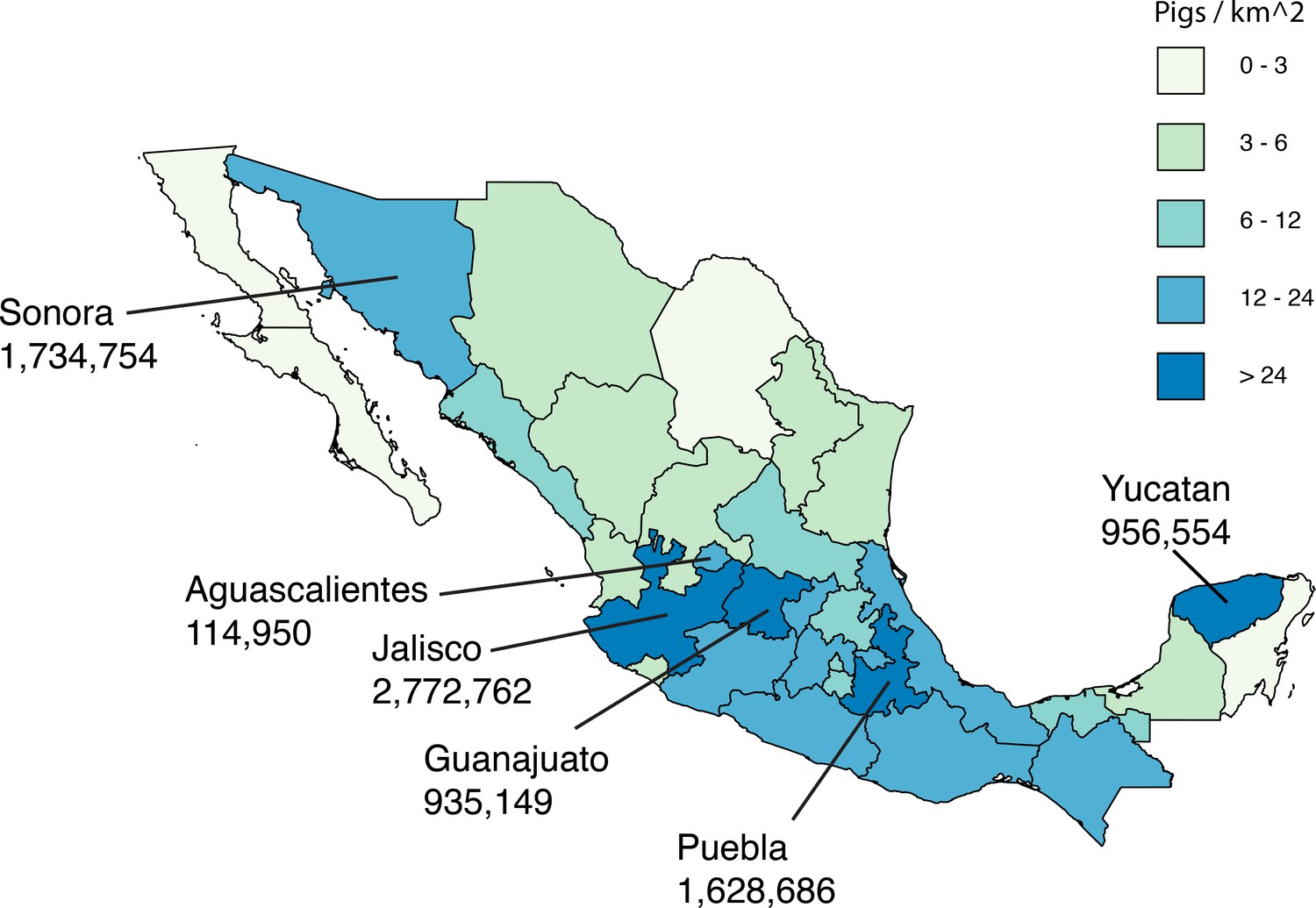
Live swine production in Mexico.
Each Mexican state is shaded according to the density of pigs (the number of pigs per square kilometer, light green = lower and dark blue = higher). The six Mexican states where influenza viruses were collected for this study are indicated, and the number of pigs in the state is provided below the state’s name. Source data from Mexico’s Secretariat of Agriculture, Livestock, Rural Development, Fisheries and Food (SAGARPA) is available in Figure 1—source data 1.
-
Figure 1—source data 1
Swine population sizes in Mexican states in 2014.
Data available from Mexico's Secretariat of Agriculture, Livestock, Rural Development, Fisheries and Food (SAGARPA) (www.siap.gob.mx).
- https://doi.org/10.7554/eLife.16777.004
Figure 2
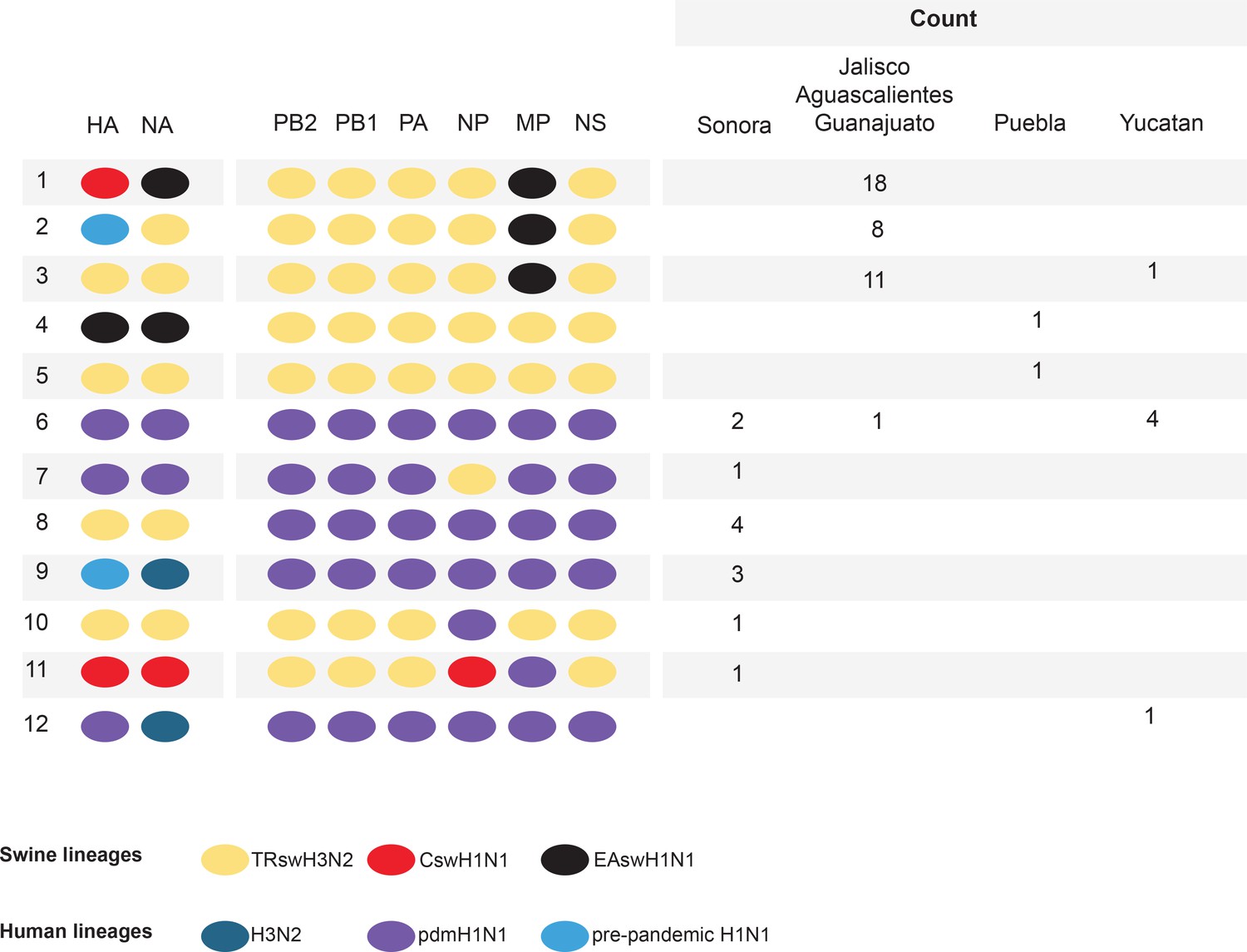
Genetic diversity of IAVs in Mexican swine, 2010–2014.
