Structures of TorsinA and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia
- Massachusetts Institute of Technology, United States
- Whitehead Institute for Biomedical Research, United States
Figures
Figure 1 with 5 supplements
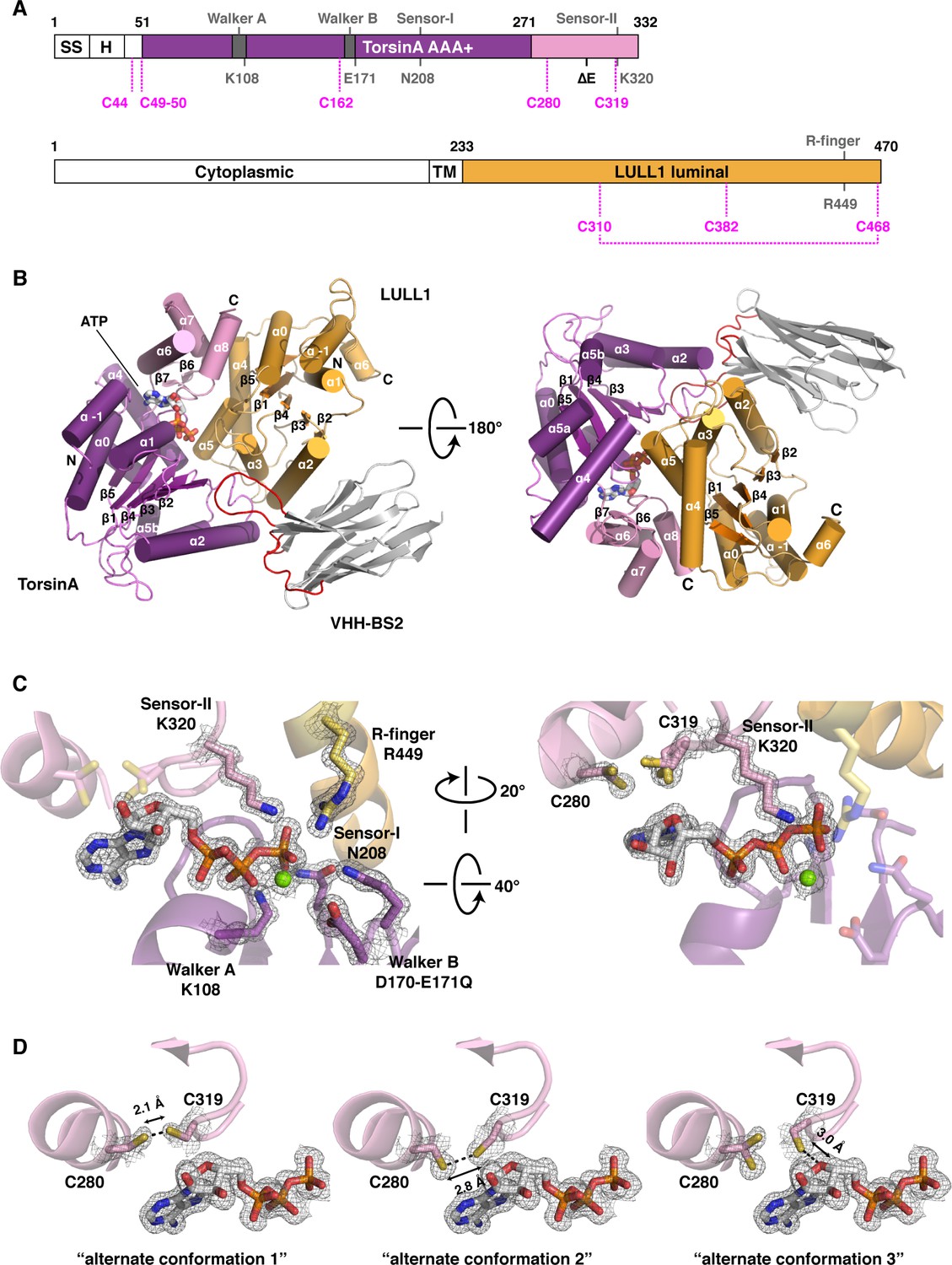
Architecture of the TorsinA-LULL1 complex.
(A) Schematic diagrams of TorsinA and LULL1. Important residues and sequence motifs are indicated. The colored areas mark the crystallized segments. Large and small domains of TorsinA are colored in purple and pink, respectively. SS, signal sequence; H, hydrophobic region; TM, transmembrane helix. (B) Cartoon representation of the TorsinA-LULL1 complex in two orientations. Color-coding as in (A). A nanobody (VHH-BS2, grey; complementarity determining regions, red) was used as a crystallization chaperone. Numbers refer to secondary structure elements. (C) Close-up of the ATP binding site. Key residues are labeled. 2Fo−Fc electron density contoured at 2σ displayed as grey mesh. (D) Close-up of the proximal cysteines 280 and 319 next to the adenine base of the bound ATP. 2Fo−Fc electron density is contoured at 1σ. The cysteine pair adopts three alternate conformations, but remains reduced in all of them.
