Proteolytic maturation of α2δ represents a checkpoint for activation and neuronal trafficking of latent calcium channels
- University College London, United Kingdom
Abstract
The auxiliary α2δ subunits of voltage-gated calcium channels are extracellular membrane-associated proteins, which are post-translationally cleaved into disulfide-linked polypeptides α2 and δ. We now show, using α2δ constructs containing artificial cleavage sites, that this processing is an essential step permitting voltage-dependent activation of plasma membrane N-type (CaV2.2) calcium channels. Indeed, uncleaved α2δ inhibits native calcium currents in mammalian neurons. By inducing acute cell-surface proteolytic cleavage of α2δ, voltage-dependent activation of channels is promoted, independent from the trafficking role of α2δ. Uncleaved α2δ does not support trafficking of CaV2.2 channel complexes into neuronal processes, and inhibits Ca2+ entry into synaptic boutons, and we can reverse this by controlled intracellular proteolytic cleavage. We propose a model whereby uncleaved α2δ subunits maintain immature calcium channels in an inhibited state. Proteolytic processing of α2δ then permits voltage-dependent activation of the channels, acting as a checkpoint allowing trafficking only of mature calcium channel complexes into neuronal processes.
https://doi.org/10.7554/eLife.21143.001Introduction
The α2δ subunits of voltage-gated calcium channels (CaV) have been identified to be key proteins in synaptic function and synaptogenesis (Dickman et al., 2008; Kurshan et al., 2009; Hoppa et al., 2012; Eroglu et al., 2009; Saheki and Bargmann, 2009). Therefore an understanding of their basic mechanism(s) of action is of paramount importance. Although CaVα1 subunits form the pore and determine the main functional and pharmacological attributes of the channels (Catterall, 2000), the CaV1 and CaV2 channels are associated with auxiliary β and α2δ subunits (Flockerzi et al., 1986; Witcher et al., 1993; Takahashi et al., 1987; Liu et al., 1996). The α2δ subunits increase CaV currents by a mechanism that is less well understood than that of the β subunits, which have a chaperone function (Van Petegem et al., 2004; Buraei and Yang, 2010; Dolphin, 2012; Zamponi et al., 2015). The topology of the α2δ proteins was initially determined for skeletal muscle α2δ-1, but is likely to generalize for all α2δ subunits (Brickley et al., 1995; Gurnett et al., 1996, 1997). A single gene encodes each α2δ protein, which undergoes several post-translational processing steps, including proteolytic cleavage into disulfide-linked α2 and δ (Davies et al., 2010; Jay et al., 1991; Ellis et al., 1988; De Jongh et al., 1990). Both α2 and δ have been shown previously to be important for the function of α2δ-1 to increase CaV currents and influence the biophysical properties of the currents (Gurnett et al., 1996, 1997). The structure of the skeletal muscle CaV1.1 complex has recently been determined at 3.6 Å by cryo-electron microscopy (Wu et al., 2016). It shows the interaction of the α2δ-1 with several extracellular linkers in Domains I-III of CaV1.1. In particular the metal ion adhesion site (MIDAS) motif of the von Willebrand factor-A (VWA) domain interacts with the extracellular loop between transmembrane segments 1 and 2 in Domain I.
Regarding the mechanism of action of α2δ subunits, we have recently shown that α2δ-1 increases the density of CaV2.2 channels inserted into the plasma membrane by about two-fold in undifferentiated neuro2A (N2A) cells (Cassidy et al., 2014); however the increase in currents directly attributable to α2δ-1 is much greater than this (Hoppa et al., 2012). In the present study, we therefore examined whether there was an additional step responsible for promoting CaV2.2 calcium current function by α2δ subunits, in addition to their demonstrated effect on calcium channel trafficking (Cassidy et al., 2014). We have specifically tested the hypothesis that this step involves the proteolytic processing of α2δ-1. Our results show that proteolytic maturation of α2δ subunits is not only a prerequisite for voltage-dependent activation of calcium channels, but is also an essential checkpoint for trafficking of these mature calcium channels into neuronal processes.
Results
Mutation of six amino acids flanking the cleavage site in α2δ-1 prevents proteolytic processing of α2δ-1 into α2 and δ
We identified the proteolytic cleavage site in rat α2δ-1 to be between A and V in the sequence LEA//VEM, by homology with the published data in rabbit (Jay et al., 1991; De Jongh et al., 1990) (Figure 1a,b). This sequence is strongly conserved in mammals (Figure 1—figure supplement 1). We initially manipulated the cleavage site in α2δ-1 (Figure 1b), in order to determine the extent of mutation required across this site required to completely prevent proteolytic processing. Transient expression in cell lines of wild type (WT) α2δ-1 resulted in only partial cleavage into α2 and δ subunits, clearly observed only following deglycosylation (Figure 1c, lanes 1 and 3), compared to the complete proteolytic cleavage observed in brain (Figure 1c, lanes 2 and 4). Replacing LEAVEM in α2δ-1 with a hexavaline (V6) sequence (Figure 1b) completely prevented its proteolytic processing, as shown by the absence of cleaved α2-1 on reducing gels of whole cell lysate (WCL) (Figure 1d). More conservative mutations did not effectively prevent the cleavage (data not shown). However, we found that the lack of proteolytic cleavage did not affect the ability of α2(V6)δ-1 to bind 3H-gabapentin (Figure 1e). Furthermore, the α2(V6)δ-1, like wild-type (WT) α2δ-1, was found to be concentrated in detergent-resistant membranes (DRMs; Figure 1—figure supplement 2), and the KD for 3H-gabapentin binding in DRMs was unchanged, being 76.2 ± 7.8 nM for WT α2δ-1 and 85.0 ± 13.7 nM for α2(V6)δ-1 (Figure 1e), indicating that the uncleaved pro-form of α2δ-1 is likely to be correctly folded.
Figure 1 with 3 supplements see all
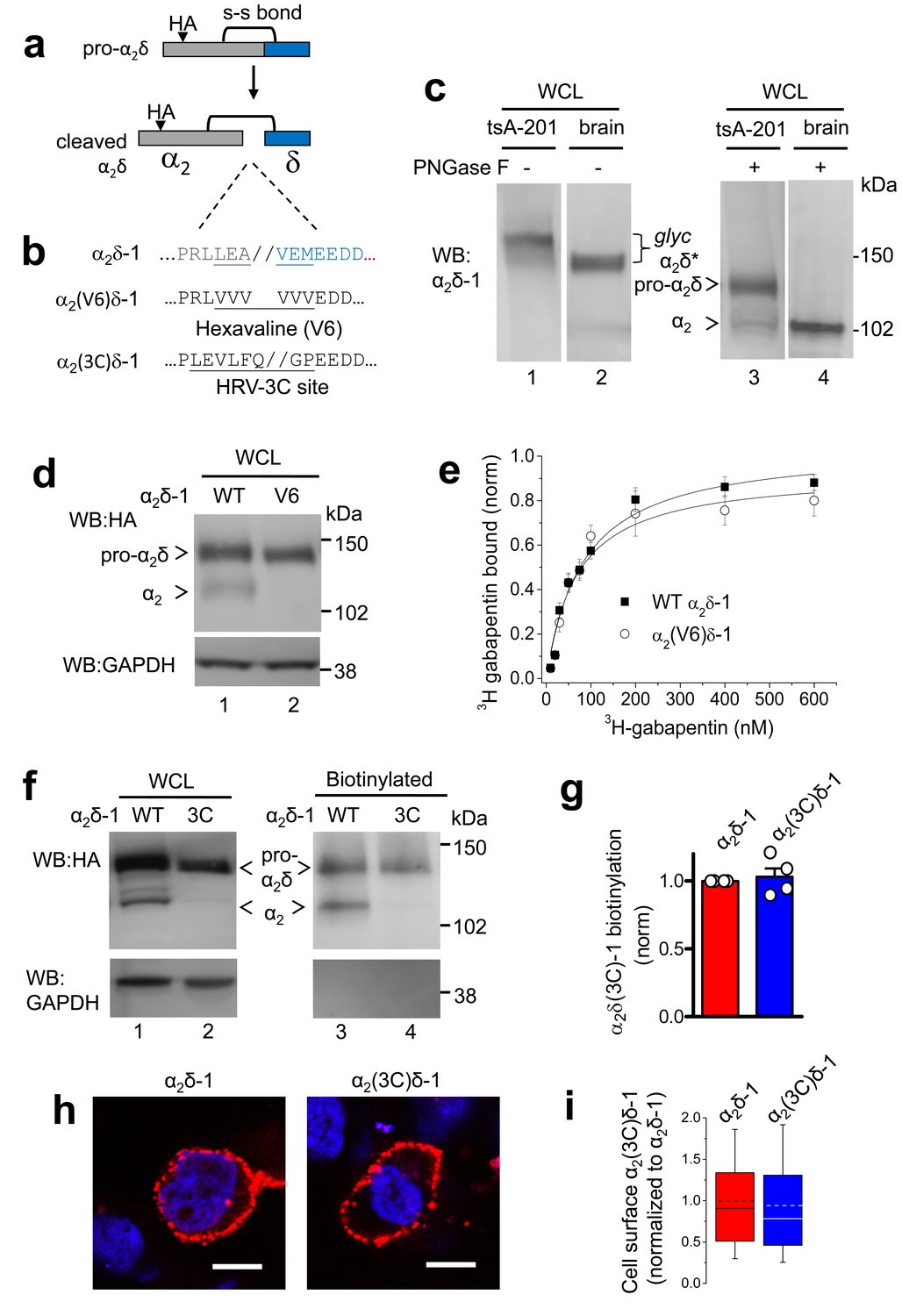
Effect of mutation of α2δ-1 to disrupt the proteolytic cleavage site.
(a) Cartoon of uncleaved pro-α2δ-1 and cleaved α2δ-1, showing the approximate position of inserted HA tag and disulfide bonds between α2 (grey) and δ (blue). (b) Rat α2δ-1 sequence at the identified cleavage-site. The underlined sequence (including LEA//VEM) in α2δ-1 was mutated to a V6 or 3C-protease motif. (c) Comparison of glycosylated (lanes 1, 2) and deglycosylated α2δ-1 (lanes 3, 4), expressed in tsA-201 cells (lanes 1, 3) or present in the brain (lanes 2, 4), showing resolution between pro-α2δ-1 and the cleaved form of α2δ after deglycosylation. α2δ* indicates glycosylated species, and pro-α2δ and α2 indicate deglycosylated proteins. Uncleaved pro-α2δ-1 is observed in transfected cells, but not brain. Proteins visualized with α2-1 Ab. (d) α2δ-1-HA (left) and α2(V6)δ-1-HA (right) expressed in tsA-201 cells; proteins deglycosylated with PNGase-F. Upper panel: HA-blots, lower panel: endogenous GAPDH loading control. (e) Normalized binding curves, using DRM fractions from transfected tsA-201 cells, for 3H-gabapentin binding to WT α2δ-1 (■, n = 4) and α2(V6)δ-1 (○, n = 4). Mean (± SEM) data are fit by hyperbolae with KD of 82.3 and 68.3 nM, respectively. (f) Imunoblot analysis of deglycosylated α2δ-1-HA and α2(3C)δ-1-HA. WCL input (lanes 1, 2) and cell-surface biotinylated material (lanes 3, 4) are shown. Upper panel: HA-blot, lower panel: endogenous GAPDH. (g) Mean ± SEM (and individual data points) of α2(3C)δ-1 (blue) cell surface levels, measured as a proportion of biotinylated: total protein, normalized to control (WT α2δ-1; red), for 4 experiments including that shown in (f). p=0.5057, one sample t test. (h) Cell-surface expression of α2δ-1-HA (left) and α2(3C)δ-1-HA (right) in non-permeabilized tsA-201 cells, using HA Ab (red). Nuclei visualized with DAPI (blue). Scale bars 20 μm. (i) Box and whisker plots for quantification of α2δ-1 on cell-surface, from HA fluorescence, for experiments including (h), for WT α2δ-1 (red) and α2(3C)δ-1 (blue). N = 290 and 317 cells, respectively, from 3 separate transfections, normalized to WT α2δ-1 in each experiment. p=0.259, Student’s t test.
Prevention of proteolytic processing does not impair trafficking of α2δ-1 to the plasma membrane
We then decided to control the proteolytic processing of α2δ-1 by inserting a Human Rhinovirus (HRV) 3C-protease site (Cordingley et al., 1990) into α2δ-1, in place of the endogenous cleavage motif, to form α2(3C)δ-1 (Figure 1b). This substitution completely prevented its proteolytic cleavage (Figure 1f). However, α2(3C)δ-1 was still expressed on the cell surface (Figure 1f–i) and in DRMs (Figure 1—figure supplement 3), at similar levels to WT α2δ-1.
We have previously shown that the amount of proteolytically-processed α2δ-1 is increased on the cell surface compared to the WCL in transfected tsA-201 cells (Kadurin et al., 2012). However, the present results show that this processing of α2δ-1 is not essential for its trafficking to the plasma membrane.