Twelve genotypes were identified by surveillance in Mexican swien herds during 2010–2014. Each oval represents one of the eight segments of the viral genome. The surface antigens HA and NA are listed first, followed by the six internal gene segments. The shading of each oval corresonds to the genetic lineage of IAVs found in humans and swine globally. The number of swIAVs with a given genotype is indicated for each region in Mexico : Sonora (northern Mexico), Jalisco/Aguascalientes/Guanajuato (central-west Mexico), Puebla (central-east Mexico), and Yucatan (eastern Mexico). The genotype and additional characteristics of each of the 58 swIAVs collected and sequenced from Mexico for this study are provided in Figure 2—source data 1.
-
Figure 2—source data 1
Characteristics of the 58 svIAVs collected in Mexico for this study.
Viruses are listed in order of date of collection. Mexican state is provided (see Figure 1 for a map of Mexico). The genotype (Figure 1B) and lineage and clade Figure 2) of each segment for each virus is provided. Internal genes are categorized as triple reassortant internal genes (trig), classical (NP segment of AVX-30), pdmH1N1 (pdm) or Eurasian (eur). HA segments are categorized as classical swine (H1c), human seasonal H1N1 (H1hu), H3, pdmH1N1 (pdm), or Eurasian (eur). NA segments are categorized as classical swine (N1c), N2 (1998 or 2002 lineage), pdmH1N1 (pdm), or Eurasian (eur). Within these lineages, clades are differentiated by location: Jalisco (J), Sonora (S), Yucatan (Y), Puebla (P), Guanajuato (G). Singleton viruses are indicated (sing).
- https://doi.org/10.7554/eLife.16777.006
Figure 3 with 1 supplement
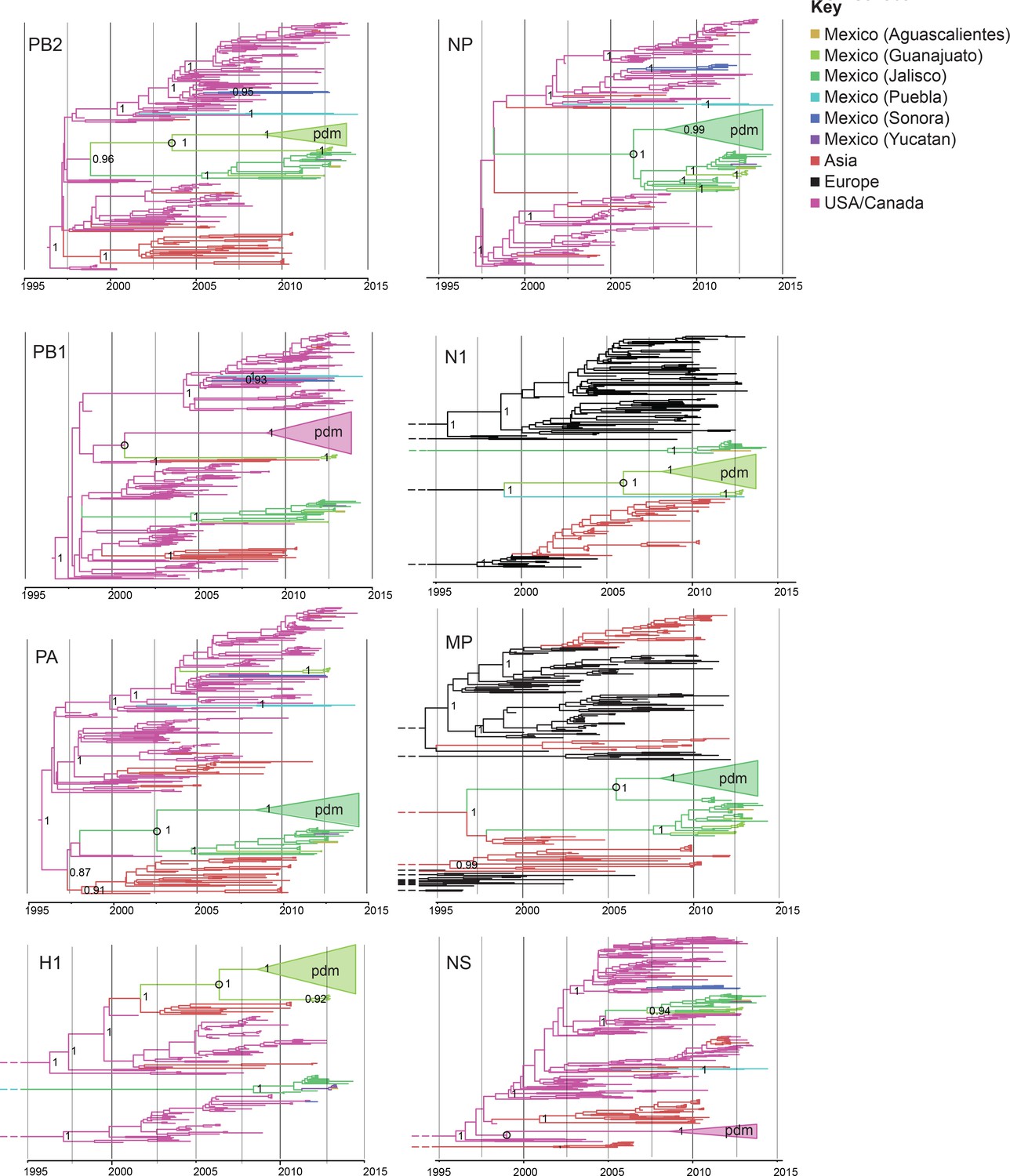
Evolutionary relationships between swIAVs collected in Mexico and pdmH1N1.
Time-scaled Bayesian MCC trees inferred for the eight segments of the IAV genome, for the lineages found in pdmH1N1: TRIG (PB2, PB1, PA, NP, and NS), classical (H1), and avian-like Eurasian (N1 and MP). Trees include the 58 swIAVs collected in Mexico for this study, representative pdmH1N1 viruses, and other related swIAVs collected globally. The color of each branch indicates the most probable location state. For clarity, the pdmH1N1 clade is depicted as a triangle, the color of which represents the inferred location state of the node representing the inferred common ancestor of pdmH1N1 and the most closely related swIAVs (indicated by an open circle). Posterior probabilities are provided for key nodes. More detailed phylogenies including tip labels are provided in Figure 3—source data 1. along with trees inferred for lineages not shown here. Similar phylogenies inferred using maximum likelihood methods are provided in Figure 3—source data 2. Similar phylogenies that use genotype instead of geographic location as a trait are provided in Figure 3—source data 3. Nine Mexican swIAVs were excluded from the phylogenetic analysis because they were outliers in root-to-tip divergence (Figure 3—figure supplement 1). More detailed phylogenies of the pdmH1N1 viruses reveal multiple independent introductions from humans into swine in Mexico (Figure 3—source data 4).