Figure 1—figure supplement 1
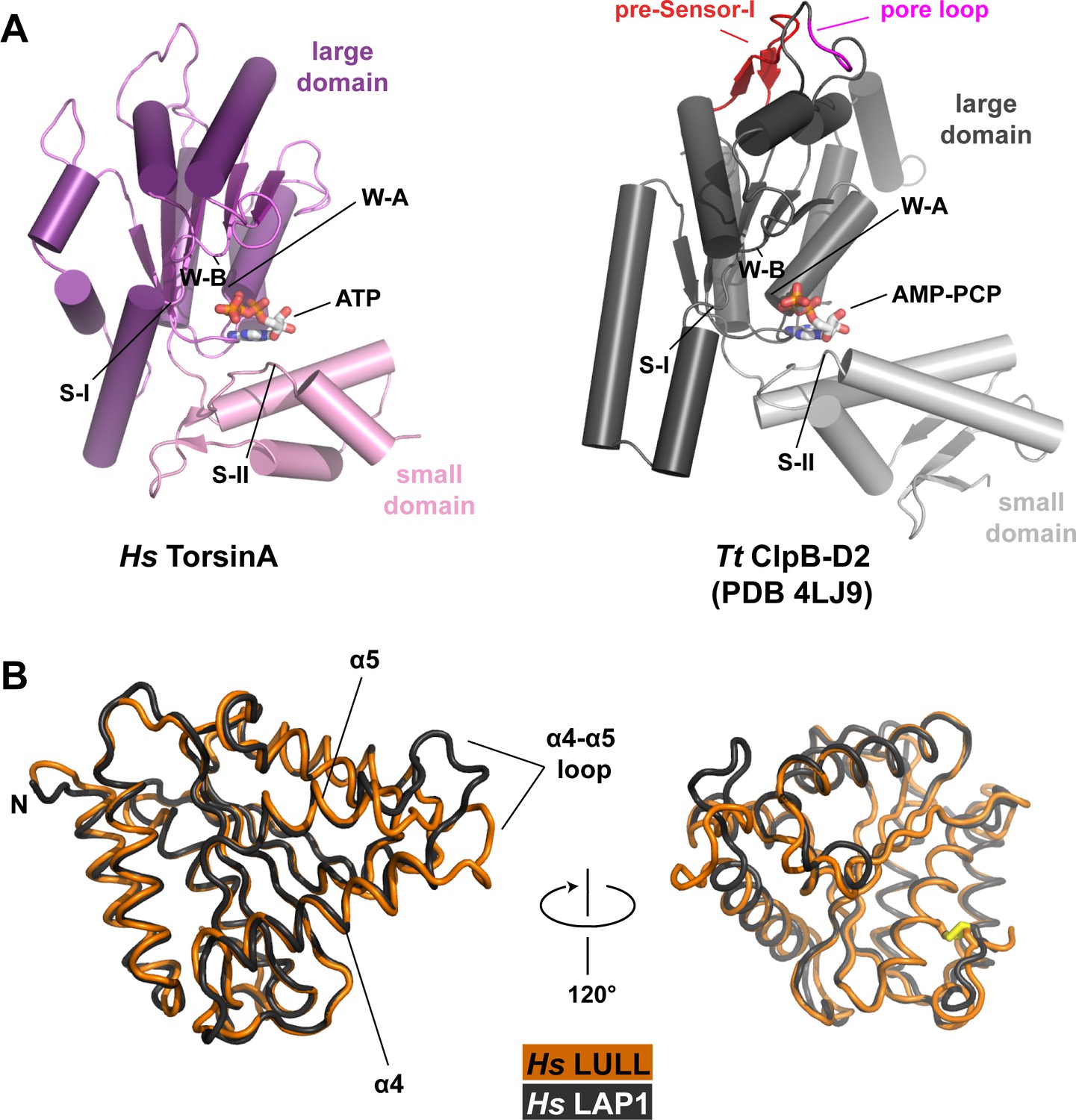
Structural comparisons.
(A) Human TorsinA-ATP (left) displayed as a cartoon, compared to the D2 domain of the double-ringed AAA+ ATPase ClpB-AMPPCP (right) from Thermus thermophilus (Zeymer et al., 2014) (PDB code 4LJ9) in the same orientation. Important structure motifs are labeled. (B) Human LULL1 (orange) superposed on human LAP1 (grey, PDB code 4TVS), shown in two orientations. The one region of major structural difference is labeled (left). The disulfide bridge within LAP1/LULL1 is in yellow (right).
