Cooperative and acute inhibition by multiple C-terminal motifs of L-type Ca2+ channels
- Tsinghua University, China
- Columbia University, United States
Figures
Figure 1 with 1 supplement
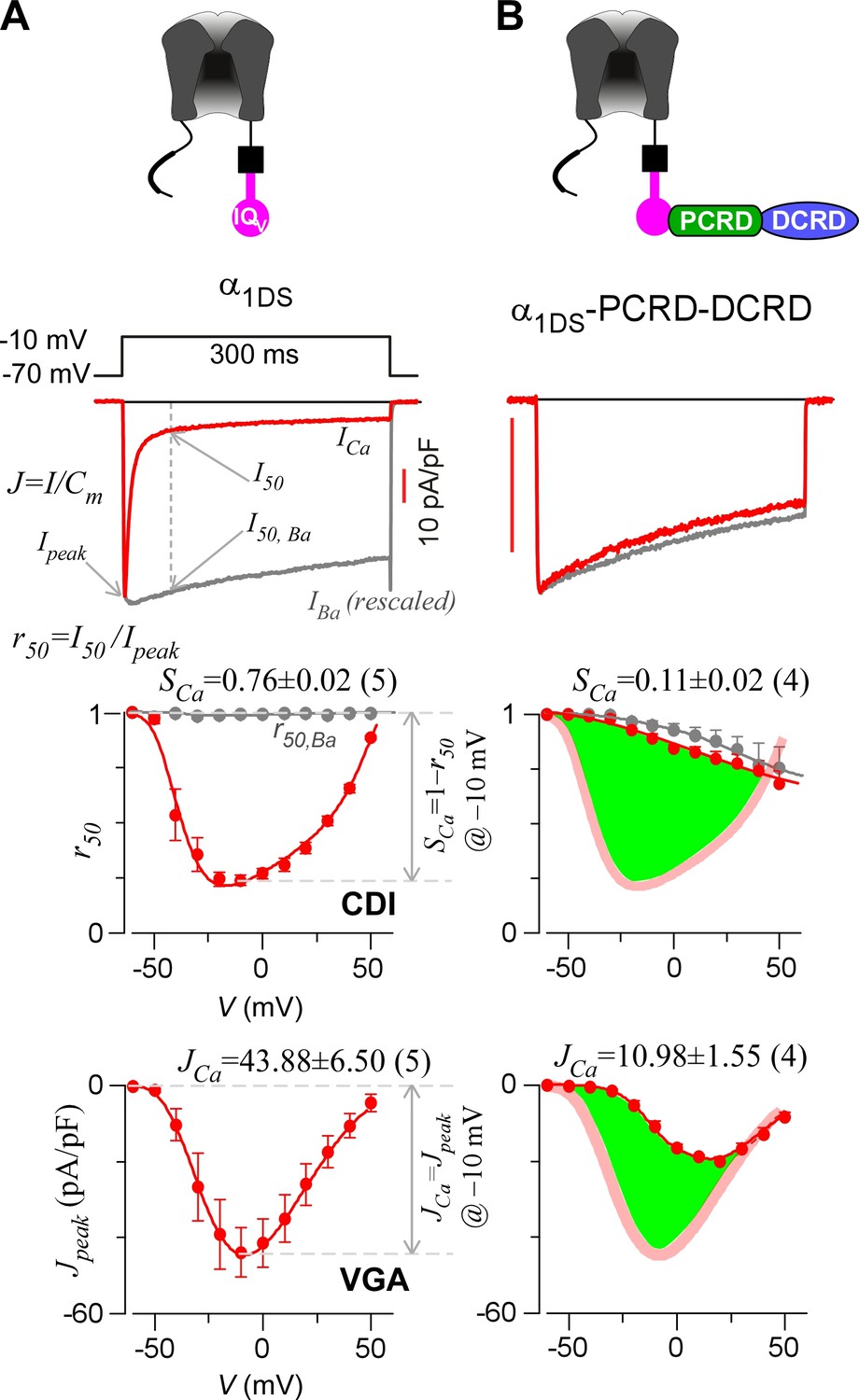
Inhibition of CaV1.3 gating by carboxyl terminal motifs.
(A) Parameters and indices illustrated by the control group of short CaV1.3 channels. Representative current exemplars (Ca2+ current ICa in red, with the scale bar in red; Ba2+ current IBa in gray, rescaled) were shown for α1DS at the membrane potential (V) of −10 mV, with the amplitudes measured at the time of peak (Ipeak) and 50 ms (I50). Inactivation profiles across the full range of V for IBa and ICa were quantified by the remaining current at 50 ms (r50), in ratio between I50 and Ipeak. The CDI strength was quantified by 1−r50,Ca or SCa, serving as one of the major indices. Based on Ca2+ current density normlized to the cell capacitance (Cm), VGA in Ca2+ was profiled by Jpeak (in pA/pF), with the Jpeak amplitude at −10 mV or JCa as the other major index. (B) In contrast to α1DS channels with pronounced CDI (SCa) and strong VGA (JCa), α1DS-PCRD-DCRD incorporating all the three motifs of IQV, PCRD and DCRD exhibited strong inhibitions on both CDI and VGA (less pronounced U-shape or V-shape), indexed with SCa and JCa (smaller values) respectively. Thick semi-transparent lines in red depict the CDI and VGA profiles of the α1DS control group (A); and the differences in profiles (green areas as visual cues) illustrate the potency of CMI.
Figure 1—figure supplement 1
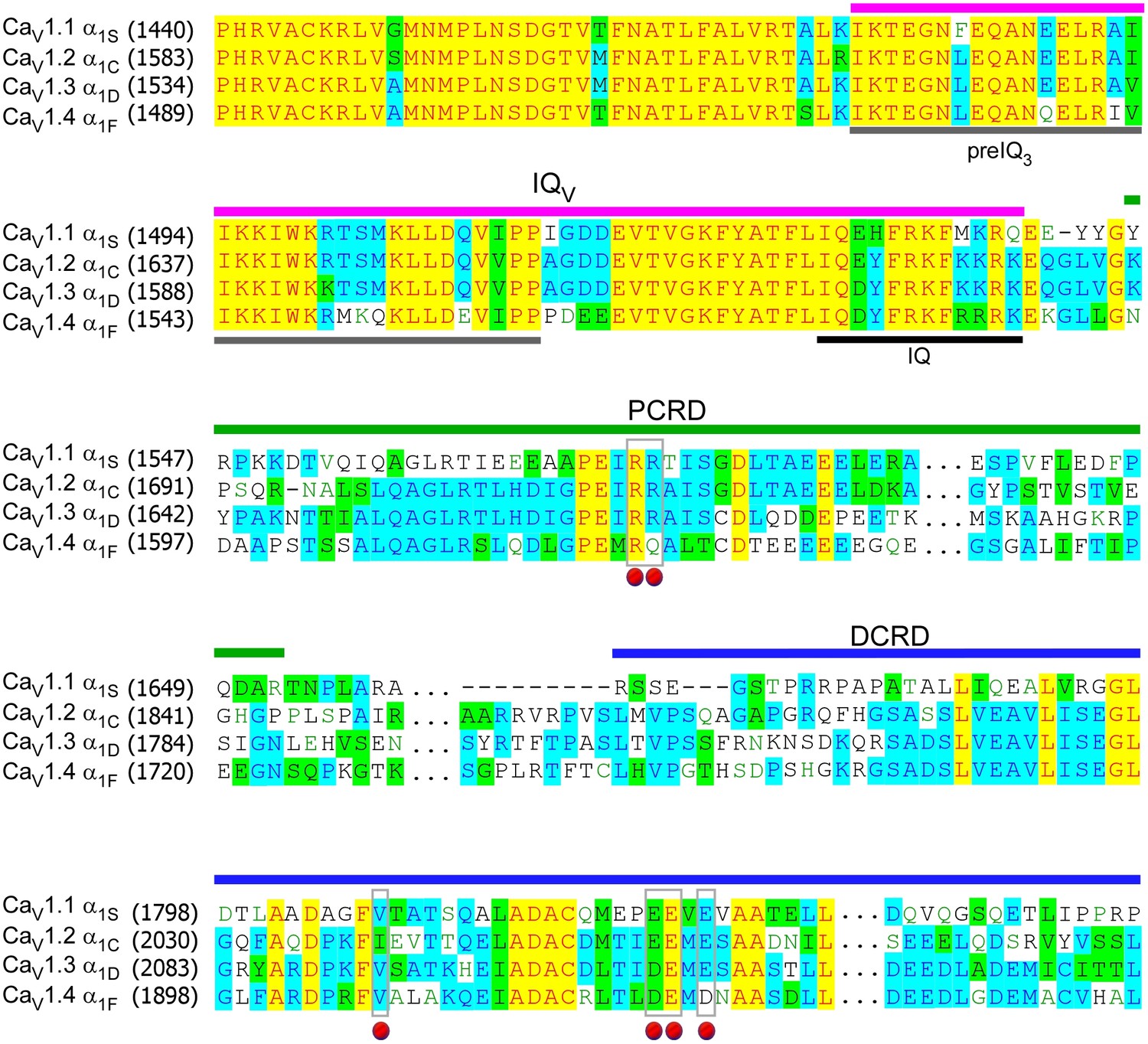
Sequence alignment of carboxyl termini for L-type CaV1 channels.
The sequences of α1 subunits were aligned for CaV1.1 (α1S, XM_983862.1), CaV1.2 (α1C, NM_199460.3), CaV1.3 (α1D, NM_000720) and CaV1.4 (α1F, NP005174), with GenBank accession numbers in parentheses. Key domains of pre-IQ3, IQ, IQV, PCRD, and DCRD were underlined with different colors, and the conjunction domain (…) between PCRD and DCRD was skipped for clarity. Identical (yellow and blue), similar (green) and gapped (−) sequences were indicated. Key residues for potential interactions were marked by red dots, including valine (V) on DCRD, and residues of positively charged arginine (R) or negatively charged aspartic acid (D) or glutamine (E) on PCRD or DCRD, respectively.
