Structure of the malaria vaccine candidate antigen CyRPA and its complex with a parasite invasion inhibitory antibody
- Swiss Tropical and Public Health Institute, Switzerland
- University of Basel, Switzerland
- Small Molecule Research, Roche Innovation Center Basel, F Hoffmann-La Roche Ltd., Switzerland
Figures
Figure 1 with 1 supplement

Binding of anti-PfCyRPA mAbs to fragments of PfCyRPA.
Binding of 16 mAbs to PfCyRPA fragments (black bars) expressed on the cell surface of HEK cells as assessed by Western blotting analysis and live-cell immunofluorescence staining. (x) indicates staining and (–) no staining; (a) indicates no reactivity in Western blot analysis of HEK cell lysates. Expression on the surface of the HEK cells has been demonstrated for all PfCyRPA fragments by immunofluorescence analysis using anti-Histidine tag HIS-6/9 mAb (Figure 1—figure supplement 1). For reference, the 17 residues constituting the epitope on PfCyRPA identified from the complex crystal structure with the Fab of mAb c12 is shown in all constructs as red bars. According to their reactivity pattern, anti-PfCyRPA mAbs were assigned to different epitope groups: A: c10, SB2.5; B: c02, c06, c08, c09, c12, SB3.7; C: c04; D: c05; E: c13, SB3.9; F: SB1.6, SB2.1, SB2.3, SB3.3.
Figure 1—figure supplement 1
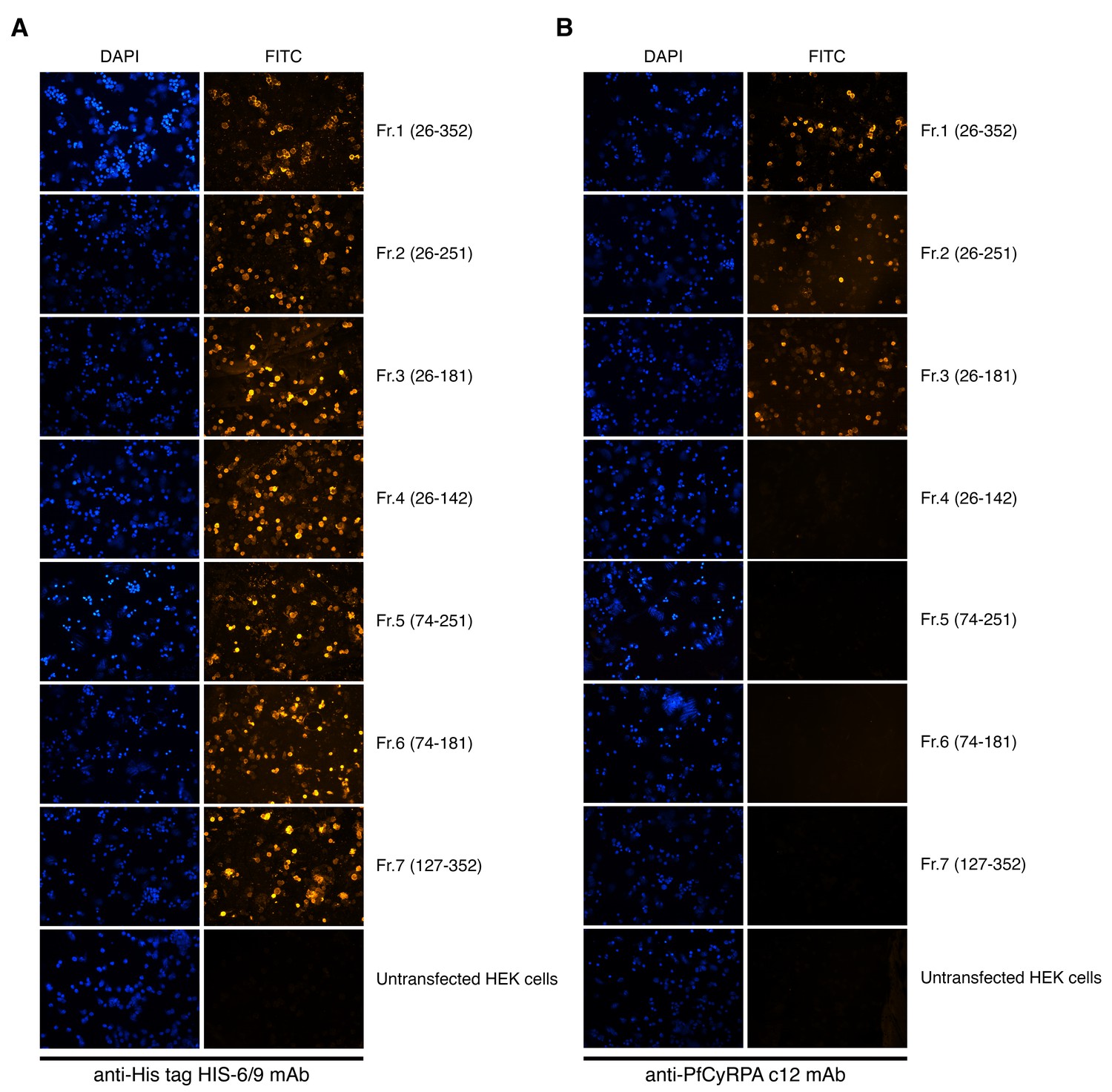
Cell-surface expression of PfCyRPA fragments on transiently transfected HEK cells.
Fluorescence staining of HEK cells expressing PfCyRPA fragments on their surface after staining with (A) anti-His tag HIS-6/9 mAb or (B) anti-PfCyRPA c12 mAb and FITC-labelled anti-mouse IgG antibodies. Nuclei were stained with DAPI. Untransfected HEK cells served as negative control.
Figure 2 with 1 supplement

Anti-PfCyRPA and anti-PfRH5 mAbs have both in vitro and in vivo an additive inhibitory effect on parasite growth.
(A) Growth inhibition in vitro. Synchronized P. falciparum 3D7 blood-stage parasites were cultured for one cycle of merozoite invasion (48 hr) in the presence of anti-PfCyRPA c12 mAb, anti-PfRH5 BS1.2 mAb, and their combinations. An isotype-matched, malaria-unrelated mAb (NR4.2) (Rose et al., 2016) was used as negative control. Inhibitory and non-inhibitory anti-MSP-1 mAbs (12.10 and 2F10, respectively) were also included as reference (Blackman et al., 1990, 1994). Percent parasite growth inhibition was calculated against the parasitemia of PBS control wells. Each bar represents the mean of a triplicate experiment, and error bars indicate the standard deviation (SD). Differences in parasite growth inhibition between mAbs c12 and BS1.2 alone and their combinations are statistically significant (unpaired t test with Welch’s correction, 95% confidence interval, two-tailed p value). (B) Growth inhibition in vivo. NODscidIL2Rγnull mice received purified anti-PfCyRPA c12 mAb and/or anti-PfRH5 BS1.2 mAb by i.v. injections. Mice were then infected with P. falciparum 3D7 and parasitemia was monitored over 6 days. Values represent the mean parasitemia in human erythrocytes in peripheral blood of three mice per group. Error bars indicate the SD. PBS and an unrelated control mAb were used as negative control.