Figure 1—figure supplement 2
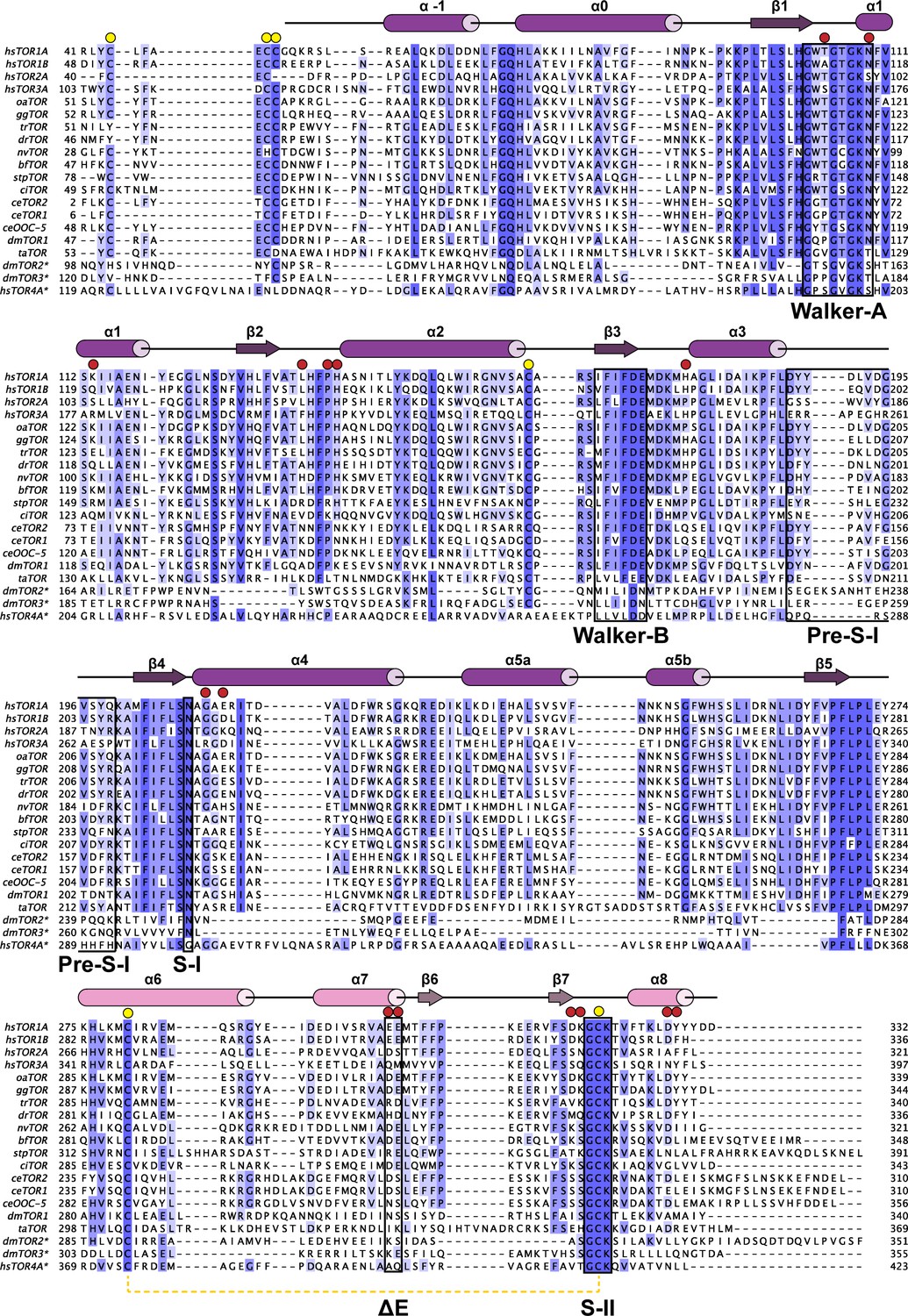
Phylogenetic analysis of Torsins.
Maximally diverged torsins are aligned. Secondary structure elements of human TorsinA are displayed above the alignment. Important sequence motifs are boxed. LULL1 contacts, red circles, conserved cysteines, yellow circles. Proximal cysteines 280 and 319 connected with a dashed yellow line. Asterisk denotes putative torsin homologs based on sequence analysis. hs, Homo sapiens; oa, Ornithorhynchus anatinus; gg, Gallus gallus; tr, Takifugu rubripes; dr, Danio rerio; nv, Nematostella vectensis; bf, Branchiostoma floridae; stp, Strongylocentrotus purpuratus; ci, Ciona intestinalis; ce, Caenorhabditis elegans; dm, Drosophila melanogaster; ta, Trichoplax adherens.
Figure 1—figure supplement 3

Phylogenetic analysis of LAP1/LULL1.
Maximally diverged LAP1 and LULL1 sequences are aligned. If not experimentally confirmed, sequences were assigned as LAP1 or LULL1 based on the presence of an N-terminal, extraluminal domain with basic signature, characteristic of LAP1. Secondary structure elements of human LULL1 are displayed above the alignment. The strictly conserved Arg-finger is boxed. TorsinA contacts, red circles, conserved cysteines, yellow circles. Disulfide bridge depicted as a yellow line. hs, Homo sapiens; oa, Ornithorhynchus anatinus; gg, Gallus gallus; tr, Takifugu rubripes; dr, Danio rerio; nv, Nematostella vectensis; bf, Branchiostoma floridae; stp, Strongylocentrotus purpuratus; ci, Ciona intestinalis; ce, Caenorhabditis elegans; dm, Drosophila melanogaster; ta, Trichoplax adherens.