Figure 2 with 1 supplement
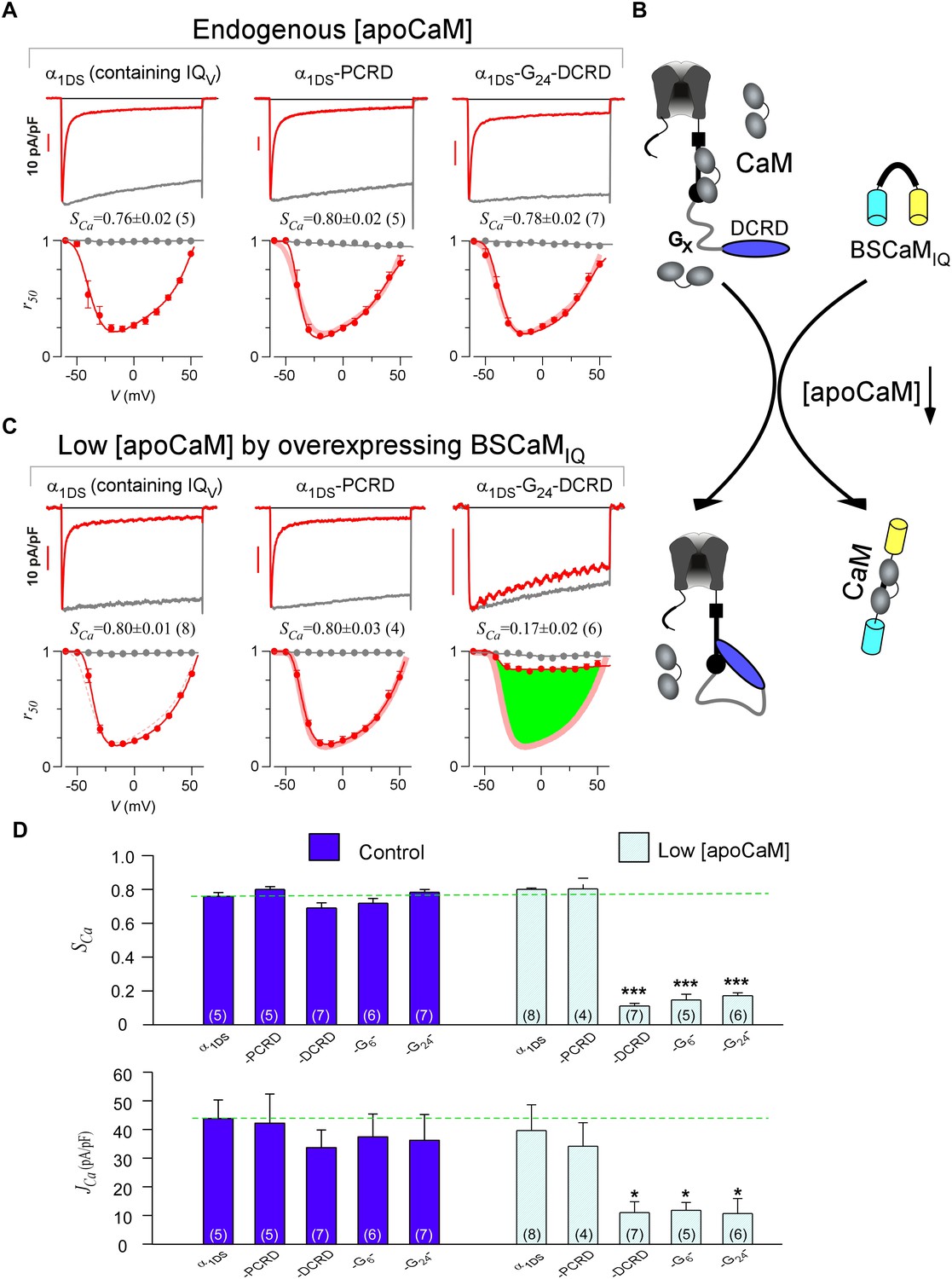
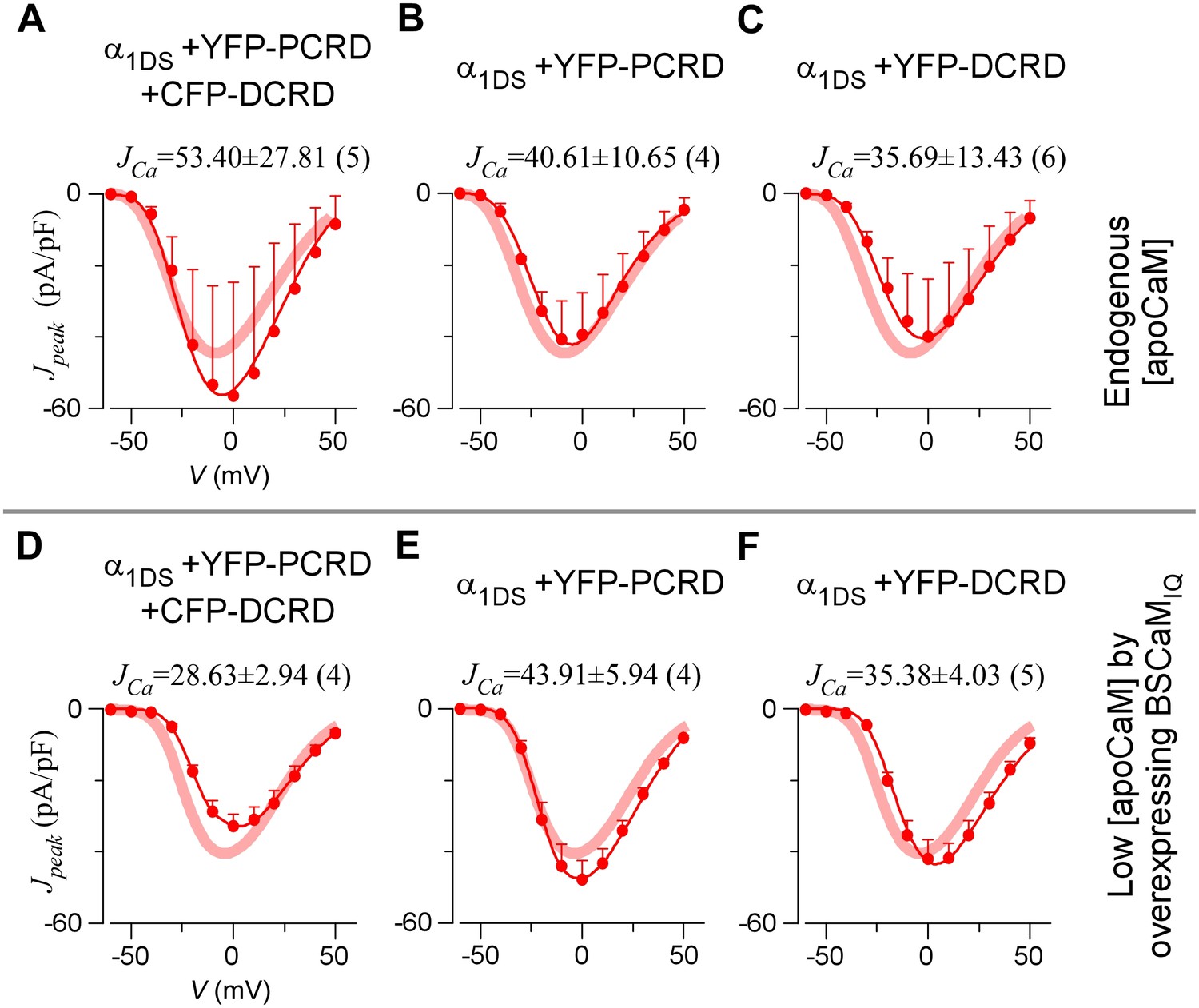
DCRD is indispensable and sufficient to induce CMI in low [apoCaM].
(A) All the channel variants including α1DS, α1DS-PCRD and α1DS-G24-DCRD exhibited strong CDI, indexed with SCa. (B) Schematic illustration for the strategy to explore the minimum requirement of CMI. For α1DS-GX-DCRD containing glycine linkers (GX) of different length (0, 6 or 24), BSCaMIQ that binds apoCaM was overexpressed to downregulate endogenous [apoCaM], in hope to promote the binding of DCRD with the channel (IQV). (C) With low [apoCaM] by overexpressing BSCaMIQ, α1DS-G24-DCRD channels exhibited much attenuated CDI, evidenced by ICa trace, SCa value and r50 profile (green area indicating the potency), in contrast to ultrastrong CDI of α1DS or α1DS-PCRD, both lacking DCRD. (D) Statistical summary of CDI (SCa) and VGA (JCa) in endogenous (control) or low [apoCaM] for all channel variants of α1DS, α1DS-PCRD and α1DS-GX-DCRD, with additional information in Figure 2—figure supplement 1. Notably, for the three α1DS-GX-DCRD variants, both CDI and VGA were concurrently and significantly attenuated. Statistical significance was evaluated and indicated (p<0.001, ***; p<0.05, *).
Figure 2—figure supplement 1
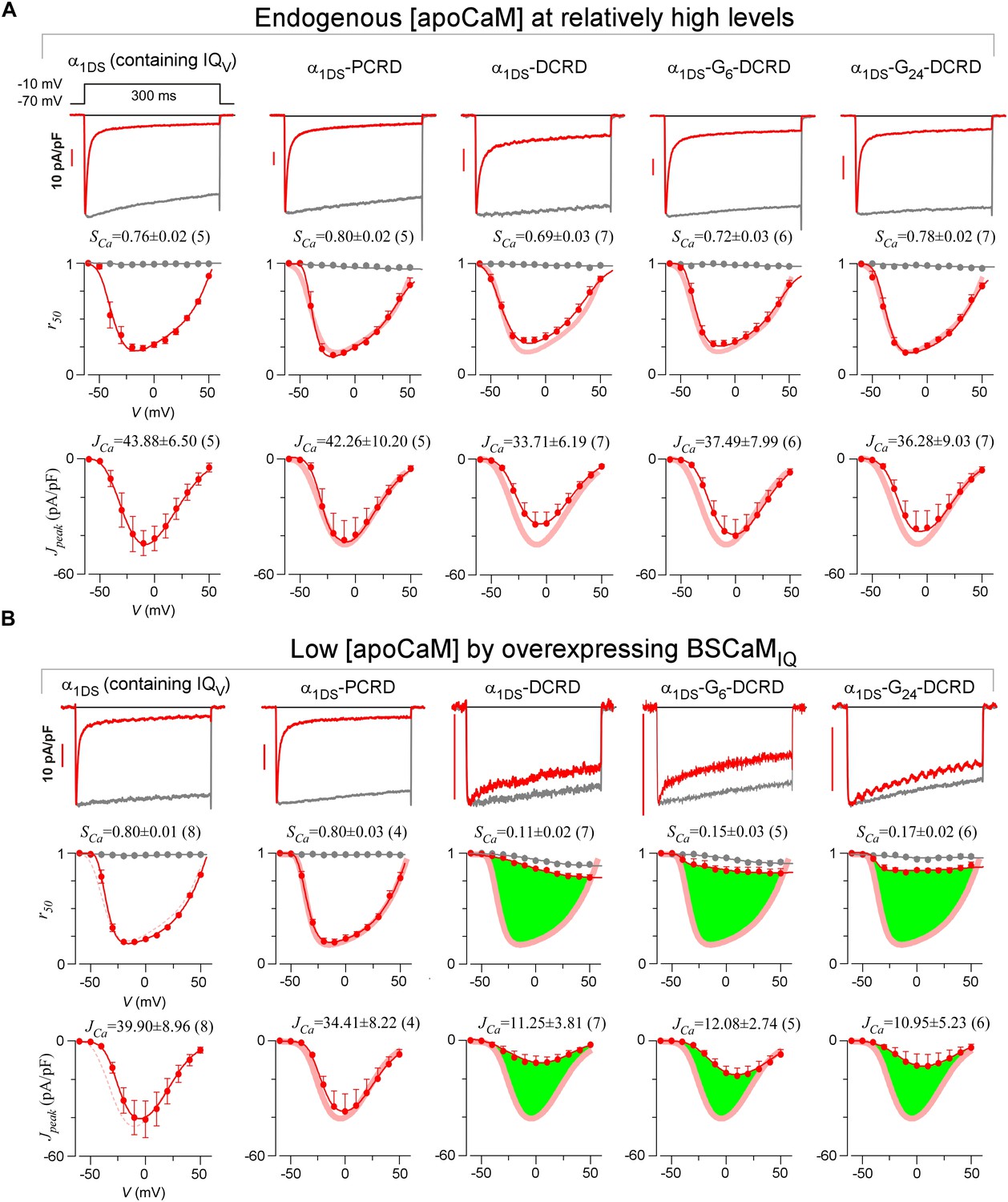
Full CDI and VGA profiles of different channel variants in endogenous and low [apoCaM].