Prevention of proteolytic processing of α2δ-1 abolishes CaV2.2 current enhancement
It has previously been established that co-expression of WT α2δ-1 results in an enhancement of CaV2.2/β1b currents in heterologous systems (for review see [Dolphin, 2012]). We could confirm this result using β1b-GFP to ensure that all cells examined contain β1b; WT α2δ-1 produced an 8.6-fold increase of CaV2.2/β1b currents at +10 mV (Figure 2a,b). Strikingly, no increase was observed when α2(3C)δ-1 was co-expressed with CaV2.2/β1b (Figure 2a,b; Figure 2—figure supplement 1). This is in contrast to the ability of α2(3C)δ-1 to reach the plasma membrane itself. In agreement with this, another uncleavable construct used, α2(V6)δ-1, was also unable to increase CaV2.2 currents (Figure 2—figure supplement 2).
Figure 2 with 3 supplements see all

Effect of mutation of α2δ-1 cleavage site to an HRV-3C site on cell-surface expression and functional properties of CaV2.2.
(a) Example traces (−30 to +10 mV in 5 mV steps) for CaV2.2/β1b-GFP and no α2δ (black traces), WT α2δ-1 (red traces) or α2(3C)δ-1 (blue traces). Charge carrier 1 mM Ba2+. Scale bars refer to all traces. (b) Mean (± SEM) IV curves for CaV2.2/β1b-GFP and no α2δ (black open circles, n = 14), WT α2δ-1 (red squares, n = 34) or α2(3C)δ-1 (blue triangles, n = 21). Gmax: 0.25 ± 0.04, 1.91 ± 0.30 and 0.20 ± 0.03 nS/pF, respectively. V50,act: 2.85 ± 0.68, 3.33 ± 0.46 and 3.89 ± 0.53 mV, respectively. (c) tsA-201 cells transfected with GFP-CaV2.2 (lanes 1 and 3) or GFP (lanes 2 and 4), plus β1b, and either WT α2δ-1 (lanes 1 and 2) or α2(3C)δ-1 (lanes 3 and 4). Immunoprecipitation of GFP-CaV2.2 with anti-GFP Ab; WB with CaV2.2 II-III loop Ab (upper panels, lanes 1 and 3) produced co-immunoprecipitation (lower panels) of WT α2δ-1 (lane 1) and α2(3C)δ-1 (lane 3), revealed by α2δ-1 mAb. Right panels: WCL input for lanes 1–4: upper panels, CaV2.2-GFP input; lower panels, α2δ-1 input. All samples deglycosylated. (d) Immunocytochemical detection of cell-surface expression of CaV2.2-BBS, with β1b, and empty vector (left), WT α2δ-1-HA (middle) or α2(3C)δ-1-HA (right) in N2A cells. Upper panel: CaV2.2-BBS cell-surface staining prior to permeabilization (grey-scale); lower panel: total CaV2.2 after permeabilization (II-III loop Ab, red). Scale bar 5 µm. (e) Quantification of CaV2.2-BBS cell-surface expression (box and whisker plots) with empty vector (open bar, n = 206), WT α2δ-1 (red bar, n = 191) or α2(3C)δ-1 (blue bar, n = 181). Statistical differences: ANOVA and Bonferroni post-hoc test; ***p<0.001, compared to no α2δ. (f) Resting membrane potential of control N2A cells (white bar, n = 16) and following expression of TASK3 (gray bar, n = 12). Box and whisker plots; ***p<0.0001, Student’s t test. (g) Quantification of CaV2.2-BBS cell-surface expression in N2A cells co-expressing TASK3, with empty vector (gray bar, n = 70), WT α2δ-1 (pink bar, n = 73) or α2(3C)δ-1 (pale blue bar, n = 81). Box and whisker plots, statistical differences: ANOVA and Bonferroni post-hoc test, compared to no α2δ; ***p<0.001. (h) Cartoon showing the ability of ‘latent’ CaV2.2 (cyan) plus α2(3C)δ-1 (grey α2, blue δ), to traffic to the plasma membrane (PM).
Proteolytic processing of α2δ-1 is not required for the interaction with CaV2.2, and trafficking of the complex to the plasma membrane in cell lines
We next examined whether α2(3C)δ-1 is impaired in its interaction with CaV2.2, which would explain its inability to increase CaV2.2 currents. However, we found that CaV2.2 co-immunoprecipitates with α2(3C)δ-1 to the same extent as WT α2δ-1 (Figure 2c). Furthermore, using extracellularly-tagged CaV2.2 to quantify the channels inserted into the plasma membrane (Cassidy et al., 2014), we found that in undifferentiated N2A cells, the uncleaved α2(3C)δ-1 remains capable of increasing the cell surface density of CaV2.2 channels (123% increase compared to CaV2.2/β1b alone), by a similar extent to WT α2δ-1 (140% increase; Figure 2d,e). α2(3C)δ-1 also increased cell surface expression of CaV2.2 in the tsA-201 cells used for electrophysiology (Figure 2—figure supplement 3). We then asked whether the depolarized resting membrane potential of N2A cells might influence their ability to traffic CaV2.2. We therefore co-expressed the leak K+ channel, TASK3 (Kim et al., 2000), to hyperpolarize their membrane potential (Figure 2f), in order to determine whether this would differentially affect CaV2.2 trafficking supported by WT α2δ-1 and α2(3C)δ-1. This was not the case, as a similar increase in CaV2.2 surface expression was observed in the presence of TASK3 for CaV2.2, with either WT α2δ-1 or α2(3C)δ-1 (Figure 2g).
Thus, when heterologously expressed in cell lines, α2(3C)δ-1 cause an increase of CaV2.2 surface expression, but does not result in an increase of CaV2.2 currents (see cartoon in Figure 2h), strongly suggesting α2(3C)δ-1 may play an inhibitory role on CaV2.2 function.
Proteolytic processing of α2δ-1 is essential for voltage-dependent activation of CaV2.2 Currents
In order to determine whether α2(3C)δ-1 could be cleaved at the inserted 3C-protease site, we then co-expressed an ER-lumen-targeted 3C-protease together with α2(3C)δ-1 (Figure 3a). This resulted in efficient proteolytic cleavage of α2(3C)δ-1 into α2 and δ, as shown by the appearance of a band of the size of the α2 moiety, which reached the cell surface (Figure 3b; Figure 3—figure supplement 1a). As expected, co-expression of an inactive form of 3C-protease with a mutation in the catalytic site (C147V) failed to affect α2(3C)δ-1 (Figure 3—figure supplement 1b).
Figure 3 with 2 supplements see all
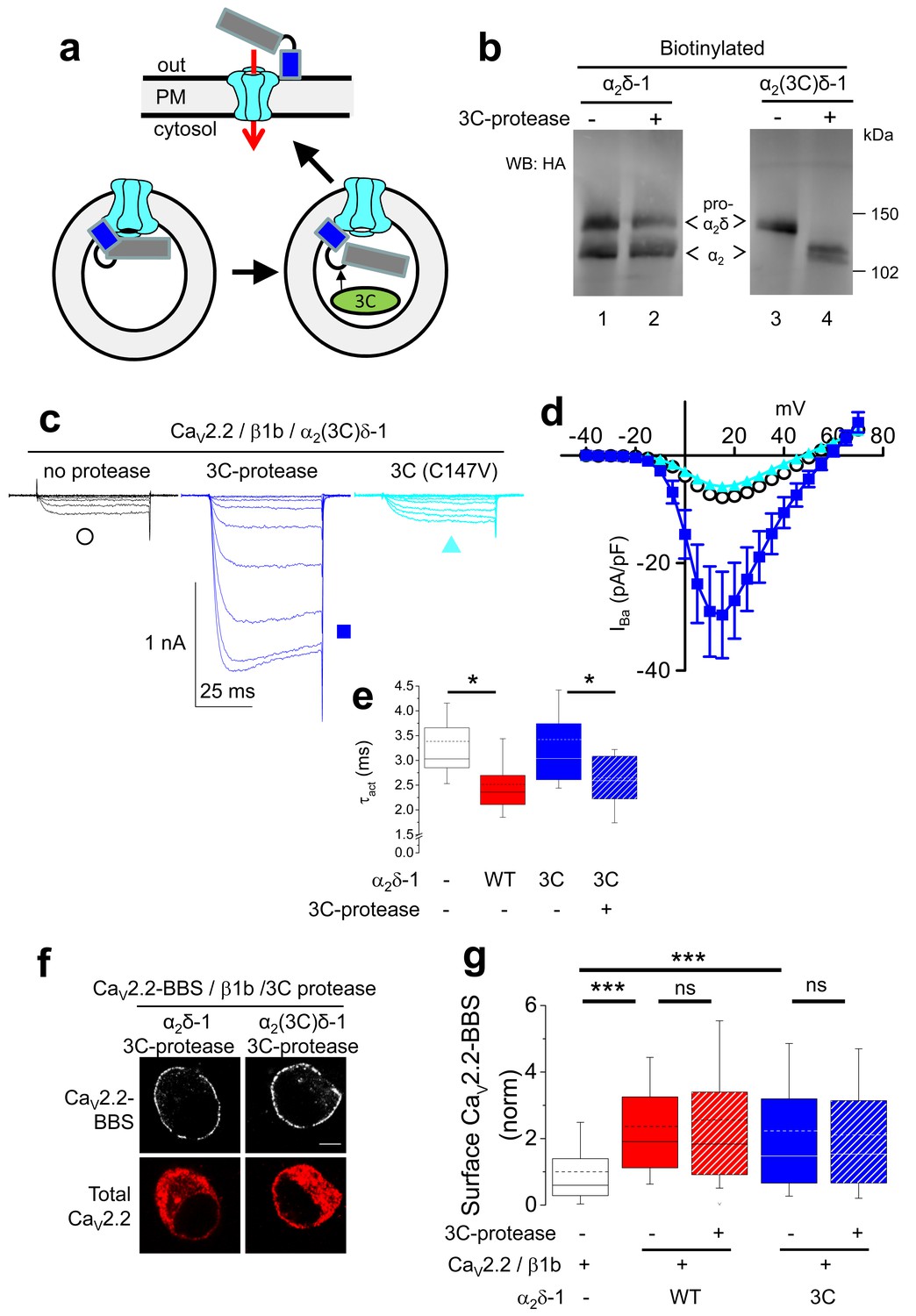
Effect of proteolytic cleavage of α2δ-1 containing an HRV-3C cleavage site on cell-surface expression and functional properties of CaV2.2.
(a) Cartoon showing intracellular cleavage by 3C-protease (green) of α2(3C)δ-1 (gray/blue) associated with CaV2.2 (cyan). (b) Deglycosylated, cell-surface biotinylated fractions for α2δ-1-HA (lanes 1 and 2) and α2(3C)δ-1-HA (lanes 3 and 4), expressed in tsA-201 cells without (lanes 1 and 3) or with (lanes 2 and 4) 3C-protease. Representative of n = 4 experiments. WCL in Figure 3—figure supplement 1 (c) Example traces (−30 to +10 mV steps) for CaV2.2/β1b-GFP/α2(3C)δ-1-HA and no protease (black traces), 3C-protease (blue traces) or inactive mutant 3C-(C147V) protease (cyan traces). Charge carrier 1 mM Ba2+. Scale bars refer to all traces. (d) Mean (± SEM) IV curves for CaV2.2/β1b-GFP/α2(3C)δ-1-HA and no protease (black open circles, n = 26), 3C-protease (blue squares, n = 22) or 3C-(C147V)-protease (cyan triangles, n = 23). Gmax: 0.26 ± 0.04, 0.90 ± 0.22 and 0.22 ± 0.03 nS/pF, respectively. Gmax values in the presence of the active 3C-protease were greater than in the absence of protease or presence of 3C-(C147V)-protease (Kruskal-Wallis test with Dunn’s multiple comparison post-hoc test, p<0.05). V50,act: 6.05 ± 0.82, 5.18 ± 0.74 and 6.20 ± 1.20 mV, respectively. (e) Time constant of activation (τact) for IBa at +10 mV for CaV2.2/β1b-GFP with no α2δ-1 (open bar, n = 14), WT α2δ-1-HA (red bar, n = 25), α2(3C)δ-1-HA (blue bar, n = 21) or α2(3C)δ-1-HA + 3C-protease (blue hatched bar, n = 17). Box and whisker plots. Statistical significance determined by ANOVA and Bonferroni’s post-hoc test (*p<0.05). (f) Immunocytochemical detection of cell-surface CaV2.2-BBS, plus β1b, and α2δ-1-HA (left panel) or α2(3C)δ-1-HA (right panel), with 3C-protease. Upper panel: CaV2.2-BBS cell-surface staining (grey-scale), lower panel total CaV2.2 (II-III loop staining). Scale bar 5 µm. (g) Lack of effect of 3C-protease (hatched bars) on cell-surface expression of CaV2.2-BBS following expression of CaV2.2-BBS/β1b in N2A cells, with no α2δ (open bar, n = 206), WT α2δ-1 (red bar, n = 212), WT α2δ-1 and 3C-protease (red hatched bar, n = 192), α2(3C)δ-1 (blue bar, n = 181) or α2(3C)δ-1 and 3C-protease (blue hatched bar, n = 200). Box and whisker plots. Statistical differences determined by ANOVA and Bonferroni’s post-hoc test (***p<0.001; ns: p>0.05).