Figure 1—figure supplement 4
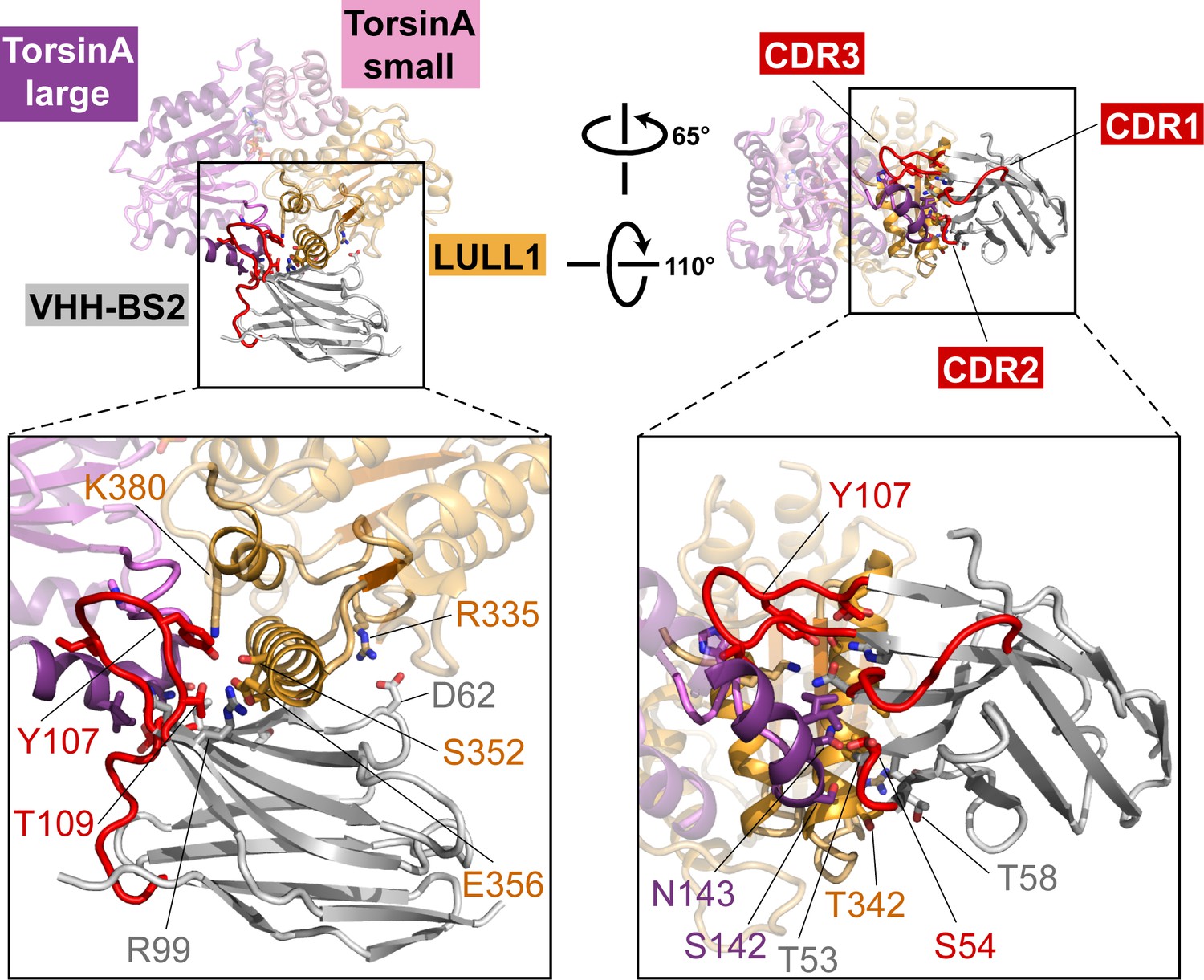
Nanobody interaction.
The heterotrimeric TorsinA(ATP)-LULL1-VHH-BS2 complex is shown in two orientations. Nanobody and interacting secondary structure elements of TorsinA and LULL1 are shown in full color, non-interacting elements in faded colors. Complementarity determining region (CDR) loops in red. Insets show close-ups with important interacting residues labeled.
