Biogenesis of phased siRNAs on membrane-bound polysomes in Arabidopsis
- Chinese Academy of Agricultural Sciences, China
- University of California, Riverside, United States
- Shenzhen University, China
- Institute of Genetics and Developmental Biology, China
- Nanjing Agricultural University, China
Figures
Figure 1 with 2 supplements

Enrichment of miRNAs and a set of endogenous siRNAs in the microsomal (M) and membrane-bound polysome (MBP) fractions.
(A) A brief scheme of microsome and MBP isolation. (B) Size (in nucleotides) distribution of sRNAs in total extract (T), microsome (M), and an ER pull-down (ER-IP). RPMR, reads per million 45S rRNA (see Materials and methods). Error bar indicates standard deviation from three biological replicates. (C) Composition of sRNAs in three size classes in various samples. TP, total polysome; MBP, membrane-bound polysome. Each column represents a biological replicate. (D) Size (in nucleotides) distribution of sRNAs in T, TP, and MBP. (E) Abundance of annotated miRNAs and miRNA*s in MBP and TP. (F) Abundance of siRNAs from TAS1, 2, 3, and four loci in TP and MBP. All siRNAs mapping to the TAS loci were sampled in the four size classes. (G–H) A set of endogenous siRNAs was enriched in the MBP fraction. The genome was tiled into 100 bp windows and the abundance of 21-nt (G) or 22-nt (H) sRNAs in these windows were compared between MBP and TP samples. Regions showing enrichment (DEG region) are in green.
Figure 1—figure supplement 1
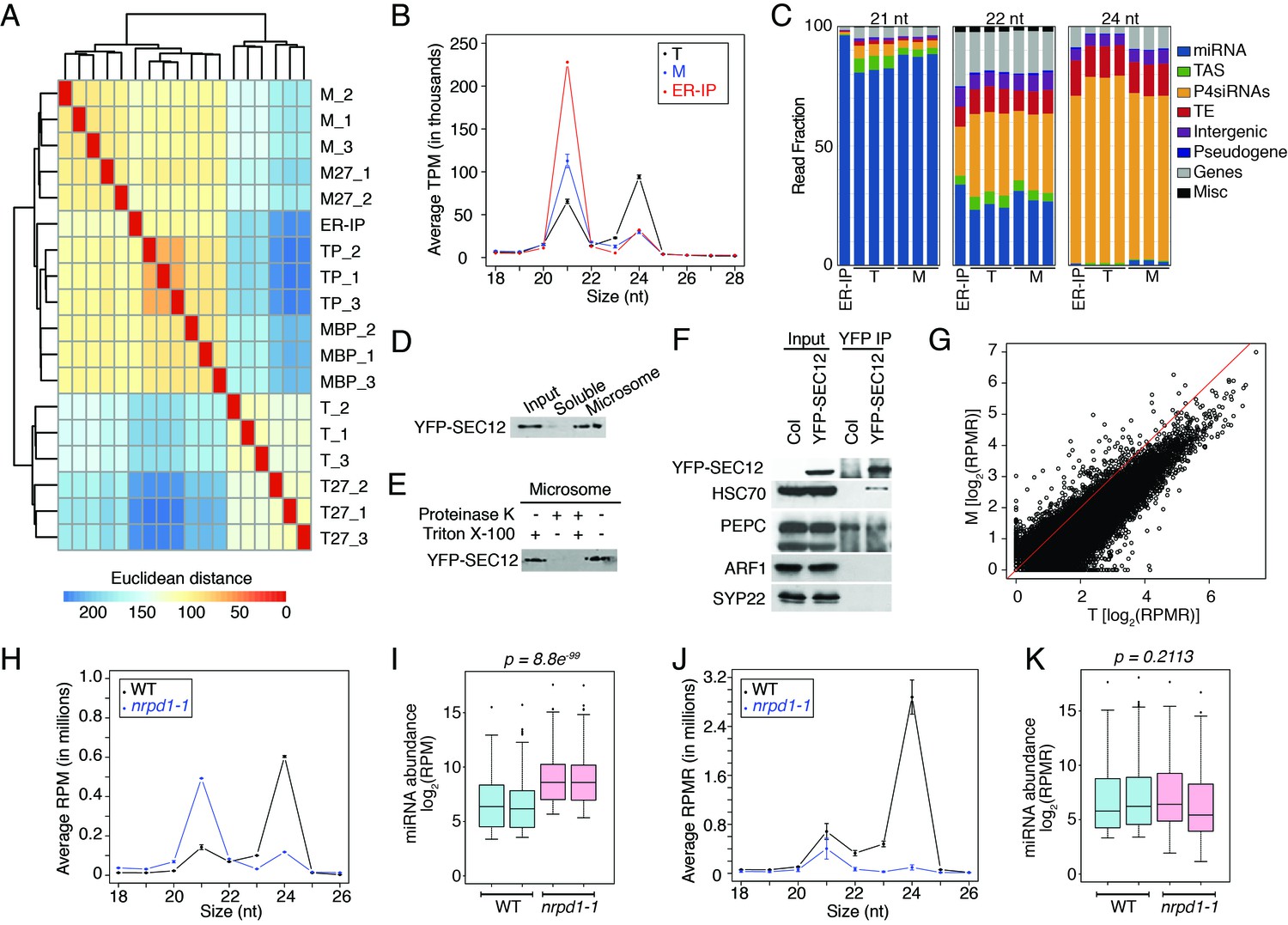
Analyses of sRNA-seq libraries.