Next we addressed the important question of whether proteolytic cleavage by 3C-protease could restore the currents through CaV2.2 channels containing α2(3C)δ-1 subunits. In order to do this, we co-expressed 3C-protease with CaV2.2/β1b/α2(3C)δ-1, and found that α2δ-1-mediated CaV2.2 current enhancement was robustly rescued by the active protease, whereas the inactive mutant 3C-protease (C147V) had no effect (Figure 3c–e). This was not accompanied by any change in CaV2.2 trafficking (Figure 3f,g). The peak IBa increase due to the 3C-protease was 5.4-fold at +10 mV, compared to inactive protease (Figure 3c,d). Co-expression of 3C-protease with α2(3C)δ-1 also resulted in an increased activation rate of the CaV2.2 currents, to the same extent as WT α2δ-1 (Figure 3e). As expected, when α2(3C)δ-1 was not present, there was no increase in CaV2.2 currents attributable to 3C-protease (Figure 3—figure supplement 2).
In summary, expression of active, but not the inactive form of 3C-protease results in cleavage of α2(3C)δ-1 and rescues enhancement of CaV2.2 currents by α2(3C)δ-1, without any effect on trafficking, providing evidence that proteolytic processing of α2δ-1 is required to promote voltage-dependent activation CaV2.2 channels.
Replacement of the endogenous proteolytic site in WT α2δ-3 with a 3C-protease site allows controlled processing of α2δ-3 to rescue CaV2.2 currents
In order to further distinguish between the effects of α2δ subunits on CaV2.2 channel trafficking and voltage-dependent activation, we also examined the behavior of α2δ-3, because we surmised it might have a less prominent trafficking effect, as it contains an incomplete MIDAS motif (Whittaker and Hynes, 2002) in its VWA domain, which we have shown is essential for the trafficking and function of α2δ-1 and α2δ-2 (Hoppa et al., 2012; Cassidy et al., 2014; Canti et al., 2005). Indeed, we found that α2δ-3 produced a much smaller increase than α2δ-1 (31%, compared to ~140%) on CaV2.2 cell surface expression (Figure 4a,b), which we attribute to the absence of a complete MIDAS motif in α2δ-3 (Whittaker and Hynes, 2002).
Figure 4 with 3 supplements see all
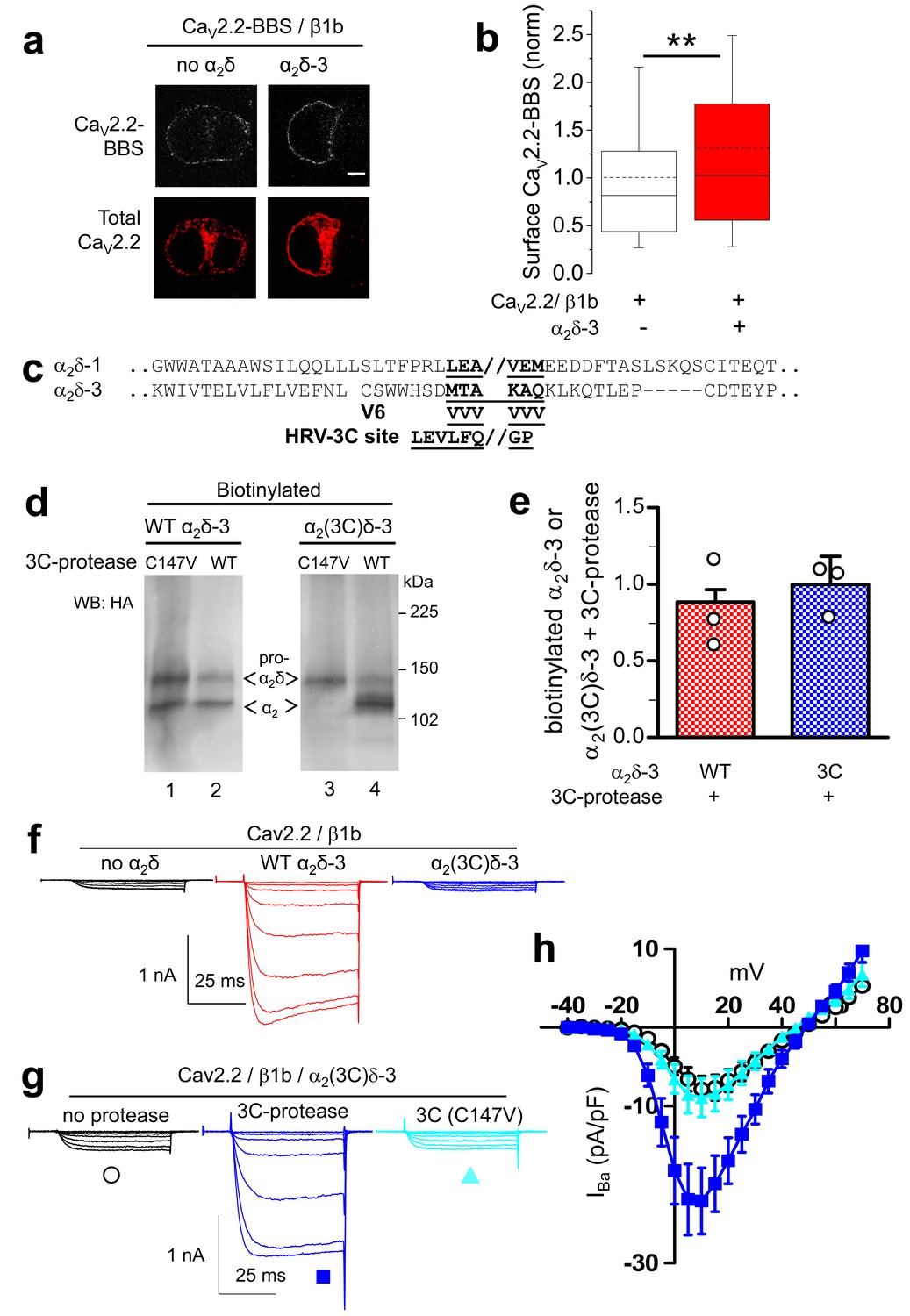
Effect of proteolytic cleavage of α2δ-3 containing an HRV-3C cleavage site on cell-surface expression and functional properties of CaV2.2.
(a) Images showing cell-surface CaV2.2-BBS (upper row, white), and total CaV2.2 (II-III loop Ab, lower row, red), for CaV2.2-BBS/β1b in N2A cells, with empty vector (panel 1) or α2δ-3-HA (panel 2). Scale bar 5 µm. (b) Quantification (box and whisker plots) of effect α2δ-3 on cell-surface CaV2.2-BBS following expression of CaV2.2-BBS/β1b with empty vector (open bar, n = 188) or WT α2δ-3 (red bar, n = 188). Statistical difference determined by Student’s t test, **p=0.0028. (c) Alignment of α2δ-3 sequence around the predicted cleavage site with α2δ-1, showing weak homology. Underlined sequence (MTAKAQ) mutated to V6 or HRV-3C cleavage motif. (d) α2δ-3-HA (lanes 1, 2) and α2(3C)δ-3-HA (lanes 3, 4) expressed in tsA-201 cells, with either inactive (C147V, lanes 1, 3) or WT 3C-protease (WT, lanes 2, 4), cell-surface biotinylated and deglycosylated. Full WB and corresponding WCL in Figure 4—figure supplement 3 (e) Quantification of cell-surface expression of WT α2δ-3-HA (red speckled bar) and α2(3C)δ-3-HA (blue speckled bar), with 3C-protease, normalized relative to inactive 3C-protease (C147V) for n = 3 experiments. Data are mean (± SEM) and individual data points: p=0.4721 for WT α2δ-3-HA and p=0.9513 for α2(3C)δ-3 (1 sample t-test compared to respective control). (f) Example traces (−30 to +5 mV steps) for CaV2.2/β1b-GFP with no α2δ (black traces), WT α2δ-3 (red traces) or α2(3C)δ-3 (blue traces). Gmax: 0.24 ± 0.03, 1.46 ± 0.22 and 0.21 ± 0.03 nS/pF respectively. V50,act: 0.91 ± 1.015, 1.01 ± 0.85 and 4.03 ± 1.04 mV, respectively. (g) Example traces (−30 to +10 mV steps) for CaV2.2/β1b-GFP/α2(3C)δ-3 and no protease (black traces), 3C-protease (blue traces) or inactive 3C-protease (C147V) (cyan traces). For (f) and (g), charge carrier 1 mM Ba2+, scale bars refer to all traces. (h) Mean (± SEM) IV curves for CaV2.2/β1b-GFP/α2(3C)δ-3 without protease (black open circles, n = 28), with 3C-protease (blue squares, n = 29) or inactive 3C-(C147V)-protease (cyan triangles, n = 24). Gmax: 0.28 ± 0.05, 0.70 ± 0.11 and 0.30 ± 0.08 nS/pF, respectively. Gmax for 3C-protease condition larger than other two conditions (Kruskal-Wallis test with Dunn’s post-hoc test, p<0.05). V50, act: 4.0 ± 0.7, 0.3 ± 0.7 and 1.5 ± 0.6 mV, respectively.
The site of proteolytic cleavage in α2δ-3 has not been previously determined, although it is fully cleaved into α2 and δ in vivo (Davies et al., 2010), and enhances CaV2.2 currents (Davies et al., 2010). We therefore first identified a potential cleavage site in α2δ-3 by sequence alignment (Figure 4c), and then showed by mutational analysis that a V6 mutation across the predicted cleavage site prevented proteolytic cleavage, determined by the appearance of the δ-3 moiety in a reducing gel (Figure 4—figure supplement 1), and also prevented the functional effects of α2δ-3 on CaV2.2 currents (data not shown). We then replaced this sequence in α2δ-3 with the HRV-3C motif, to form α2(3C)δ-3 (Figure 4c). We found that α2(3C)δ-3 was expressed on the cell surface to a similar extent to WT α2δ-3 (Figure 4d), despite the complete absence of proteolytic cleavage (Figure 4d, lane 3). Importantly, when the 3C-protease was co-expressed, we found that α2(3C)δ-3 was still present on the cell surface, and was almost completely cleaved at the inserted HRV-3C site (Figure 4d, lane 4), although this had no effect on WT α2δ-3 (Figure 4d,e; Figure 4—figure supplement 2a,b). As we also found for α2(3C)δ-1, α2(3C)δ-3 did not increase CaV2.2 currents, whereas WT α2δ-3 produced a 6.6-fold increase (Figure 4f). However, inducing proteolytic cleavage of α2(3C)δ-3 by 3C-protease substantially rescued the enhancement of CaV2.2 currents, whereas the mutant protease 3C (C147V) did not (Figure 4g,h). The increase in peak IBa due to the 3C protease was 2.7-fold at +10 mV, compared to inactive protease.
In summary WT α2δ-3 has a much smaller effect on CaV2.2 cell surface trafficking than WT α2δ-1, but proteolytic cleavage of α2(3C)δ-3 still produces a substantial increase in CaV2.2 currents.
Proteolytic processing of α2δ subunits on the cell surface leads to rescue of CaV2.2 Currents
In order to further probe the role of proteolytic processing of the α2δ subunits on calcium channel currents, independent of their trafficking effects, we next turned to application of extracellular protease. However, we found that incubation of cells expressing α2(3C)δ-1 or α2(3C)δ-3 with purified 3C-protease did not result in their proteolytic cleavage at the plasma membrane (Figure 4—figure supplement 3, and data not shown). As an alternative approach, we inserted a thrombin cleavage site into α2δ-3 to produce α2(Th)δ-3 (Figure 5a,b). Importantly, we first showed that extracellular thrombin did not cleave WT α2δ-3 (Figure 5—figure supplement 1), and we found that α2(Th)δ-3 reached the cell surface as an uncleaved protein (Figure 5c,d; Figure 5—figure supplement 1). Furthermore, application of thrombin in the extracellular medium then produced marked cleavage of α2(Th)δ-3 on the cell surface (Figure 5c), and the optimal incubation period was about 60 min (Figure 5—figure supplement 1). This experiment was not attempted for α2δ-1 as preliminary studies revealed that α2δ-1 contained an ectopic thrombin cleavage site (data not shown).