-
Figure 3—source data 1
MCC trees presenting the evolutionary relationships between swIAVs collected in Mexico and swIAVs and human IAVs collected globally, for each IAV segment as well as for each IAV lineage found in Mexican swine: PB2 (TRIG/pdmH1N1), PB1 (TRIG/pdmH1N1), PA (TRIG/pdmH1N1), H1 (classical/pdmH1N1), H1 (avian-like Eurasian), H1 (human seasonal/human- like swine), H3 (human seasonal/human-like swine), NP (TRIG/classical/pdmH1N1), N1 (classical), N1 (avian-like Eurasian/pdmH1N1), N2 (human seasonal/human-like swine), MP (TRIG/classical), MP (avian-like Eurasian/pdmH1N1), NS (TRIG/classical/pdmH1N1).
The color of each branch indicates the most probable location state, similar to Figure 3. Clades of Mexican viruses are labeled. Posterior probabilities >80 are provided for key nodes. The 95% HPD values for the estimated tMRCA also are provided for key nodes with light blue bars. Viruses with genotype 1 similar to pdmH1N1 are indicated with green stars on the PB2 tree; a more detailed presentation of the evolution of genotypes in Mexico in all trees is provided in Figure 3—source data 3.
- https://doi.org/10.7554/eLife.16777.008
-
Figure 3—source data 2
Maximum likelihood trees with tip labels.
Evolutionary relationships of pdmH1N1 and swIAVs sampled in Mexico and globally for the eight segments of the IAV genome. Phylogenies and color scheme are similar to Figure 3, except the trees are inferred using maximum likelihood methods and all horizontal branch lengths are drawn to scale (nucleotide substitutions per site). Trees are midpoint rooted for clarity and bootstrap values >70 are provided for key nodes.
- https://doi.org/10.7554/eLife.16777.009
-
Figure 3—source data 3
Time spent in a genotype using Markov rewards.
Similar phylogenies as presented in Figure 3—source data 1, but in this case Mexican swine viruses are specified by genotype instead of geography. Non-Mexican viruses remain specified by geographical location. Markov rewards representing the time spent in a genotype between state transitions are provided in the upper right. Genotype 1, which is similar to pdmH1N1 (Figure 2), is shaded gold.
- https://doi.org/10.7554/eLife.16777.010
-
Figure 3—source data 4
Detailed phylogenetic analysis of pdmH1N1 in Mexican swine.
Evolutionary relationships of pdmH1N1 viruses collected from humans and swine, 2009–2014 for the representative PB2 and HA segments. Separate viral introductions from humans into Mexican swine are indicated.
- https://doi.org/10.7554/eLife.16777.011
Figure 3—figure supplement 1
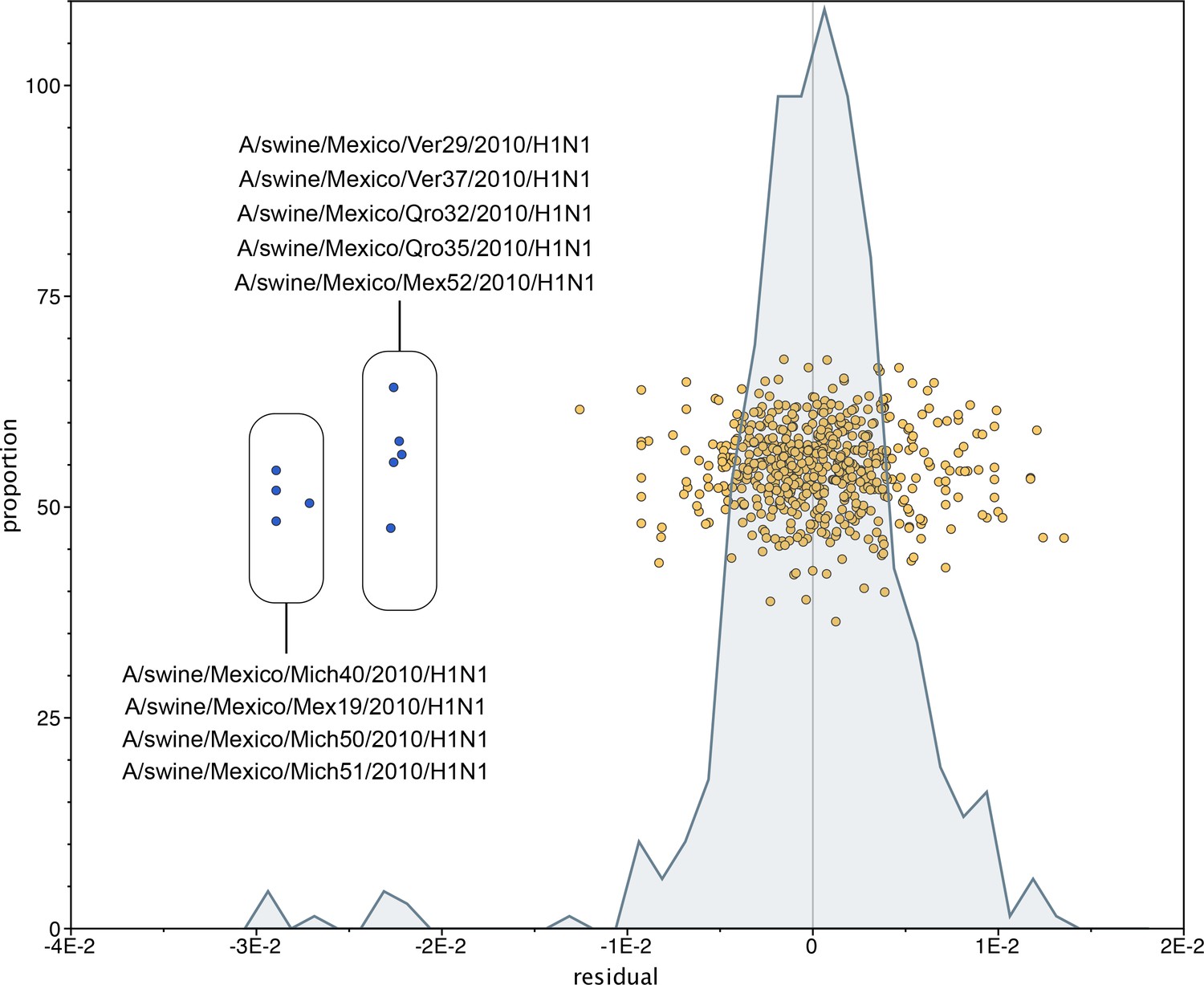
Mexican swIAVs excluded from the phylogenetic analysis.
A residual analysis was used to identify outliers from the linear regression line describing the relationship between the date of collection and genetic distances of sequences in our analysis. Nine swIAVs from Mexico were identified as outliers and removed from our analysis. These viruses were collected in 2010 but are closely related to reference triple reassortant viruses from 1998–1999.