Figure 2—figure supplement 1
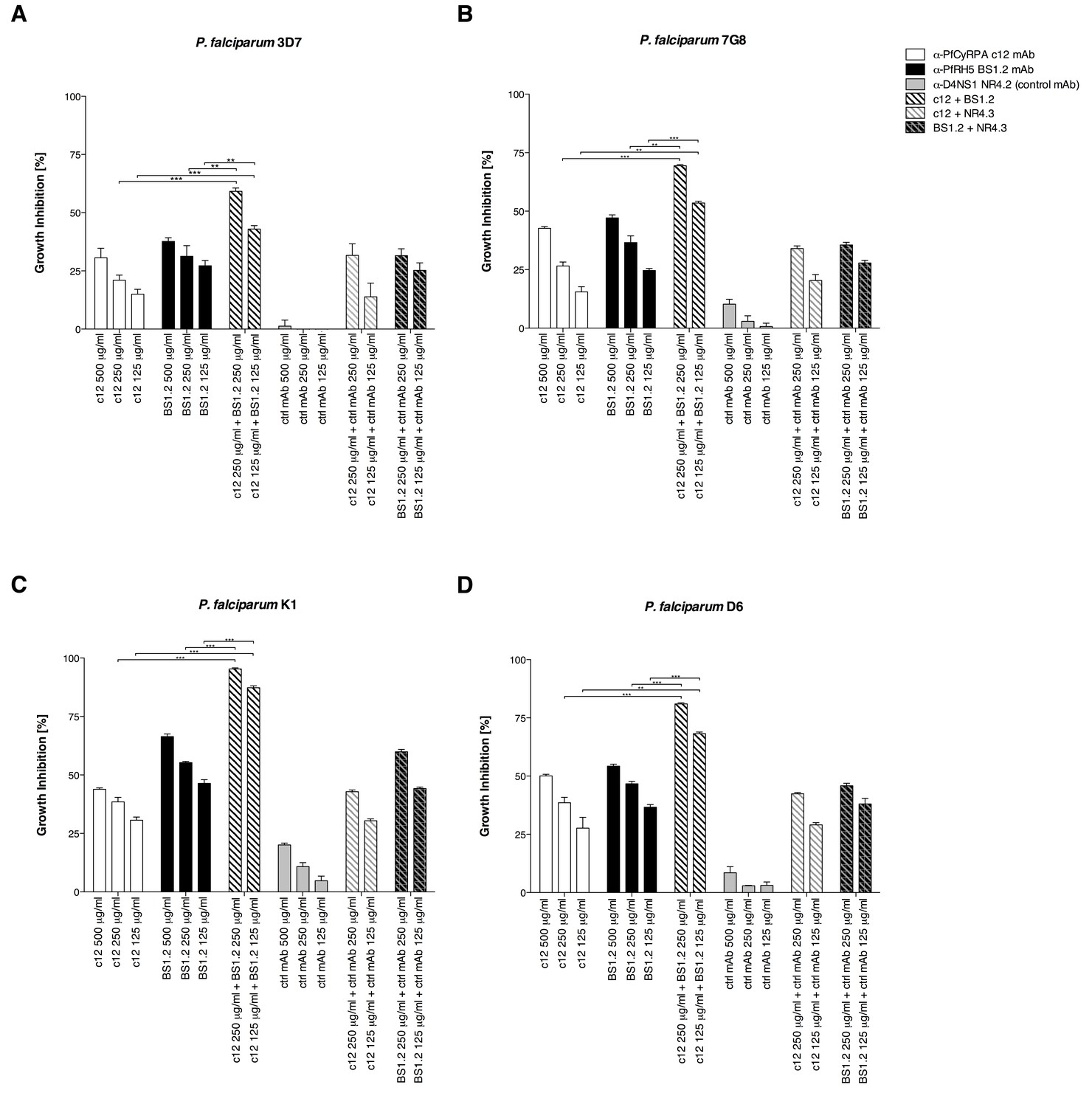
Anti-PfCyRPA and anti-PfRH5 mAbs inhibit parasite growth of various P. falciparum strains.
Synchronized P. falciparum 3D7, 7G8, K1 and D6 (A, B, C, and D, respectively) blood-stage parasites were cultured for one life cycle in the presence of anti-PfCyRPA c12 mAb, anti-PfRH5 BS1.2 mAb, and their combinations. An isotype-matched, malaria-unrelated control mAb (NR4.2 mAb) was used as negative control. Percent parasite growth inhibition was calculated against the parasitemia of PBS control wells. Each bar represents the mean of a triplicate experiment, and error bars indicate the standard deviation (SD). Differences in parasite growth inhibition between mAbs c12 and BS1.2 alone and their combinations are statistically significant (unpaired t test with Welch’s correction, 95% confidence interval, two-tailed p value).
Figure 3 with 3 supplements

PfCyRPA adopts the neuraminidase fold.
(A) Structure-sequence relationship of PfCyRPA. Indicated are an Actinase E cleavage site at Asp189 (red), a sialidase-typical Asp-box (dotted underlined), the two Trp residues (dotted underlined), and the sequential disulfide bonds (connected by lines and in same color). β-strands are shown as arrows colored according to the blade they form. The epitope recognized by mAb c12 is underlined in bold. (B) Cross-eyed stereo view of the ribbon representation of a superposition of the two PfCyRPA molecules in the asymmetric unit with the blades numbered 1–6 from the N-terminus and colored individually. Blade one is made up of an N-terminal (black) and three C-terminal β-strands (red). One protomer is shown with white, the other with black loop regions, which may differ substantially (arrows in blade 5). The Trp and Cys residues are drawn as stick models. (C) The same orientation of the catalytic domain of Vibrio cholerae sialidase (PDB-ID 1w0o), the next structural homolog of PfCyRPA with a DALI score of 18 (Z < 5 is structurally dissimilar). Sialic acid and residues in the Vibrio enzyme are displayed as balls and sticks. Structural Ca2+ ions are marked as magenta spheres. None of the residues necessary for metal ion binding, substrate binding, or catalysis is present in PfCyRPA. (D) Superposition of PfCyRPA with the V. cholerae sialidase. While both proteins are 6-bladed β-propellers, the blades have very different angles, extents, and loop lengths and conformations connecting the β-strands. The four Asp-boxes in the bacterial sialidase (grey) are colored black. PfCyRPA (orange) has only a single Asp-box connecting the third and fourth β-strands in blade 3 (colored blue). Other β-strand connections are made by sequences unrelated to the Asp-box motif, in agreement with poor conservation of the Asp-box in other, e.g. viral, sialidases. The view in (D) is rotated by 180° about the horizontal axis compared to (B) and (C).
Figure 3—figure supplement 1

Sequence alignment of CyRPA orthologs.
Sequence alignment of CyRPA orthologs from P. falciparum (P. fal.), P. vivax (P. viv.; PVX_090240), P. knowlesi (P. kno.; PKNH_0515800), P. cynomolgi (P. cyn.; PCYB_053730), and P. reichenowi (P. rei.; PRCDC_0421000). Full-length protein sequences (PlasmoDB; plasmodb.org/plasmo) were aligned with Clustal O (1.2.1). Asterisks indicate conserved positions, colons indicate strong biophysical conservation, and periods indicate weak biophysical conservation. Ten of the twelve Cys residues are conserved (same color code as in Figure 3). The predicted PfCyRPA secretion signal sequence (M1–C28) is indicated in boldface, and the predicted GPI-anchor motif (I353-E362) is underlined. The N-glycosylation sites that are used in human cells (N145, N322, and N338) are shown in lowercase and the non-synonymous SNPs (D73, D110, V165, P168, R174, F187, D236, N270, V292, N338, R339, L341, N352) are colored in red.