(A) Hierarchical clustering analysis showing the degree of similarity among the sRNA-seq libraries. Sample-to-sample distances were calculated based on log-transformed normalized read counts. The biological replicates of each sample type were highly reproducible. T: total extract; M: microsome; TP: total polysome; MBP: membrane-bound polysome; T27: sRNA-seq from total extracts in ago1-27; M27: sRNA-seq from microsomes in ago1-27. The other samples were from wild type. The numbers after the underscore represent biological replicates. (B) Size distribution of sRNAs in the indicated samples. T: total extract; M: microsome; ER-IP: ER immunoprecipitation. Note that normalization was performed against total r/t/snRNA-depleted reads. (C) The composition of sRNAs in each of three size classes. Each column represents a biological replicate. Note that normalization was performed against total r/t/snoRNA-depleted reads. (D) Western blotting using anti-GFP antibodies to show that YFP-SEC12 was in the microsomal fraction. (E) Topology mapping showing that the YFP tag of the ER transmembrane protein YFP-SEC12 was on the cytosolic side. (F) Western blotting showing that ER immunoprecipitation (using anti-GFP antibodies with the YFP-SEC12 transgenic line) contained two ER markers (YFP-SEC12 and HSC70) but not markers for Golgi (ARF1) or endosomes (SYP22). PEPC is a soluble protein. Col, wild type (without the YFP-SEC12 transgene). (G) A scatter plot showing the depletion of Pol IV-dependent siRNAs (P4siRNAs) from the microsomal fraction. Each dot represents a 100 bp window that generates P4siRNAs. Note that normalization was performed using rRNA fragments as internal controls. (H–K) Wild type and nrpd1-1 sRNA-seq samples that differ greatly in sRNA compositions were used as a proof-of-concept for the rRNA fragment-based normalization method. (H–I) Normalization using total r/tRNA-depleted, genome-matched reads led to an exaggeration of the 21-nt peak (H) and an apparent increase in the abundance of miRNAs (I) in nrpd1-1, which lacks most 24-nt siRNAs. (J–K) Normalization using rRNA fragments resulted in similar abundance of 21-nt siRNAs (J) and miRNAs (K) between wild type and nrpd1-1.
Figure 1—figure supplement 2
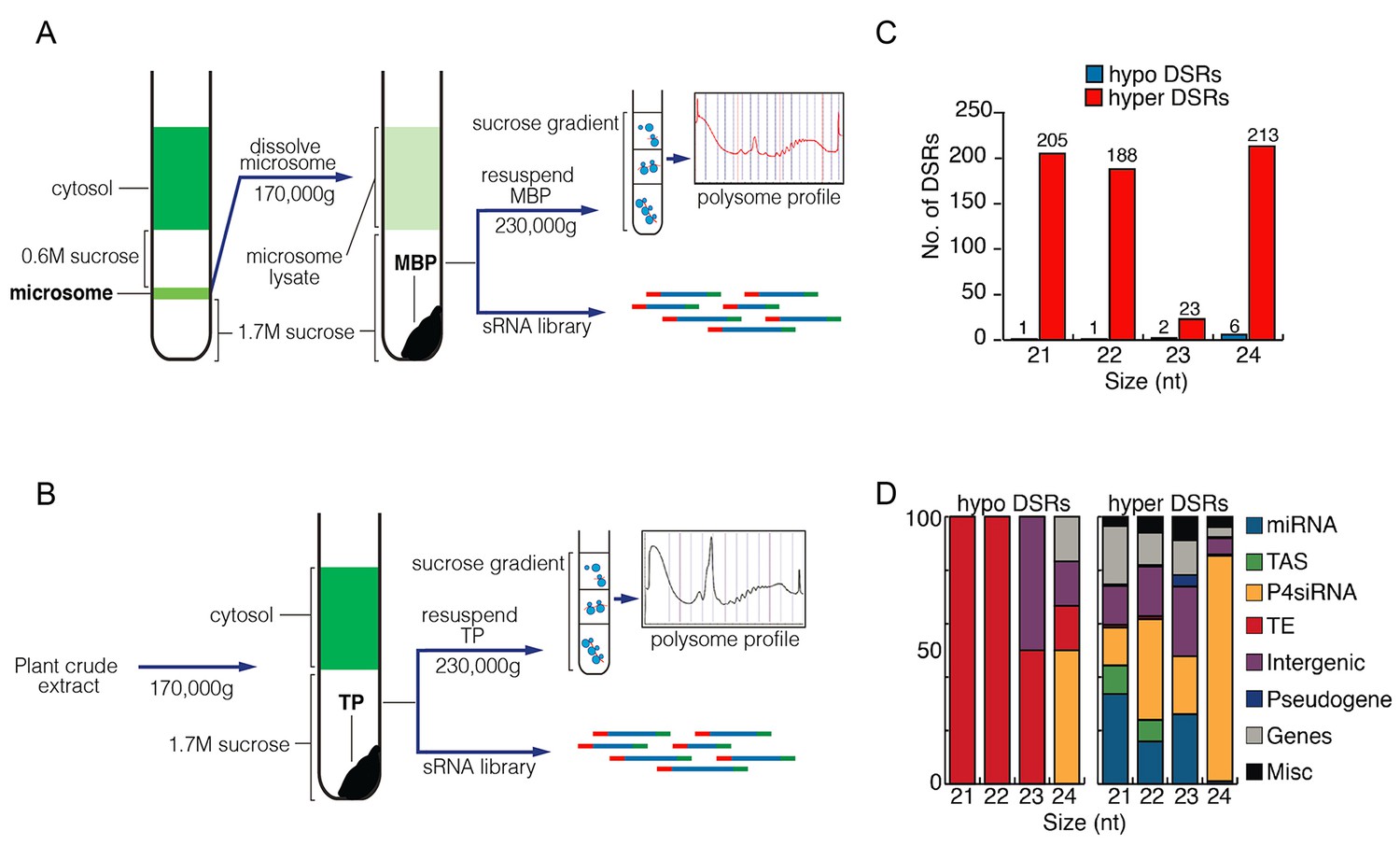
Comparison of MBP and TP sRNAs.