Figure 4 with 2 supplements
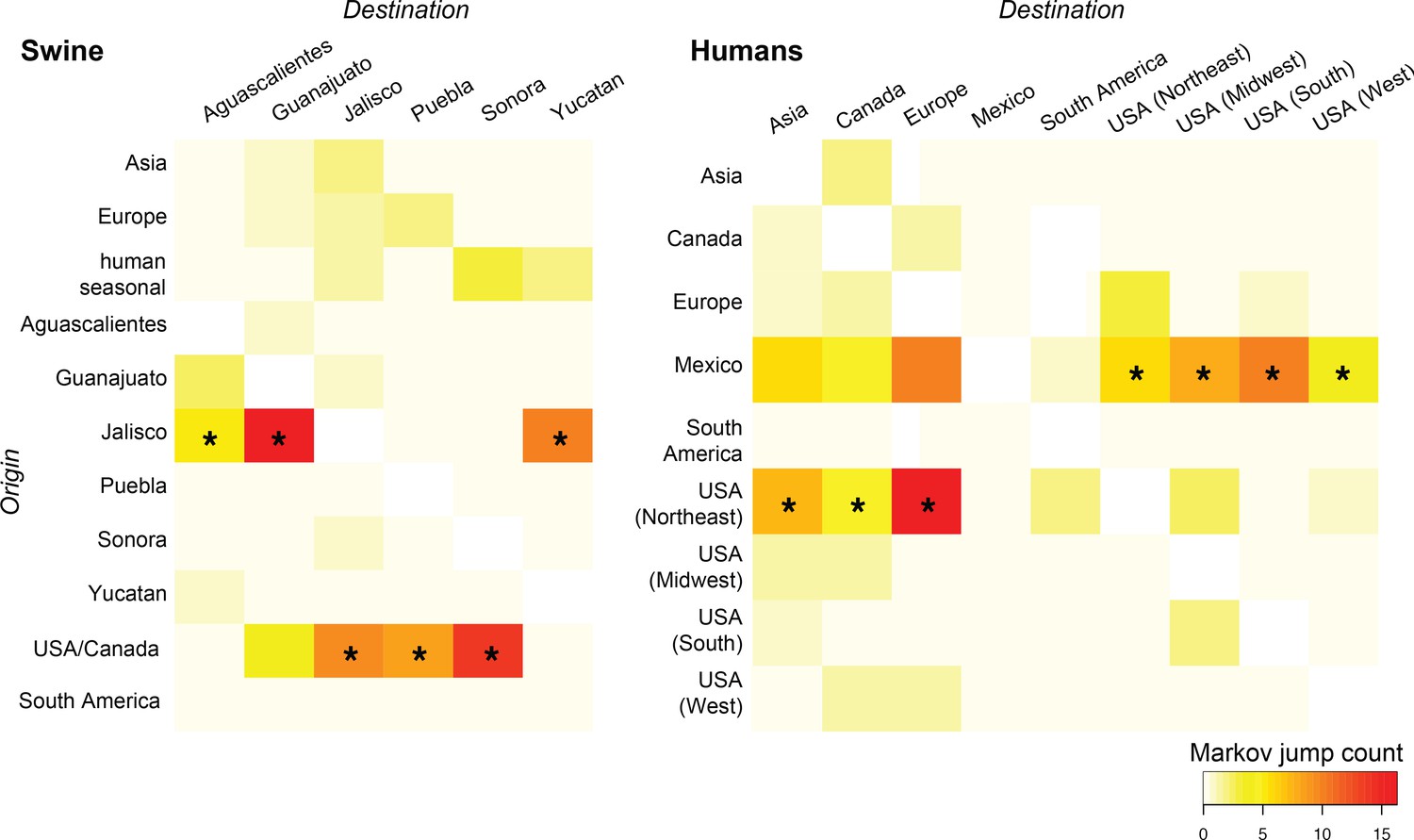
Heat-maps of IAV gene flow between locations.
'Markov jump' counts measure the number of inferred transitions, modeled by a continuous-time Markov chain process, that occur along the branches of the phylogeny, providing a measure of gene flow. The intensity of the color (red = high; white = low) reflects the number of Markov jump counts from a swine population in a location of origin (y-axis) to swine in one of six Mexican states (destination, x-axis; asymmetrical, summarized across all lineages and segments), and between human populations in Mexico, the United States, and globally during the early spatial dissemination of pdmH1N1 in humans during March–May 2009. Asterisks indicate the geographical source (y-axis) of the highest number of Markov jump counts for a particular destination (x-axis). Similar spatial linkages were observed using a Bayes factor (BF) test (Figure 4—figure supplement 1). A phylogenetic tree depicting the evolutionary relationships between human pdmH1N1 viruses is provided in Figure 4—figure supplement 2. Source data for both heat-maps is provided in Figure 4—source data 1.
-
Figure 4—source data 1
Expected number of location state transitions ('Markov jump' counts) along the branches of inferred phylogenies, summarized for (a) all segments and lineages identified in Mexican swine and (b) human pdmH1N1.
- https://doi.org/10.7554/eLife.16777.014
Figure 4—figure supplement 1
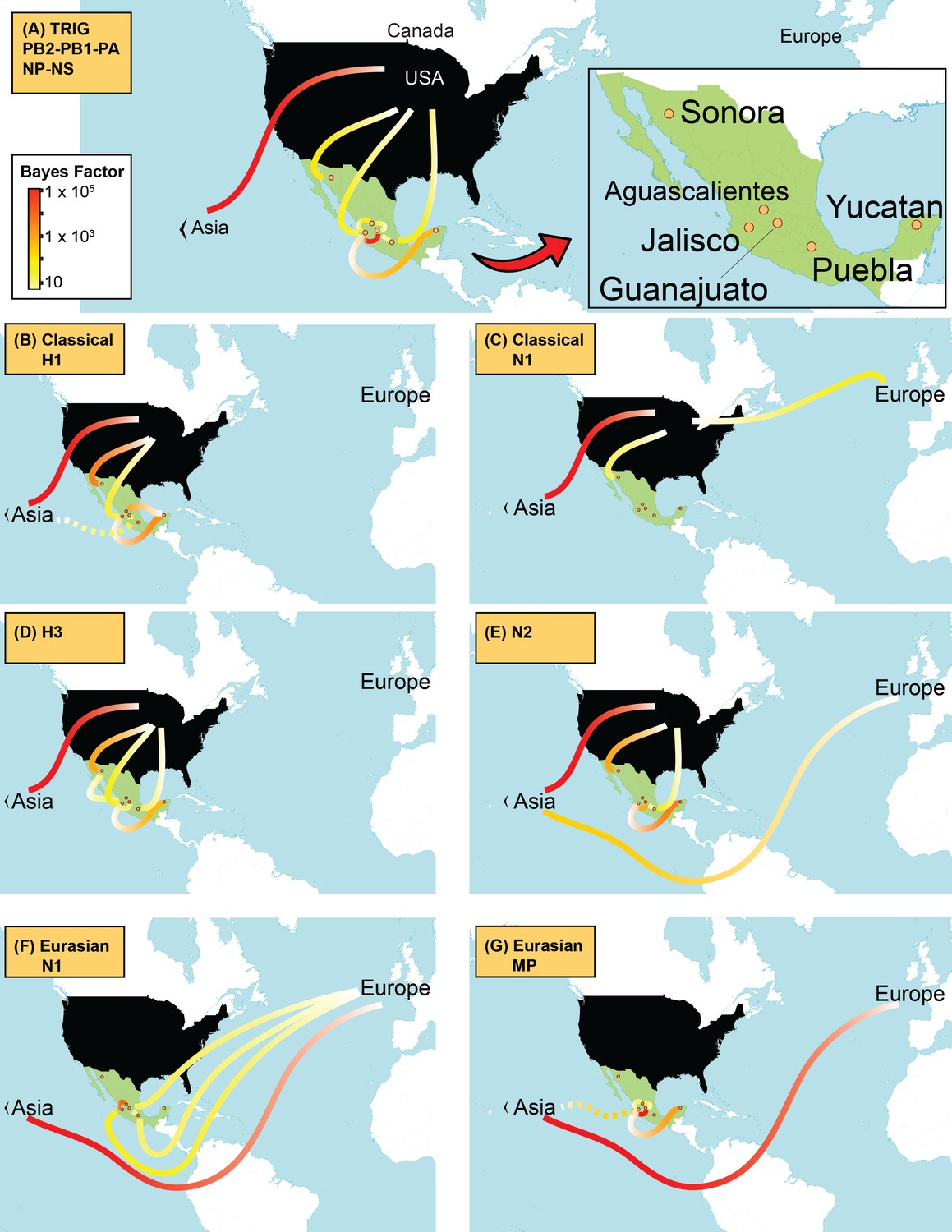
Supported rates of viral migration.