Figure 5 with 1 supplement see all
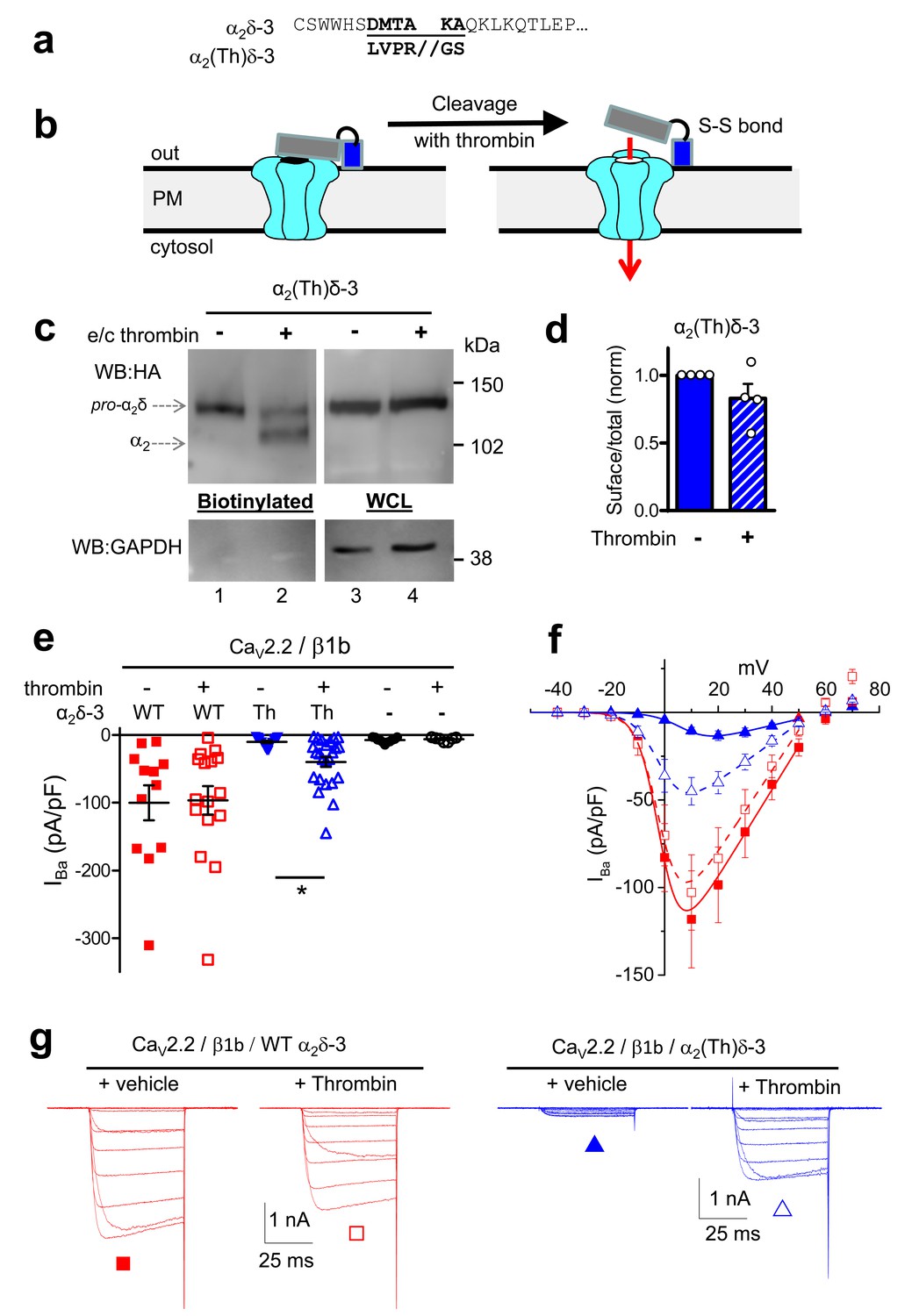
Effect of thrombin on the properties and function of α2δ-3 containing a thrombin proteolytic cleavage site.
(a) Sequence at α2δ-3 cleavage site mutated to a thrombin cleavage site. (b) Cartoon of thrombin cleavage of α2δ-3 on cell-surface. (c) Cell-surface biotinylation (left panel) shows efficient cleavage of cell-surface α2(Th)δ-3-HA (lane 2), with no effect on total α2(Th)δ-3-HA in WCL (right panel, lane 4). Samples were deglycosylated prior to loading. (d) Quantification of cell surface biotinylation experiments such as those shown in (c), indicating that thrombin does not decrease the amount of α2(Th)δ-3-HA on the cell surface (hatched blue bar), normalized to vehicle application in each experiment (solid blue bar). Mean (± SEM) and individual data points for n = 4; p=0.2105, 1 sample t test. (e) Mean (± SEM) and individual data points of peak IBa at +10 mV, for CaV2.2/β1b with WT α2δ-3 (red squares), α2(Th)δ-3 (blue triangles) or no α2δ (black circles) and either no protease (closed symbols), or 60 min thrombin incubation (open symbols). The charge carrier was 2 mM Ba2+. For data without or with thrombin, respectively, n = 12, 16 for WT α2δ-3; 15, 24 for α2(Th)δ-3 and 11, 7 without α2δ, from at least 3 independent transfections. Statistical difference between thrombin and vehicle determined by Kruskal-Wallis ANOVA and Dunn’s multiple comparison post-hoc test, *p<0.05. (f) Mean (± SEM) full IV curves for the same conditions as in (e) (excluding the no α2δ data), fitted with a modified Boltzmann equation to +50 mV. Gmax values (nS/pF) were 2.80 ± 0.61 (WT α2δ-3; n = 10), 2.60 ± 0.55 (WT α2δ-3 plus thrombin, n = 15), 0.43 ± 0.08 (α2(Th)δ-3; n = 14), 1.26 ± 0.21 (α2(Th)δ-3 plus thrombin, n = 21). V50,act values (mV) were +0.44 ± 1.71 (WT α2δ-3), + 0.55 ± 1.28 (WT α2δ-3 plus thrombin), +9.34 ± 0.08 (α2(Th)δ-3), +0.87 ± 1.38 (α2(Th)δ-3 plus thrombin). (g) Example Ba2+ currents (from −50 to +60 mV) for the four conditions shown in (f).
In line with previous results, uncleaved α2(Th)δ-3 failed to mediate CaV2.2 current enhancement (Figure 5e). However, when thrombin was added to cells expressing CaV2.2/β1b and α2(Th)δ-3, prior to Ba2+ current recording, it resulted in an acute increase in IBa amplitude (Figure 5e–g), clearly demonstrating that cleavage of α2(Th)δ-3 has a direct effect to rescue CaV2.2/β1b/α2(Th)δ-3 currents. In contrast, extracellular application of thrombin had no effect on CaV2.2/β1b/WT α2δ-3 or CaV2.2/β1b currents (Figure 5e–g). Therefore, the functional role of α2δ-3 on CaV2.2 channels can be acutely restored at the plasma membrane via specific proteolytic cleavage of extracellular α2(Th)δ-3.
Identification of the subcellular location of physiological cleavage of α2δ-1 subunits in DRG neurons
The α2δ-1 protein is extensively up-regulated in DRG neurons, following experimental peripheral nerve injury, such as spinal nerve ligation (SNL), and is trafficked to presynaptic terminals along the axon in intracellular trafficking vesicles (Bauer et al., 2009). This up-regulation allowed us to obtain sufficient protein to follow the processing of α2δ-1. Following dissection of DRGs and associated axons, we observed that α2δ-1 was fully glycosylated in the cell bodies of the DRGs, but remained only partially cleaved into α2 and δ (Figure 6a, tissue segment 3). We have shown previously by electron microscopy that within the cell body, α2δ-1 is present both in the endoplasmic reticulum and at the plasma membrane, whereas in axons it is entirely associated with intracellular transport vesicles (Bauer et al., 2009). In the axons, we observed that proteolytic cleavage of α2δ-1 was complete, both in the spinal nerve (Figure 6a, tissue segments 1, 2), and in the dorsal roots (Figure 6a, tissue segments 4, 5). This finding implies that all α2δ-1 is proteolytically processed prior to intracellular trafficking into DRG axons. Indeed, our biochemical and cell fractionation data indicate that proteolytic cleavage occurs mainly during trafficking to the plasma membrane (Kadurin et al., 2012) (data not shown). However, this result opens the possibility that uncleaved α2δ-1 may reach the cell surface in DRGs cell bodies and have a functional role there.
Figure 6
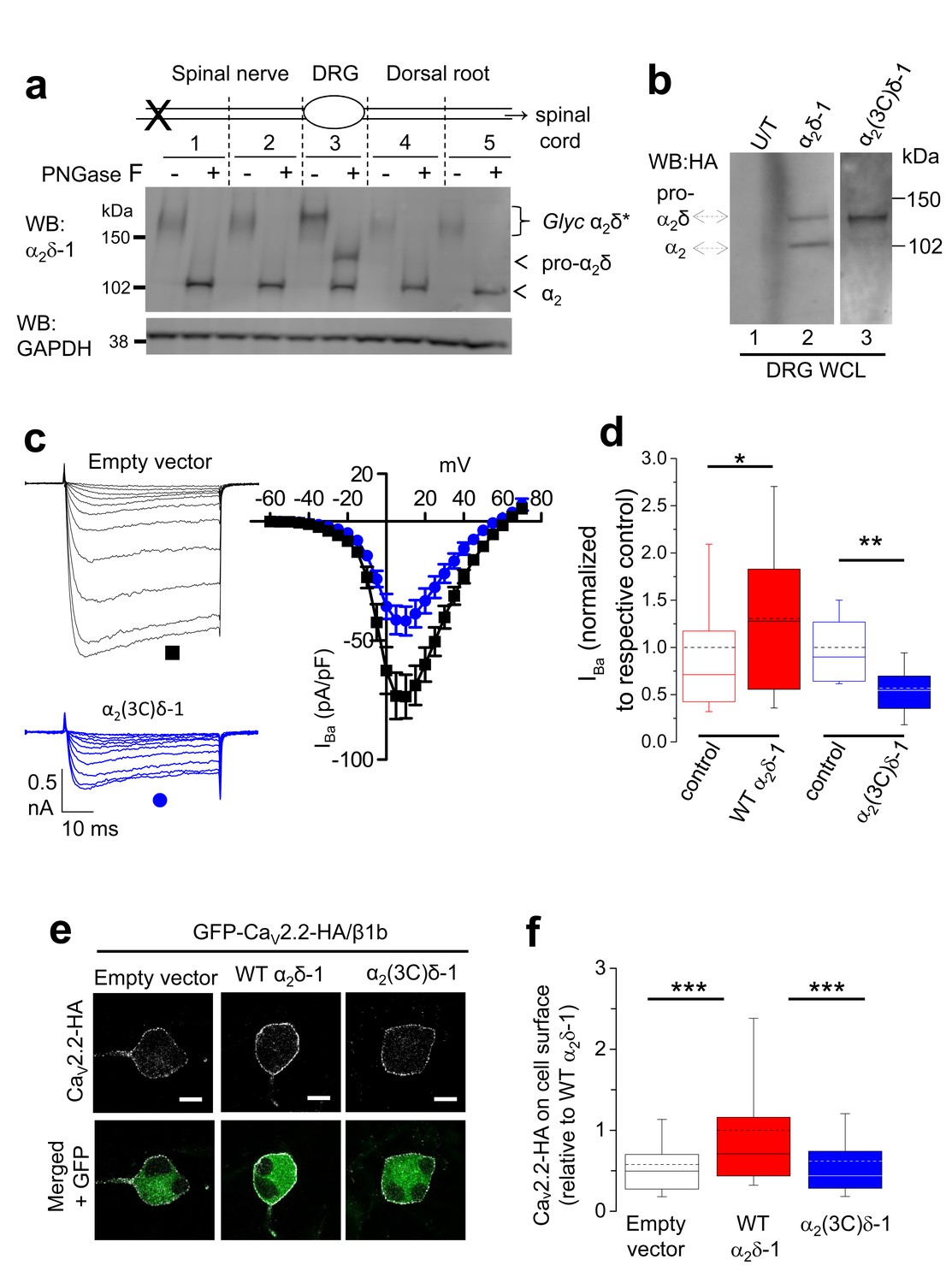
Proteolytic processing of endogenous α2δ-1 and effect of exogenous expression of α2δ-1 and α2(3C)δ-1 in DRGs.
(a) DRGs, spinal nerves and dorsal roots from rats, 4 days after SNL, were dissected and segmented according to the diagram. X marks site of ligation. Tissue was pooled from 4 rats. It was either treated or not with PNGase-F as indicated, and reduced with DTT; deglycosylation allows resolution of two α2-immunoreactive bands. Unprocessed α2δ-1 is present only in the cell body compartment (segment 3) and is distinct from processed α2-1 (indicated by arrows). Lower blot is GAPDH loading control. (b) WCL for empty vector-transfected DRGs (U/T, lane 1); WT α2δ-1-HA-transfected DRGs (lane 2); α2(3C)δ-1-HA transfected DRGs (lane 3). WB: anti-HA. (c) Left: Example traces (−45 to +5 mV steps) for control (empty vector-transfected) DRG neurons (black traces, top) and DRGs transfected with α2(3C)δ-1 (blue traces, bottom). The charge carrier is 1 mM Ba2+. The scale bars refer to all traces. Right: Mean (± SEM) IV curves for control DRG neurons (black squares, n = 12) and DRGs transfected with α2(3C)δ-1 (blue circles, n = 14), from 3 independent experiments. Gmax values were 2.20 ± 0.30 and 1.26 ± 0.14 nS/pF, respectively; p=0.0061 (Student’s t test). V50, act values were −1.5 ± 1.2 and −1.5 ± 0.7 mV, respectively. (d) Comparison of normalized peak IBa in control DRGs (open red bar, n = 55) and WT α2δ-1-transfected DRGs (closed red bar, n = 54) including data from Figure 2d in, or comparison of control DRGs (open blue bar, n = 12) with α2(3C)δ-1 transfected DRGs (closed blue bar, n = 14). Box and whisker plots. Statistical differences, Student’s t test: *, p=0.048, **p=0.0067 compared to respective control. (e) Confocal optical sections (1 µm) showing GFP-CaV2.2-HA in non-permeabilized DRG neurons (top, white), when co-transfected with β1b and either empty vector (left), WT α2δ-1 (middle) or α2(3C)δ-1 (right). GFP fluorescence is shown in the merged lower panel. Scale bars: 10 µm. (f) Box and whisker plot of cell surface HA fluorescence density as a ratio of internal GFP density for CaV2.2-HA expression in DRG somata, transfected with empty vector (open bar, n = 81), WT α2δ-1 (red bar, n = 133) or α2(3C)δ-1 (blue bar, n = 159). ***p<0.001, 1 way ANOVA and Bonferroni post hoc test.