(A–B) Schemes of MBP (A) and TP (B) isolation. The MBPs and TPs were resolved on sucrose gradients to ensure that the expected profiles were seen. (C) Differential small RNA regions (DSRs) for each sRNA size class between MBP and TP samples. The genome was tiled into 100 bp windows and sRNA abundance was compared between the MBP and TP samples for each window. Hypo: MBP<TP; Hyper: MBP>TP. (D) The genomic features of the hypo and hyper DSRs in (C) according to TAIR10 annotation.
Figure 2

The production and MBP association of 22-nt miRNA isoforms.
(A) Many miRNAs exist as both 21-nt and 22-nt isoforms. A heatmap showing the abundance of 21-nt and 22-nt isoforms of 178 miRNAs that were at levels > 1 RPMR in the 12 sRNA-seq samples (three samples each for T, M, MBP and TP from wild type). Each horizontal line represents a miRNA. The annotated sizes are indicated on top. The actual sizes are indicated to the left. (B) 22-nt miRNAs are enriched in MBP relative to TP. The abundance of 22-nt miRNAs in MBP and TP samples is shown. Only a few miRNAs (red dots) were from loci annotated to produce 22-nt miRNAs; most were 22-nt isoforms (cyan dots) from loci that produce predominantly 21-nt miRNAs. (C) Origins of 22-nt miRNA isoforms. The positions of the 5’ and 3’ ends of the 22-nt isoforms are defined relative to the annotated miRNA 5’ and 3’ ends using a scheme shown in the diagram of miR408-3p. This miRNA exists predominantly as a 21-nt form. ‘m_-1_0’ represents a 22-nt isoform with an extra nucleotide on the 5’ end. ‘m_1_2’ represents a 22-nt isoform that is shifted relative to the 21-nt isoform by 1-nt and 2-nt at the 5’ and 3’ ends, respectively. A heatmap showing the abundance of 22-nt isoforms that fall into the various categories. The annotated sizes of the miRNAs are shown on the top. The various mechanisms to generate a 22-nt isoform are indicated to the right. 3’ extension is the predominant mechanism that generates 22-nt isoforms from loci that predominantly produce 21-nt or 20-nt miRNAs. (D) DCL1 generates 22-nt miRNA isoforms. The abundance of 22-nt miRNA isoforms was determined using published sRNA-seq from wild type (WT) and various dcl mutants (GSE6682) (Fahlgren et al., 2007). The various mechanisms of 22-nt miRNA production are as defined in (C). TPM, transcript per million.