Bayes factor (BF) test for significant non-zero rates of swIAV migration in Mexico. Rates supported by a BF greater than 5 are indicated. The color of the line represents the relative strength of support for a rate; red lines indicate strong support and pale yellow lines indicate weaker support. For the 5 TRIG segments (PB2, PB1, PA, NP, and NS), the average BF is provided. The direction of viral flow is indicated by the line’s gradient, with the white end indicating the source location. The dotted lines indicate that rates between Asia and Mexico likely arise from gaps in sampling in Eurasian swine prior to 1995, and are unlikely to represent actual viral movements from Asia to Mexico.
Figure 4—figure supplement 2
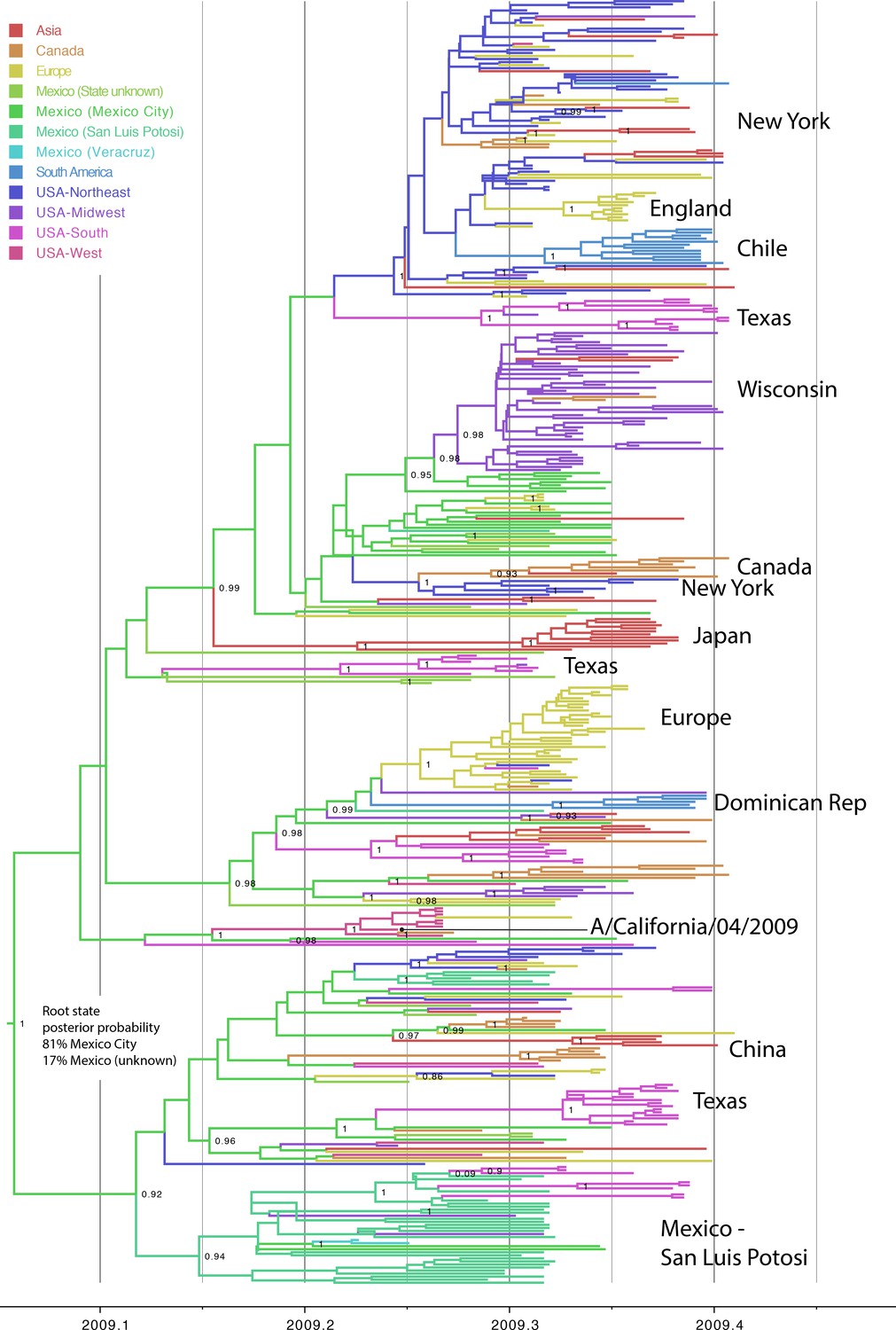
Phylogeography of pdmH1N1 in humans.
Evolutionary relationships between pdmH1N1 viruses collected in humans globally during March 1, 2009–May 31, 2009, inferred for 422 concatenated genome sequences. The color of each branch indicates the most probable location state: red = Asia; orange = Canada; yellow = Europe; light green = Mexico, for which the state of location was not known; green = Mexico City, Mexico; greenish blue = San Luis Potosi, Mexico; light blue = Veracruz, Mexico; blue = South America; bluish purple = Northeastern United States, including New York; purple = Midwestern United States, including Wisconsin; pink = Southern United States, including Texas; and coral = Western United States, including California. Select spatial clusters are labeled. Posterior probabilities >80 are provided for key nodes.
