Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles
- Yale University School of Medicine, United States
- Howard Hughes Medical Institute, Yale University School of Medicine, United States
- Yale Center for Genome Analysis, United States
- Craniosynostosis and Positional Plagiocephaly Support, United States
- The Rockefeller University, United States
Figures
Figure 1
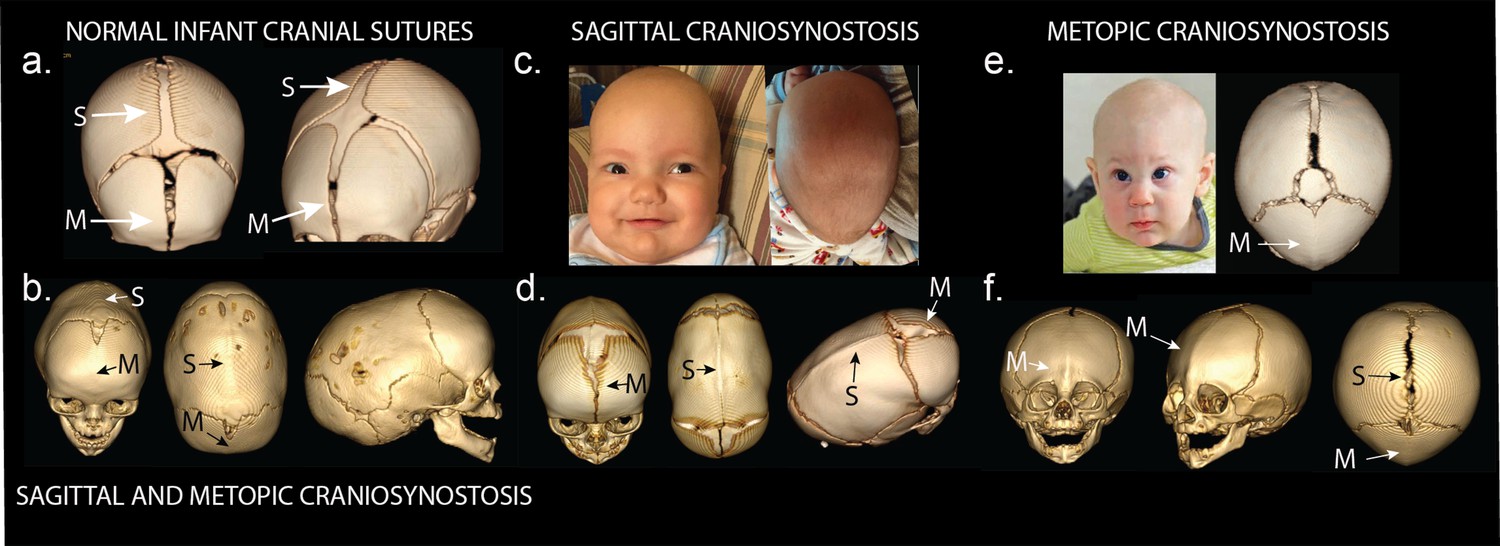
Phenotypes of midline craniosynostosis.
(a) Normal infant skull with patent sagittal (S) and metopic (M) sutures. (b) Three-dimensional reconstruction of computed tomography (3D CT) demonstrating premature fusion of both the sagittal and metopic sutures. (c) A three-month-old boy with sagittal craniosynostosis featuring scaphocephaly (narrow and elongated cranial vault), and frontal bossing. (d) 3D CT reconstruction of a one-month-old boy found to have sagittal craniosynostosis. (e) A six-month-old boy presenting with trigonocephaly (triangulation of the cranial vault, with prominent forehead ridge resulting from premature fusion of the metopic suture) and hypotelorism (abnormally decreased intercanthal distance, also a result of premature fusion of the metopic suture). 3D CT reconstruction demonstrated metopic craniosynostosis. (f) 3D CT reconstruction demonstrating premature fusion of the metopic suture with characteristic trigonocephaly and hypotelorism.
Figure 2 with 2 supplements
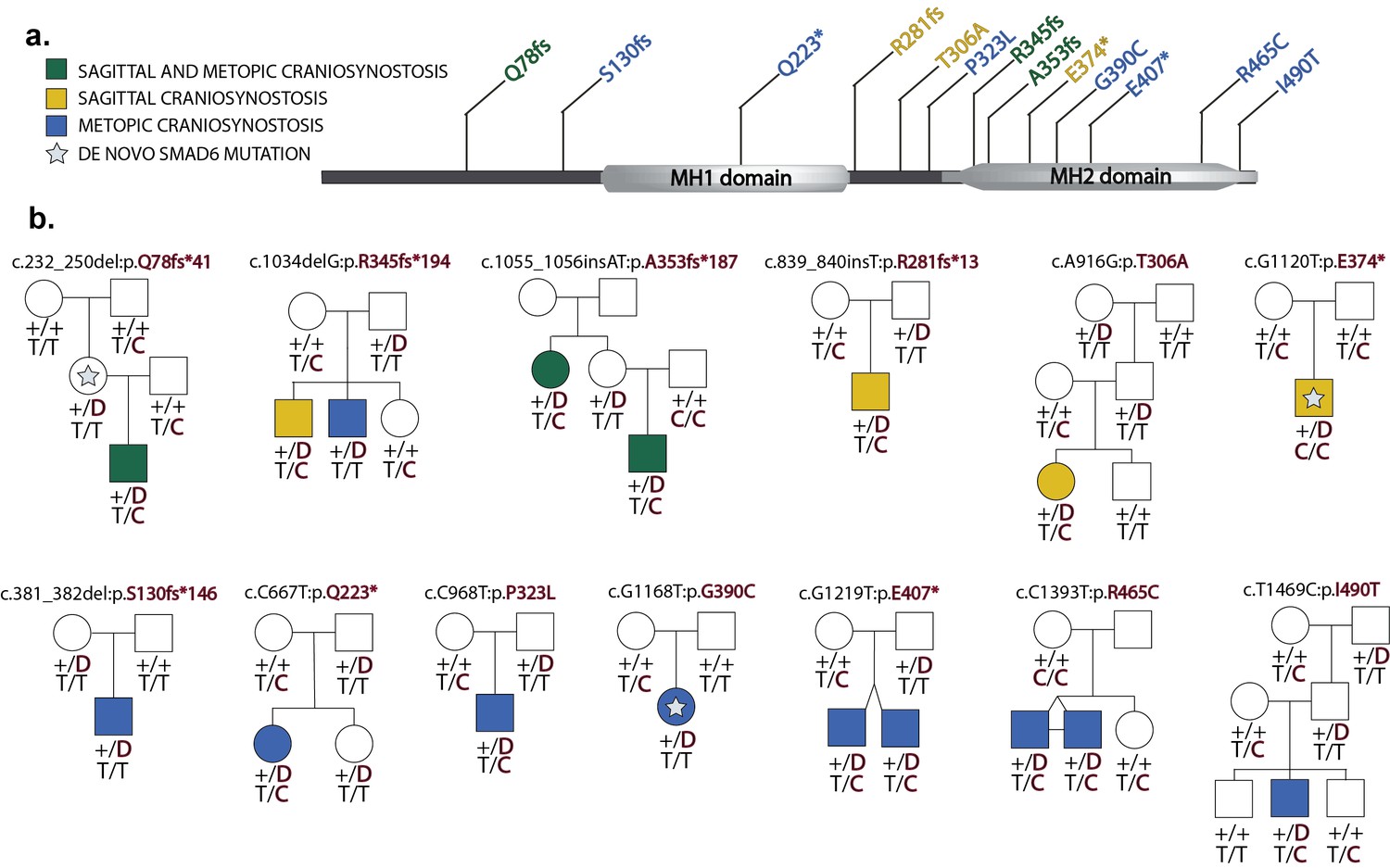
Segregation of SMAD6 mutations and BMP2 SNP genotypes in pedigrees with midline craniosynostosis.