We then expressed WT α2δ-1 in cultured DRG neurons in order to mimic its upregulation in the neuropathic state, and detected a significant amount of pro-α2δ-1 in WCL (Figure 6b, lane 2). As expected, transfection of α2(3C)δ-1 into DRG neurons resulted in abundant expression of the pro-α2(3C)δ-1 form in WCL (Figure 6b, lane 3).
Uncleaved pro-α2δ-1 plays an inhibitory role in the calcium channel function
We then investigated whether uncleaved α2δ-1 was inactive, or whether its presence might play an inhibitory role, as suggested by the fact that α2(3C)δ-1 increases cell surface expression of CaV2.2 by about 2.5-fold in cell lines, but does not support any increase of CaV2.2 currents. We therefore examined the effect of α2(3C)δ-1 expressed in cultured DRG neurons on native calcium channel currents (Figure 6c). We have previously shown that expression of WT α2δ-1 in cultured DRG neurons caused an increase of native calcium currents (D'Arco et al., 2015) (shown with additional data in Figure 6d). Here, we found that expression of α2(3C)δ-1 produced a marked reduction in endogenous calcium channel currents in DRG neurons (by 43.2% at +10 mV; Figure 6c,d), supporting the hypothesis that pro-α2δ-1 inhibits endogenous DRG calcium currents.
Furthermore, when CaV2.2-HA was transfected with β1b and WT α2δ-1 into DRG neurons, there was a clear increase in CaV2.2-HA on the cell surface of the DRG cell bodies after 2 days, compared to CaV2.2-HA/β1b alone (Figure 6e,f). In contrast, for co-expression with α2(3C)δ-1, the amount of CaV2.2-HA on the plasma membrane was the same as without exogenous α2δ, rather than showing any inhibition (Figure 6e,f). This suggests that the inhibitory effect of α2(3C)δ-1 on DRG calcium currents (Figure 6c,d) is not due to inhibition of channel trafficking to the somatic plasma membrane.
Proteolytic cleavage of α2δ-1 is required for trafficking of CaV2.2 Into neuronal processes
In view of our finding that all endogenous α2δ-1 is proteolytically cleaved in axons but not cell bodies (Figure 6a), we then asked whether proteolytic processing was required for trafficking either of α2δ-1 itself, or of associated CaV2.2, into neuronal processes. For this purpose, we used cultured hippocampal neurons which can be transfected after the establishment of extensive neurite outgrowth in culture. We found the surprising result that, when we co-transfected CaV2.2/β1b with either WT α2δ-1, α2(3C)δ-1 or the corresponding empty vector, WT α2δ-1 was essential for trafficking of CaV2.2 into the processes of hippocampal neurons, whereas in the presence of α2(3C)δ-1, or without α2δ, there was virtually no trafficking of CaV2.2 out of the soma (Figure 7a,b). Strikingly, the inability of α2(3C)δ-1 to drive CaV2.2 into neuronal processes was reversed by co-expression of 3C-protease (Figure 7c,d).
Figure 7 with 1 supplement see all
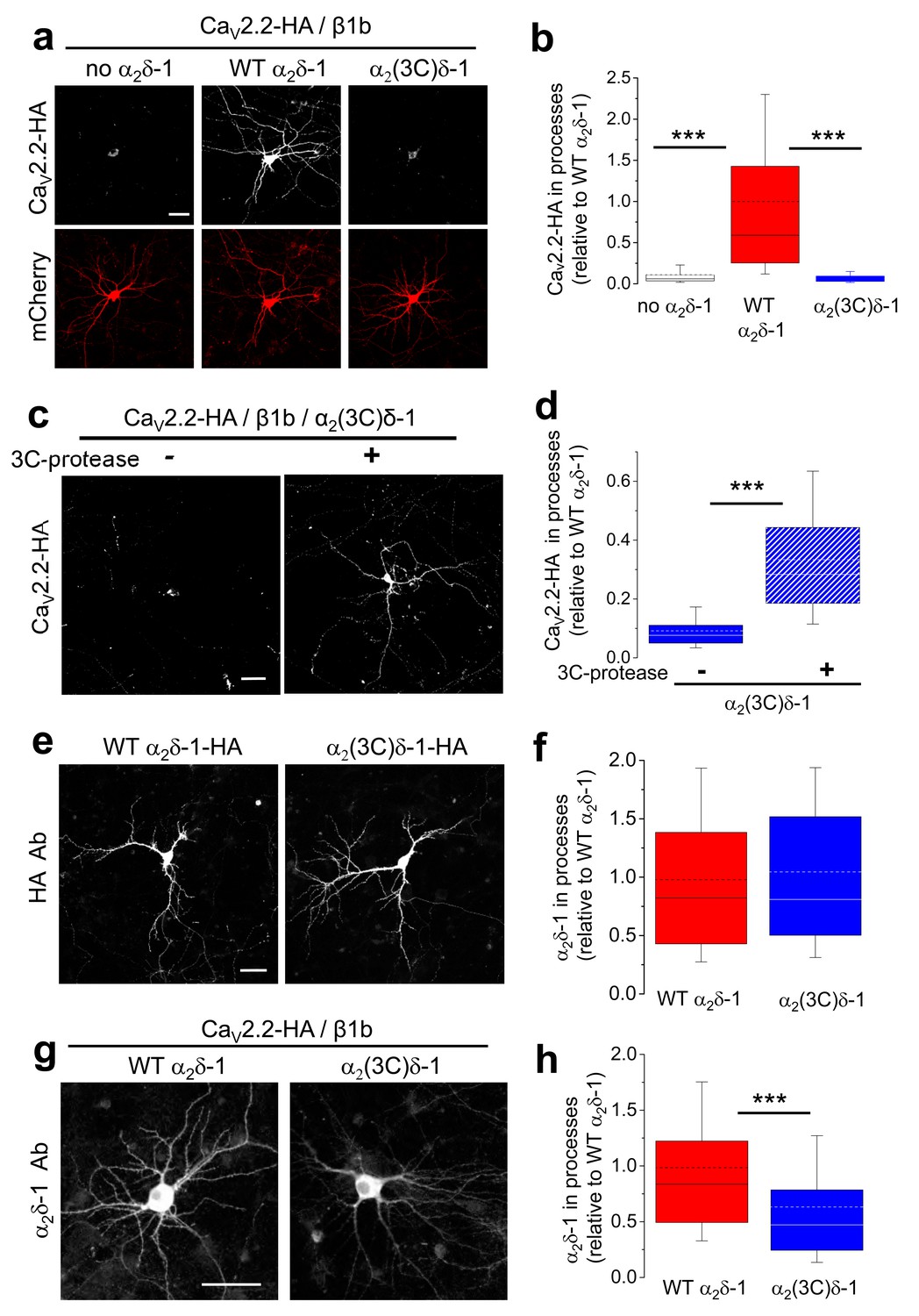
Effect of α2δ-1 and proteolytic cleavage of α2(3C)δ-1 on trafficking of CaV2.2 into hippocampal neurites.
(a) Images showing CaV2.2-HA in permeabilized hippocampal neurons (top, white), when co-transfected with β1b, mCherry (bottom, red) and either no α2δ (left), WT α2δ-1 (middle) or α2(3C)δ-1 (right). Scale bar: 50 µm applies to all images. (b) Box and whisker plots for CaV2.2-HA expression in processes without α2δ (open bar, n = 136 processes from 29 cells), with α2δ-1 (red bar, n = 147 processes from 27 cells) or with α2(3C)δ-1 (blue bar, n = 109 processes from 22 cells). ***p<0.001, 1 way ANOVA and Bonferroni post hoc test. (c) Images showing CaV2.2-HA in permeabilized hippocampal neurons (white), when co-transfected with β1b, α2(3C)δ-1 and mCherry (transfection marker, not shown), either without (left) or with (right) 3C-protease. Scale bar: 50 µm applies to both images. (d) Box and whisker plots for CaV2.2-HA expression in processes with α2(3C)δ-1, transfected without (solid blue bar, n = 191 processes), or with 3C-protease (blue hatched bar, n = 187 processes). ***p<0.001, 1 way ANOVA and Bonferroni post hoc test. (e) Images showing WT α2δ-1-HA (left) or α2(3C)δ-1-HA (right) expressed in permeabilized hippocampal neurons (white), co-transfected only with mCherry (transfection marker, not shown). Antigen retrieval was used prior to the HA Ab. Scale bar: 50 µm applies to both images. (f) Box and whisker plots for expression in the processes of WT α2δ-1-HA (red bar, n = 248 processes from 52 cells) and α2(3C)δ-1 (blue bar, n = 263 processes from 51 cells). (g) Images showing α2δ-1 in hippocampal neurons (white), when transfected with CaV2.2-HA, β1b, mCherry (transfection marker, not shown) and either WT α2δ-1 (left) or α2(3C)δ-1 (right). Antigen retrieval was used prior to the α2δ-1 mAb. Scale bar: 50 µm applies to both images. (h) Box and whisker plots for α2δ-1 expression in hippocampal processes, for WT α2δ-1 (red bar, n = 221 processes) and α2(3C)δ-1 (blue bar, n = 184 processes). ***p<0.0001, Student’s t test.
The failure of CaV2.2 to traffic into the neurites in the presence of α2(3C)δ-1 was not primarily due to a defect in the trafficking of the uncleaved α2δ subunit itself, since, when the α2δ subunits were transfected alone, WT α2δ-1 and α2(3C)δ-1 were both able to extensively access the hippocampal neurite compartment (Figure 7e,f). In contrast, when the localization of the α2δ subunits was examined following co-transfection with CaV2.2/β1b, the trafficking of α2(3C)δ-1 into neurites was 37% lower than that of WT α2δ-1 (Figure 7g,h), suggesting it has been retained by interaction with CaV2.2. Similar results to those with α2δ-1 were obtained when comparing the ability of WT α2δ-3 and α2(3C)δ-3 to traffic CaV2.2 into neurites, which was also reversed by 3C-protease (data not shown). This further confirmed that the association of CaV2.2 with mature processed α2δ subunits is essential for trafficking of the complex into hippocampal neurites (Figure 7—figure supplement 1).
Proteolytic cleavage of α2(3C)δ-1 enhances Ca2+ entry into hippocampal boutons
We then examined the effect of expression of α2(3C)δ-1 alone in hippocampal neurons on action potential (AP)-induced Ca2+ entry into presynaptic boutons. These were identified by co-expressed vesicle-associated membrane protein-mOrange2 (VAMP-mOr2), and Ca2+ transients were measured using synaptophysin-GCaMP6f (sy-GCaMP6f, Figure 8a). The uncleaved α2(3C)δ-1 reduced Ca2+ entry in response to single AP stimulation in all experiments performed (Figure 8b,c), to 65.8 ± 9.1% (n = 7, p=0.0093, 1 sample t test) of control. The inhibitory effect of α2(3C)δ-1 on the response to a single AP was reversed by co-expression of 3C-protease in all experiments performed (Figure 8d,e), an increase of 81.5 ± 16.1% (n = 8, p<0.0001, 1 sample t test).
Figure 8
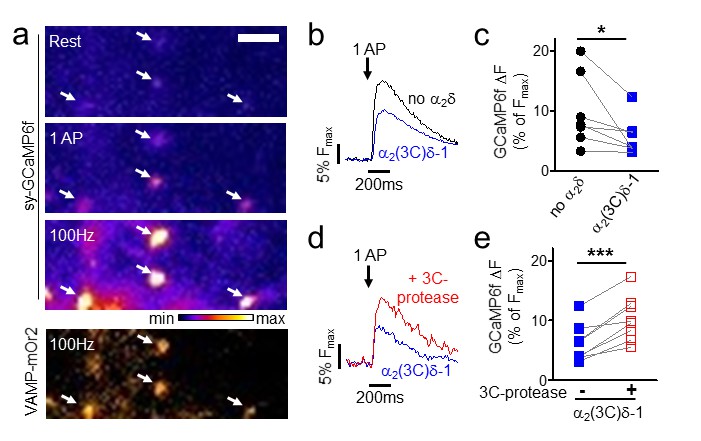
Effect of proteolytic cleavage of α2(3C)δ-1 on Ca2+ influx in presynaptic terminals of hippocampal neurons.