(A) Under normal [apoCaM], channel variants of α1DS, α1DS-PCRD and α1DS-GX-DCRD (the number of glycine X equals 0, 6 and 24, respectively) exhibited no significant changes in exemplar ICa traces (top row), CDI (middle row) and VGA (bottom row) profiles with respective indices of SCa and JCa. Small changes in VGA profiles for α1DS-GX-DCRD were within the experimental variations at least partially due to expression fluctuations in transient transfections; meanwhile, CDI, supposedly concurrently changing with VGA, was very close to the α1DS control (thick semitransparent red lines). (B) When [apoCaM] was substantially lower down by overexpressing the apoCaM buffer of BSCaMIQ, α1DS-GX-DCRD exhibited potent attenuations on both CDI and VGA (green areas indicating the differences from the control group), but not for α1DS or α1DS-PCRD containing no DCRD. Thick red lines (semitransparent) represent the r50 and Jpeak profiles of the control group (α1DS under low [apoCaM], left) for direct comparison. Dotted lines in the control group (left column) were replicated from α1DS in normal [apoCaM] (A), essentially no difference.
Figure 3 with 2 supplements

Individual DCRD peptides expressed separately from IQV are unable to induce CMI.
(A) PCRD and DCRD tagged with fluorescent proteins were coexpressed with α1DS as separate peptides. The presence of both YFP-PCRD and CFP-DCRD peptides in the same cell was confirmed under a fluorescence microscope (Figure 3—figure supplement 1). Under normal [apoCaM] in cells, no CMI effect was observed from ICa trace exhibiting CDI similarly as α1DS control, confirmed by comparable SCa values and indistinguishable r50 profiles. (B and C) Experiments and analyses were performed in normal [apoCaM] similarly as (A), except that only YFP-PCRD (B) or only YFP-DCRD (C) was expressed with α1DS channels. Both resulted into strong CDI, with SCa and r50 indistinguishable from α1DS control. (D−F) When free [apoCaM] was substantially reduced by overexpressing BSCaMIQ (apoCaM buffers), the above three cases in (A−C) were re-examined. Exemplar ICa traces exhibited strong CDI as quantified by SCa values and r50 profiles, similarly as the control group of α1DS in low [apoCaM] (semitransparent line in red).
Figure 3—figure supplement 1
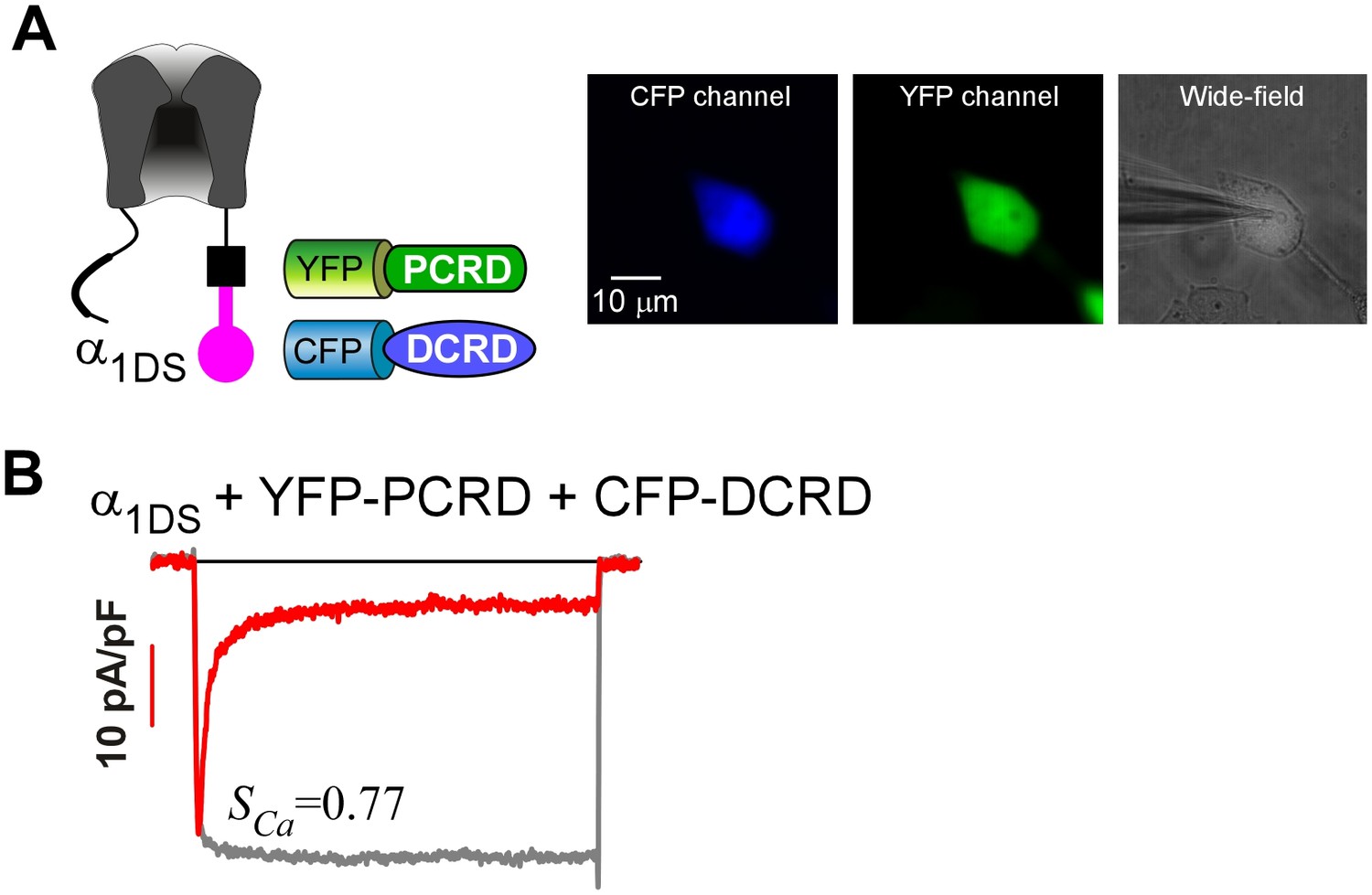
To confirm the presence of both PCRD and DCRD peptides in single cells.
(A) YFP-PCRD and CFP-DCRD were coexpressed with α1DS (left). For one single cell under patch recording (right image), the epi-fluorescence of high intensity via CFP (left image) and YFP (middle image) channels was observed to confirm that both peptides were well expressed. (B) The exemplar recording of ICa from that particular cell in (A) exhibited strong CDI (SCa), within the range of standard SCa for α1DS (0.76 ± 0.02, n = 5).
Figure 3—figure supplement 2
PCRD and DCRD as separate peptides are unable to inhibit VGA.
(A–C) In normal [apoCaM], when both PCRD and DCRD were expressed but as separate peptides (A), the VGA profile of α1DS (control, red thick semitransparent lines) was unaltered as indexed by JCa. Similarly, no inhibition of VGA was observed when only PCRD (B) or only DCRD (C) was transfected. (D−F) When [apoCaM] was reduced by overexpressing BSCaMIQ, for all the three cases in (A−C), their VGA profiles were indistinguishable from the control group (red thick semitransparent lines), also confirmed by similar JCa values across different groups.
Figure 4 with 2 supplements
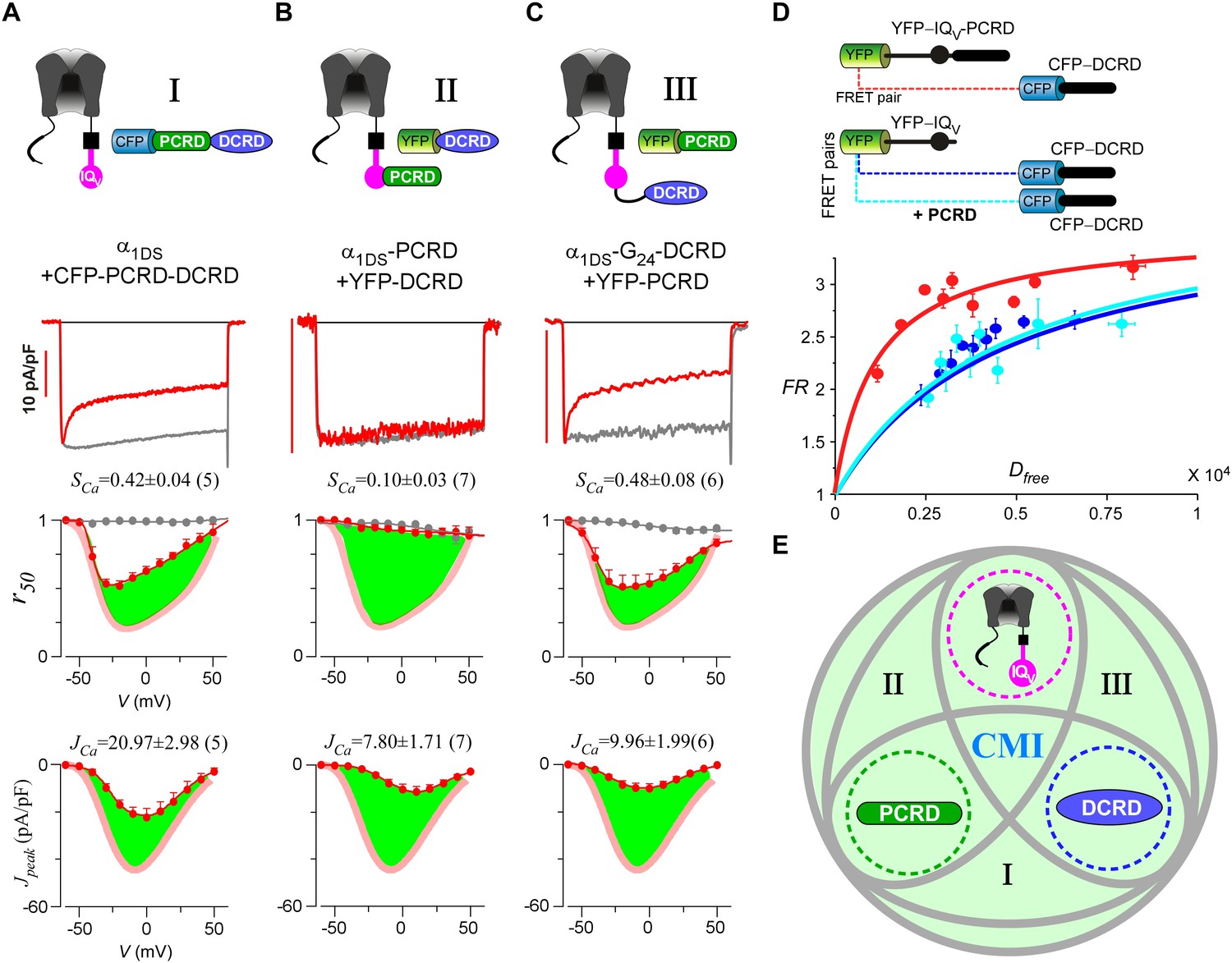
Cooperation by PCRD, DCRD and IQV to induce CMI.