(a) Domain structure of SMAD6 showing location of the MH1 and MH2 domains. The MH1 domain mediates DNA binding and negatively regulates the functions of the MH2 domain, while the MH2 domain is responsible for transactivation and mediates phosphorylation-triggered heteromeric assembly with receptor SMADs. De novo or rare damaging mutations identified in craniosynostosis probands are indicated. Color of text denotes suture(s) showing premature closure. (b) Pedigrees harboring de novo (denoted by stars within pedigree symbols) or rare transmitted variants in SMAD6. Filled and unfilled symbols denote individuals with and without craniosynostosis, respectively. The SMAD6 mutation identified in each kindred is noted above each pedigree. Below each symbol, genotypes are shown first for SMAD6 (with 'D' denoting the damaging allele) and for rs1884302 risk locus downstream of BMP2, (with 'T' conferring protection from and 'C' conferring increased risk of craniosynostosis). All 17 subjects with craniosynostosis have SMAD6 mutations, and 14/17 have also inherited the risk allele at rs1884302, whereas only 3 of 16 SMAD6 mutation carriers without the rs1884302 risk allele have craniosynostosis.
-
Figure 2—source data 1
Variants identified in SMAD6.
Highlighted variants indicate de novo mutations; 'D' and 'T' respectively denote damaging and tolerated missense variants called by MetaSVM.
- https://doi.org/10.7554/eLife.20125.007
-
Figure 2—source data 2
PCR primer sequences for Sanger sequencing of reported variants.
Source Data for Figure 2—figure supplement 2.
- https://doi.org/10.7554/eLife.20125.008
Figure 2—figure supplement 1
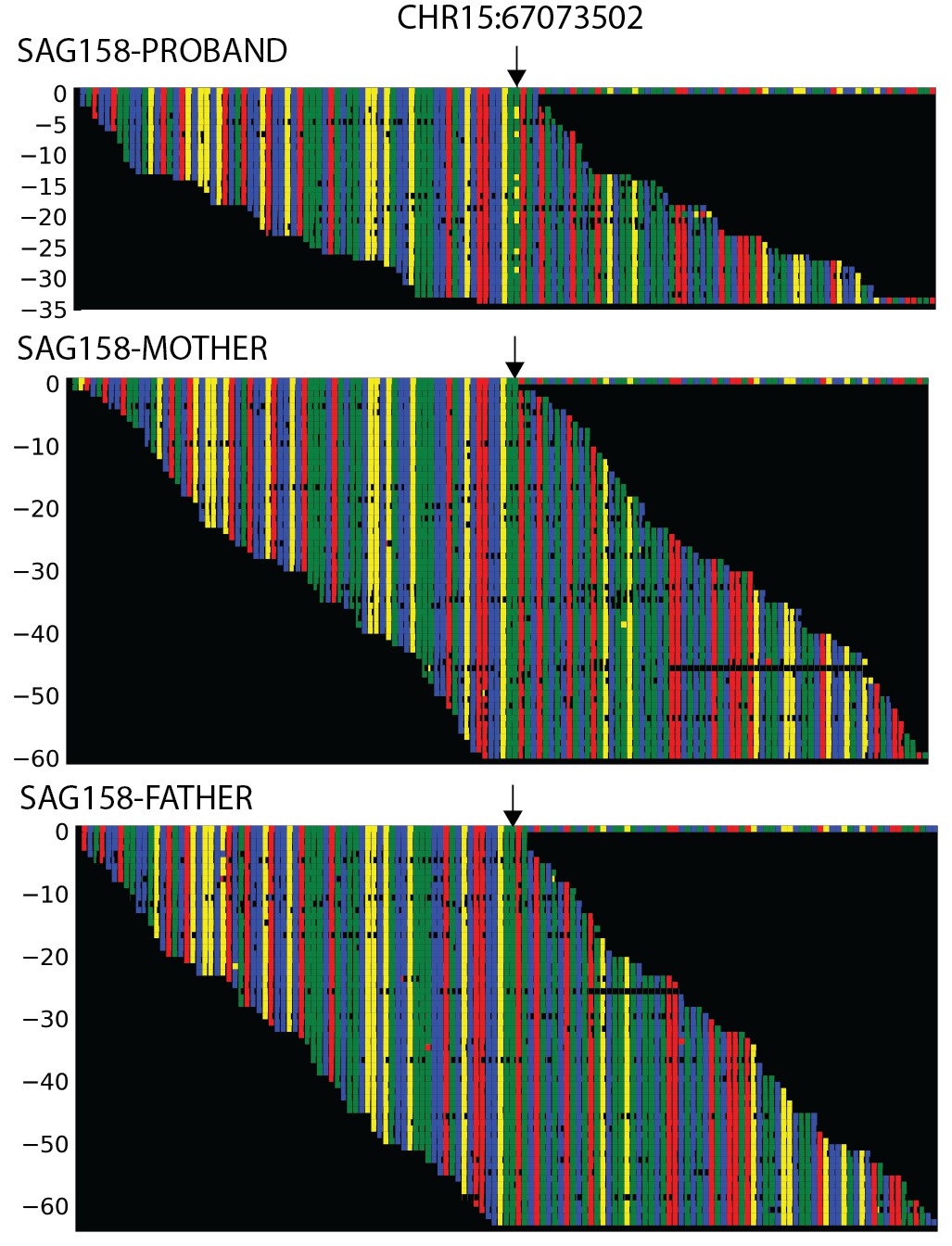
Plots of independent Illumina sequencing reads in a parent-offspring trio showing de novo SMAD6 mutation.