Figure 1—figure supplement 5
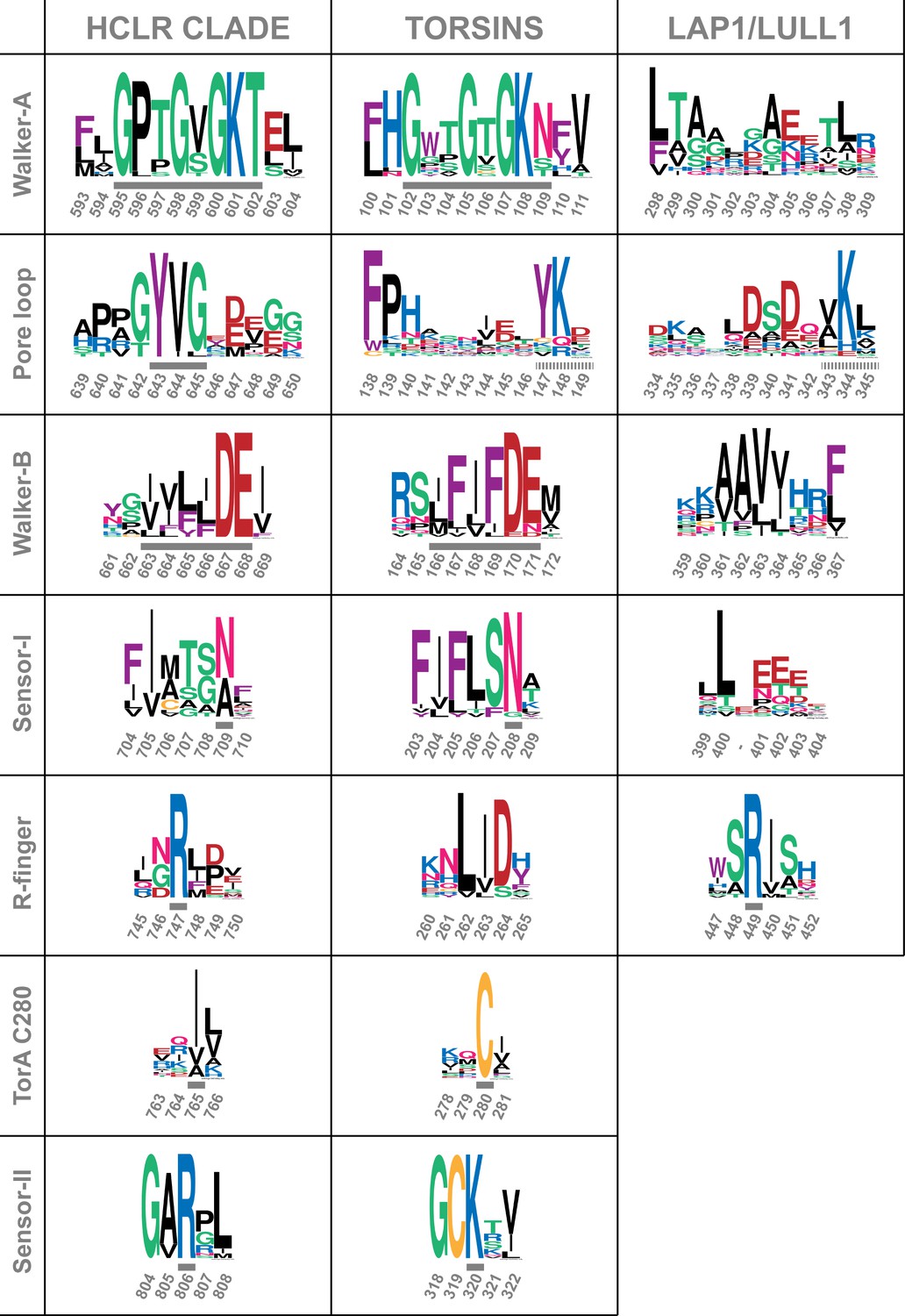
Comparison of sequence motifs of AAA+ ATPases.
Torsins and LAP1/LULL1 sequences are compared to the HCLR clade, the most similar branch within the AAA+ ATPase family (Erzberger and Berger, 2006; Iyer et al., 2004). Sequential elements characteristic for each of the 3 groups are displayed as WebLogos (Crooks et al., 2004). Numbering refers to ClpB-D2 from Thermus thermophilus for the HCLR class, human TorsinA for Torsins, and human LULL1 for LAP1/LULL1. Grey bars indicate the characteristic motif or residue, surrounded by a few adjacent residues to emphasize the distinct conservation. All three groups have elements that can be used to distinguish them among each other. Since Torsins and LAP1/LULL1 lack a pore loop consensus sequence φφG (where φ denotes a bulky hydrophobic residue), putative pore loop areas have been determined structurally. Dashed grey bars indicate residues which can be structurally aligned to the pore loop motif of the closest HCLR AAA+ clade members.