(A) Both CDI and VGA of α1DS channels were attenuated by pre-linked PCRD-DCRD illustrated in the scheme (top), shown in the exemplar traces (middle) and voltage-dependent r50 and Jpeak profiles (lower two panels). The potency of CMI was indexed by SCa and JCa values and also illustrated by the green areas. Profiles of α1DS control were indicated by thick semitransparent lines in red (lower two panels). (B) Both CDI and VGA of α1DS-PCRD were strongly prohibited by DCRD. (C) Both CDI and VGA of α1DS-G24-DCRD were attenuated by PCRD. (D) FRET 2-hybrid assays demonstrated that pre-linked IQV-PCRD motif (YFP tagged) exhibited strong binding with CFP-DCRD, with higher binding affinity (Kd = 1135, units in donor-cube fluorescence intensity) than the binding affinity (Kd = 4700) between CFP-DCRD and YFP-IQV itself (without PCRD being fused). Additional PCRD peptides did not make any appreciable change (Kd = 4624) for the binding between CFP-DCRD and YFP-IQV, unable to rescue the low affinity back to the high level that the constitutive PCRD fusion (YFP-IQV-PCRD) could achieve. FRmax values for all these binding curves were similar (3.50—3.87). Each data point represented the FR (y-axis, FRET ratio) and Dfree (x-axis, free donor concentration) values averaged over five adjacent individual cells sorted by Dfree. (E) Working model to illustrate the collaboration among three components of PCRD, DCRD and IQV (embedded within α1DS) for CMI induction. Grey circles represent the effective combinations, which require that at least two (out of three) components are closely engaged (e.g., fusion) to form the trio complex incorporating the third (separate) component. The three key combinations are I (IQV+PCRD-DCRD), II (IQV-PCRD+DCRD) and III (IQV-DCRD+PCRD), where ‘–’ denotes fusion or spatial closeness and ‘+’ indicates the separate peptide to be coexpressed. In addition, the positive control (IQV-PCRD-DCRD) also represents the native long variant α1DL; and the three components (dotted circles in green, pink and blue) completely separate to each other (IQV+PCRD+DCRD) produce no CMI effect, serving as one negative control.
Figure 4—figure supplement 1
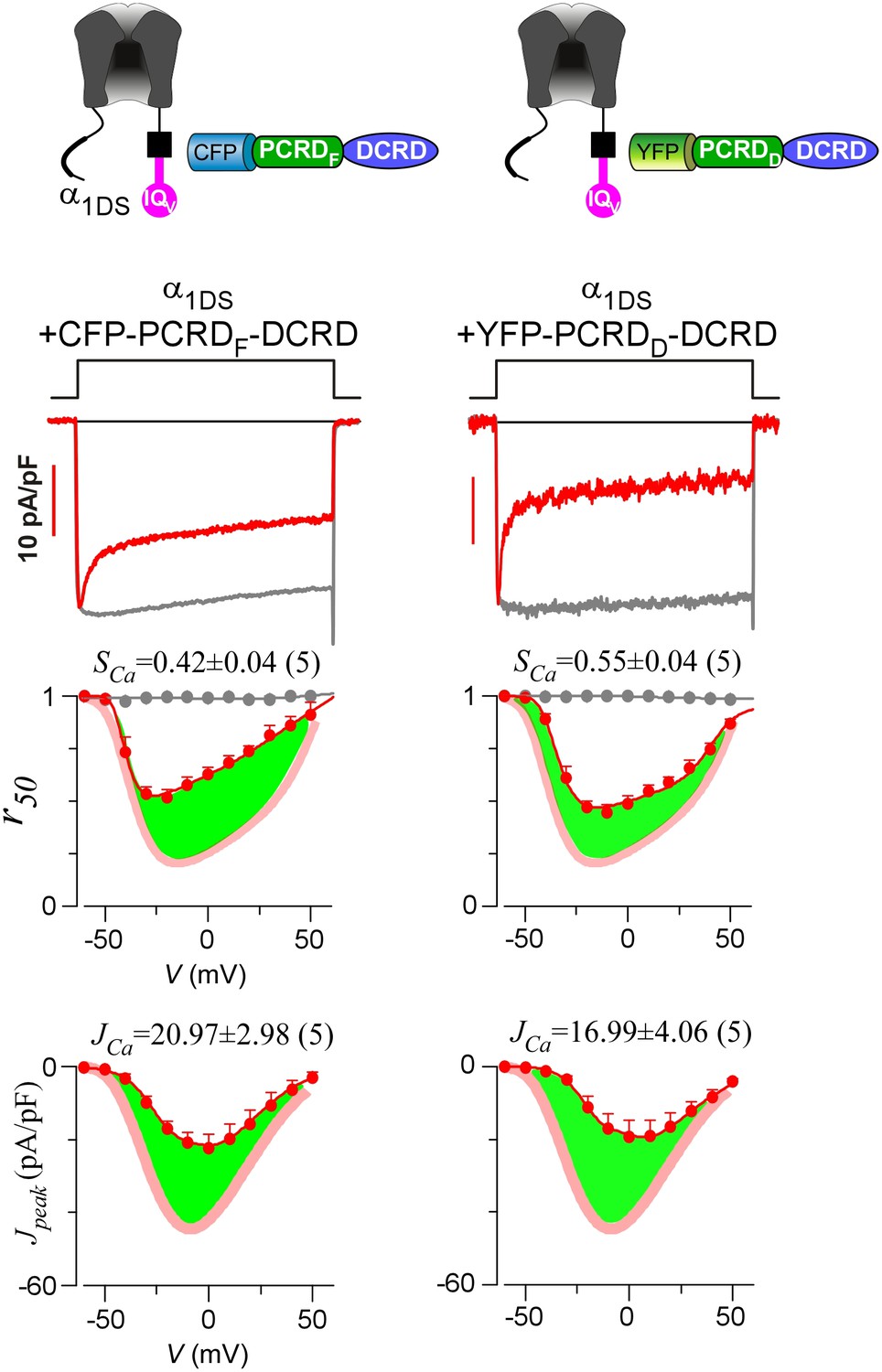
Functional similarities between PCRDF and PCRDD.
The pronounced CDI (SCa) and strong VGA (JCa) of α1DS channels (indicated by red thick semitransparent lines in r50 and Jpeak plots) were similarly attenuated by PCRD-DCRD peptides made from either PCRDF from CaV1.4 (left column) or PCRDD from CaV1.3 (right column). Representative ICa traces (top), CDI and VGA profiles (panels in the two bottom rows) across the full voltage range were shown for the two types of peptides, with no significant difference. Green areas represent the potency of attenuation, supporting that PCRDF and PCRDD were interchangeable for CMI in this study as we assumed.
Figure 4—figure supplement 2
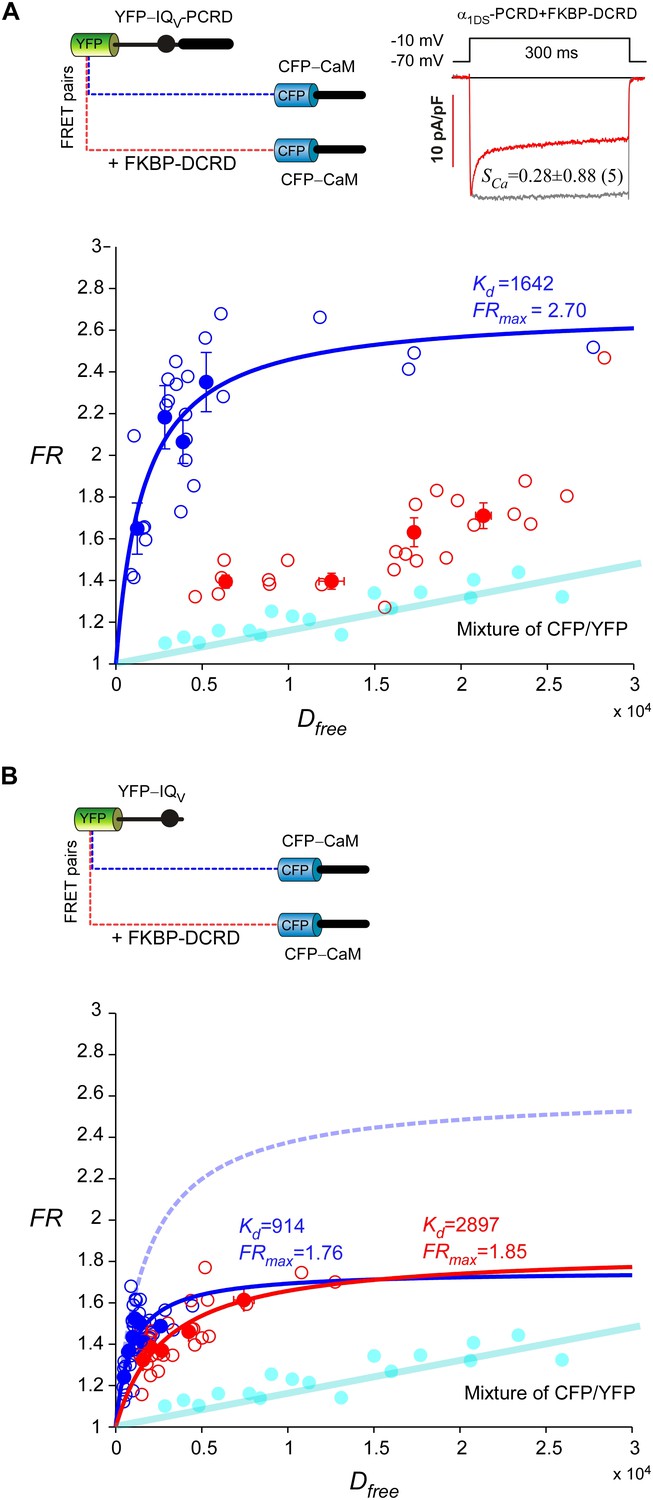
Cooperative perturbation of apoCaM/IQV binding by DCRD and PCRD.