The reference sequence of a segment of SMAD6 that includes base 15:67073502 (denoted by arrow) is shown in the top row, with red, blue, green and yellow squares representing A, C, G, T, respectively. Below, all independent reads that map to this interval are shown. The results show that the proband has 23 reads of reference ‘G’, and 10 reads of non-reference ‘T’. Only the reference ‘G’ is seen in both parents, providing evidence of a de novo mutation.
Figure 2—figure supplement 2
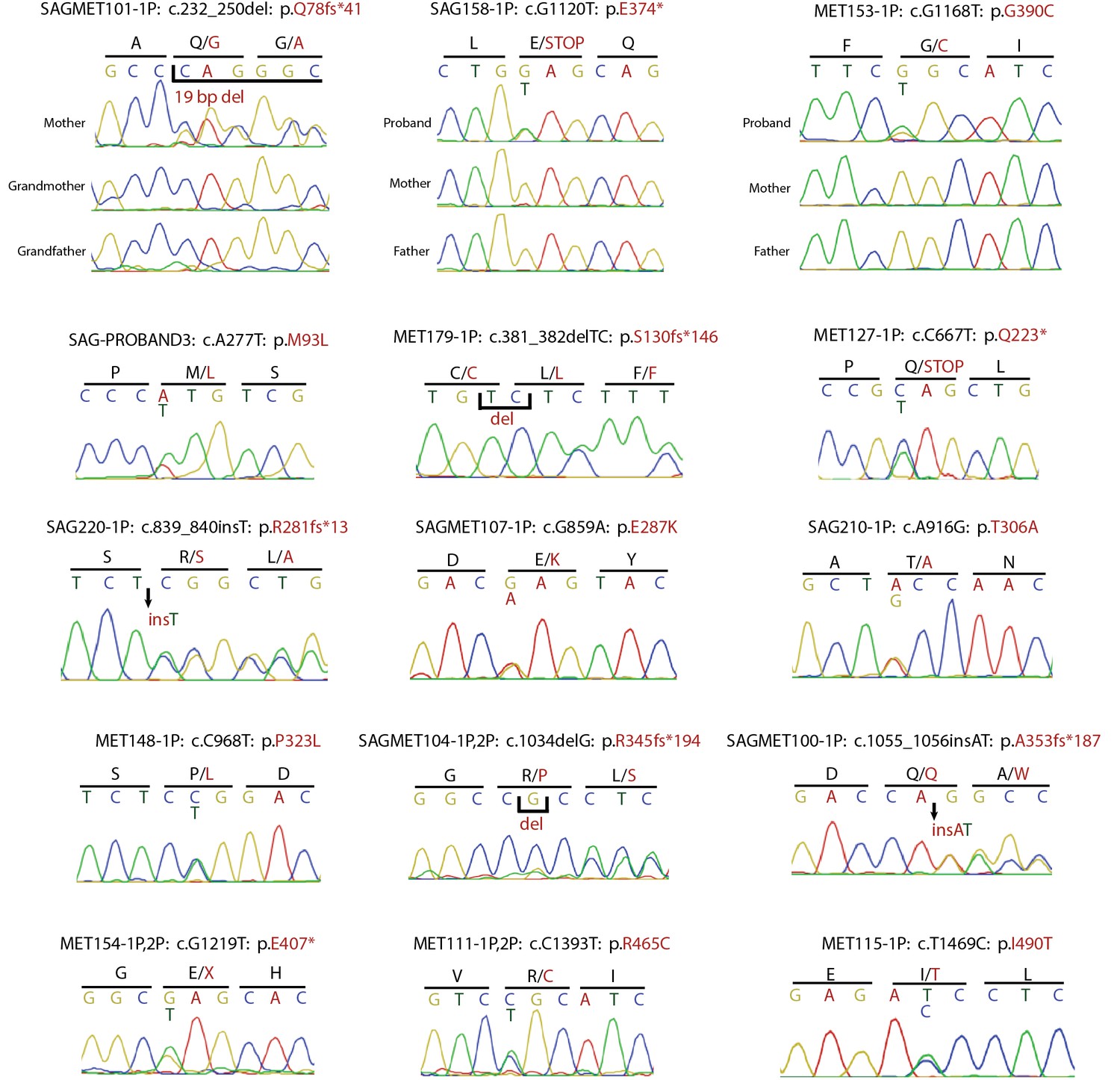
Confirmation of SMAD6 mutations by Sanger sequencing of PCR products.