Figure 2 with 1 supplement
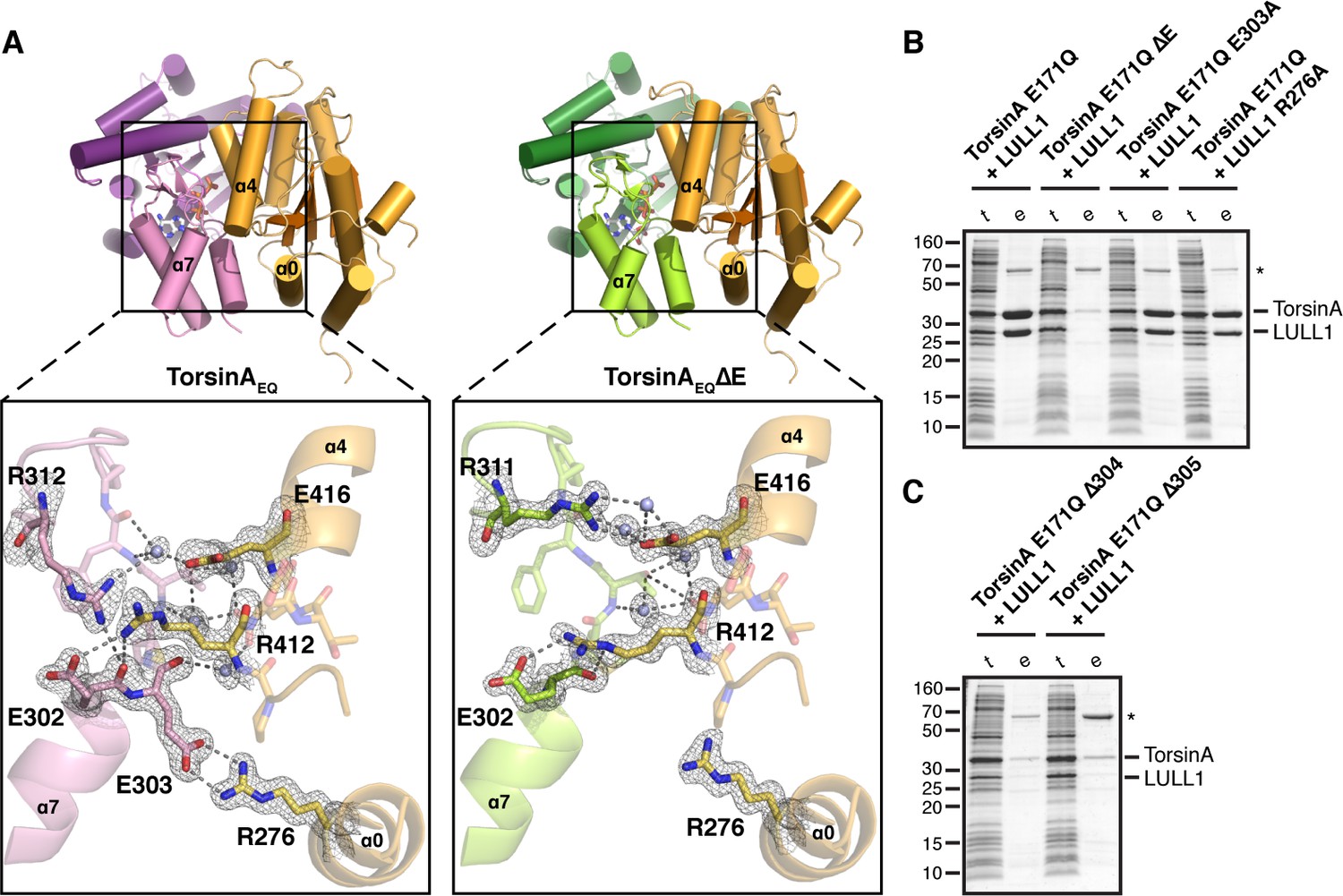
Analysis of the TorsinA-LULL1 interface.
(A) Side-by-side comparison of TorsinA-ATP-LULL1 (left) and TorsinAΔE-ATP-LULL1 (right). Zoomed insets show the atomic details of the interactions between TorsinA/TorsinAΔE and LULL1, with a focus on the ΔE303 area. (B and C) Mutational analysis of the TorsinA-LULL1 interface. Substitution or deletion of residues involved in TorsinA-LULL1 binding were probed using a Ni-affinity co-purification assay with recombinant, bacterial-expressed protein. Only TorsinA is His-tagged. SDS-PAGE analysis is shown. Lack of binding is observed by the absence of complex (uncomplexed His-tagged TorsinA is insoluble). t, total lysate, e, Ni eluate. Asterisk denotes an unrelated contaminant.