Figure 3—figure supplement 2
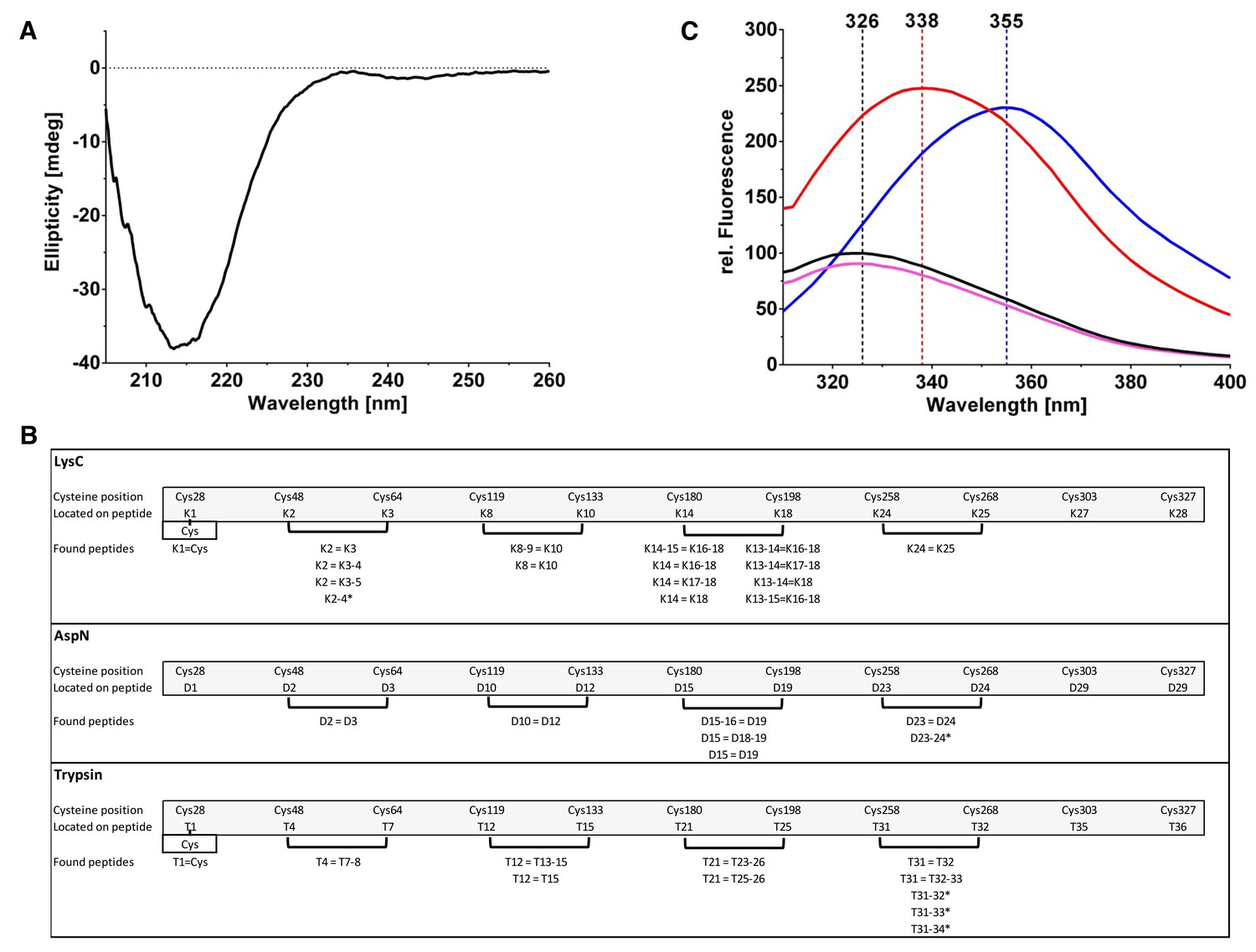
Biophysical analysis of PfCyRPA.
(A) The CD spectrum of PfCyRPA shows a prominent minimum around 215 nm, consistent with a high content of β-secondary structure. The minimum is remote from the minima expected for helical (208 nm and 222 nm) and random coil (200 nm) structures. (B) Identification of disulfide bonds in PfCyRPA. The peptide fragments are labeled K (LysC cleavage), D (AspN cleavage), and T (trypsin cleavage). A cut-off of 2% signal intensity was applied to separate highly populated from less populated fragments. Masses that correspond to two proteolytic peptides connected by disulfide bonds were repeatedly found (parentheses) when using different proteases. An equal sign (=) signifies a disulfide bond and a minus sign (− ) signifies a skipped cleavage. For instance, the first disulfide bond between Cys48 and Cys64 was identified four times in the LysC hydrolysate: K2 = K3 corresponds to the mass of the second and third expected proteolytic fragment linked by a disulfide bond. K2 = K3–4 is the second expected LysC peptide (K2) disulfide-linked to a larger peptide (K3–K4) that still contains a LysC cleavage site but this site was not recognized by the enzyme. The same reasoning applies to K2 = K3–5, where two LysC cleavage sites were not recognized. An asterisk (*) denotes a disulfide bond in a peptide. The K2-4* fragment has a mass corresponding to an un-cleaved and disulfide-linked peptide with two intact LysC cleavage sites that were not recognized. The last disulfide bond ion PfCyRPA between Cys303/327 was not identified by any protease, possibly due to low abundance of the proteolytic fragments containing it. (C) The fluorescence emission spectrum of native PfCyRPA (black) shows a maximum at 326 nm that shifts to 338 nm when heated to 70°C (red) or 355 nm when unfolded by 9 M urea (blue). Addition of 50 mM DTT (magenta) has no significant effect on the fluorescence, suggesting that the disulfide bonds are not solvent-accessible. The unusual finding that the Trp fluorescence quantum yield of the unfolded state is higher than in the native state indicates that the excited state of Trp is efficiently quenched by nearby residues in the native structure (Chen and Barkley, 1998).