(a) Fluorescence changes in presynaptic terminals of hippocampal neurons expressing sy-GCaMP6f and VAMP-mOr2 in response to electrical stimulation. White arrows point to transfected boutons. Top three panels show sy-GCaMP6f fluorescence: at rest (top), after 1 AP (middle) and after 100 Hz stimulation for 1 s (bottom). The bottom panel shows VAMP-mOr2 fluorescence after 100 Hz stimulation for 1 s. Scale bar 5 µm. The pseudocolour scale is shown below the third panel. (b) Mean example traces from the same experiment of sy-GCaMP6f fluorescence changes in response to 5 single APs from individual presynaptic terminals of neurons co-transfected with either empty vector (black trace) or α2(3C)δ-1 (blue trace). (c) Sy-GCaMP6f fluorescence changes (expressed as % of Fmax) in response to 1 AP from boutons co-transfected with either empty vector (black filled circles) or α2(3C)δ-1 (blue filled squares) (n = 7 independent experiments, *p=0.049, paired t test). (d) Mean example traces of sy-GCaMP6f fluorescence changes in response to 5 single APs from presynaptic terminal of neurons co-transfected with either α2(3C)δ-1 (blue trace) or α2(3C)δ−1 + 3C-protease (red trace). (e) Sy-GCaMP6f fluorescence changes (expressed as % of Fmax) in response to 1 AP from boutons co-transfected with either α2(3C)δ-1 (blue filled squares) or α2(3C)δ−1 + 3C-protease (red open squares) (n = 8 independent experiments, ***p=0.0005, paired t test).
Discussion
In this study, we have identified key roles for proteolytic processing of pro-α2δ subunits. Our main findings are: firstly that proteolytic cleavage of pro-α2δ represents an essential step for the expression of mature functional calcium channels. Here we show, using a combination of techniques, that the appearance of functional CaV2.2 channels associated with the proteolytic processing of α2δ subunits, occurs independently of changes in channel trafficking to the plasma membrane in undifferentiated cell lines. A key experiment is the demonstration that acute restoration of voltage-dependent activation by proteolytic cleavage of pro-α2δ can be induced at the cell surface by extracellularly applied protease. Secondly, we find that lack of proteolytic cleavage of α2δ-1 represents a very significant barrier to trafficking of CaV2.2 channels into cultured hippocampal neuronal processes. Trafficking of the channel complex into neurites is dependent on an effect of mature cleaved α2δ on CaV2.2, rather than due primarily to the differential ability of mature α2δ to be trafficked and uncleaved pro-α2δ to be retained in the soma. This is clearly shown by the fact that uncleaved α2(3C)δ-1 is fully able to traffic alone into neurites. Thirdly, we provide evidence for an inhibitory role for the pro-form of α2δ-1. It is highly likely that proteolytic cleavage of α2δ could induce a conformational change, which would impact on its interaction with the α1subunit. The recent structure of the CaV1.1 complex resolves the domain structure of the α2δ-1 subunit to contain the VWA domain and four tandem cache domains (Wu et al., 2016). The δ subunit contributes part of the fourth cache domain, and it is therefore possible that the domain organization would be modified by proteolytic cleavage of α2δ.
In a previous study, we showed that WT α2δ-1 increases the amount of CaV2.2 on the cell surface in N2A cells, by about two-fold (Cassidy et al., 2014). We have also found that heterologously-expressed α2δ subunits are only partially processed into α2 and δ in all expression systems examined, although proteolytic cleavage was much more marked at the plasma membrane (Davies et al., 2010; Kadurin et al., 2012). The incomplete cleavage of heterologously expressed WT α2δ is in contrast to the complete processing of muscle and brain α2δ-1 (Jay et al., 1991; Patel et al., 2013) and cerebellar α2δ-2 (Davies et al., 2006), and likely represents saturation of the processing enzyme(s). Thus, it was clear that determining the role of proteolytic cleavage of α2δ on calcium channel function would require additional strategies. A previous study found that various mutations around the cleavage site reduced, but did not abolish, calcium current enhancement by α2δ-1 subunits (Andrade et al., 2007), leaving the role of proteolytic cleavage of α2δ-1 an open question. Here we show conclusively that for both α2δ-1 and α2δ-3, mutations that prevent their cleavage into α2 and δ do not prevent the appearance of pro-α2δ on the cell surface. Furthermore, in the cell lines examined here, but not in neurons, CaV2.2 trafficking to the plasma membrane was enhanced by an uncleaved pro-form of α2δ-1 to a similar extent as for WT α2δ-1, utilising a mechanism that is independent of the plasma membrane potential. However, this led to the increased cell surface expression of a calcium channel complex which appeared to be non-functional, since the uncleaved pro-α2δ did not enhance CaV2.2 calcium currents. Thus trafficking of CaV2.2 to the plasma membrane by α2δ-1 subunits in cell lines can be uncoupled from the functional effects of α2δ subunits on voltage-dependent activation of the channels.
Previous in vitro studies have examined the effect of α2δ subunits on calcium channel currents resulting from several combinations of CaVα1 and β subunits. In whole-cell current recordings, α2δ subunits increase the maximum conductance from 3 to 10-fold, depending on subunit combination and conditions used (Canti et al., 2005; Mori et al., 1991; Klugbauer et al., 1999; Hobom et al., 2000; Gao et al., 2000; Barclay et al., 2001; Hendrich et al., 2008). However, they have no effect on single channel conductance, and for CaV2 channels there are only minor effects of α2δ subunits on most parameters relating to open probability (Hoppa et al., 2012; Barclay et al., 2001; Brodbeck et al., 2002; Wakamori et al., 1999; Meir and Dolphin, 1998), which would be unlikely to account for the large increase in whole-cell conductance. By contrast, a finding that is consistent with the increase in whole-cell current, is that the fraction of null traces was markedly reduced by α2δ-1 in unitary current recordings from oocytes expressing CaV2.2/β1b/α2δ-1 (9% null traces), a 3–4-fold decrease, compared with those expressing CaV2.2 alone (39% null traces) or CaV2.2/β1b (28% null traces) (Wakamori et al., 1999). This observation suggests that α2δ shifts the equilibrium towards active modes of the channel, from an inactive null mode represented by the long closed state. Furthermore, it has recently been shown that α2δ-1 promotes voltage sensor movement of CaV1.2 (Savalli et al., 2016), thus hyperpolarizing channel activation (Savalli et al., 2016). Although the interaction of α2δ subunits with CaV2 channels may differ, as there is not such a clear shift in the voltage-dependence of activation (Kadurin et al., 2012; Canti et al., 2005), it is tempting to speculate that the association of the channels with uncleaved pro-α2δ might interfere with voltage sensor movement.
We have found previously that the MIDAS motif in the VWA domains of α2δ-1 and α2δ-2 subunits is key to increasing calcium channel function (Hoppa et al., 2012; Cassidy et al., 2014; Canti et al., 2005). The recent structure of CaV1.1 indicates that the VWA domain MIDAS motif of α2δ-1 closely associates with the loop between S1 and S2 in Domain I of the α1S subunit (Wu et al., 2015). The VWA domain is present in all α2δ subunits, but it only has a perfect MIDAS motif in α2δ-1 and α2δ-2 (Dolphin, 2013). Thus α2δ-3 has an incomplete MIDAS motif, which is predicted to have reduced function (Whittaker and Hynes, 2002), and indeed α2δ-3 has less effect on cell surface expression of CaV2.2 than α2δ-1 in the present study (compare Figures 2c and 4b).
In agreement with the hypothesis that pro-α2δ is an inhibitory gate-keeper for calcium channel function, we observed here that an uncleaved α2δ-1 construct (α2(3C)δ-1) had an inhibitory effect on endogenous DRG neuron calcium currents, and on presynaptic Ca2+ entry in hippocampal neuronal boutons, which could be reversed by co-expression of the 3C-protease. This is in contrast to a previous study in which overexpression of WT α2δ subunits, which will form a mixture of both cleaved and uncleaved α2δ (Figure 6b), reduced Ca2+ entry at synapses (Hoppa et al., 2012). Our result indicates that proteolytic processing of α2δ-1 represents an essential functional checkpoint to allow channel activation by depolarization, and the pro-α2δ species inhibits function.
Furthermore, we show here that native pro-α2δ-1 can be observed in the cell bodies of DRG neurons, but in the axons it is completely processed to α2 and δ; this is despite the fact that it is still present in intracellular trafficking vesicles (Bauer et al., 2009). It is of interest in this regard that although α2δ-1 is elevated in all DRG neurons following nerve injury (Bauer et al., 2009; Luo et al., 2001), calcium currents from DRGs extracted after nerve injury show a variable decrease in calcium currents (Hogan et al., 2000; McCallum et al., 2006, 2011). This would agree with the present finding that somatic DRG α2δ-1 upregulated after nerve injury remains in part uncleaved, and that only when in its mature processed form can it function to increase calcium currents and trafficking of calcium channels into neurites.
The functional importance of proteolytic cleavage of α2δ subunits is further emphasised by our finding that the trafficking of CaV2.2 into the processes of cultured hippocampal neurons is completely prevented by the uncleaved α2(3C)δ-1, and this is reversed by its intracellular proteolytic cleavage with the 3C protease. Thus, the proteolytic processing of α2δ represents an essential checkpoint for neuronal trafficking of calcium channels, to ensure that only mature channel complexes capable of voltage-dependent activation reach specific plasma membrane domains such as presynaptic terminals (Figure 7—figure supplement 1). We have shown previously that the small GTPase, rab11, is involved in α2δ-mediated trafficking of calcium channels (Tran-Van-Minh and Dolphin, 2010), and rab11, among many other proteins, is important for vesicular cargo transport into neurites (Villarroel-Campos et al., 2016). We have also identified the importance of adaptor protein-1 in CaV2.2 trafficking (Macabuag and Dolphin, 2015). It will be of great interest in future studies to determine the additional mechanisms present in neurons, in contrast to non-neuronal cell lines, restricting the cargo transport into neurites to activatable calcium channel complexes in which α2δ subunits are proteolytically cleaved.
Materials and methods
Molecular biology
Request a detailed protocolThe cDNAs used were: rat α2δ-1 (M86621) and mouse α2δ-3 (AJ010949), rabbit CaV2.2 (D14157 without 3' UTR), and rat β1b (Tomlinson et al., 1993). In some experiments CaV2.2 was used with an N-terminal GFP (Macabuag and Dolphin, 2015), or containing an extracellular BBS tag or HA tag (Cassidy et al., 2014). α2δ-1-HA (HA-tag sequence YPYDVPDYA inserted between Asn-549 and Asp-550) (Kadurin et al., 2012) and α2δ-3-HA (HA between Lys-595 and Arg-596) were used in all imaging and other experiments except when the subunits were co-expressed with CaV2.2-HA in hippocampal neurons, in which case untagged α2δ-1 or α2δ-3 were used. Both α2δ-HA constructs showed normal function in terms of enhancing CaV currents ([Kadurin et al., 2012] and data not shown). Proteolytic cleavage site mutations were made as indicated (1b and 4c). GFP-β1b was used in some electrophysiological experiments (Waithe et al., 2011). Human TASK3 (KCNK9) cDNA (NM_001282534) was obtained from Prof. A Mathie. The cDNAs were in the pMT2 vector for expression in tsA-201 cells, in pcDNA3 for expression in N2A cells and pcDNA3 or pCAGGS for expression in DRG or hippocampal neurons, respectively. In our hands expression of the large constructs encoding CaV2.2 and α2δ in pcDNA3 was very poor in hippocampal neurons (data not shown), whereas expression from the pCAGGS vector, containing chicken β-actin promoter, was strong and sustained. The cDNAs encoding Human Rhinovirus (HRV)−3C protease or mutated 3C (in which the active site Cys-147 was mutated to Val) were first subcloned into the pHLSec vector (Yurtsever et al., 2012; Aricescu et al., 2006), using Age I and Kpn I sites in frame with an N-terminal signal sequence, to include the signal sequence from pHLSec, and then into pMT2 and pCAGGS vectors for expression in cell lines or hippocampal neurons. VAMP-mOr2 was generated by replacing mCherry from pCAGGs-VAMPmCherry (gift from Dr. TA Ryan) with mOrange2. Sy-GCaMP6f was made by replacing GCaMP3 in pCMV-SyGCaMP3 (gift from Dr. TA Ryan) by GCaMP6f (Chen et al., 2013). The cDNA for mut2-GFP (Cormack et al., 1996) in pMT2 was used as a negative control in co-immunoprecipitation (co-IP) experiments. Site-directed mutagenesis was carried out using standard procedures, and all subcloning and mutations confirmed by sequencing.