Sanger sequencing traces of PCR amplicons containing SMAD6 mutations identified by exome sequencing are shown. Above each trace or set of traces, the kindred ID, mutation identified in the DNA sequence and its impact on SMAD6 protein is indicated. Above sequence traces, the inferred DNA sequence is shown, along with the inferred amino acid sequence (shown in single letter code). Heterozygous mutations are indicated beneath the wild-type sequence and non-reference amino acid sequences are shown in red. Deleted and inserted bases are denoted, and result in an overlap of wild-type and mutant sequences.
Figure 3 with 3 supplements
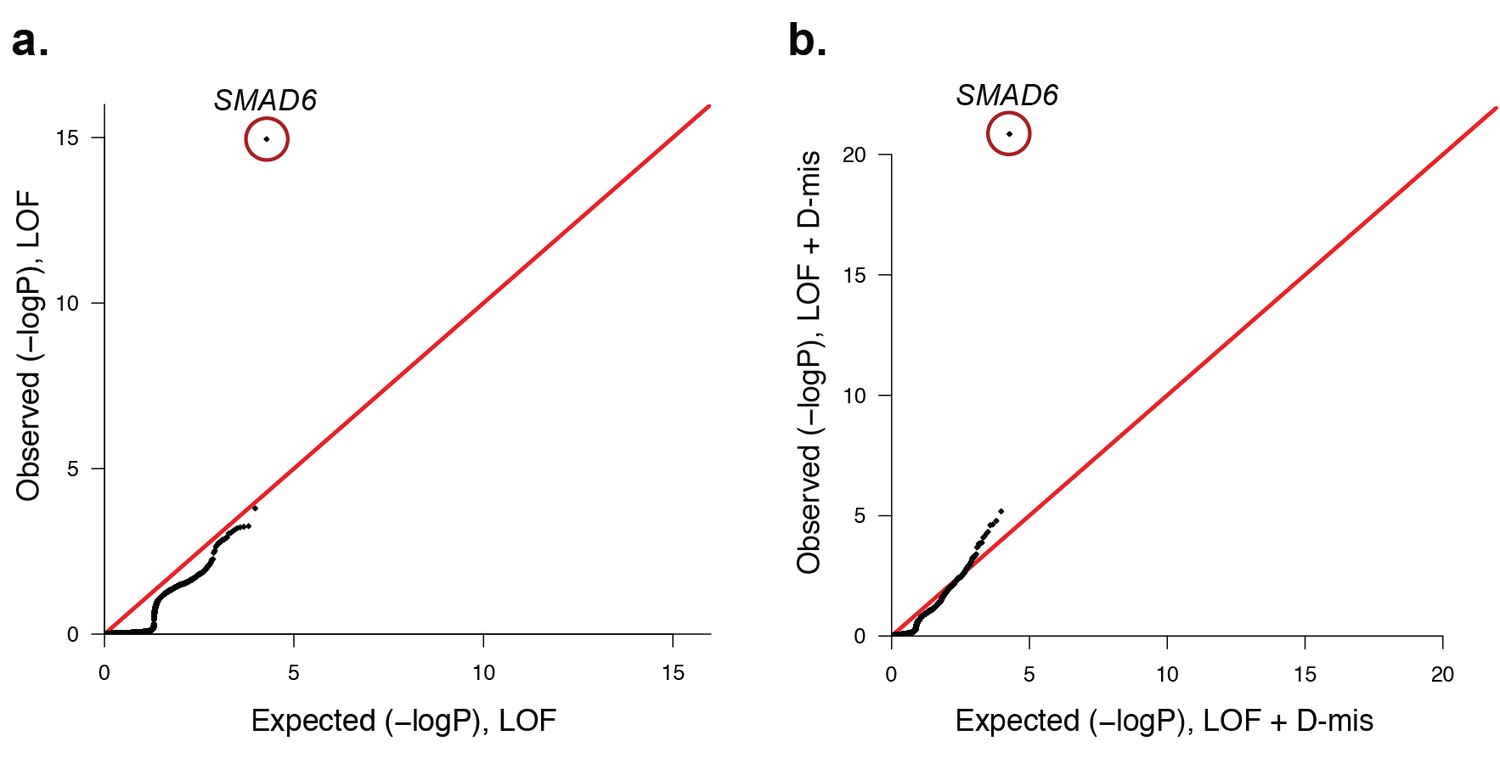
Quantile-quantile plots of observed versus expected p-values comparing the burden of rare LOF and damaging (LOF + D-mis) variants in protein-coding genes in craniosynostosis cases.
Rare (allele frequency <2 × 10–5 in the ExAC03 database) loss of function (LOF) and damaging missense (D-mis) variants were identified in 191 probands. The probability of the observed number of variants in each gene occurring by chance was calculated from the total number of observed variants and the length of the coding region of each gene using the binomial test. The distribution of observed P-values compared to the expected distribution is shown. (a) Q-Q plot for rare LOF variants in each gene from a total of 1135 LOF variants identified in probands. The distribution of observed p-values closely conforms to expectation with the exception of SMAD6, which shows p=1.1 × 10–15 and 156-fold enrichment in cases. (b) Q-Q plot for rare damaging (LOF + D-mis) variants in each gene from a total of 3156 damaging variants in probands. Again, SMAD6 deviates greatly from the expected distribution, with p<10–20 and 91-fold enrichment.
-
Figure 3—source data 1
Source data for Figure 3—figure supplement 3.