Figure 3—figure supplement 3
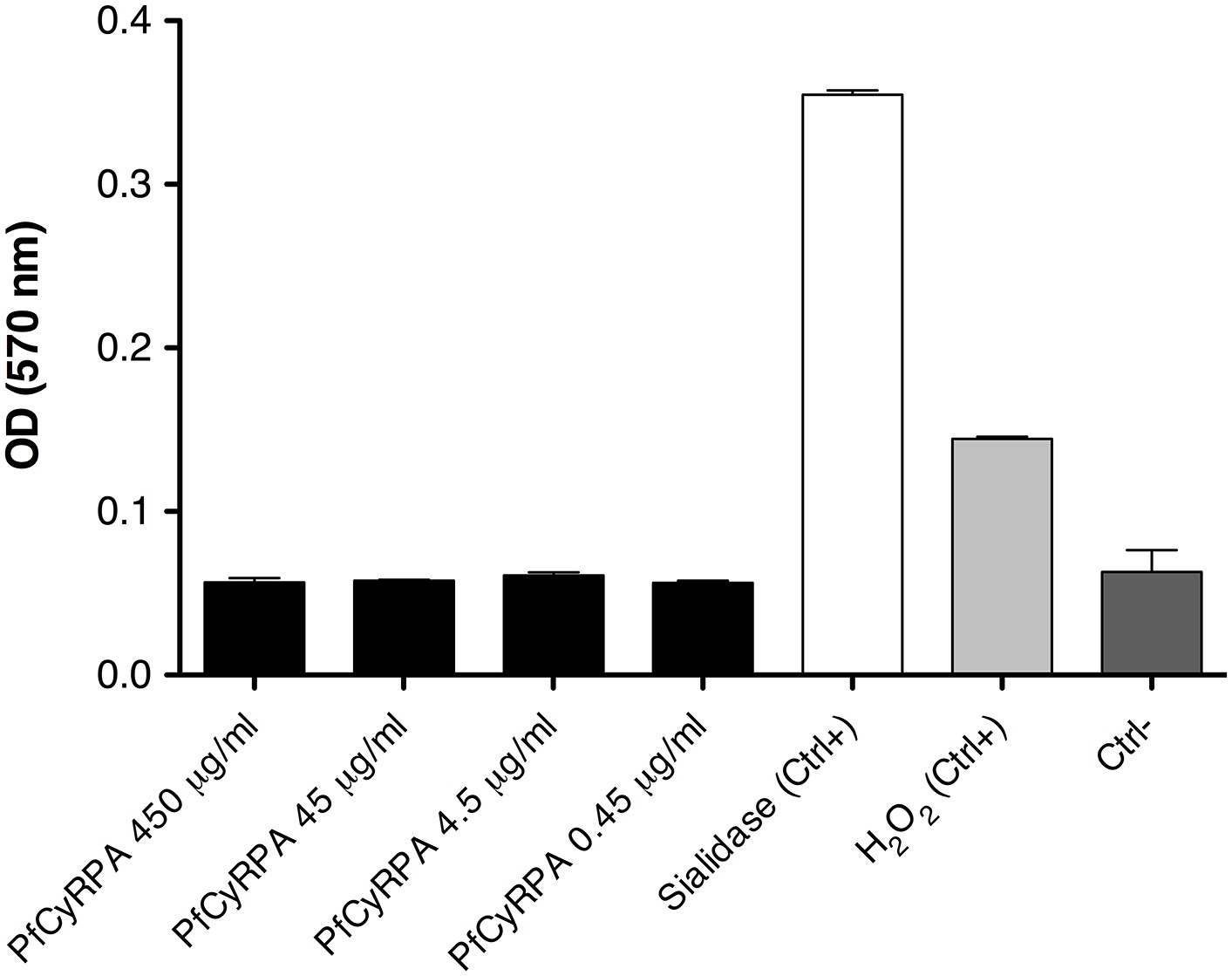
PfCyRPA lacks detectable sialidase activity in a functional assay.
Sialidase activity was estimated using the Amplex Red Neuraminidase (Sialidase) assay kit (Molecular Probes, Inc.). The assay utilizes Amplex Red to detect H2O2 generated by galactose oxidase oxidation of desialiated galactose, the end product of sialidase activity. The H2O2 in the presence of HRP reacts with Amplex Red reagent to generate resorufin, the red fluorescent oxidation product, which was detected at 570 nm using the Sunrise Absorbance Reader (Tecan). PfCyRPA was mixed 1:1 (v/v) with the Amplex Red reagent/HRP/galactose oxidase/fetuin working solution (0.2 U/mL HRP, 4 U/mL galactose oxidase, 500 µg/mL fetuin), and the mixture was incubated at 37°C for 30 min in a light-protected container. Sialidase and H2O2 (supplied with the kit) were used as positive controls (Ctrl+); a no-sialidase sample as negative control (Ctrl−).
Figure 4 with 1 supplement
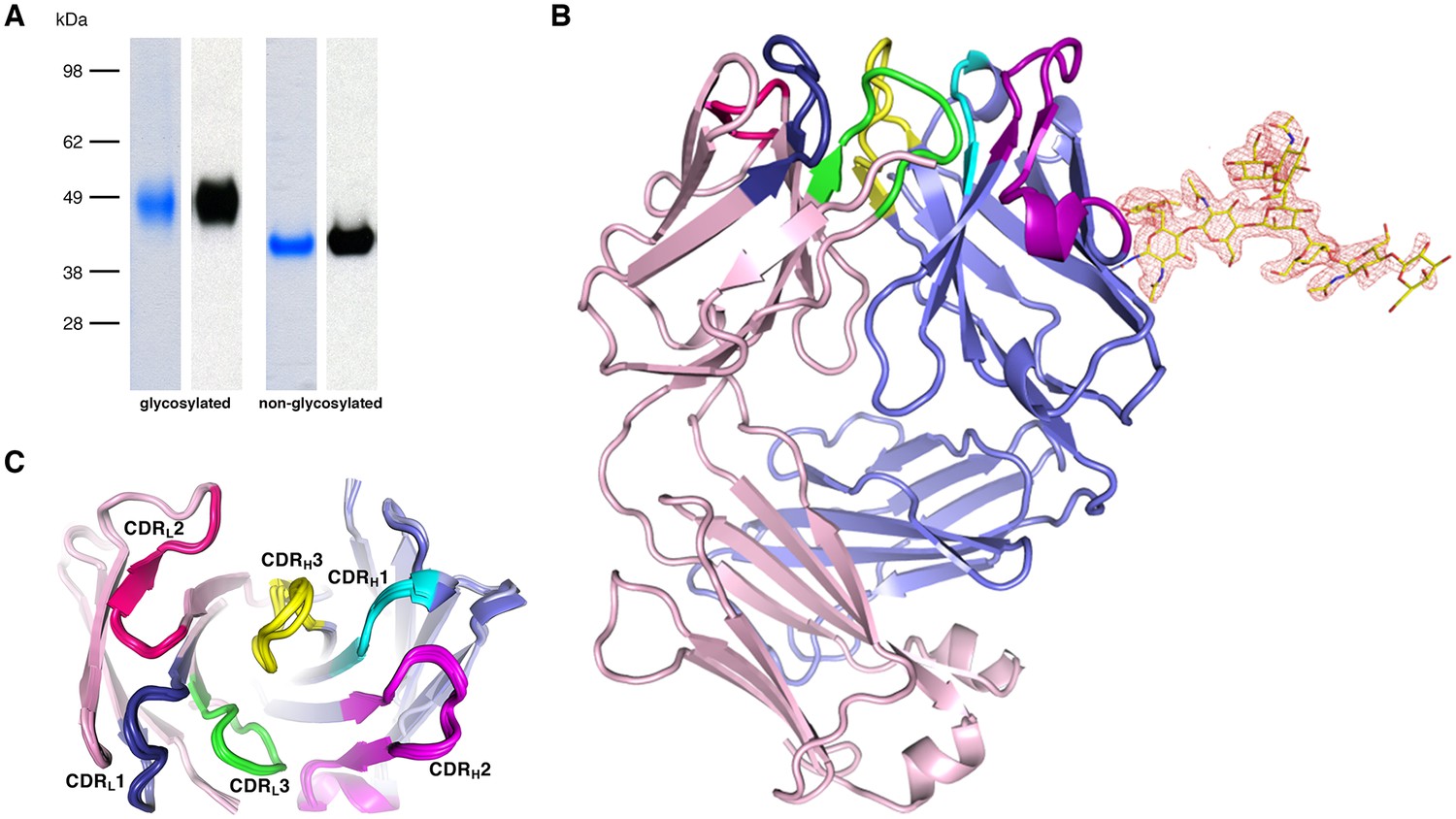
Recognition of PfCyRPA by the mAb c12 and structure of c12.