Figure 5 with 1 supplement
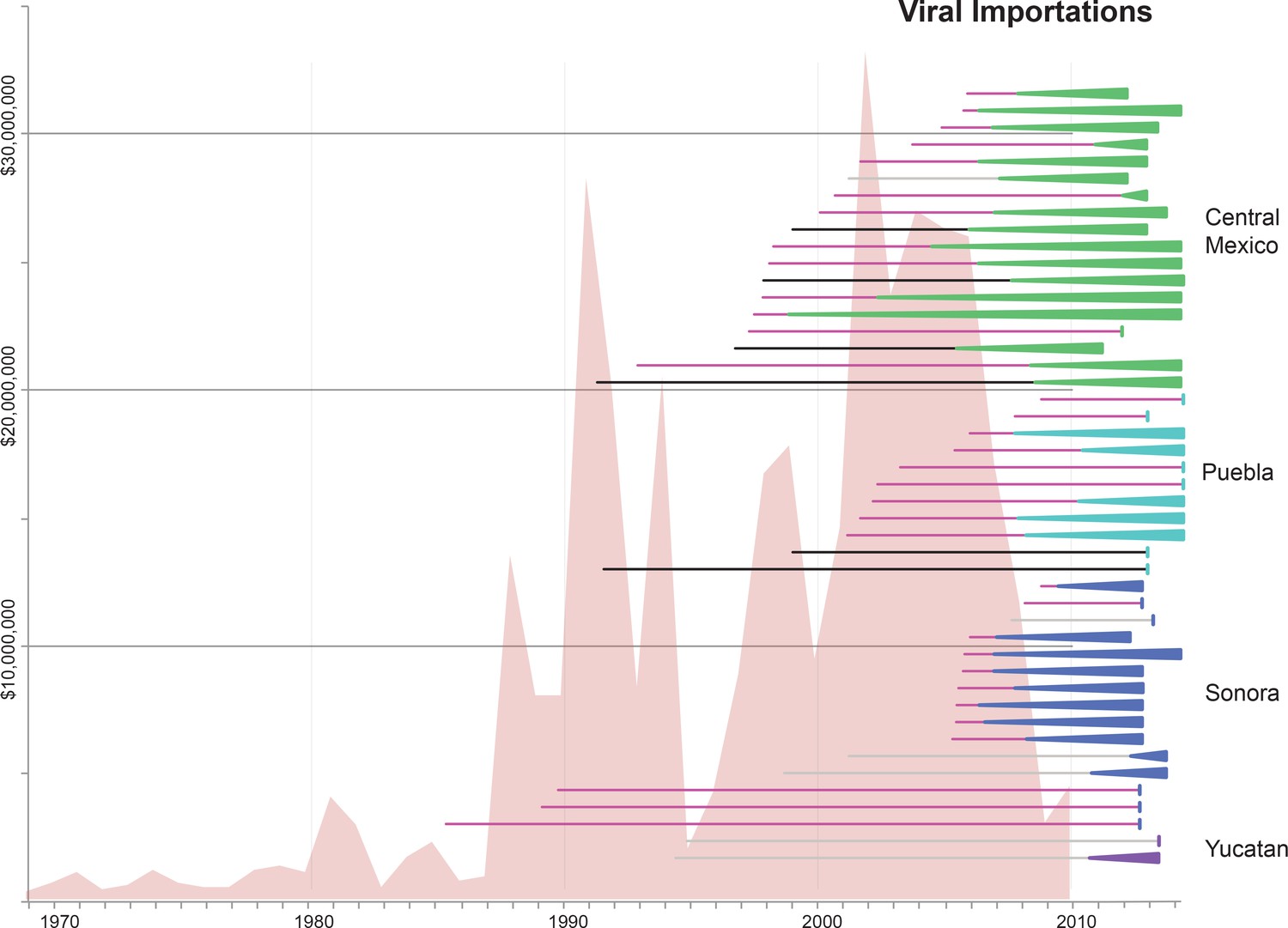
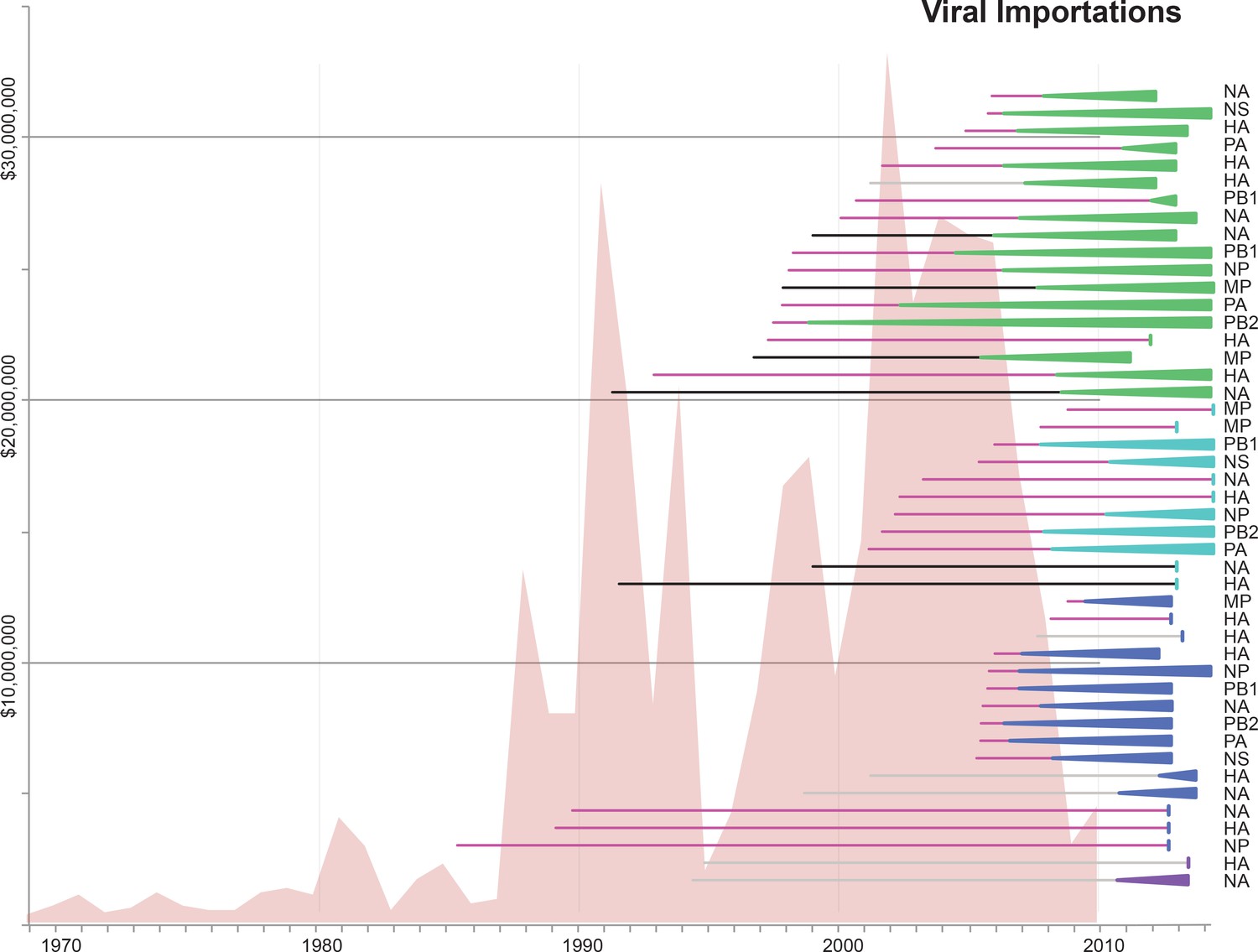
Import of live swine and IAVs into Mexico.
The value of live swine imported into Mexico (USD, y-axis, left) from all countries during 1969–2010 is presented in the background in pink, based on trade data reported to the Food and Agricultural Organization (FAO) of the United Nationals, available in Figure 5—source data 1. Each horizontal line represents an introduction of an IAV segment into Mexican swine, the timing of which is inferred from the MCC trees. The shading of each line indicates the inferred location of origin of an introduction, consistent with Figure 3: dark pink = USA/Canadian swine, black = Eurasian swine, grey = humans. The length of the line indicates uncertainty in the timing of an introduction. Triangles represent clades resulting from onward transmission in Mexico and extend forward as far as the most recently sampled virus. The shading of each triangle indicates the destination location, consistent with Figure 3: dark purple = Yucatan, dark blue = Sonora, light blue = Puebla, and green = Jalisco, Aguascalientes, and Guanajuato (central-west Mexico). Lines without triangles represent singletons. A similar figure annotated with the segment associated with each introduction is provided in Figure 5—figure supplement 1.