- https://doi.org/10.7554/eLife.20125.013
Figure 3—figure supplement 1
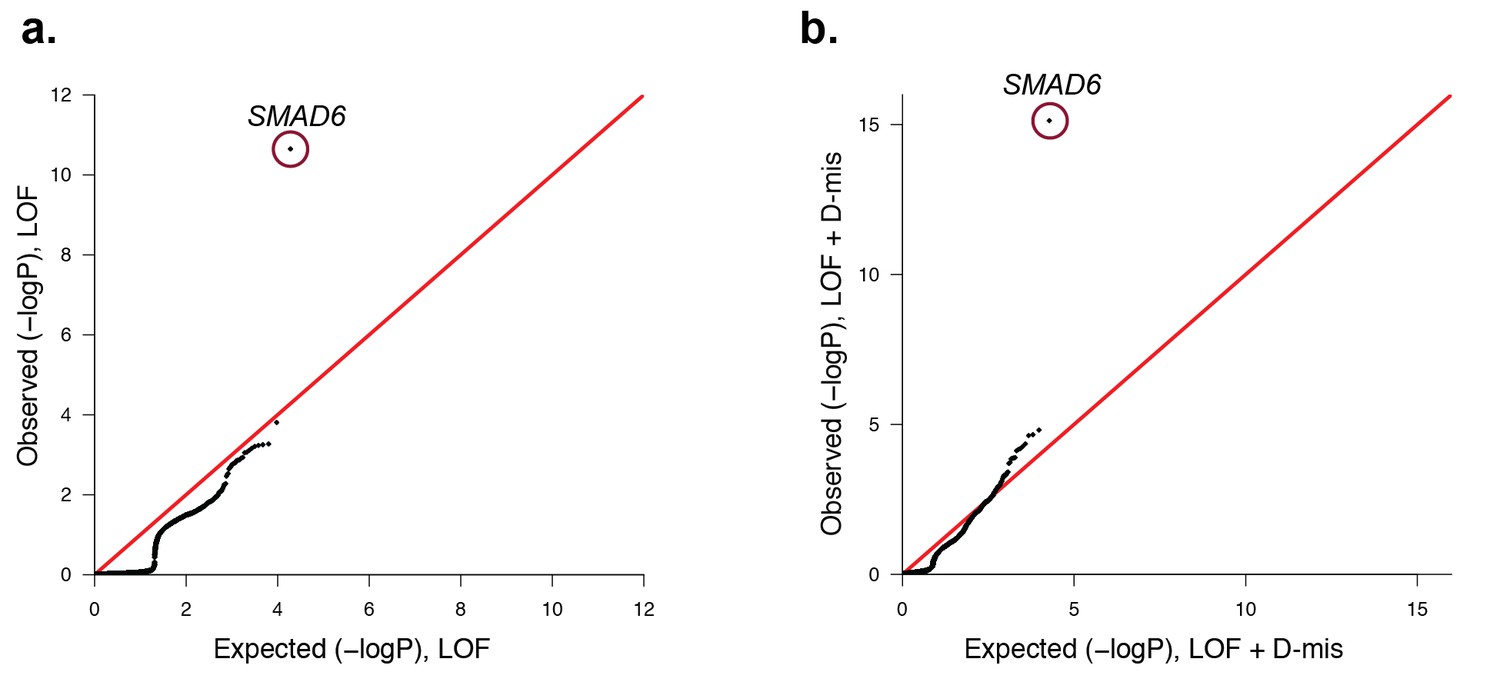
Quantile-quantile plots comparing all transmitted, damaging variants in protein-coding genes in 191 probands with midline craniosynostosis to the expected binomial distribution.
De novo variants were excluded from this analysis, leaving 1122 rare (ExAC allele frequency < 2 x10−5), transmitted LOF variants and 3115 transmitted damaging (LOF + D-mis) variants. All genes closely matched expectation, with the exception of SMAD6. (a) There were 6 transmitted SMAD6 LOF mutations, a 118-fold enrichment compared to the expected 0.05 (p=2.2 × 10–11). (b) Similarly, there were 10 transmitted damaging SMAD6 variants, a 71-fold enrichment compared to the expected 0.14 (p=7.0 × 10–16). The results demonstrate genome-wide significance of rare transmitted variants in SMAD6 independent of de novo mutations.
Figure 3—figure supplement 2
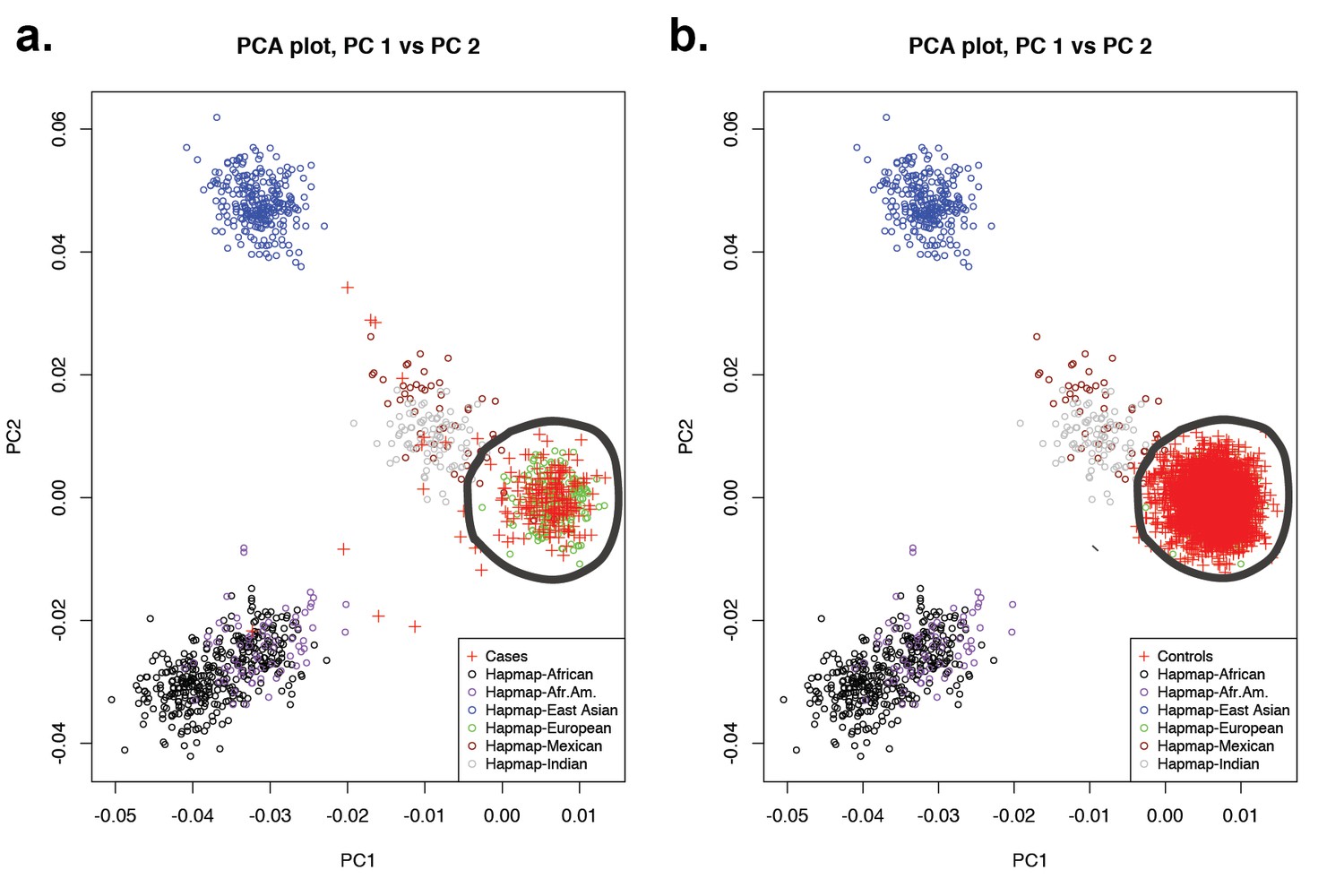
Principal-component analysis of 191 probands and 3337 European autism controls.