Antibodies and other materials
Request a detailed protocolCa channel antibodies (Abs) used were: α2δ-1 Ab (mouse monoclonal against α2-1 moeity, Sigma-Aldrich, epitope identified in [Cassidy et al., 2014]), α2-3 (71–90) Ab (rabbit; polyclonal) (Davies et al., 2010), δ-3 (1035–1049) Ab (Davies et al., 2010), anti-CaV2.2 II-III loop Ab (rabbit polyclonal) (Raghib et al., 2001). Other Abs used were anti-HA (rat monoclonal, Roche), anti-HA (rabbit polyclonal, Sigma), anti-GAPDH Ab (mouse monoclonal, Ambion), anti-flotillin-1 (mouse monoclonal, BD Biosciences), and GFP Ab (Living Colors, rabbit polyclonal; BD Biosciences). For immunocytochemistry, secondary Abs (1:500) used were anti-rabbit-Alexa Fluor 594, anti-rat-Alexa Fluor 594, anti-mouse-Alexa Fluor 647 (Life Technologies) or fluorescein isothiocyanate (FITC)-anti-rat (Sigma-Aldrich). The following secondary Abs were used for western blot: goat anti-rabbit, goat anti-rat and goat anti-mouse Abs coupled to horseradish peroxidase (HRP) (Biorad, Hemel Hempstead, UK). The signal was obtained by HRP reaction with fluorescent product (ECL 2; Thermo Scientific) and membranes were scanned on a Typhoon 9410 phosphorimager (GE Healthcare). Lyophilized active thrombin was obtained from Sigma, suspended in filter-sterilised PBS (Sigma; pH7.4) to 1000 U/ml and frozen in aliquots until use.
Cell culture, transfection and enzymatic treatment
Request a detailed protocolCell lines were plated onto cell culture flasks, coverslips or glass-bottomed dishes (MatTek Corporation, Ashland, MA), coated with poly-L-lysine, and cultured in a 5% CO2 incubator at 37°C. The human embryonic kidney tsA-201 cells (European Collection of Authenticated Cell Cultures, # 96121229), tested to be mycoplasma-free, were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 1 unit/ml penicillin, 1 μg/ml streptomycin and 1% GlutaMAX (Life Technologies, Waltham, MA). tsA-201 cells were transfected using Fugene6 (Promega, Fitchburg, WI), according to the manufacturer’s protocol. The enzymatic treatments with 30 U/ml Thrombin protease (Sigma) diluted in DMEM without supplements were done for indicated times in a 5% CO2 incubator at 37°C. Mouse neuroblastoma N2A cells (American Tissue culture collection, # CCL-131) were obtained from Professor Roger Morris, Kings College London (Parkyn et al., 2008), and were tested to be mycoplasma-free. They were cultured in 50% DMEM and 50% OPTI-MEM supplemented with 5% FBS, 1 unit/ml penicillin, 1 μg/ml streptomycin, and 1% GlutaMAX. N2A cells were transfected using PolyJet (SignaGen Laboratories, Gaithersburg, MD), according to the manufacturer’s protocol.
Neuronal culture and transfection
Request a detailed protocolDRG neurons were isolated from P10 male Sprague Dawley rats and transfected essentially as described recently (D'Arco et al., 2015) with an Amaxa Nucleofector (Lonza, Basel, Switzerland) according to the manufacturer’s protocol. Transfected neurons were plated onto coverslips coated with poly-L-lysine, and cultured in DMEM/F12 supplemented with 10% FBS, 1 unit/ml penicillin, 1 μg/ml streptomycin, 1% GlutaMAX and 100 ng/ml NGF in a 5% CO2 incubator at 37°C. Hippocampal neurons were obtained from P0 rat pups as previously described (Morales et al., 2000). Approximately 75 × 103 cells in 100 μl of plating solution (Neurobasal medium supplemented with B27 (Life Technologies; 2%), HEPES (10 mM), horse serum (5%), glutamine (0.5 mM), and 1 unit/ml penicillin, 1 μg/ml streptomycin) were seeded onto sterile poly-lysine-coated glass coverslips. After 2 hr, the plating solution was replaced with 1 ml of growth medium (serum-free Neurobasal medium supplemented with B27 (Life Technologies; 4%), 2-mercaptoethanol (25 μM), glutamine (0.5 mM), and 1 unit/ml penicillin, 1 μg/ml streptomycin), half of which was replaced every 3–4 days. At 7 days in vitro (DIV) and 2 hr before transfection, half of the medium was removed, and kept as ‘conditioned’ medium and 500 μl of fresh medium was added. The hippocampal cell cultures were then transfected with Lipofectamine 2000 according to the manufacturer’s protocol using 2 μg of cDNA mix per well (Invitrogen). After 2 hr, the transfection mixes were replaced with growth medium consisting of 50% ‘conditioned’ and 50% fresh medium. The cultures were used for immunostaining experiments at 14 DIV, or for live imaging as described below.
Cell surface biotinylation, Cell lysis, deglycosylation and immunoblotting
Request a detailed protocolThe procedures were modified from those described in more detail previously (Davies et al., 2010; Page et al., 2004). Briefly, 72 hr after transfection, tsA-201 cells were incubated for 30 min at room temperature with 0.5 mg/ml Premium Grade EZ-link Sulfo-NHS-LC-Biotin (Thermo Scientific) in PBS and the reaction was quenched with 200 mM glycine. The cells were incubated for 45 min on ice in PBS, pH 7.4 at 4°C containing 1% Igepal; 0.1% SDS and protease inhibitors (PI, cOmplete, Roche; used according to manufacturer’s instructions), to allow cell lysis. The WCL was then centrifuged at 18,000 ×g for 20 min at 4°C and the pellet discarded. The supernatant was assayed for total protein (Bradford assay, Biorad). Immunoblot analysis was performed essentially as described previously (Kadurin et al., 2012). Cleared WCL corresponding to 20–40 µg total protein was diluted with Laemmli sample buffer (Davies et al., 2010) supplemented with 100 mM dithiothreitol (DTT), incubated at 60°C for 10 min and resolved by SDS-polyacrylamide gel electrophoresis (PAGE) on 3%–8% Tris-Acetate or 4–12% Bis-Tris gels (Invitrogen) and transferred to polyvinylidene fluoride (PVDF) membrane (Biorad) by western blotting. When required the membrane was cut and incubated with different antibodies. Biotinylated lysates (adjusted to between 0.5 and 1 mg/ml total protein concentration) were applied to 40 µl prewashed streptavidin-agarose beads (Thermo Scientific) and rotated overnight at 4°C. The beads were then washed 3 times with PBS containing 0.1% Igepal and, when required, the streptavidin beads were re-suspended in PNGase-F buffer (PBS, pH 7.4, supplemented with 75 mM β-mercaptoethanol, 1% Igepal, 0.1% SDS, and PI) and deglycosylated for 3 hr at 37°C with 1 unit of PNGase-F (Roche Applied Science) added per 10 μl volume. The samples were then resuspended in an equal volume of 2 × Laemmli buffer with 100 mM DTT, followed by 10 min incubation at 60°C. The eluted protein was analysed by immunoblotting, as described above. GAPDH is ~39 kDa and does not resolve well in 3–8% Tris-Acetate gels. Therefore, in cases when 3–8% gels were used to resolve high MW proteins, which required GAPDH as negative control for biotinylated fractions, the same samples were re-loaded on a 4–12% Bis-Tris gel to analyse separately.
Co–immunoprecipitation
Request a detailed protocolThe protocol was adapted from a procedure described previously (Gurnett et al., 1997). Briefly, a tsA-201 cell pellet derived from one confluent 75-cm2 flask was resuspended in a co-IP buffer (20 mM HEPES (pH 7.4), 300 mM NaCl, 1% Digitonin and PI), sonicated for 8 s at 20 kHz and rotated for 1 hr at 4°C. The samples were then diluted with an equal volume of 20 mM HEPES (pH 7.4), 300 mM NaCl with PI (to 0.5% final concentration of Digitonin), mixed by pipetting and centrifuged at 18,000 ×g for 20 min. The supernatants were collected and assayed for total protein (Bradford assay; Biorad). 1 mg of total protein was adjusted to 2 mg/ml with co-IP buffer and incubated overnight at 4°C with anti-GFP polyclonal antibody (1:200; BD Biosciences). 30 μl A/G PLUS Agarose slurry (Santa Cruz) was added to each tube and further rotated for 2 hr at 4°C. The beads were then pelleted by 500 ×g centrifugation at 4°C and washed three times with co-IP buffer containing 0.2% Digitonin. The beads were then resuspended in an equal volume of PNGase-F buffer and proteins were deglycosylated with PNGase-F as described above. Aliquots of the initial WCL prior to co-IP were also deglycosylated in parallel; 2 × Laemmli buffer with 100 mM DTT was added and samples were analysed by SDS-PAGE and western blotting.
Preparation of triton X-100-insoluble membrane fractions (DRMs)
Request a detailed protocolThe protocol was similar to that described previously (Davies et al., 2010; Kadurin et al., 2012). All steps were performed on ice. Confluent tsA-201 cells from two 175-cm (Kurshan et al., 2009) flasks were taken up in Mes-buffered saline (MBS, 25 mm Mes, pH 6.5, 150 mm NaCl, and PI) containing 1% (v/v) Triton X-100 (TX-100) (Thermo Scientific), and left on ice for 1 hr. An equal volume of 90% (w/v) sucrose in MBS was then added to a final concentration of 45% sucrose. The sample was transferred to a 13 ml ultracentrifuge tube and overlaid with 10 ml of discontinuous sucrose gradient, consisting of 35% (w/v) sucrose in MBS (5 ml) and 5% (w/v) sucrose in MBS (5 ml). The sucrose gradients were ultra-centrifuged at 140,000 ×gavg (Beckman SW40 rotor) for 18 hr at 4°C. 1 ml fractions were subsequently harvested from the top to the bottom of the tube and aliquots of 10 μl from each fraction were analysed by SDS-PAGE and western blotting to obtain DRM profiles. When necessary, DRMs (combined peak fractions identified by the presence of flotillin-1) from the gradient were washed free of sucrose by dilution into 25 volumes of cold PBS (pH 7.4) and pelleted by ultracentrifugation at 150,000 ×g (Beckman Ti 70 rotor) for 1 hr at 4°C. TX-100-insoluble protein was resuspended in appropriate buffers as described for 3H-gabapentin binding or for deglycosylation as described above.
Immunocytochemistry, imaging and analysis
Request a detailed protocolThe procedure in tsA-201 and N2A cells was performed essentially as described previously with minor modifications (Davies et al., 2010; Kadurin et al., 2012). Briefly, 48 hr post-transfection the cells were fixed with 4% paraformaldehyde (PFA) in PBS, pH7.4 at 20°C for 5 min, and then incubated for PBS for 15 min, which contained 0.1% TX-100 if permeabilization was applied. Blocking was performed for 1 hr at 20°C in PBS containing 20% goat serum and 5% bovine serum albumen (BSA). The indicated primary antibodies were then applied (diluted in PBS with10% goat serum and 2.5% BSA) overnight at 4°C or for 1 hr at 20°C. In live-labelling experiments, cells were washed with Krebs Ringer HEPES (KRH) buffer, labelled with α-bungarotoxin (BTX)-AF 488 (Invitrogen; 1:100 in KRH buffer) at 17°C for 30 min, then washed with KRH and fixed as described above. The indicated secondary antibodies were applied (1:500 dilution in PBS, containing 2.5% BSA and 10% goat serum) at 20°C for 1 hr. Cell nuclei were stained with 0.5 µM 4’,6’-diamidino-2-phenylindole (DAPI) in PBS for 5 min. The coverslips were mounted onto glass slides using VECTASHIELD mounting medium (Vector Laboratories, Peterborough, UK). Cultures of transfected hippocampal neurons were fixed after 14 DIV in PBS containing 4% PFA/4% sucrose for 5 min at 20°C, and then the procedure was as described above. In some cases, where stated, an antigen retrieval step was performed between the fixation and blocking steps: the cells were incubated for 10 min at 95°C in 10 mM citrate buffer (pH 6) containing 0.05% Tween 20.
Imaging was performed on Zeiss LSM 780 confocal microscope as described in more detail elsewhere (Davies et al., 2010; Kadurin et al., 2012). Images were obtained at fixed microscope settings for all experimental conditions of each experiment. Images of N2A and tsA-201 cells were obtained using a 63 × objective at a resolution of 1024 × 1024 pixels and an optical section of 0.8–1 μm. After choosing a region of interest containing transfected cells, the 3 × 3 tile function of the microscope allowed imaging of a larger area selected without bias. Every cell identified as transfected was included in the measurements, to ensure lack of bias.
Images of tsA-201 and N2A cells were analyzed using imageJ (imagej.net) using a modification of the procedure described previously (Davies et al., 2010; Kadurin et al., 2012). Surface labelling in non-permeabilized or total staining in permeabilized cell bodies was measured using the freehand line tool and manually tracing the surface of the cell or drawing around the cell (omitting the nucleus) respectively. The value of the mean pixel intensity in different channels was measured separately and background was subtracted by measuring the intensity of an imaged area without transfected cells. All data were then normalized to the appropriate positive control for each experiment before combining experiments.
Hippocampal neurons were imaged using a 20 × objective with a 5 μm optical section. Large tiles were manually selected following all processes expressing mCherry. The fluorescence intensity along neuronal projections of hippocampal neurons was assessed in FIJI as follows: a circle of 100 μm diameter was drawn around each neuronal cell body. A free-hand line (2 μm thick; ~ 30 μm long) was drawn along the neurite extending beyond the circle and the mean grey intensity of all the pixels within the line was measured in both channels corresponding to the fluorescence of HA or α2δ-1 immunostaining and mCherry.