Figure 2—figure supplement 1
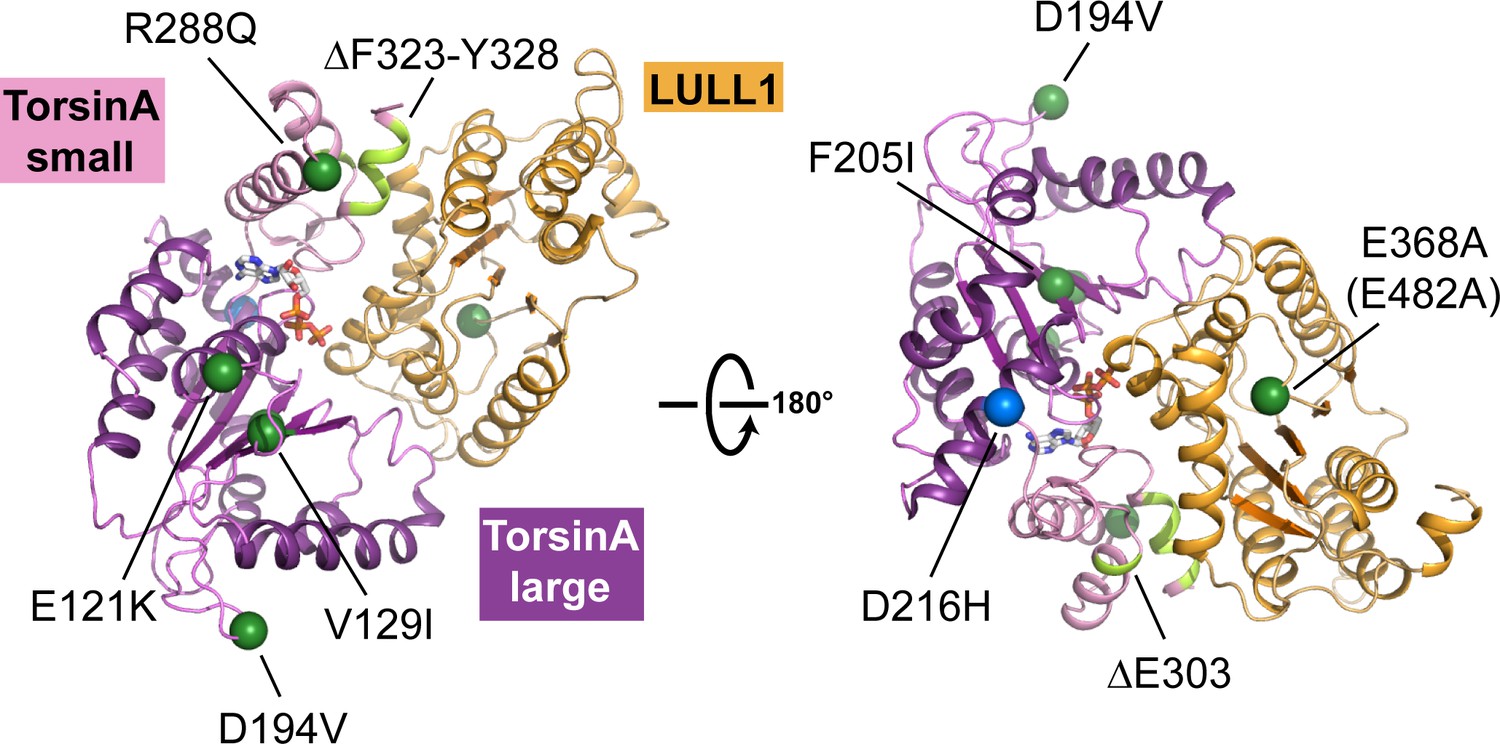
Structural mapping of mutations causing dystonia.
All known point mutations and deletions that lead to dystonia are marked as green dots and shown in light green color, respectively, on the TorsinA-ATP-LULL1 structure. A modifier TorsinA mutation, D216H, is marked as a blue dot. The structural equivalent of the LAP1 missense mutation (E482A) is LULL1 E368A, marked as a green dot. See Table 2 for an explanation of the likely structural consequence.
Figure 3
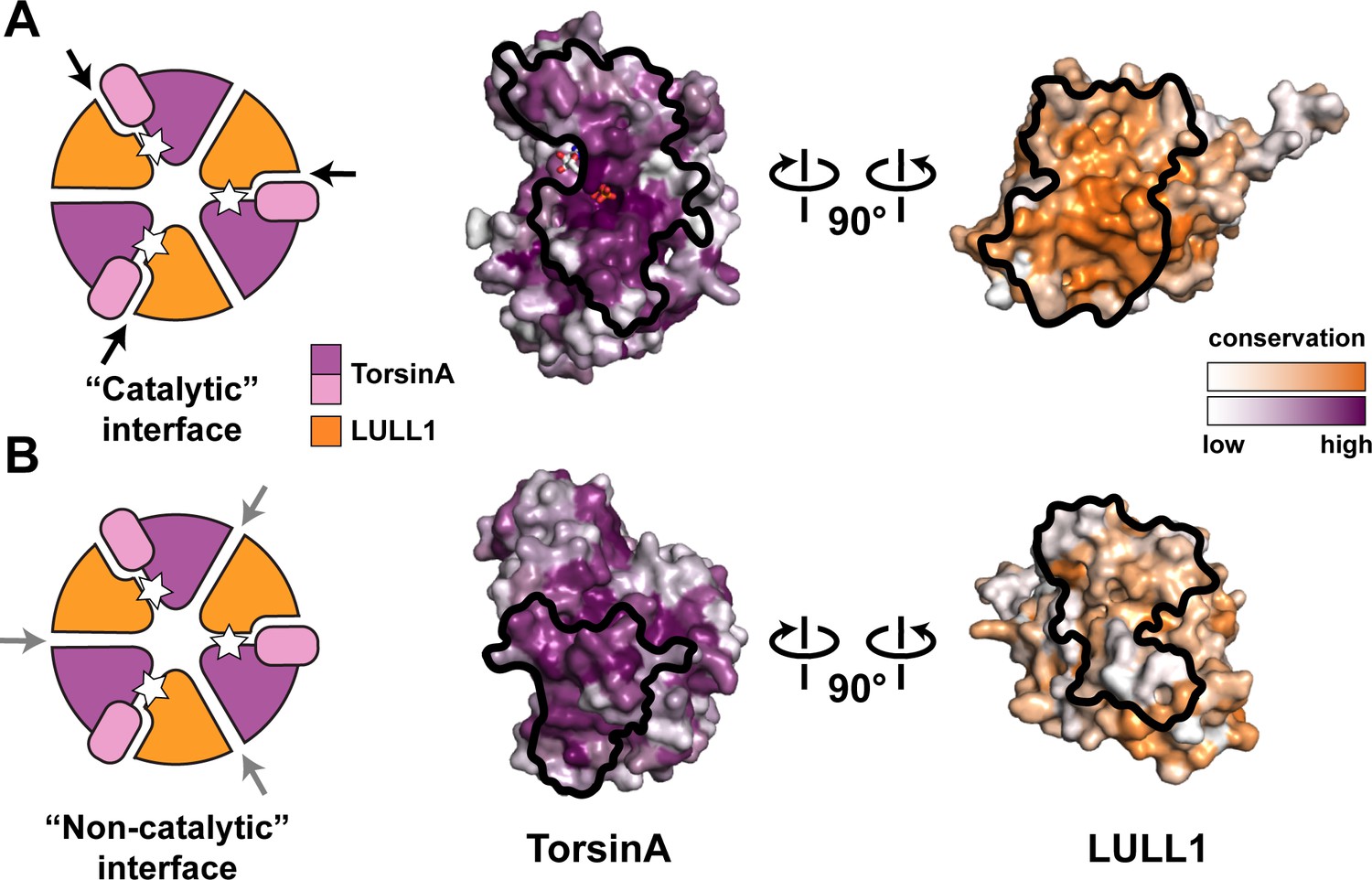
Oligomerization of TorsinA-LULL1.