(a) Principal component analysis of exome sequence genotypes from 191 probands with sagittal, metopic, or combined sagittal and metopic craniosynostosis clustered along with HapMap subjects. Results identify 172 craniosynostosis subjects that cluster with HapMap European subjects. (b) Principal component analysis of genotypes from exome sequencing data of European autism parent controls (n = 3337) showing clustering with HapMap subjects. In both panels, subjects considered to be of European ancestry are circled.
Figure 3—figure supplement 3
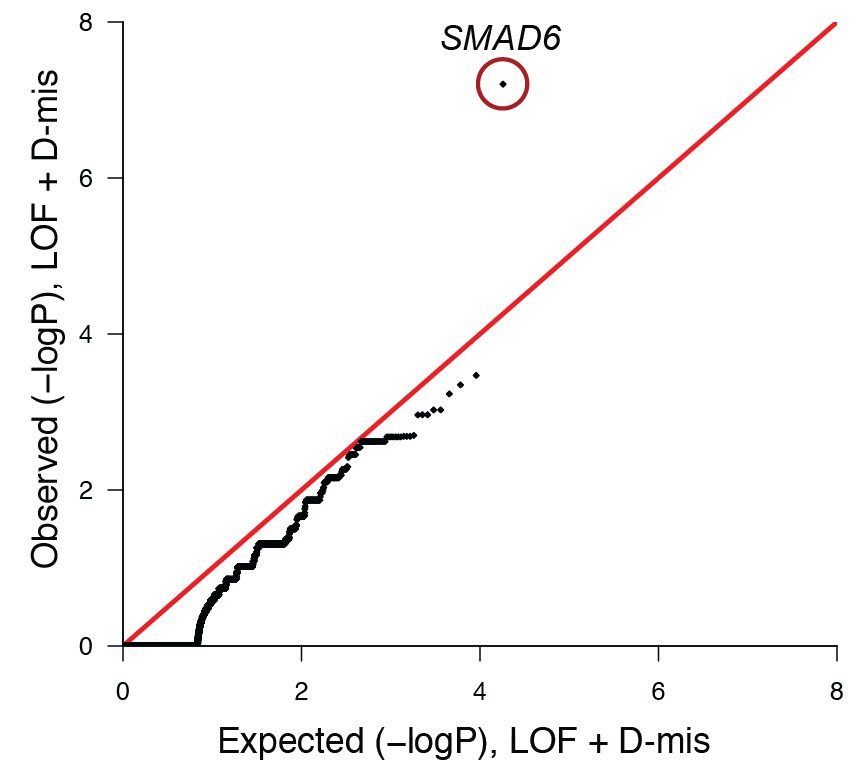
Quantile-quantile plot of observed versus expected p-values comparing the burden of damaging (LOF + D-mis) variants in protein-coding genes in craniosynostosis cases and controls.
The frequency of rare (allele frequency < 2 × 10–5 in the ExAC03 database) loss of function and D-mis variants in each gene was compared in 172 European probands with midline craniosynostosis and 3337 European controls. The distribution of observed p-values conforms to expectation with the exception of SMAD6, which deviates significantly from expectation. Because exon 1 of SMAD6 was poorly captured using the V2 capture reagent (used in control samples), 3 damaging variants in exon 1 in cases were excluded from this analysis.