(A) Reducing SDS-PAGE of glycosylated (left) and non-glycosylated (right) PfCyRPA detected by Coomassie-staining (blue) and Western blotting with mAb c12 (black). Recognition by c12 is independent of the glycosylation. (B) Overview of the c12 structure with a glycan located at heavy-chain Asn37. mFo-DFc electron density for the glycan is shown as a red mesh drawn at the three rmsd level. The light and heavy chains are colored light pink and light blue, respectively. Heavy chain CDR1-3 are colored cyan, magenta, and yellow, and light chain CDR1-3 are drawn in dark blue, pink, and green. (C) Comparison of the four c12 structures shows little conformational variability of the CDR loops. The four c12 molecules superimpose onto their variable VHVL di-domains with an average rmsd of 0.35 Å, which reveals a minor spread of the elbow angles between 133.1° and 135.8°. The high structural congruence indicates that the CDR conformations are genuine and not dominated by crystal contacts. The view is from above on top of the CDR loops.
Figure 4—figure supplement 1
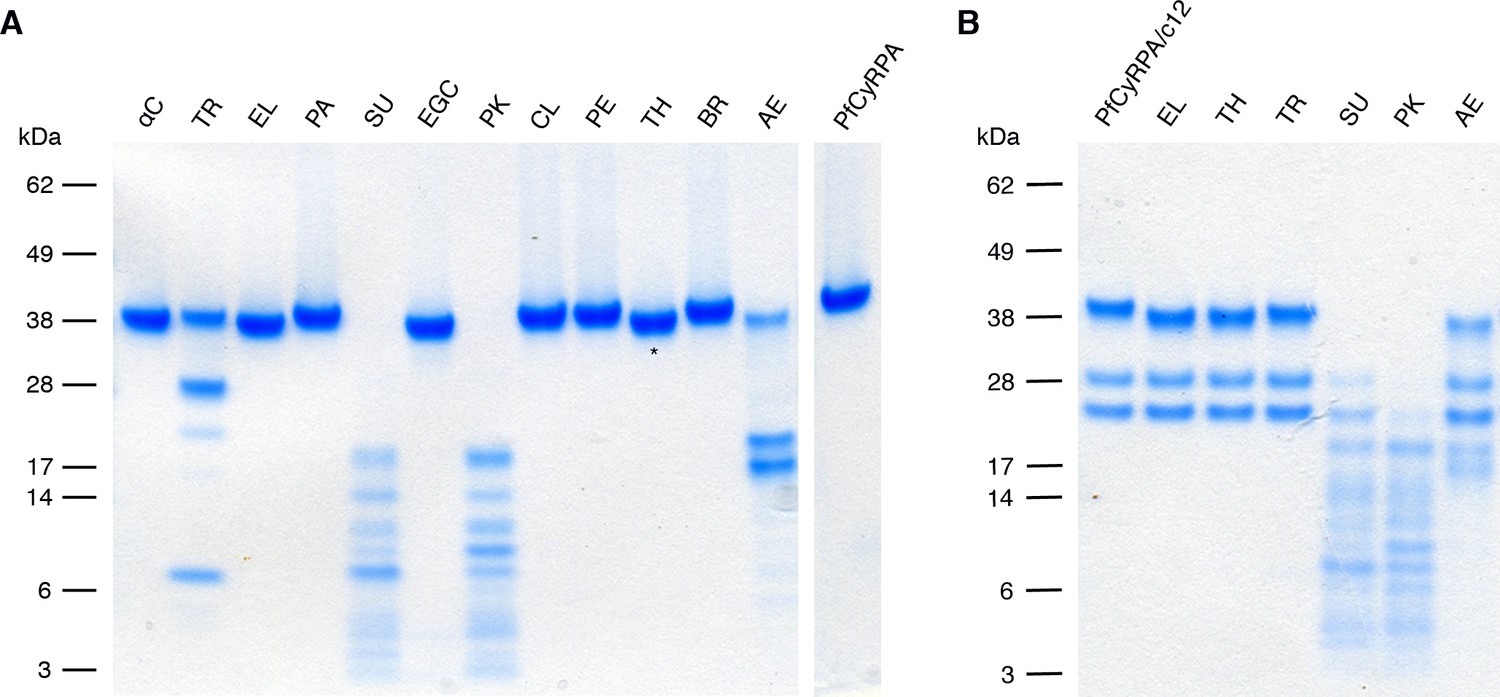
Limited proteolysis of PfCyRPA and the PfCyRPA/c12 complex.
(A) PfCyRPA. (B) PfCyRPA/c12 complex. Proteases are abbreviated as: αC: α-Chymotrypsin; TR: Trypsin; EL: Elastase; PA: Papain; SU: Subtilisin; EGC: Endoproteinase Glu-C; PK: Proteinase K; CL: Clostripain; PE: Pepsin; TH: Thermolysin; BR: Bromelain; AE: Actinase E. The two bands around 28 kDa in (B) correspond to the Fab. Actinase E affects PfCyRPA irrespective of bound c12, yielding two major proteolysis products of apparent molecular weight 17 kDa and 20 kDa.
Figure 5 with 2 supplements
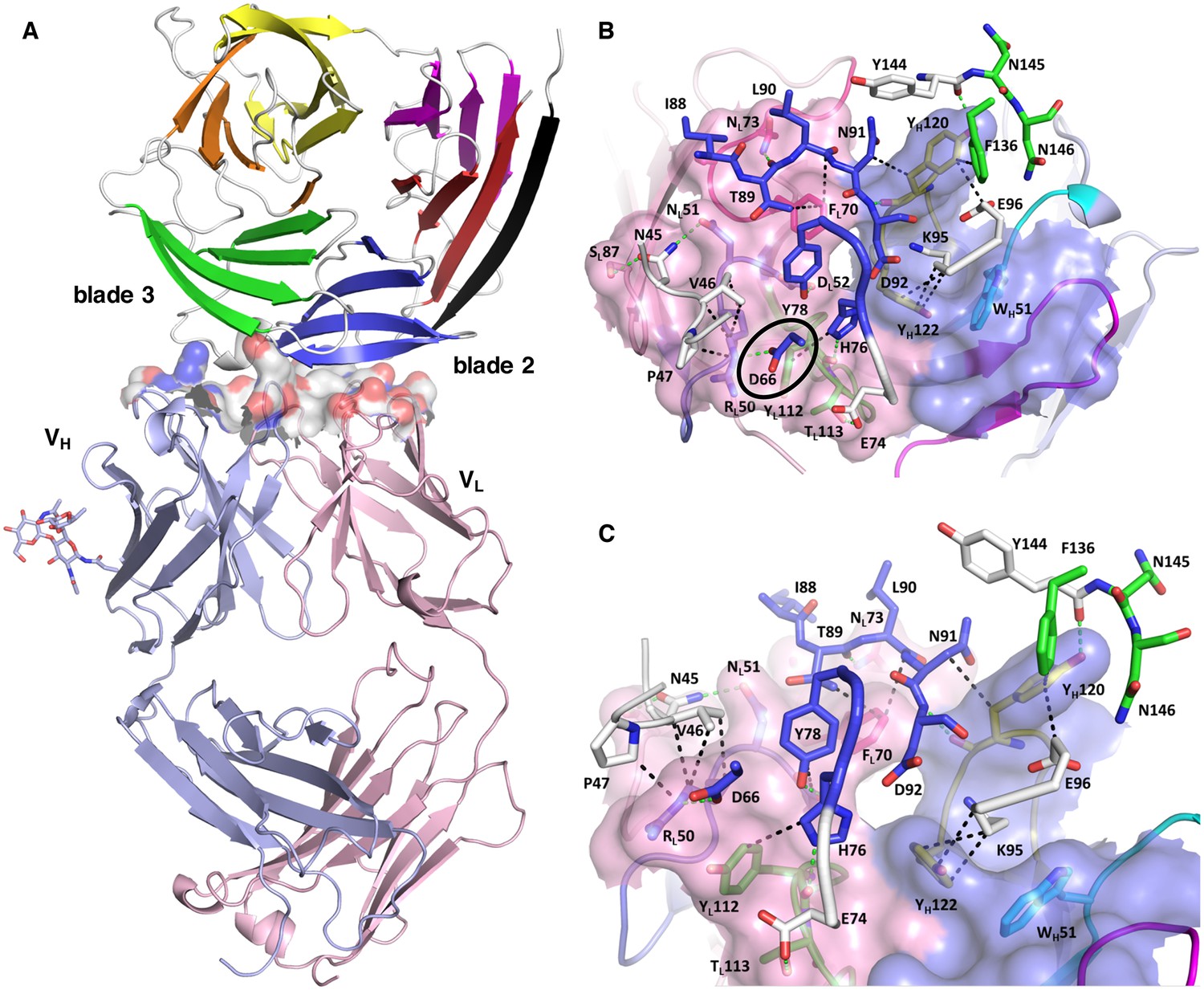
Structure of the PfCyRPA/c12 complex.