Analysis of α2δ-1 in dorsal root ganglion (DRG) neurons and axons
Request a detailed protocolImmunoblotting of DRG tissue and associated nerves was performed as described previously (Bauer et al., 2009). Tissue was taken from rats 4d following a spinal nerve ligation procedure, performed in the course of a previous study (Bauer et al., 2009), in order to increase the amount of α2δ-1 protein in the harvested tissue. Tissue from DRGs, sections of spinal nerve and dorsal roots were pooled from 4 rats, and stored at −80°C until use in this study. For deglycosylation, protein samples were diluted in PBS + 1% Igepal + 1% β-mercaptoethanol and treated with 1 unit of PNGase F overnight at 37°C.
3H gabapentin binding assay
Request a detailed protocolBinding of 3H-gabapentin to DRM preparations was carried out, essentially as previously described (Lana et al., 2014), in a final volume of 250 µl at room temperature for 45 min. DRM fractions (4 µg of protein per tube) were incubated with various concentrations of [3H]-gabapentin (specific activity 36 Ci/mmol, American Radiolabeled Chemicals, St. Louis, MO, USA) in 10 mM HEPES/KOH pH 7.4, then rapidly filtered through GF/B filters, pre-soaked with 0.3% polyethyleneimine. Filters were washed three times with 3 ml ice-cold 50 mM Tris/HCl, pH 7.4 and counted on a scintillation counter. Concentrations of [3H]-gabapentin greater than 50 nM were achieved by adding non-radioactive gabapentin and correcting the specific binding by the dilution factor (Canti et al., 2005; Lana et al., 2014). Non-specific binding was determined in the presence of 20 µM non-radioactive gabapentin. Data points were determined in triplicate for each experiment, and data for each experiment were analysed by fitting specific binding to the Hill equation (Lana et al., 2014).
Electrophysiology
Request a detailed protocolCalcium channel currents in transfected tsA-201 cells were investigated by whole cell patch clamp recording, essentially as described previously (Berrow et al., 1997). The patch pipette solution contained in mM: Cs-aspartate, 140; EGTA, 5; MgCl2, 2; CaCl2, 0.1; K2ATP, 2; Hepes, 10; pH 7.2, 310 mOsm with sucrose. The external solution for recording Ba2+ currents contained in mM: tetraethylammonium (TEA) Br, 160; KCl, 3; NaHCO3, 1.0; MgCl2, 1.0; Hepes, 10; glucose, 4; BaCl2, 1,or 2 as indicated, pH 7.4, 320 mosM with sucrose. Unless otherwise stated, 1 mM extracellular Ba2+ was the charge carrier. Pipettes of resistance 2–4 MΩ were used. An Axopatch 1D or Axon 200B amplifier was used, and whole cell voltage-clamp recordings were sampled at 10 kHz frequency, filtered at 2 kHz and digitized at 1 kHz. 70–80% series resistance compensation was applied and all recorded currents were leak subtracted using P/8 protocol. For DRG neurons, whole cell voltage clamp experiments were performed in small (<19 pF) and medium (20–38 pF) neurons. Membrane potential was held at –80 mV for experiments in tsA-201 cells and −90 mV for DRG experiments. Cells were accepted where the access resistance was less than 5 MΩ, the inward current was > −3 pA/pF at +10 mV, a complete IV relationship was obtained and there was no evidence of poor voltage clamp. Analysis was performed using Pclamp 9 (Molecular Devices) and Origin 7 (Microcal Origin, Northampton, MA). IV relationships were fit by a modified Boltzmann equation as follows: I=Gmax*(V−Vrev)/(1+exp(−(V−V50, act)/k)) where I is the current density (in pA/pF), Gmax is the maximum conductance (in nS/pF), Vrev is the apparent reversal potential, V50, act is the midpoint voltage for current activation, and k is the slope factor. Recordings of resting membrane potential were performed as previously described (Margas et al., 2016).
Live cell imaging
Request a detailed protocolHippocampal neurons were transfected with VAMP-mOr2 and sy-GCaMP6f, together with the other cDNAs used at 7 DIV. Neurons were imaged after 14–21 DIV. Coverslips were mounted in a laminar-flow perfusion and stimulation chamber (Warner Instruments) on the stage of an epifluorescence microscope (Axiovert 200 M, Zeiss). White and 470 nm LEDs served as light sources (Cairn Research, UK). Fluorescence excitation and collection was performed through a 40 × 1.3 NA Fluar Zeiss objective using 450/50 nm excitation and 510/50 nm emission and 480 nm dichroic filters, and a 545/25 nm excitation and 605/70 nm emission and 565 nm dichroic filters (for mOrange2). Live cell images were acquired as previously described with minor modifications (Margas et al., 2016; Ferron et al., 2014) with an Andor iXon+ (model DU-897U-CS0-BV) back-illuminated EMCCD camera. Fluorescence was collected at 100 Hz over a 512 × 266 pixel area (7 ms integration time). Cells were perfused (0.5 ml min−1) in a saline solution at 22°C containing (in mM) 119 NaCl, 2.5 KCl, 2 CaCl2, 2 MgCl2, 25 HEPES (buffered to pH 7.4), 30 glucose, 10 µM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 50 µM D,L-2-amino-5-phosphonovaleric acid (AP5). Neurons were stimulated by passing 1 ms current pulses through the field stimulation chamber via platinum electrodes. Neurons expressing syn-GCaMP6f were identified by stimulating the preparation at 33 Hz for 180 ms every 4 s. Subsequently, single stimulations of 1 ms (mimicking a single AP) were repeated 5 times at 45 s intervals. Functional synaptic boutons were identified by the increase of fluorescence of VAMP-mOr2 in response to a 100 Hz stimulation for 1 s. Changes in GCaMP6f fluorescence were normalized to the maximum fluorescence (Fmax) measured in the presence of 5 µM ionomycin and 2 mM Ca2+ in the perfusion solution. Analysis was performed with ImageJ (http://rsb.info.nih.gov/ij), using a custom-written plugin (http://rsb.info.nih.gov/ij/plugins/time-series.html). Regions of interest (ROI) of 2 µm diameter were selected according to their responsiveness to a 100 Hz stimulation for 1 s (on average, 20 to 100 ROIs were analyzed per field of view). Peak fluorescence in response to mean of 5 single AP stimuli was determined by averaging 5–10 points of the plateau phase and subtracting the average of 10 points of the baseline before stimulation.
Data analysis
Request a detailed protocolData are given as mean ± SEM, or scatter plots, or box (25–75%) and whisker (10–90%) plots with mean and median (dashed and solid lines). Statistical comparisons were performed using either Student’s t test, paired t test, 1-sample t test, ANOVA with appropriate post-hoc test, or Krushkal-Wallis test with appropriate post-hoc test, as stated, using Graphpad Prism 5. Details of tests are given in Supplementary statistics file.
References
-
Proteolytic cleavage of the voltage-gated Ca2+ channel alpha2delta subunit: structural and functional featuresEuropean Journal of Neuroscience 25:1705–1710.https://doi.org/10.1111/j.1460-9568.2007.05454.x
-
Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cellsJournal of Neuroscience 21:6095–6104.
-
Properties of cloned rat alpha1A calcium channels transiently expressed in the COS-7 cell lineEuropean Journal of Neuroscience 9:739–748.https://doi.org/10.1111/j.1460-9568.1997.tb01422.x
-
The ß subunit of voltage-gated Ca2+ channelsPhysiological Reviews 90:1461–1506.https://doi.org/10.1152/physrev.00057.2009
-
Structure and regulation of voltage-gated Ca2+ channelsAnnual Review of Cell and Developmental Biology 16:521–555.https://doi.org/10.1146/annurev.cellbio.16.1.521
-
Substrate requirements of human rhinovirus 3C protease for peptide cleavage in vitroThe Journal of Biological Chemistry 265:9062–9065.
-
The upregulation of α2δ-1 subunit modulates activity-dependent Ca2+ signals in sensory neuronsJournal of Neuroscience 35:5891–5903.https://doi.org/10.1523/JNEUROSCI.3997-14.2015
-
Subunits of purified calcium channels. Alpha 2 and delta are encoded by the same geneThe Journal of Biological Chemistry 265:14738–14741.
-
Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyondNature Reviews Neuroscience 13:542–555.https://doi.org/10.1038/nrn3317
-
The α2δ subunits of voltage-gated calcium channelsBiochimica Et Biophysica Acta (BBA) - Biomembranes 1828:1541–1549.https://doi.org/10.1016/j.bbamem.2012.11.019
-
Functional properties of a new voltage-dependent calcium channel alpha(2)delta auxiliary subunit gene (CACNA2D2)Journal of Biological Chemistry 275:12237–12242.https://doi.org/10.1074/jbc.275.16.12237
-
Extracellular interaction of the voltage-dependent Ca2+ channel alpha2delta and alpha1 subunitsThe Journal of Biological Chemistry 272:18508–18512.https://doi.org/10.1074/jbc.272.29.18508
-
Neuronal distribution and functional characterization of the calcium channel alpha2delta-2 subunitEuropean Journal of Neuroscience 12:1217–1226.https://doi.org/10.1046/j.1460-9568.2000.01009.x
-
Structural characterization of the dihydropyridine-sensitive calcium channel alpha 2-subunit and the associated delta peptidesThe Journal of Biological Chemistry 266:3287–3293.
-
Calcium currents are enhanced by α2δ-1 lacking its membrane anchorThe Journal of Biological Chemistry 287:33554–33566.https://doi.org/10.1074/jbc.M112.378554
-
TASK-3, a new member of the tandem pore K(+) channel familyJournal of Biological Chemistry 275:9340–9347.https://doi.org/10.1074/jbc.275.13.9340
-
Molecular diversity of the calcium channel alpha2delta subunitJournal of Neuroscience 19:684–691.
-
Identification of three subunits of the high affinity omega-conotoxin MVIIC-sensitive Ca2+ channelThe Journal of Biological Chemistry 271:13804–13810.https://doi.org/10.1074/jbc.271.23.13804
-
Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerve-injured ratsJournal of Neuroscience 21:1868–1875.
-
Effect of knockout of α 2 δ-1 on action potentials in mouse sensory neuronsPhilosophical Transactions of the Royal Society B: Biological Sciences 371:20150430.https://doi.org/10.1098/rstb.2015.0430
-
Single channel properties and influence of auxiliary subunits on calcium channel α1 subunits expressed in COS7 cellsJournal of Physiology 509:38P.
-
LRP1 controls biosynthetic and endocytic trafficking of neuronal prion proteinJournal of Cell Science 121:773–783.https://doi.org/10.1242/jcs.021816
-
Dominant-negative synthesis suppression of voltage-gated calcium channel Cav2.2 induced by truncated constructsJournal of Neuroscience 21:8495–8504.
-
Presynaptic CaV2 calcium channel traffic requires CALF-1 and the alpha(2)delta subunit UNC-36Nature Neuroscience 12:1257–1265.https://doi.org/10.1038/nn.2383
-
The α2δ-1 subunit remodels CaV1.2 voltage sensors and allows Ca2+ influx at physiological membrane potentialsThe Journal of General Physiology 148:147–159.https://doi.org/10.1085/jgp.201611586
-
Functional properties of a neuronal class C L-type calcium channelNeuropharmacology 32:1117–1126.https://doi.org/10.1016/0028-3908(93)90006-O
-
Beta-subunits promote the expression of Ca(V)2.2 channels by reducing their proteasomal degradationJournal of Biological Chemistry 286:9598–9611.https://doi.org/10.1074/jbc.M110.195909
-
Self-cleavage of human CLCA1 protein by a novel internal metalloprotease domain controls calcium-activated chloride channel activationJournal of Biological Chemistry 287:42138–42149.https://doi.org/10.1074/jbc.M112.410282
Article and author information
Author details
Funding
Wellcome Trust (098360/Z/12/Z)
- Ivan Kadurin
- Laurent Ferron
- Simon W Rothwell
- Wojciech Margas
- Manuela Nieto-Rostro
Medical Research Council (G0901758)
- Laurent Ferron
- Leon R Douglas
- Claudia S Bauer
Medical Research Council (G0801756)
- Laurent Ferron
- Leon R Douglas
- Claudia S Bauer
Medical Research Council (MR/J013285/1)
- Laurent Ferron
- Leon R Douglas
- Claudia S Bauer
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported by a Wellcome Trust Investigator award to ACD (098360/Z/12/Z) and Medical Research Council (UK) (grants G0901758 and G0801756). We thank Kanchan Chaggar for tissue culture. We thank Prof. AH Dickenson and Dr. Wahida Rahman for the collaboration Bauer et al., 2009 in which tissue for Figure 6a was collected. We thank Dr. Matthew Gold for HRV-3C protease cDNA, Prof. Alistair Mathie for TASK3 cDNA and Prof Tim Ryan for VAMP-mCherry and Synaptophysin-GCaMP3 cDNAs.
Copyright
© 2016, Kadurin et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,707
- views
-
- 456
- downloads
-
- 49
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 49
- citations for umbrella DOI https://doi.org/10.7554/eLife.21143
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Proteolytic maturation of α2δ represents a checkpoint for activation and neuronal trafficking of latent calcium channels
eLife 5:e21143.
https://doi.org/10.7554/eLife.21143




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}