Figure 4
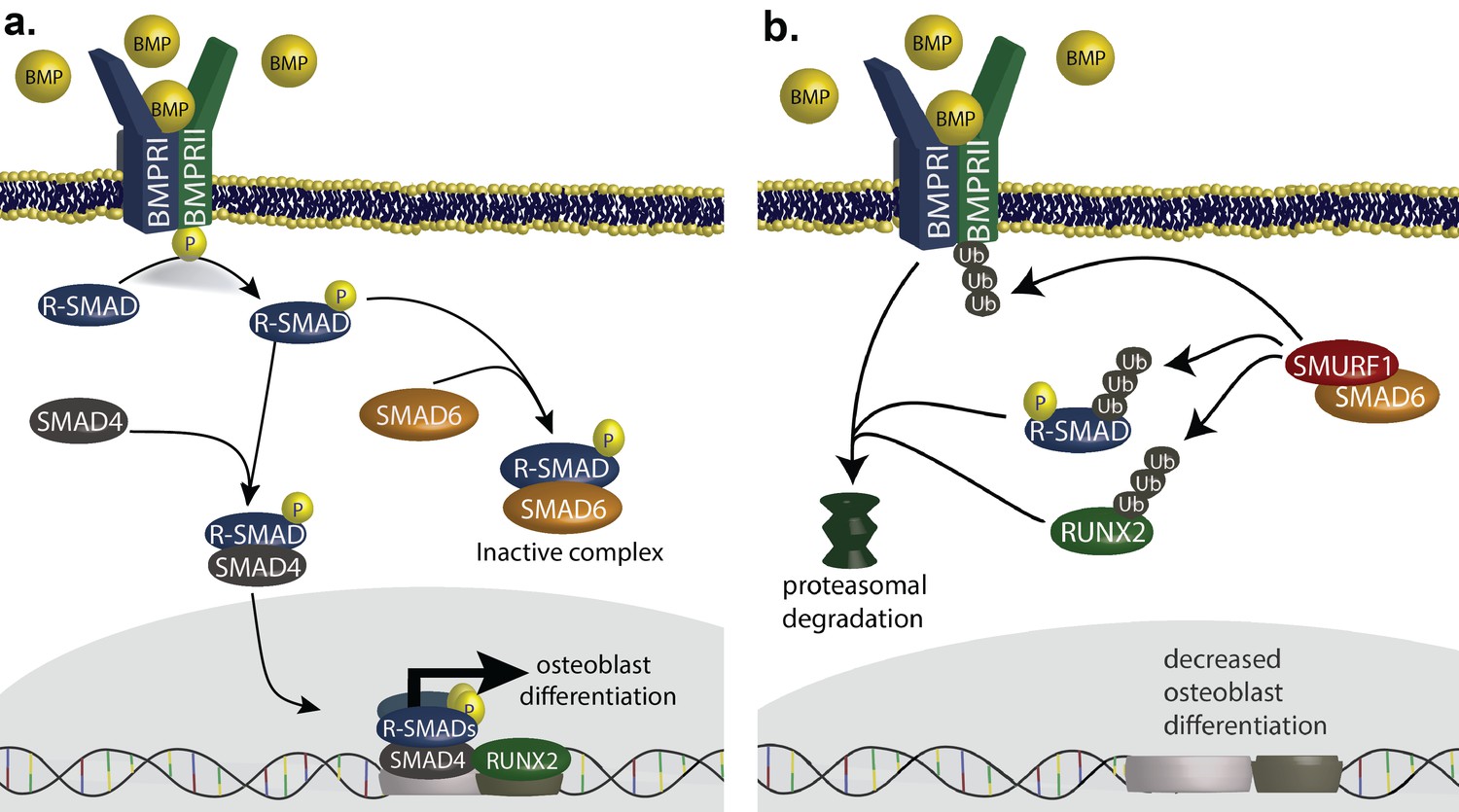
SMAD6 inhibits osteoblast differentiation by inhibiting BMP-mediated SMAD signaling (Salazar et al., 2016).
(a) BMP ligands activate BMP receptors, leading to phosphorylation of receptor-regulated SMADs (R-SMADs), which complex with SMAD4 and enter the nucleus, cooperating with RUNX2 to induce osteoblast differentiation. SMAD6 inhibits this signal by competing with SMAD4 for binding to R-SMADs, preventing nuclear translocation. (b) SMAD6 also cooperates with SMURF1, an E3 ubiquitin ligase, to induce ubiquitin-mediated proteasomal degradation of R-SMADs, BMP receptor complexes, and RUNX2.
Figure 5
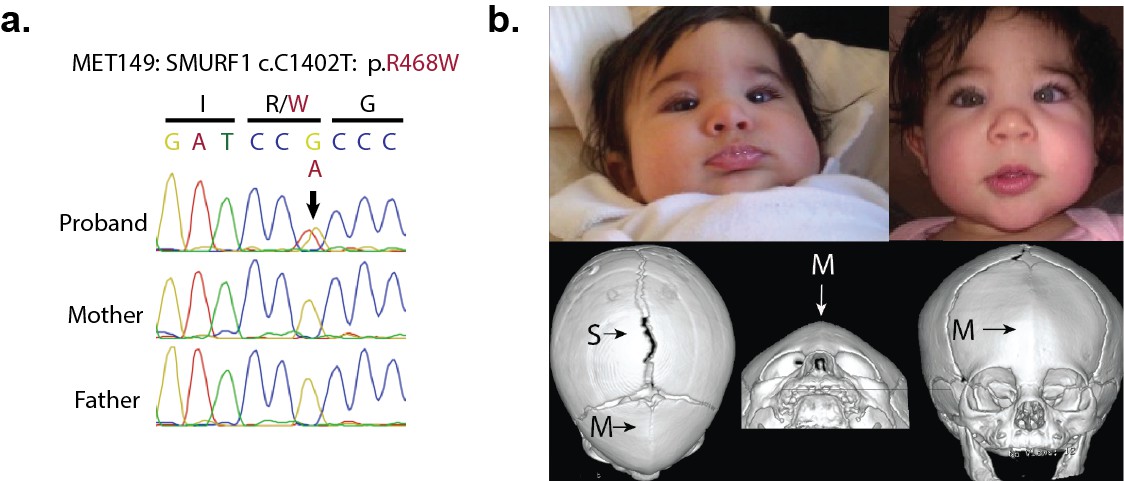
A de novo variant identified in SMURF1.
(a) Sanger sequence electropherogram of a PCR product amplified from the genomic DNA of a proband with metopic craniosynostosis, confirming a de novo R468W mutation in SMURF1, a SMAD6 binding partner. (b) Patient photographs of the proband, who presented with trigonocephaly and mild orbital abnormalities. 3D CT reconstruction demonstrates metopic craniosynostosis, trigonocephaly, and a patent sagittal suture.
Figure 6

De novo loss-of-function mutations in Sprouty genes.
(a) Pedigree and Sanger sequencing traces for kindred SAG150, demonstrating a de novo nonsense mutation in SPRY4 (p.E160*) in the proband. (b) Pedigree and Sanger sequencing traces in a kindred with a de novo SPRY1 frameshift mutation (p.Q6fs*8) that was transmitted to two affected offspring.