(A) Overview showing that the majority of the interface is made by interactions between the light chain of c12 and blade 2 of PfCyRPA. (B) Details of the interface viewed from top onto the CDR loops. The light and heavy chain surfaces buried by PfCyRPA are colored pink and blue, respectively. Possible hydrogen bonds and van der Waals interactions are indicated by dashed green and black lines. The CDR loops are color-coded as in Figure 4. The Asp66-Arg50 salt bridge is circled. (C) Close-up of (B).
Figure 5—figure supplement 1
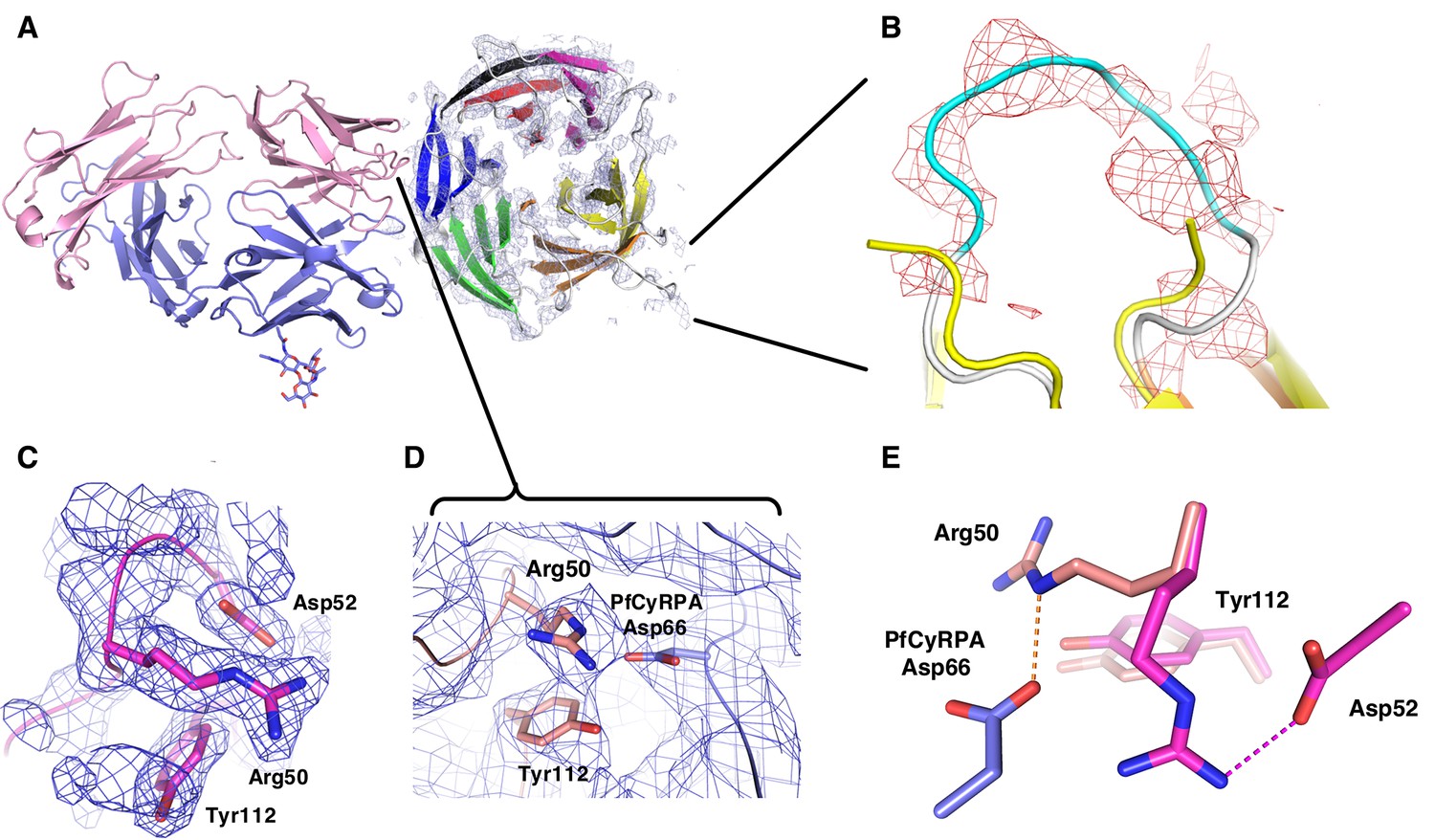
Electron density of the PfCyRPA/c12 complex.
(A) Initial electron density at a resolution of 3.6 Å contoured at the one rmsd level for PfCyRPA after molecular replacement phasing of the PfCyRPA/c12 complex data with c12 as the search model. The light and heavy chains of c12 are colored pink and cyan, respectively. The final model of PfCyRPA (blades colored in the same pattern as in Figure 3A) is superimposed for reference but could not be traced using this map. This map fragment was cut out and used as starting model for molecular replacement of the PfCyRPA structure. (B) Zoom of the loop region 186-KFKDDNK-192. This loop was cut at Asp189 by Actinase E. The loop opened and the sequence could not be completely traced in the isolated PfCyRPA structure (yellow). Difference electron density at a resolution of 3.6 Å contoured at the 1.8 rmsd level shows that the loop is intact in the PfCyRPA/c12 complex and can be traced in the electron density maps. The refined model of the complex (loop colored in cyan) is superimposed for reference. (C) Region around Arg50 in the free c12 Fab structure 5ezj. 2mFo-DFc electron density is shown at the one rmsd level. Arg50 binds to nearby Asp52. (D) This interaction is lost in the complex with PfCyRPA. Arg50 now binds to PfCyRPA residue Asp66 (light blue). 2mFo-DFc electron density is shown at the 0.8 rmsd level. (E) Superposition of the structures from panels (C) and (D) shows the large movement of Arg50 in the free c12 Fab (magenta) versus its position in the complex with PfCyRPA (pink). By contrast, the conformation of Tyr112 does not change much.