(A) By 2-hybrid FRET with the CFP- or YFP-labeled constructs illustrated on top, IQV-PCRD (YFP-tagged) and CaM (CFP-tagged, apo-state) exhibited strong binding (blue), quantified by the maximum FRET ratio (FRmax) and effective equilibrium dissociation constant (Kd) from iterative curve fitting. The values of FRmax and Kd were indicated right above the fitted curves (in the same color) when applicable. Unfilled-dots and filled-dots represent individual cells and averaged results (over five cells) respectively. The binding between apoCaM and IQV-PCRD were severely perturbed by FKBP-DCRD (with FKBP tag adopted from subsequent rapamycin-inducible CMI, but without fluorescent tags of CFP/YFP), resulted into the data points (red) approaching the baseline of CFP/YFP mixture (cyan), indicative of very weak Kd. The perturbation was presumably by way of the close cooperation between FKBP-DCRD and IQV-PCRD to compete against apoCaM. The FRET binding analyses were consistent with the patch-clamp recordings confirming that CDI (SCa) of α1DS-PCRD was attenuated in the presence of FKBP-DCRD (inset). (B) In the absence of PCRD, the perturbation by FKBP-DCRD was much less effective. Without the presence of PCRD, CFP-CaM and YFP-IQV (blue) were still able to bind. However, in contrast to the strong perturbation seen in (A), in the absence of PCRD, inclusion of DCRD peptides made no or very little difference in Kd and FRmaxvalues for CaM and IQV. The binding curve (dotted line in light blue) was replicated from CFP-CaM and YFP-IQV-PCRD (A) for comparison.
Figure 5 with 2 supplements
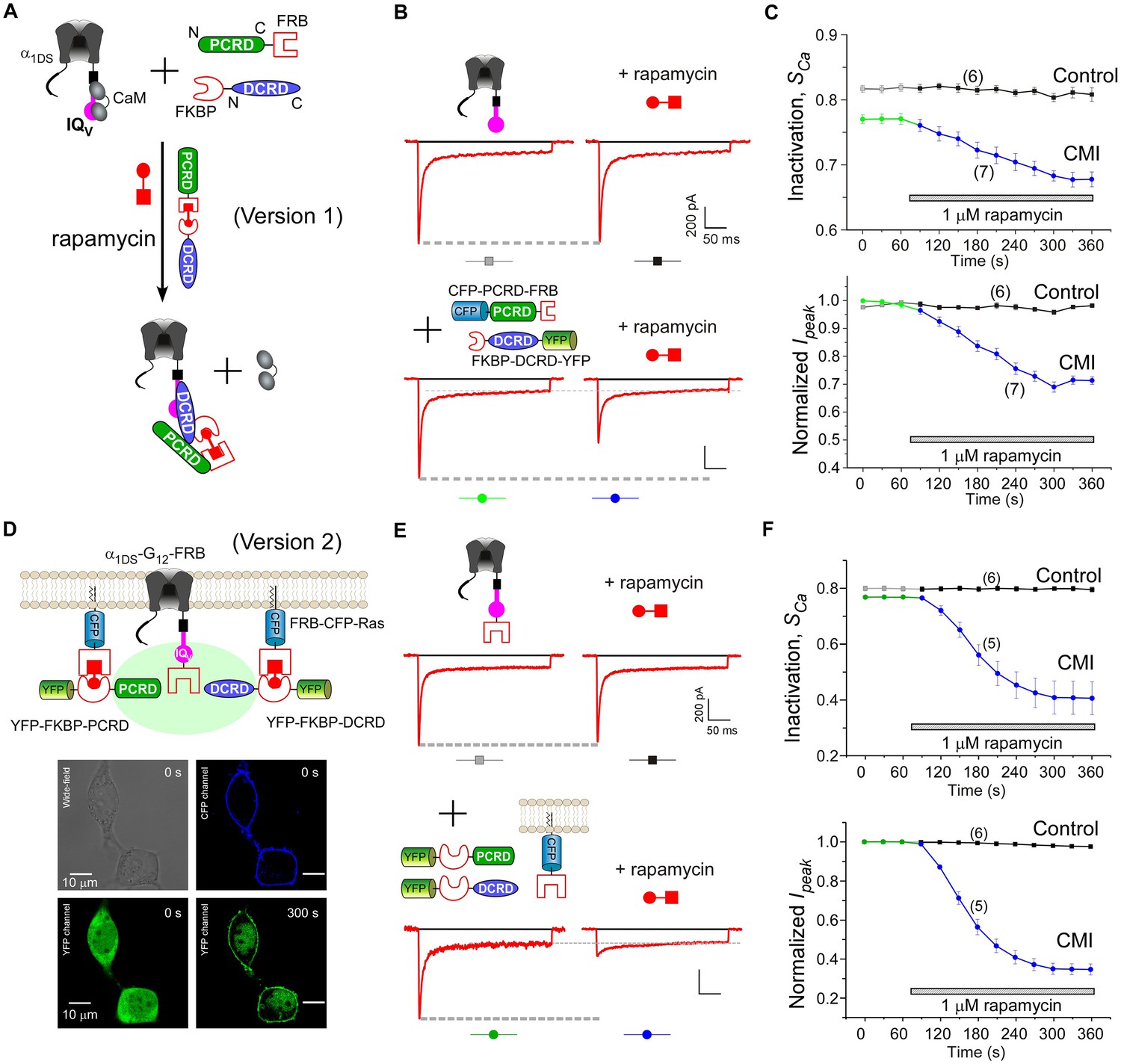
Design schemes and experimental implementations of rapamycin-inducible CMI.
(A) Design principles for chemical-inducible CMI. Rapamycin simultaneously binds one FKBP and one FRB to combine any two FRB/FKBP-tagged components (PCRD and DCRD, in this version 1) selected from the three components of PCRD, DCRD and IQV. Thus, the three components would form the combinations (combination I, in the version 1) to satisfy the requirement of cooperative CMI (Figure 4E). (B) One representative implementation of chemical-inducible CMI. According to the design of version one in (A), PCRD-FRB and FKBP-DCRD were constructed and coexpressed with α1DS. Exemplars of Ca2+ current traces demonstrated that acute effects were induced by applying 1 μM rapamycin to the multi-component system of version 1 (lower panel), in contrast to α1DS control with no appreciable changes in ICa (upper panel). (C) Statistical summary of rapamycin-induced CMI (version 1). Averaged values over multiple cells (number indicated in parentheses) for the indices SCa (upper) or normalized Ipeak (lower) demonstrated time-dependent attenuations on both CDI and VGA upon rapamycin application as compared to the control group. (D) Rapamycin perfusion induced rapid translocation of YFP-FKBP-DCRD or YFP-FKBP-PCRD (version 2) onto the membrane by linking with FRB-CFP-Ras within 5 min, shown by confocal images via wide-field, CFP, and YFP channels. The local concentrations of YFP-FKBP-DCRD and YFP-FKBP-PCRD were substantially enhanced as suggested by the condensed YFP fluorescence at the membrane (outlined by CFP fluorescence). (E and F) According to rapamycin-inducible CMI of version 2, recombinant channels of α1DS-G12-FRB were coexpressed with cytosolic YFP-FKBP-PCRD and YFP-FKBP-DCRD. To enhance local concentrations of FKBP-tagged peptides near the channels, membrane-localized FRB-CFP-Ras was also overexpressed. Exemplars of Ca2+ current traces (E) exhibited strong attenuation (lower), in contrast to stable ICa from α1DS-G12-FRB alone (upper). In contrast to the control group, temporal profiles of rapamycin-inducible CMI (version 2) indicated strong attenuations on CDI (SCa, upper) and VGA (normalized Ipeak, lower). Based on totally five trials (cells), SCa changed from 0.77 ± 0.00 to 0.41 ± 0.06 and Ipeak was reduced to 35% ± 3% of the basal levels (F).
Figure 5—figure supplement 1
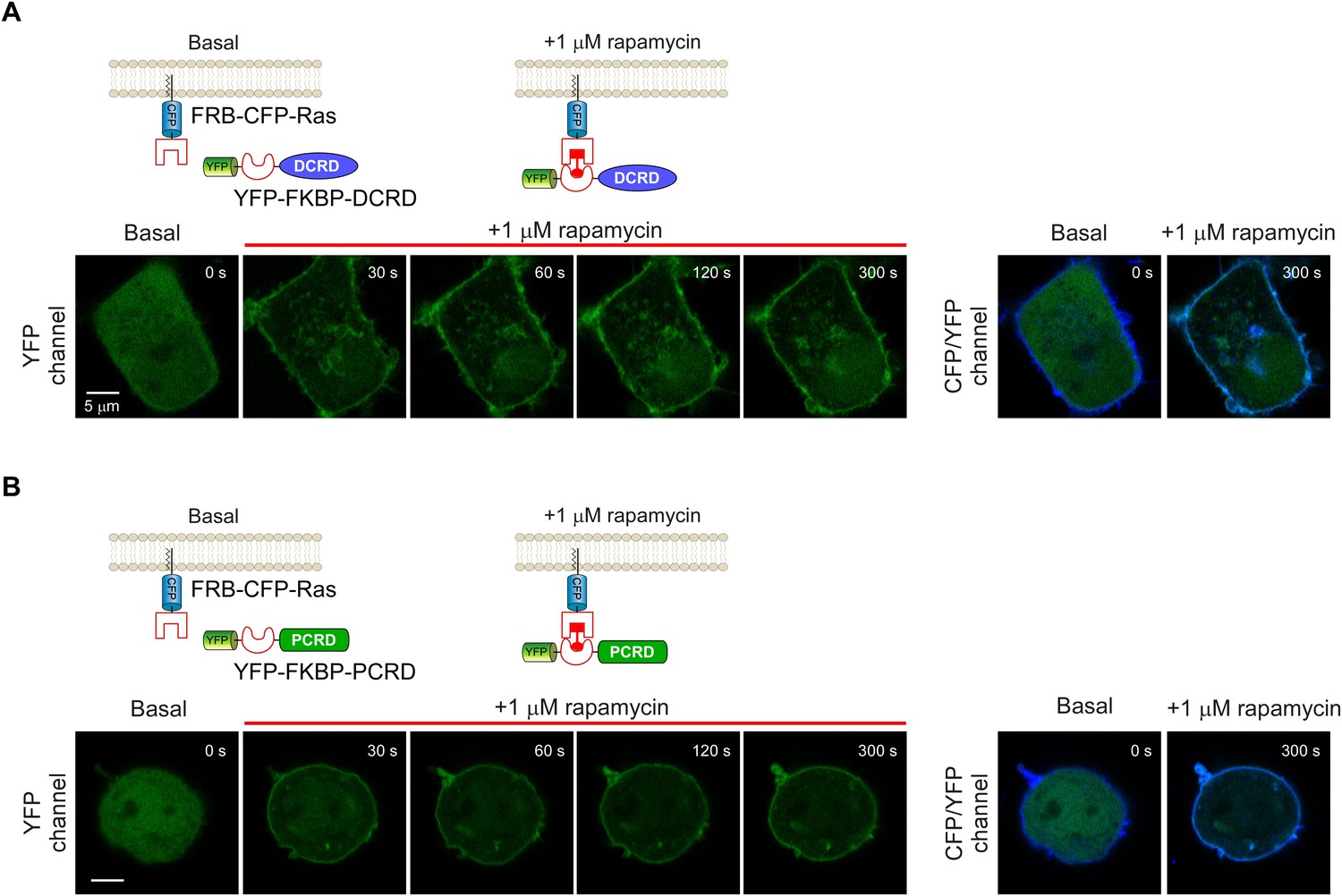
Temporal profiles of fluorescent images for rapamycin-induced membrane-targeting.