-
Figure 5—source data 1
Reported value of live swine imports into Mexico from all countries during 1969–2010.
Data available from the Food and Agriculture Organization (FAO) of the United Nations Datasets repository, http://data.fao.org/datasets.
- https://doi.org/10.7554/eLife.16777.018
Figure 5—figure supplement 1
Similar to Figure 5, but annotated with the segment associated with each introduction.
https://doi.org/10.7554/eLife.16777.019
Figure 6
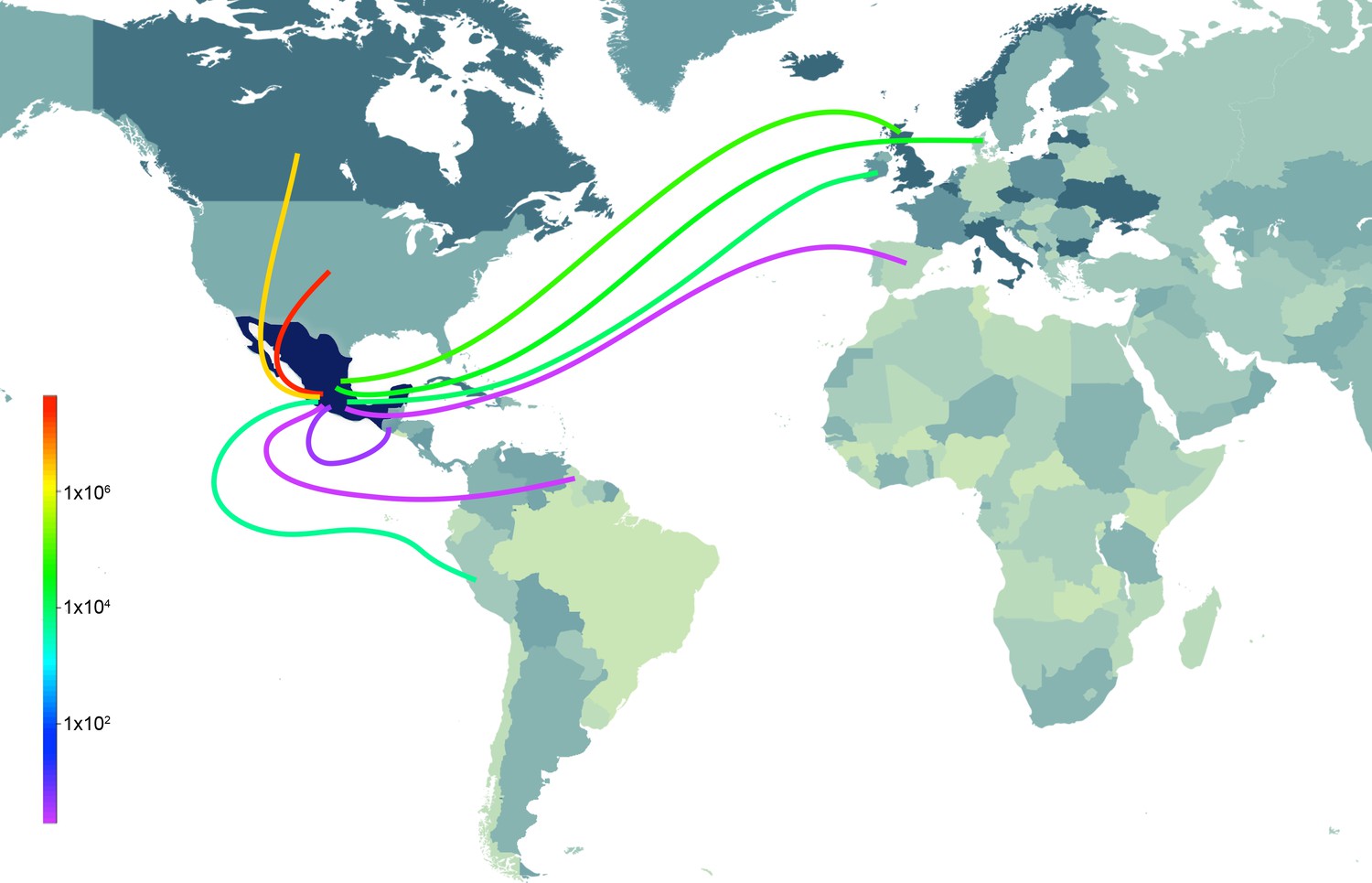
Sources of live swine imports into Mexico.
Total imports of live swine into Mexico (USD) during 1996–2012 from nine reported trade partners: United States, Canada, Ireland, United Kingdom, Denmark, Spain, Guatemala, Guyana, and Peru. The shade of the line indicates the volume of imports (red = high, purple = low). The shading countries is for purposes of clarity only. Trade data is available from the UN Commodity Statistics Database (Figure 6—source data 1).
-
Figure 6—source data 1
Pairwise information on imports of live swine from specific countries is available from 1996–2012.
Data available from the United Nations' Commodity Trade Statistics Database, http://comtrade.un.org.
- https://doi.org/10.7554/eLife.16777.021
Figure 7
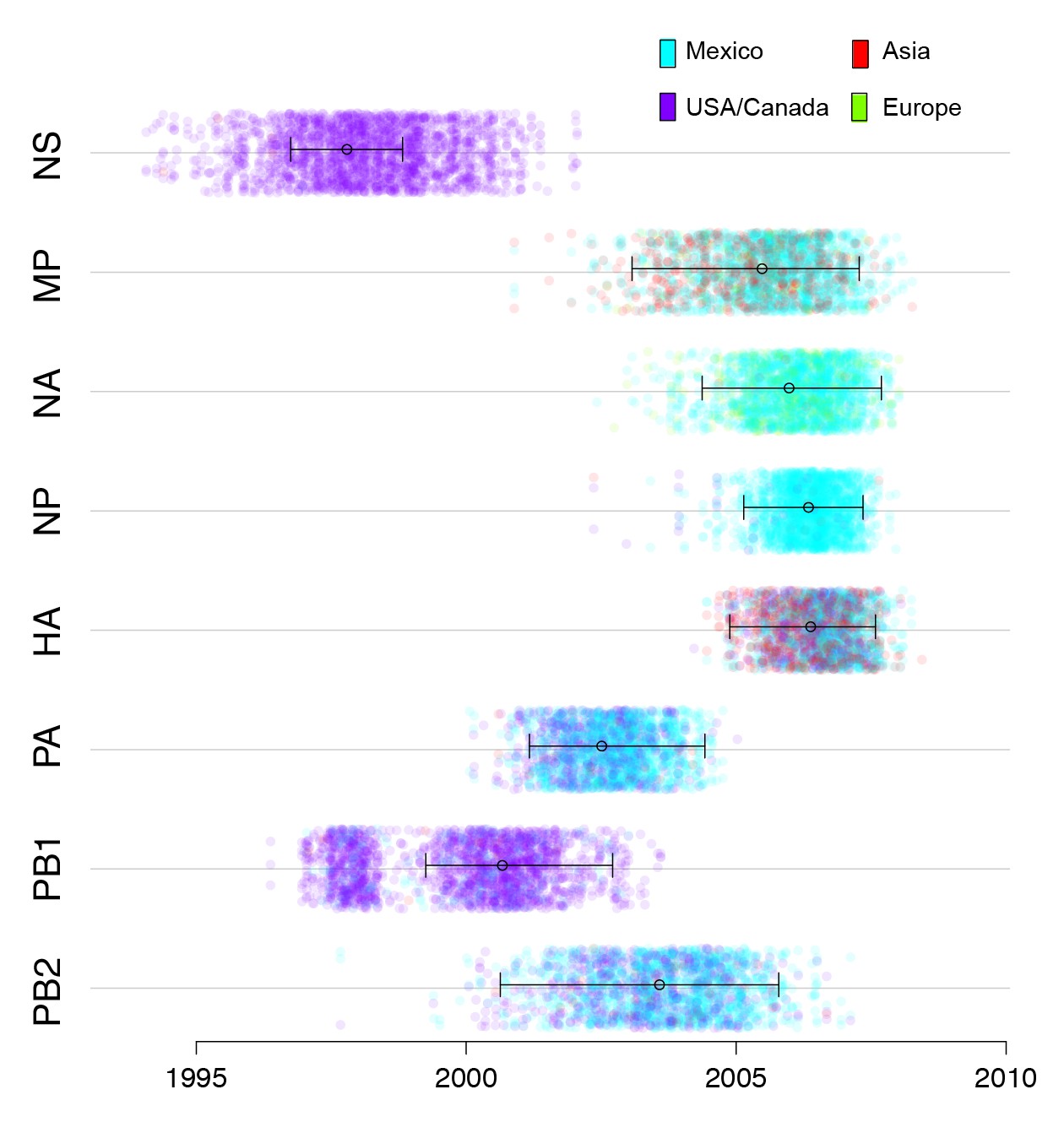
Timing and location of swine ancestors of pdmH1N1.