Figure 5—figure supplement 2
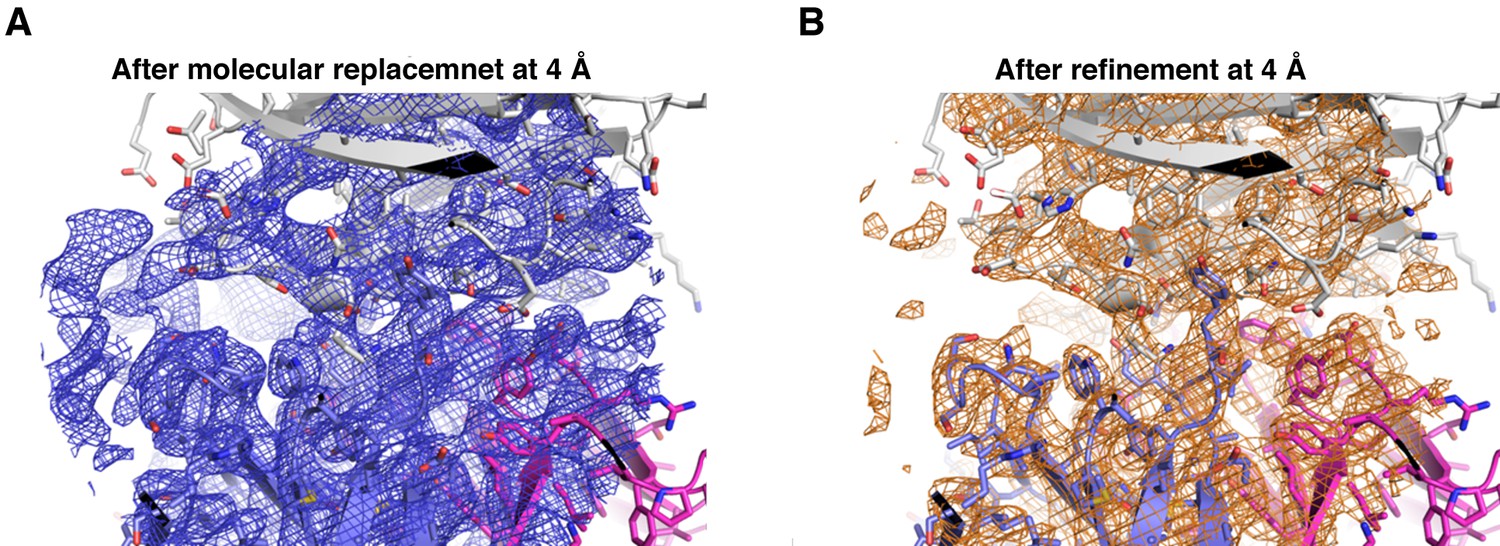
Electron density of the PfCyRPA/c12 complex at the interface.
The final model of the PfCyRPA/c12 complex is shown. PfCyRPA is at the top, shown as a grey ribbon representation. Fab c12 is at the bottom with the light and heavy chain colored magenta and blue, respectively. 2Fo-Fc electron density at 4 Å resolution is contoured at the one rmsd level at a radius of 22 Å around the center of the image. The panel on the left-hand side shows the electron density after molecular replacement (A), while the panel on the right-hand side depicts the density after refinement (B).
Figure 6
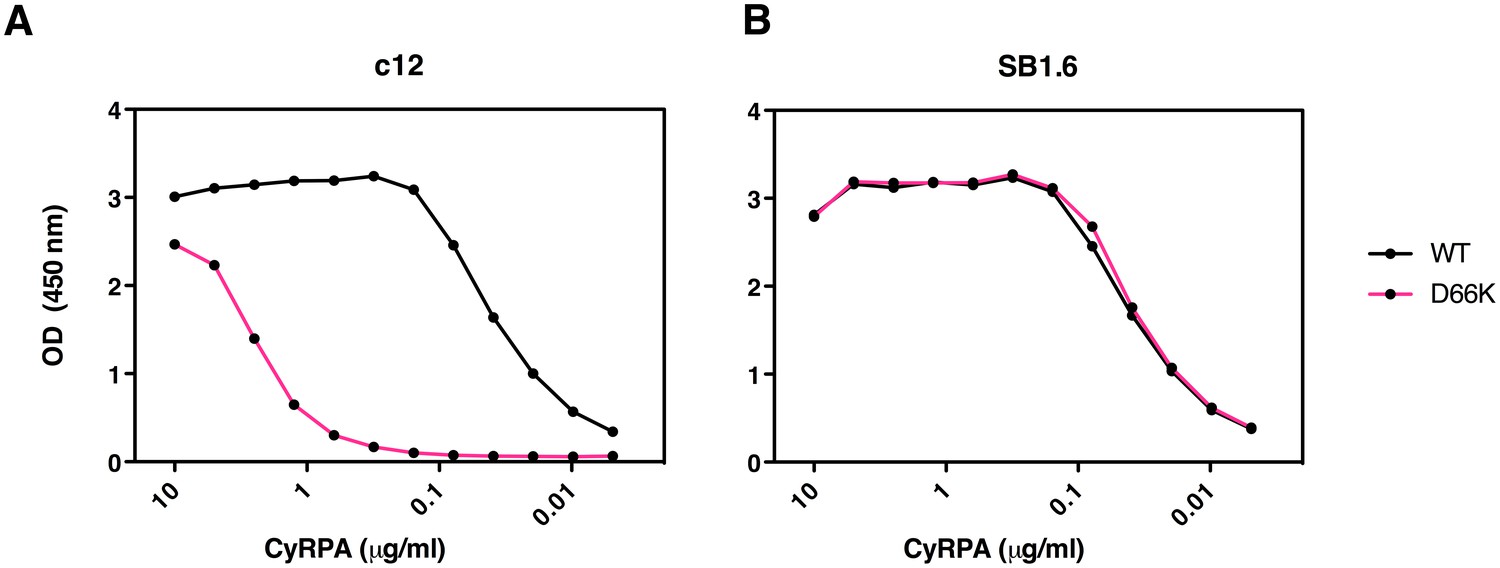
Identification of Asp66 as a key contact residue for PfCyRPA/c12 interaction.
mAb binding to purified wild-type PfCyRPA and an Asp66Lys single amino acid sequence variant was analyzed by capture-ELISA experiments. ELISA plates were coated with 10 µg/mL anti-PfCyRPA mAbs and then incubated with serial dilutions of wild-type PfCyRPA (WT) or an Asp66Lys variant (D66K); HRPO-labeled anti-Histidine tag mAbs were used as detection antibody. (A) When compared to wild-type PfCyRPA, the Asp66Lys single amino acid exchange strongly reduced the binding to mAb c12 mAb (epitope group B; Figure 1). (B) In contrast, the Asp66Lys amino acid exchange did not affect the binding of the mAb SB1.6, which belongs to epitope group F.