(A) Left, Schematic representation of a hypothetical heterohexameric (TorsinA-LULL1)3 ring model, in analogy to canonical AAA+ ATPases. White star represents ATP. Since LULL1 cannot bind a nucleotide, there would be three catalytic (nucleotide-bound) and three non-catalytic interfaces per ring. Open-book representation of the catalytic interface between TorsinA and LULL1, as seen in this study. Black line marks the outline of the interface. Color gradient marks conservation across diverse eukaryotes. (B) The same analysis as in (A), but for the hypothetical ‘non-catalytic’ interface. The interface model on the right is based on swapping the TorsinA and LULL1 positions in the TorsinA-LULL1 complex.
Tables
Table 1
X-ray data collection and refinement statistics.
| TorsinA-LULL1233-470 | TorsinAΔE-LULL1233-470 | |
|---|---|---|
| PDB Code | 5J1S | 5J1T |
| Data collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 75.7, 90.7, 105.1 | 75.4, 88.4, 105.3 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution (Å) | 61–1.40 (1.45–1.40)* | 68–1.40 (1.45–1.40) |
| Rsym | 0.06 (0.88) | 0.10 (1.98) |
| Rpim | 0.03 (0.43) | 0.03 (0.60) |
| I / σ | 33.0 (1.5) | 30.8 (1.3) |
| Completeness (%) | 94.7 (67.5) | 97.9 (96.5) |
| Redundancy | 5.7 (4.4) | 12.4 (11.3) |
| CC(1/2) | 1.00 (0.65) | 1.00 (0.58) |
| Refinement | ||
| Resolution (Å) | 61.4–1.40 | 67.7–1.40 |
| No. reflections | 132956 | 134333 |
| Rwork / Rfree | 0.143/0.188 | 0.148/0.177 |
| No. atoms | 5898 | 5927 |
| Protein | 5241 | 5244 |
| Ligand/ion | 35 | 47 |
| Water | 622 | 636 |
| B factors (Å2) | ||
| Protein | 31.3 | 24.0 |
| Ligand/ion | 23.2 | 17.2 |
| Water | 43.1 | 33.6 |
| r.m.s. deviations | ||
| Bond lengths (Å) | 0.014 | 0.017 |
| Bond angles (°) | 1.25 | 1.71 |
| Ramachandran | ||
| Favored/allowed/outliers (%) | 98.0/1.7/0.0 | 98.6/1.4/0.0 |
-
*Values in parentheses are for highest-resolution shell. One crystal was used for each dataset.
Table 2
Dystonia mutations.
| Protein | Mutation | Structural consequence | Reference |
|---|---|---|---|
| TorsinA | ∆E302/303 | Weakened LAP1/LULL1 binding | (Ozelius et al., 1997) |
| TorsinA | ∆F323-Y328 | Weakened LAP1/LULL1 binding | (Leung et al., 2001) |
| TorsinA | R288Q | Weakened LAP1/LULL1 binding | (Zirn et al., 2008) |
| TorsinA | F205I | Folding problem | (Calakos et al., 2010) |
| TorsinA | D194V | Change to the conserved, noncatalytic interface | (Cheng et al., 2014) |
| TorsinA | ∆A14-P15 | Improper cellular targeting | (Vulinovic et al., 2014) |
| TorsinA | E121K | Charge inversion at the membrane proximal interface | (Vulinovic et al., 2014) |
| TorsinA | V129I | Folding problem | (Dobričić et al., 2015) |
| TorsinA | D216H (modifier) | Surface change; consequence unclear | (Kamm et al., 2008; Kock et al., 2006) |
| LAP1 | c.186deiG (p.E62fsTer25) | Lack of the luminal activation domain of LAP1 | (Kayman-Kurekci et al., 2014) |
| LAP1 | E482A* | Improper folding | (Dorboz et al., 2014) |
-
*Assesment based on the equivalent residue in LULL1 (E368).
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structures of TorsinA and its disease-mutant complexed with an activator reveal the molecular basis for primary dystonia
eLife 5:e17983.
https://doi.org/10.7554/eLife.17983




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}