The color of each dot represents the inferred location of the node representing the common ancestor of pdmH1N1 viruses and the most closely related swine ivurses (indicated by open circles on the MCC trees in Figure 3), for a posterior distribution of ~2000 trees inferred for each segment. A high proportion of blue dots indicates a higher proportion of trees with Mexico as the inferred location state. The x-axis indicates the tMRCA of the same node, again for each tree. The 95% HPD is provided in brackets. Older tMRCAs are associated with longer phylogenetic branch lengths and gaps in sampling.
-
Figure 7—source data 1
Times to the most recent common ancestor (tMRCA) and posterior probabilities (>0.01) for the location state of the node representing the ancestor of the pdmH1N1 clade and the most closely related swine viruses (indicated with open circles in Figure 3).
- https://doi.org/10.7554/eLife.16777.023
Figure 8
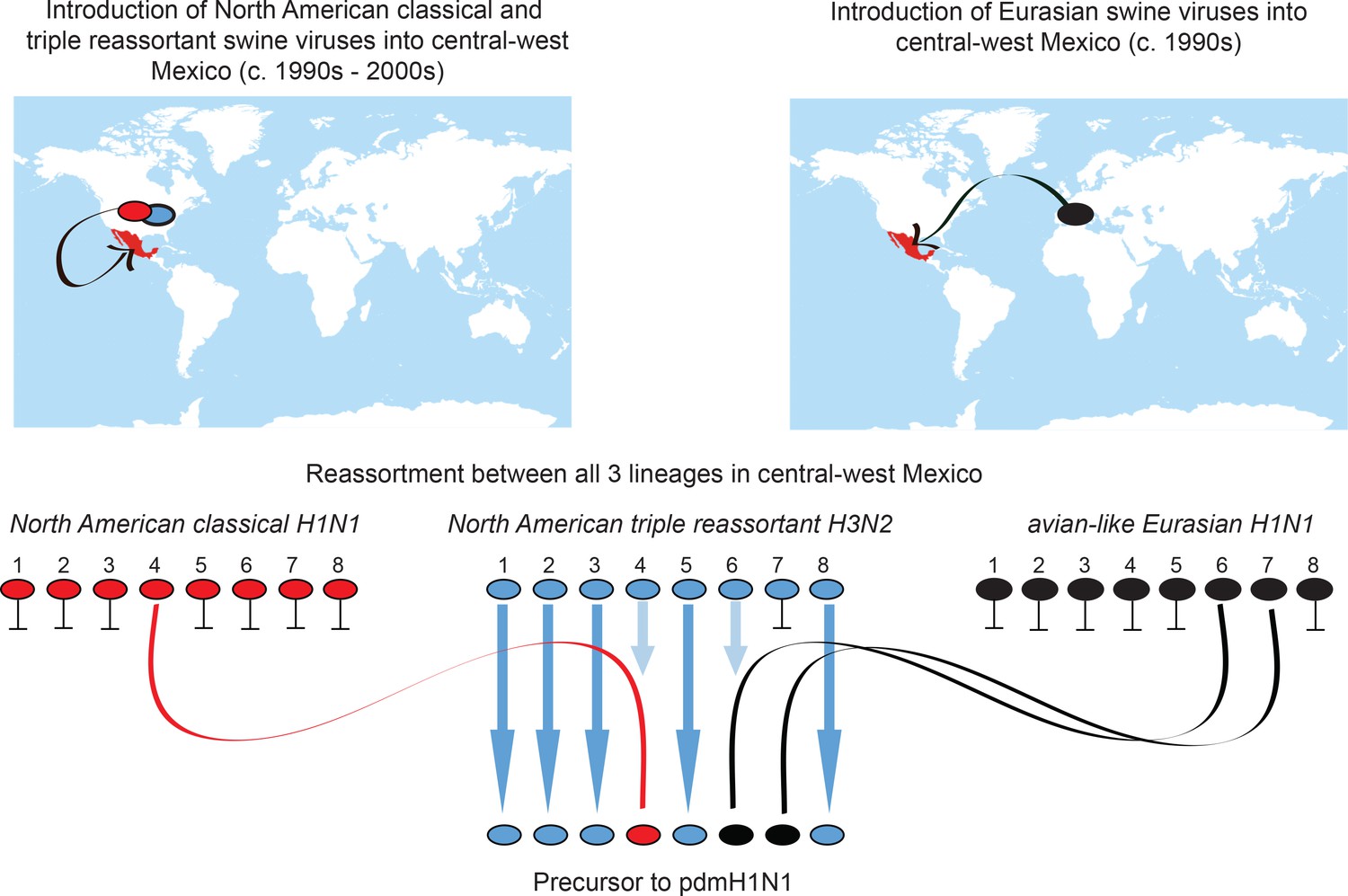
Origins of pdmH1N1.
Summary of the migration and reassortment events leading to the emergence of pdmH1N1 precursor viruses in central-west Mexican swine. Segments from classical and Eurasian viruses for which there is no evidence of onward transmission in central-west Mexican swine are indicated with short horizontal lines.
Additional files
-
Supplementary file 1
Dataset 1.
- https://doi.org/10.7554/eLife.16777.025
-
Supplementary file 2
Dataset 2.
- https://doi.org/10.7554/eLife.16777.026
-
Supplementary file 3
Dataset 3.
- https://doi.org/10.7554/eLife.16777.027
-
Reporting Standard 1
The sample and sequence data were collected according to the following CEIRS DPCC standard: CEIRS DPCC Sequence Submission FASTA Reference v2.0
- https://doi.org/10.7554/eLife.16777.028
-
Reporting Standard 2
The sample and sequence data were collected according to the following CEIRS DPCC standard: CEIRS DPCC Sequence Submission Metadata Reference v2.0
- https://doi.org/10.7554/eLife.16777.029
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Origins of the 2009 H1N1 influenza pandemic in swine in Mexico
eLife 5:e16777.
https://doi.org/10.7554/eLife.16777




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}