Tables
Table 1
Enrichment of protein-altering de novo mutations in 132 subjects with sagittal and/or metopic craniosynostosis.
| Observed | Expected | Enrichment | p-value | |||
|---|---|---|---|---|---|---|
| Class | # | #/subject | # | #/subject | ||
| All mutations | 144 | 1.09 | 142.8 | 1.08 | 1.01 | 0.47 |
| Synonymous | 21 | 0.16 | 40.4 | 0.31 | 0.52 | 3.0 × 10−4 |
| Protein altering | 123 | 0.93 | 102.4 | 0.78 | 1.17 | 0.03 |
| Total missense | 110 | 0.83 | 89.7 | 0.68 | 1.23 | 0.02 |
| T-mis | 82 | 0.62 | 75.2 | 0.57 | 1.09 | 0.23 |
| D-mis | 28 | 0.21 | 14.5 | 0.11 | 1.93 | 1.0 × 10−3 |
| Loss of function (LOF) | 13 | 0.10 | 12.7 | 0.10 | 1.03 | 0.50 |
| LOF + D-mis | 41 | 0.31 | 27.1 | 0.21 | 1.51 | 7.8 × 10−3 |
-
#, number of de novo mutations in 132 subjects; #/subject, number of de novo mutations per subject; Damaging and tolerated missense called by MetaSVM (D-mis, T-mis respectively); Loss of function denotes premature termination, frameshift, or splice site mutation. For mutation classes with enrichment compared to expectation, p-values represent the upper tail of the Poisson probability density function. For mutation classes in which we observed a paucity of mutations compared to expectation, p-values represent the lower tail.
-
Table 1—source data 1
De novo mutations in 132 trios with sagittal and/or metopic craniosynostosis.
Mutations highlighted in orange are likely loss of function mutations, those highlighted in blue are likely damaging missense mutations (D-mis) as called by MetaSVM, and those without highlight are predicted to be tolerated (T-mis) or are synonymous (syn).
- https://doi.org/10.7554/eLife.20125.005
Table 2
Probability of observed de novo mutations in SMAD6 and Sprouty genes occurring by chance in 132 subjects using gene-specific mutation probabilities.
| Gene(s) | Mutations | Number of observed mutations | Number of expected mutations | p value |
|---|---|---|---|---|
| SMAD6 | Loss of function | 2 | 0.00026 | 3.31 × 10−8 |
| SMAD6 | Missense | 1 | 0.0046 | 4.67 × 10−3 |
| SPRY1, SPRY2, SPRY3, SPRY4 | Nonsense, splice site, frameshift | 2 | 0.001193 | 7.11 × 10−7 |
-
Probabilities calculated from the Poisson distribution using DenovolyzeR. The probability of observing at least 2 LOF and 1 missense mutation in SMAD6 was 3.6 ×10−9 via Fisher’s method.
Table 3
Enrichment of de novo and transmitted damaging variants in SMAD6 in craniosynostosis.
| Observed | Expected | Enrichment | p-value | |
|---|---|---|---|---|
| De novo LOF and D-mis | 3 | 0.0049 | 612 | 3.6 × 10−9 |
| Transmitted LOF and D-mis | 10 | 0.1404 | 71.2 | 7.0 × 10−16 |
| Total | 13 | 0.1453 | 89.5 | 1.4 × 10−22 |
-
LOF, loss of function; D-mis, damaging missense variants per MetaSVM; The total number of SMAD6 variants expected in this cohort was calculated by summing the expected number of de novo and transmitted variants. P-value combining probabilities from de novo and transmitted protein damaging SMAD6 variants was determined by Fisher’s method.
Table 4
Distribution of suture involvement in kindreds with and without rare (allele frequency < 2 × 10−5) de novo and transmitted damaging (LOF + D-mis) variants in SMAD6.
| Total # kindreds | Total # SMAD6 mutations (%) | # LOF (%) | |
|---|---|---|---|
| Sagittal | 113 | 3 (2.7) | 2 (1.8) |
| Metopic | 70 | 7 (10) | 3 (3.9) |
| Sagittal and Metopic | 8 | 3 (37.5) | 3 (37.5) |
| Total | 191 | 13 (6.8) | 8 (4.2) |
Table 5
Risk of craniosynostosis in SMAD6 mutation carriers in the presence or absence of a BMP2 risk allele.
| SMAD6/BMP2 Genotypes | Craniosynostosis (+) | Craniosynostosis (−) |
|---|---|---|
| SMAD6 (+) / BMP2 risk allele (+) | 14 | 0 |
| SMAD6 (+) / BMP2 risk allele (−) | 3 | 13 |
| SMAD6 (−) / BMP2 risk allele (+) | 0 | 18 |
-
All members of kindreds found to have a mutation in SMAD6 were included. SMAD6(+) indicates the presence of a heterozygous LOF or D-mis allele. The reported BMP2 risk allele is ‘C’ at risk locus rs1884302, found within a gene desert ~345kb downstream of BMP2. p=1.4 × 10−10 by the Freeman-Halton extension of Fisher’s exact test. Odds ratio in favor of disease was incalculable due to the absence of craniosynostosis in SMAD6 (−) individuals in these kindreds.
Additional files
-
Supplementary file 1
Supplementary files for "Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles".
(A) Exome Sequencing Quality Statistics for all members of craniosynostosis kindreds (n = 455) and autism controls (n = 3337). (B) TDT of an intergenic BMP2 risk allele and intronic BBS9 risk allele in SMAD6 mutation carriers with craniosynostosis. (C) Optimized two locus and single locus parametric models of genotype specific penetrances for SMAD6 and BMP2. (D) Family specific lod scores for each kindred under the two locus and single locus models. (E) Clinical features and BMP2 genotypes in craniosynostosis patients with rare SMAD6, SMURF1, SPRY1, or SPRY4 mutations. (F) De novo mutations identified per trio.
- https://doi.org/10.7554/eLife.20125.023
-
Supplementary file 2
Exome sequencing quality statistics.
Exome sequencing quality statistics for all members of craniosynostosis kindreds (n = 455) and autism controls (n = 3337) .
- https://doi.org/10.7554/eLife.20125.024
-
Source code 1
R script for two locus and single locus linkage analyses.
- https://doi.org/10.7554/eLife.20125.025
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles
eLife 5:e20125.
https://doi.org/10.7554/eLife.20125




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}