(A and B) Top rows: cartoon depicting the design of the rapamycin-induced translocation of DCRD (A) or PCRD (B) (Version 2 of chemical-inducible CMI). FRB-CFP-Ras was constitutively anchored to the plasma membrane by incorporating membrane-targeting segment from Ras protein. Lower rows: confocal images acquired via YFP channel showing rapid translocation of YFP-FKBP-DCRD (A) or YFP-FKBP-PCRD (B) to plasma membrane upon 1 μM rapamycin perfusion (left). At the time of ~120 s or later, YFP fluorescence was substantially condensed to overlap with the stable CFP fluorescence (FRB-CFP-Ras) on the membrane (right).
Figure 5—figure supplement 2
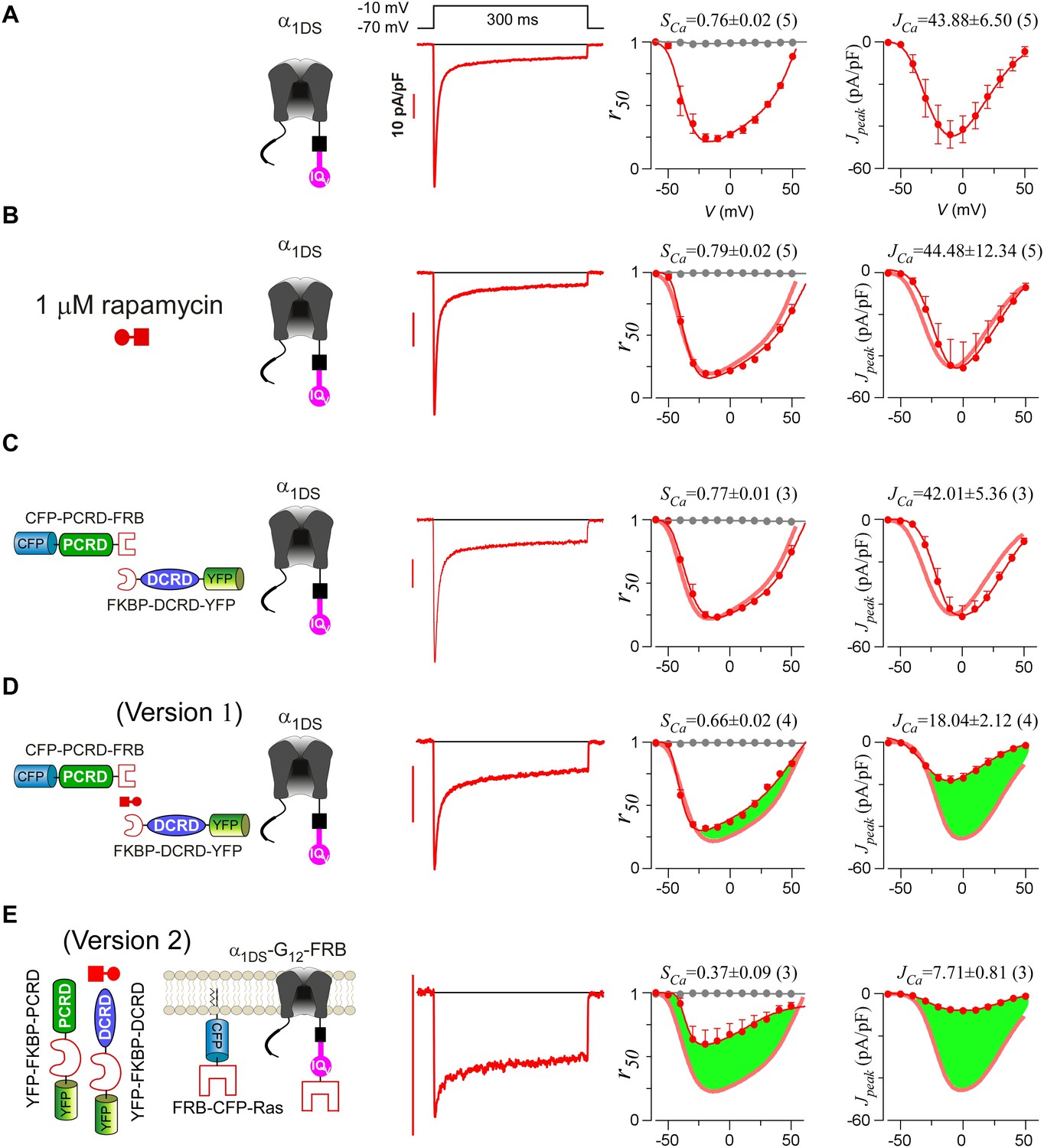
Detailed characterizations for rapamycin-induced CMI.
(A and B) Voltage-dependent profiles of CDI and VGA exhibited no difference when α1DS channels were with (B) or without (A) rapamycin. Thick lines (red semitransparent) represent the CDI and VGA profiles from the α1DS control group. (C) Coexpression of CFP-PCRD-FRB and FKBP-DCRD-YFP with α1DS representing the control conditions before rapamycin did not cause any appreciable change in CDI and VGA, confirmed by pronounced SCa and JCa comparable to the values from α1DS control. (D) When rapamycin was applied to α1DS coexpressed with CFP-PCRD-FRB and FKBP-DCRD-YFP, SCa and JCa averages over different cells were attenuated with moderate potency (illustrated by green areas) presumably by rapamycin-induced linkage between PCRD and DCRD (version 1 of inducible CMI) to form the cooperation for effective CMI. (E) In rapamycin, with enhanced local concentrations of YFP-FKBP-PCRD and YFP-FKBP-DCRD (version 2 of rapamycin-induced CMI), the potency was much improved, demonstrated by more pronounced attenuations concurrently on both CDI and VGA (larger green areas in both r50 and Jpeak profiles).
Figure 6 with 3 supplements
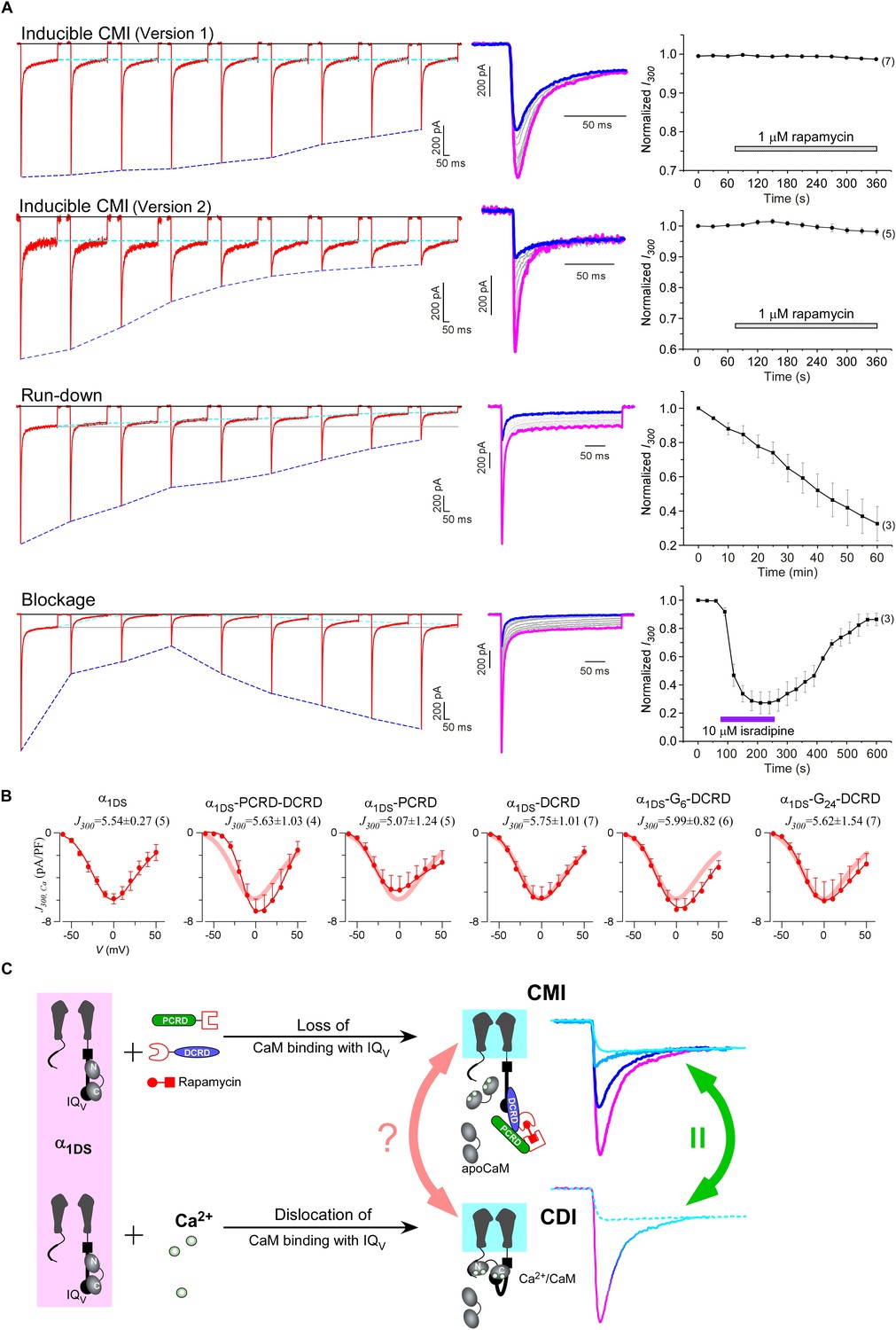
Mechanistic insights enlightened by unique features of CMI.
(A) Temporal profiles between CMI and conventional inhibitions were compared. For rapamycin-inducible CMI (two versions in the top two rows of panels), representative Ca2+ current traces in rapamycin were selected from sequential time-points (left column) and superimposed together (middle column) for comparison. The time sequence was indicated by color: the first in pink, intermediate in grey, and the last in blue. Ipeak exhibited the trend of inhibition but not for I300 (ICa at 300 ms) (right column). In contrast, for run-down process and isradipine blockage (bottom two rows), both Ipeak and I300 exhibited the declining trends indicating substantial inhibitions. (B) Ca2+ current density at 300 ms (J300,Ca) remained at the same level (indicated by the J300 values at −10 mV), for various channel variants under test, regardless of whether CMI was in effect. Thick lines (semitransparent in red) represent the J300,Ca profile of α1DS control. (C) Functional and the structural insights into the two modes of inhibition. DCT/apoCaM-dependent CMI and Ca2+/CaM-mediated CDI result in indistinguishable gating (green arrows) appearing due to similar causes, i.e., either total or partial loss of the apo-state CaM/IQV complex. That says, in CMI, apoCaM pre-association is totally lost; and in CDI, apoCaM is calcified and dislocated from the pre-association sites. Although the triggers are different for CMI vs CDI, i.e., DCT competing (off apoCaM) vs Ca2+ binding (onto apoCaM), the similarities between the two different inhibitory regulations of CMI and CDI invite the hypothesis that the core gating machinery (cyan squares) upon depolarization might step into the same scenario/mode including structural details (red arrows). A series of current traces (on the right) indicate CMI with different potency (enhanced from pink to cyan, upper), in comparison with the trace at different stages of CDI (developed from pink to cyan, lower) superimposed with the trace from ultrastrong CMI (dotted trace in cyan). These analyses point to one important notion that the lower limit of CMI is determined by end-stage CDI (the traces or the phases in cyan), and thus the dynamic changes for CMI effects on α1DS (indexed with Ipeak and SCa) are preset, i.e., from the pink (no CMI and ultrastrong CDI) to the cyan (ultrastrong CMI and no CDI).