Author response image 1
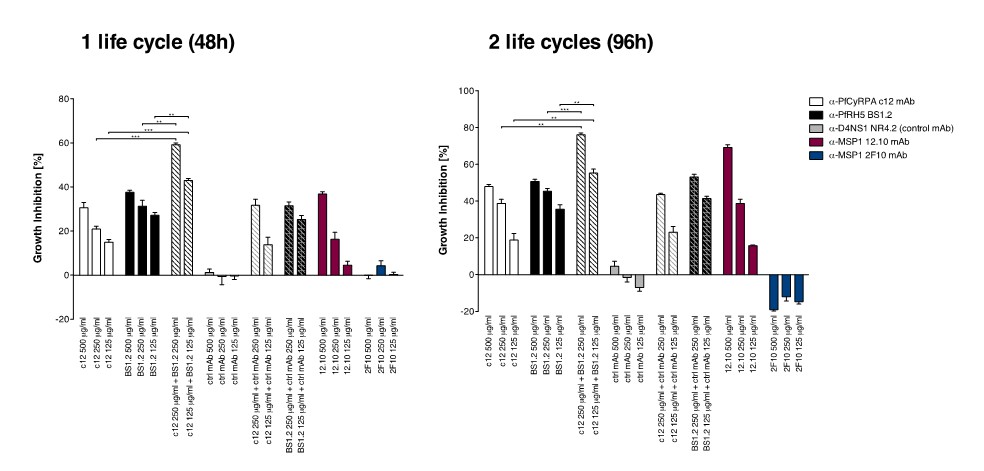
Anti-PfCyRPA and anti-PfRH5 mAbs have in vitro additive inhibitory effect on parasite growth.
Parasites were cultured for one cycle (48h) or two cycles (96h) of merozoite invasion in the presence of the anti-PfCyRPA c12 mAb with or without the anti-PfRH5 BS1.2 mAb at concentrations of 500, 250, and 125 μg/ml. Either mAb showed potent inhibitory activity, reducing parasite growth by ≥30% when tested at a concentration of 500 μg/ml. Inhibitory anti-MSP1 12.10 mAb and non-inhibitory anti-MSP-1 2F10 mAb were included in the assay as reference mAbs. When combining the anti-PfCyRPA c12 mAb with the anti-RH5 BS1.2 mAb, a significantly enhanced inhibitory activity was observed. The functional activity of both mAbs was not enhanced by the addition of the malaria-unrelated control mAb NR4.2. Differences in parasite growth inhibition between mAbs c12 and BS1.2 alone and their combinations are statistically significant (unpaired t test with Welch’s correction, 95% confidence interval, two-tailed p value); differences between mAbs c12 and BS1.2 alone and their combinations with the control mAb NR4.2 are not statistically significant.
Author response image 2
Author response image 3
Author response image 4

Author response image 5
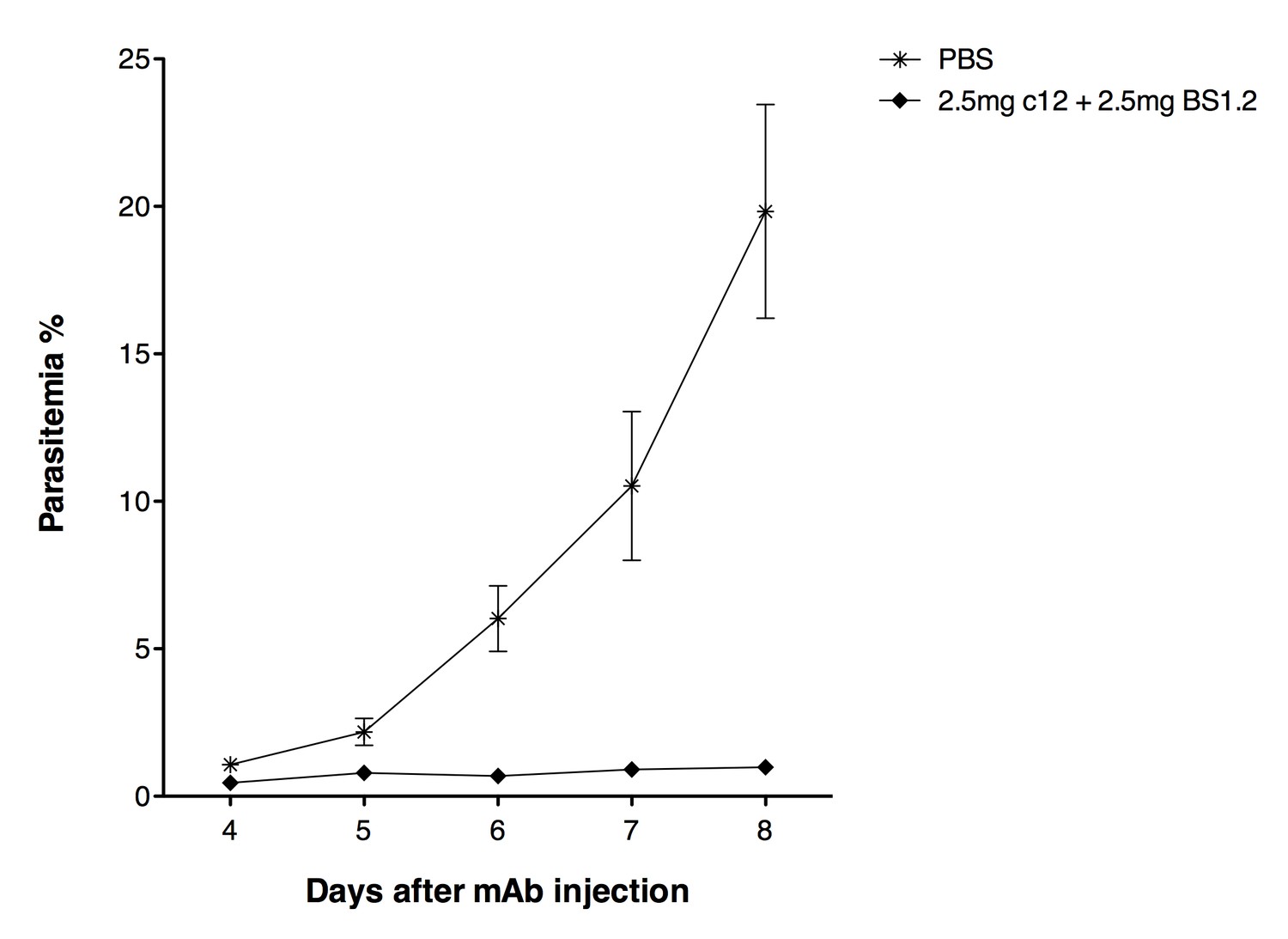
NODscidIL2Rγnullmice received purified 2.5 mg anti-PfCyRPA c12 mAb and 2.5 mg anti-PfRH5 BS1.2 mAb by i.v. injections.
PBS was used as negative control. Mice were then infected with P. falciparum 3D7 and parasitemia was monitored over five days. Values represent the mean parasitemia in human erythrocytes in peripheral blood.
Additional files
-
Supplementary file 1
Data collection and refinement statistics.
- https://doi.org/10.7554/eLife.20383.017
-
Supplementary file 2
Interactions between PfCyRPA and the c12 Fab.
Predicted interactions between PfCyRPA and the CDRs of mAb c12. The amino acid residues involved and the nature of the interactions are listed.
- https://doi.org/10.7554/eLife.20383.018
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structure of the malaria vaccine candidate antigen CyRPA and its complex with a parasite invasion inhibitory antibody
eLife 6:e20383.
https://doi.org/10.7554/eLife.20383




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}