Figure 6—figure supplement 1
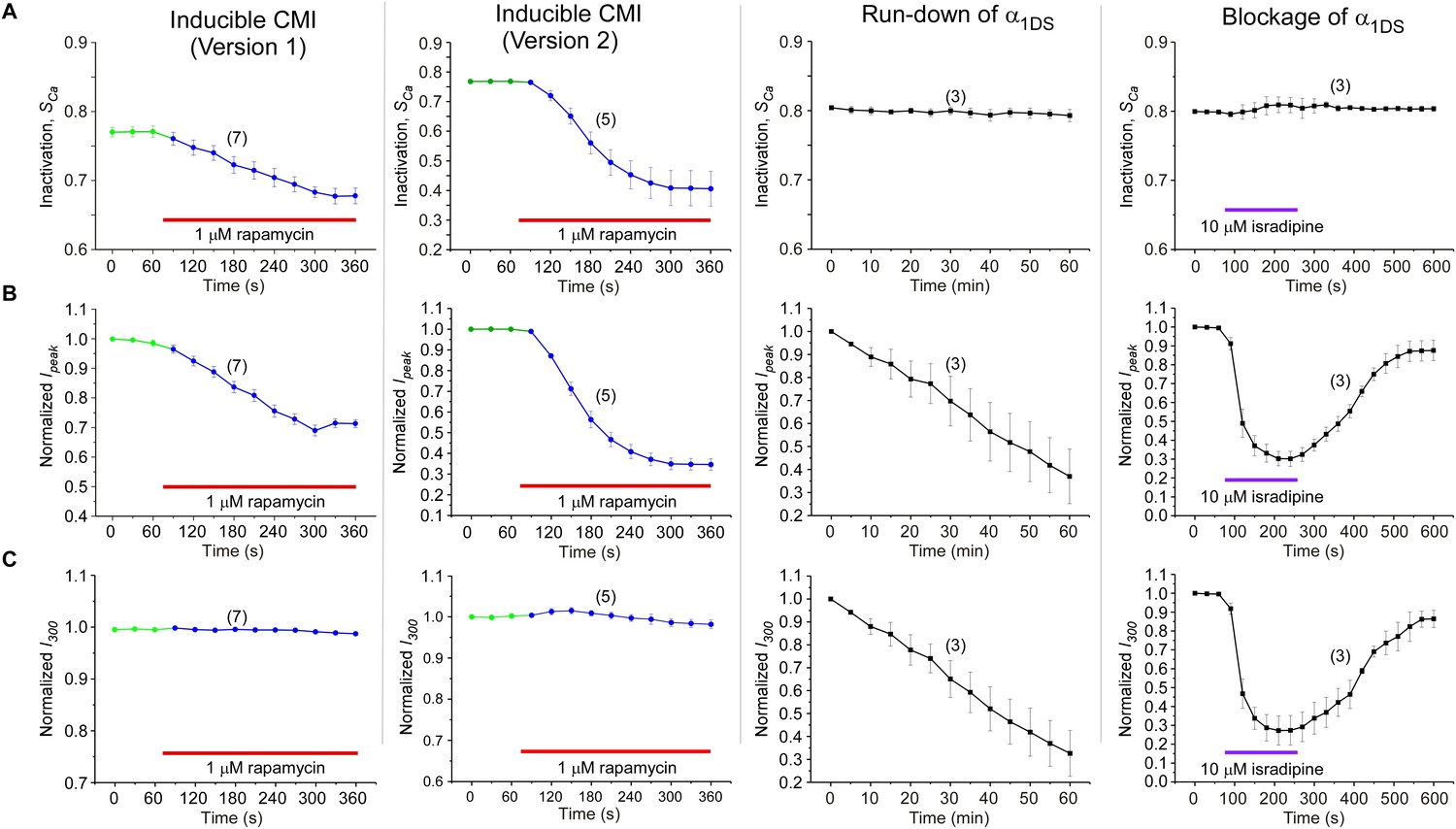
Detailed comparisons among CMI, run-down and blockage.
CMI was compared with conventional inhibitions/inhibitors including channel blockage (by isradipine) and run-down process (reduction of channel numbers), indexed with SCa, Ipeak and I300. Both rapamycin-induced CMI and run-down/blockage exhibited time-dependent decreases in Ipeak, as expected from effective ICa inhibitions (B). In contrast, CDI (SCa) did not change in run-down/blockage; whereas for CMI SCa exhibited rapamycin-dependent attenuation, similar to Ipeak (A). Moreover, the plateau of ICa (I300) remained constant in spite of Ipeak attenuation in CMI, whereas I300 changed (decreased or recovered) in a similar time-course to that of Ipeak during run-down and blockage (C). Since SCa is in proportion to Ipeak/I300, the two major types of inhibitions (CMI vs conventional inhibitions) were distinct within this context: for CMI, both Ipeak and SCa changed but with I300 being kept constant; in contrast, for conventional inhibitions, both Ipeak and I300 changed, but SCa remained unaltered.
Figure 6—figure supplement 2
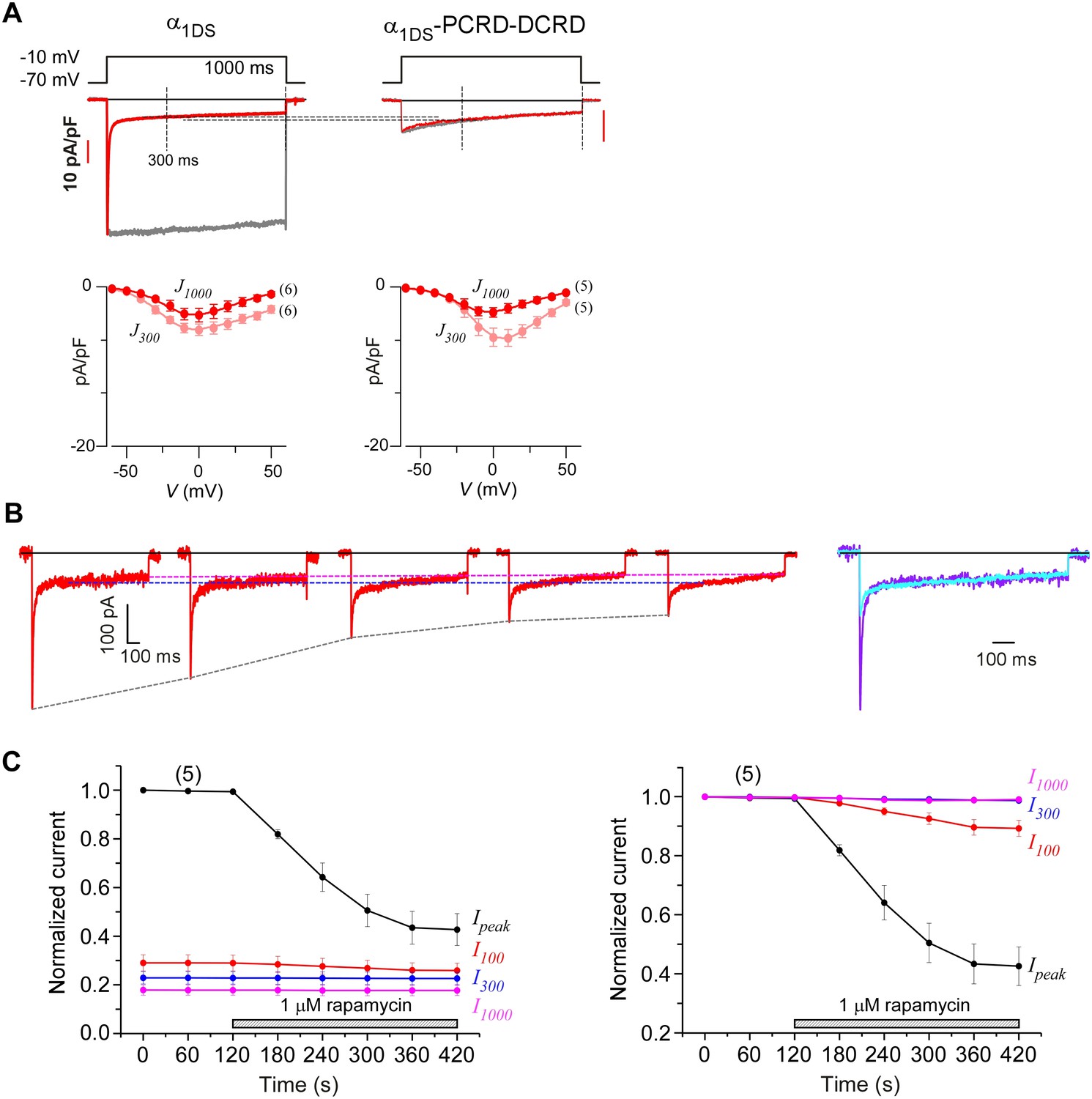
Indices at different time points of ICa to quantify CMI and end-stage CDI.
(A) Analyses at both 300 ms and 1000 ms were able to reliably unveil that channels subject to CMI were essentially tuned to approach ICa levels in end-stage CDI. Upper panel, we set I1000 (ICa at 1000 ms) of both exemplar traces to the same level for α1DS and α1DS-PCRD-DCRD (in principle, they should be the same); and the levels of ICa at 300 ms (I300) turned out to be nearly the same, similarly as the analyses with I1000. At 300 ms, the kinetic difference between IBa (normalized to the peak of ICa) and ICa was very little, indicative of end-stage CDI (upper right). Full profiles of Ca2+ current density (indexed with J300 or J1000) were indistinguishable for both groups of α1DS and α1DS-PCRD-DCRD, further supporting that both are qualified as reliable indices for CMI analysis. (B) For rapamycin-inducible CMI (version 2) by supplying PCRD, DCRD and α1DS with membrane-targeting and rapamycin-inducible mechanisms, representative ICa traces were selected from sequential time-points (left), and the first (purple) and the last (cyan) traces were superimposed together for comparison (right). Ipeak exhibited a trend of time-dependent decrease; meanwhile, both I300 and I1000 remained rather constant (dotted outlines in black, blue and pink respectively). (C) Statistical summary of ICa amplitudes for rapamycin-induced CMI (version 2). ICa values were measured at different time points, i.e., Ipeak, I100, I300 and I1000, each of which was either rescaled to Ipeak (left) or to its own (right). Ipeak exhibited robust inhibition due to CMI, and a trend of moderate decline (right) was noticeable from I100 (ICa at 100 ms); whereas I300 and I1000 clearly remained constant throughout the full time-course, thus reliably representing the end stage or steady state of CDI. In line with our previous analyses, the current level of I300 or I1000 determined the lower limit of ICa (measured by Ipeak) subject to CMI of the maximum potency.
Figure 6—figure supplement 3
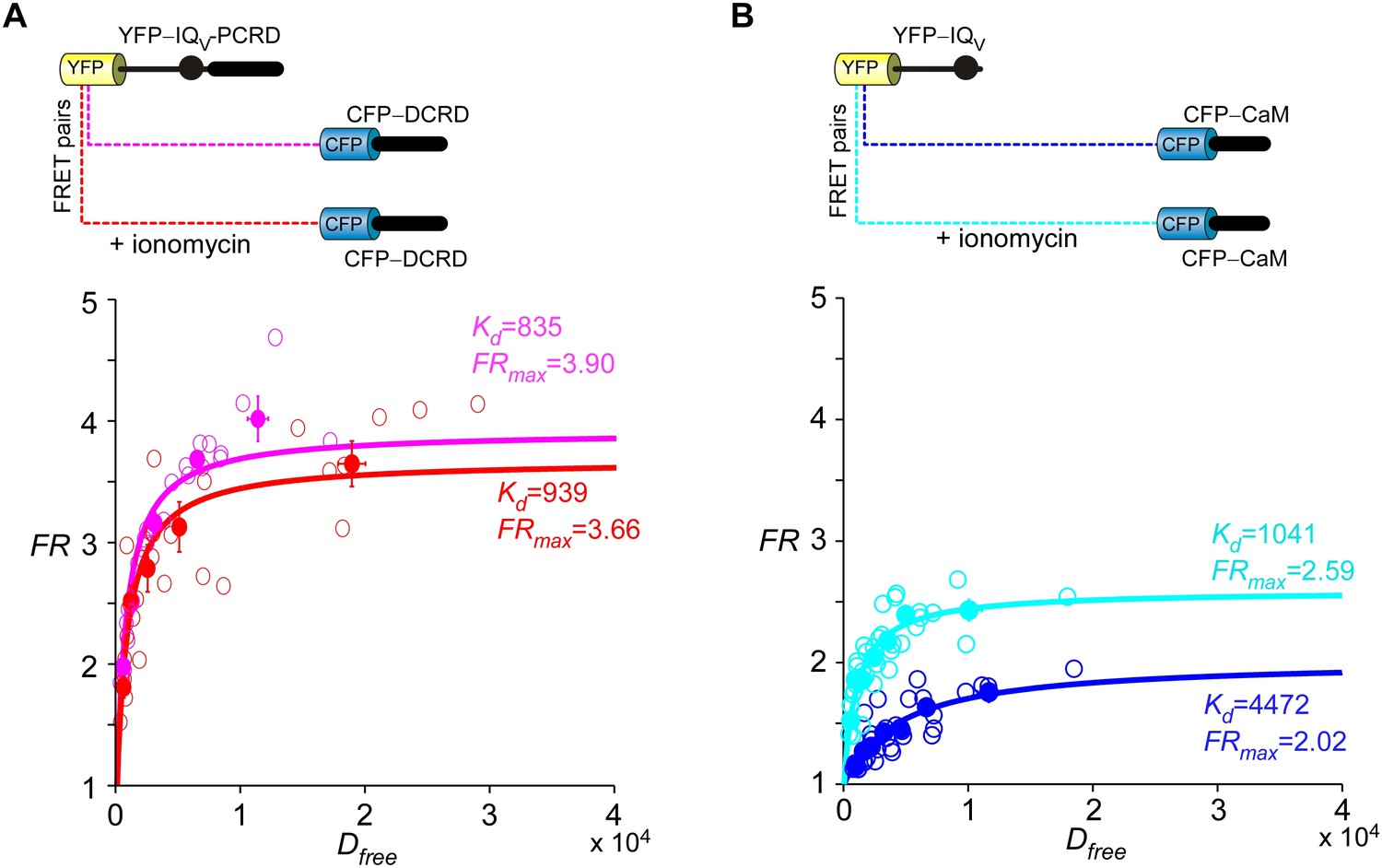
The high-affinity binding between DCRD and the channel was not perturbed by Ca2+/CaM.
(A) Upon applying ionomycin, an ionophore massively raising intracellular Ca2+ levels, endogenous CaM should switch from Ca2+-free state (apoCaM) to Ca2+-bound state (Ca2+/CaM). However, similar Kd values were obtained by fitting the FRET binding curves before and after ionomycin application (pink, before ionomycin; red, in ionomycin), indicating that Ca2+/CaM was unable to perturb the strong binding between DCRD and IQV-PCRD. In the FR-Dfree plots, unfilled-dots and filled-dots represent individual cells and averaged results (over five cells) respectively. (B) Following ionomycin administration, the indices (Kd and FRmax) of the binding between CaM and IQV were clearly strengthened consistent with the IQV/CaM interactions previously established.
Figure 7
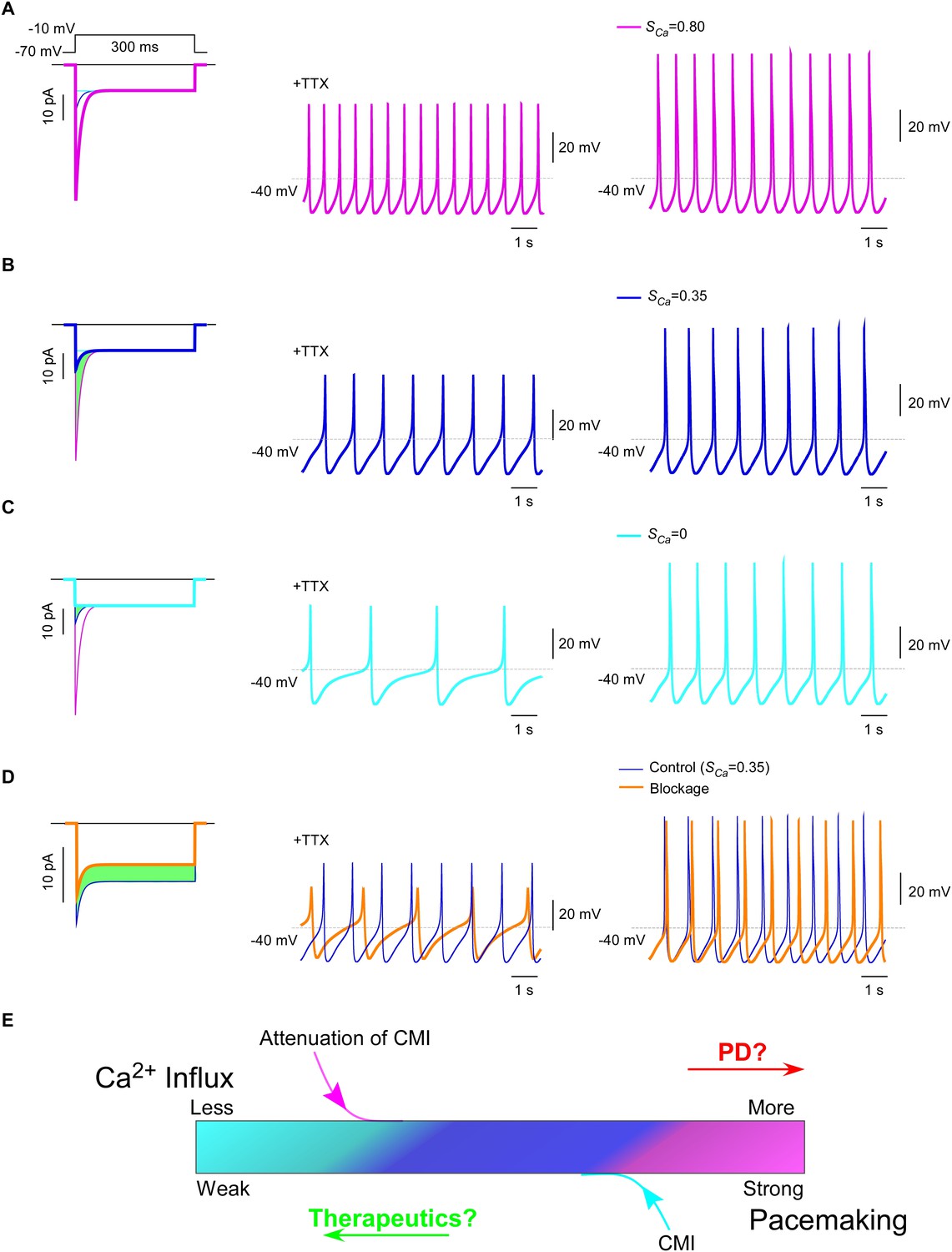
Inhibition of SNc oscillation and pacemaking by CMI.
(A and B) Oscillation and pacemaking of SNc neurons are mediated by CaV1.3, presumably mixed with both short (α1DS) and long (α1DL) splice variants. Lack of CMI, α1DS (pink) exhibited large Ipeak and strong SCa (A), altogether resulted into more Ca2+ influx than α1DL (blue), as indicated by the green area (B). Side-by-side comparison demonstrated that CaV1.3-dependent oscillation (middle column) and pacemaking (right column) were slowed down, presumably by intrinsic CMI of α1DL in comparison to α1DS. (C) Effects of ultrastrong CMI (cyan) on SNc neurons. Under the CMI of the maximum potency (left), Ipeak was reduced to the lower limit, i.e., the level of I300, and SCa was approaching 0. Accompanied with reduction in Ca2+ influx (green area), oscillation (middle) and pacemaking (right) rates were also further reduced compared with α1DS (A) or α1DL (B). (D) Effects of conventional blockage. Conventional inhibition by blockers (orange) attenuated ICa (28% reduction) and Ca2+ influx (green area) but kept SCa unaltered (left), by which oscillation (middle) and pacemaking (right) rates were slowed down compared with the α1DL control (B). (E) The potential regulatory scheme for CaV1.3-mediated pacemaking in SNc neurons. The actual gating of ICa (similar to B) could fall into intermediate levels between two extreme conditions of rather weak CMI (apoCaM-bound and capable of strong CDI) (A), or ultrastrong CMI (apoCaM-off and abolishment of CDI) (C). Hence, both CaV1.3 and pacemaking behaviors are potentially bidirectionally regulated, e.g., apoCaM and apoCaM-binding proteins, to maintain the homeostatic balance of Ca2+ influx. Pathological dysregulations of CaV1.3 and the resulted Ca2+ imbalance might underlie PD and other diseases, as the rational to develop therapeutic perturbations based on CMI.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cooperative and acute inhibition by multiple C-terminal motifs of L-type Ca2+ channels
eLife 6:e21989.
https://doi.org/10.7554/eLife.21989




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}