Figure 3 with 1 supplement
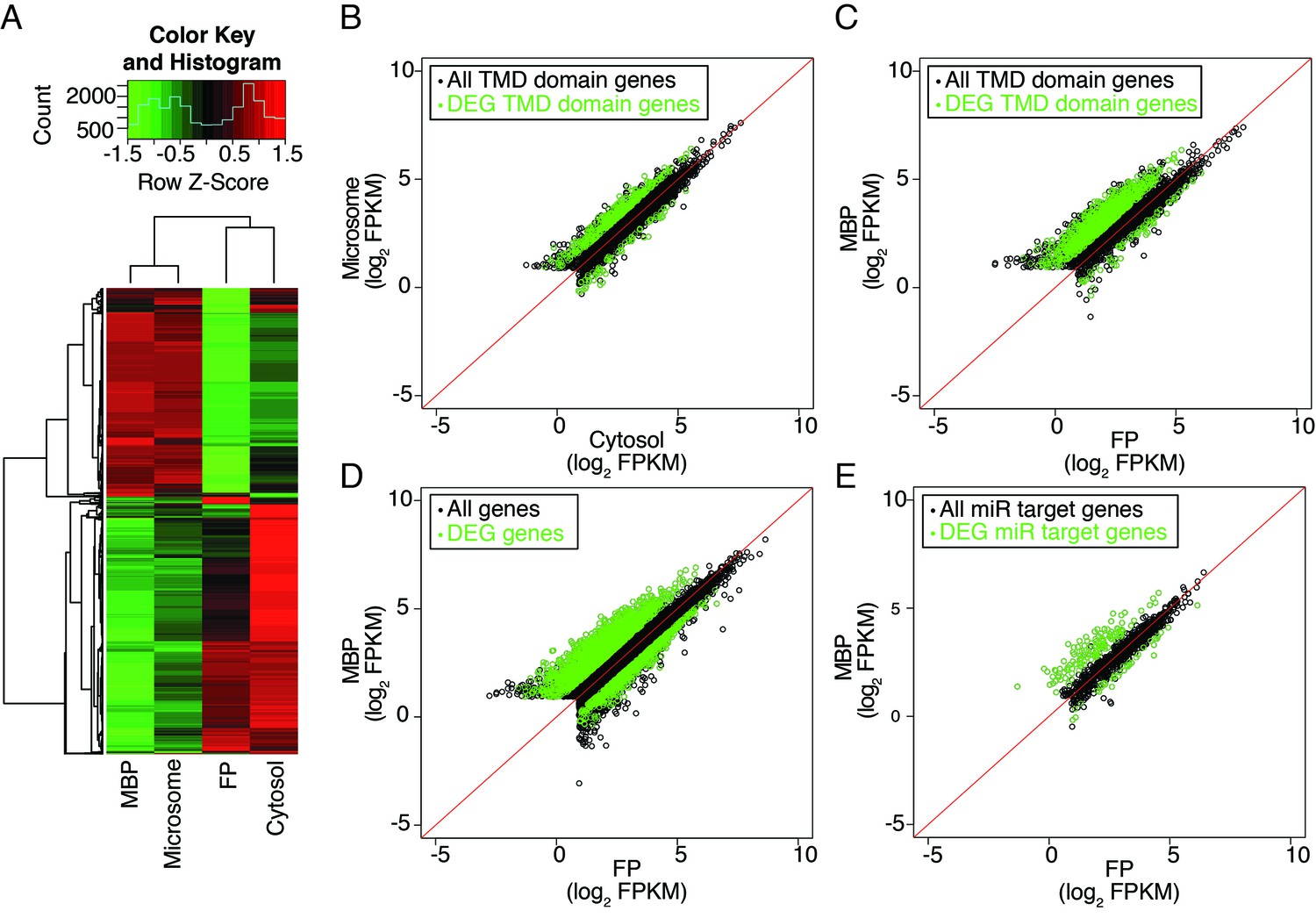
Distribution of cellular mRNAs in microsome versus cytosol and MBP versus FP.
RNA-seq was performed in three biological replicates from polyA+ RNA isolated from microsome, cytosol, MBP and FP. (A) Clustering analysis of the four sample types with the 5000 top varying transcripts. (B–E) partitioning of various groups of transcripts between microsome and cytosol or MBP and FP. FPKM, Fragments Per Kilobase of transcript per Million mapped reads. Enriched and depleted transcripts (DEG transcripts; FC>2; FDR < 0.05) are highlighted in green. (B–C) Abundance of transcripts encoding transmembrane domain (TMD) proteins in microsome vs. cytosol (B) and MBP vs. FP (C). (D–E) The partitioning of all transcripts (D) or miRNA target transcripts (E) between MBP and FP. The miRNA target transcripts were predicted with psRNATarget (Dai and Zhao, 2011) with the maximum expectation score ≤ 3.
Figure 3—figure supplement 1
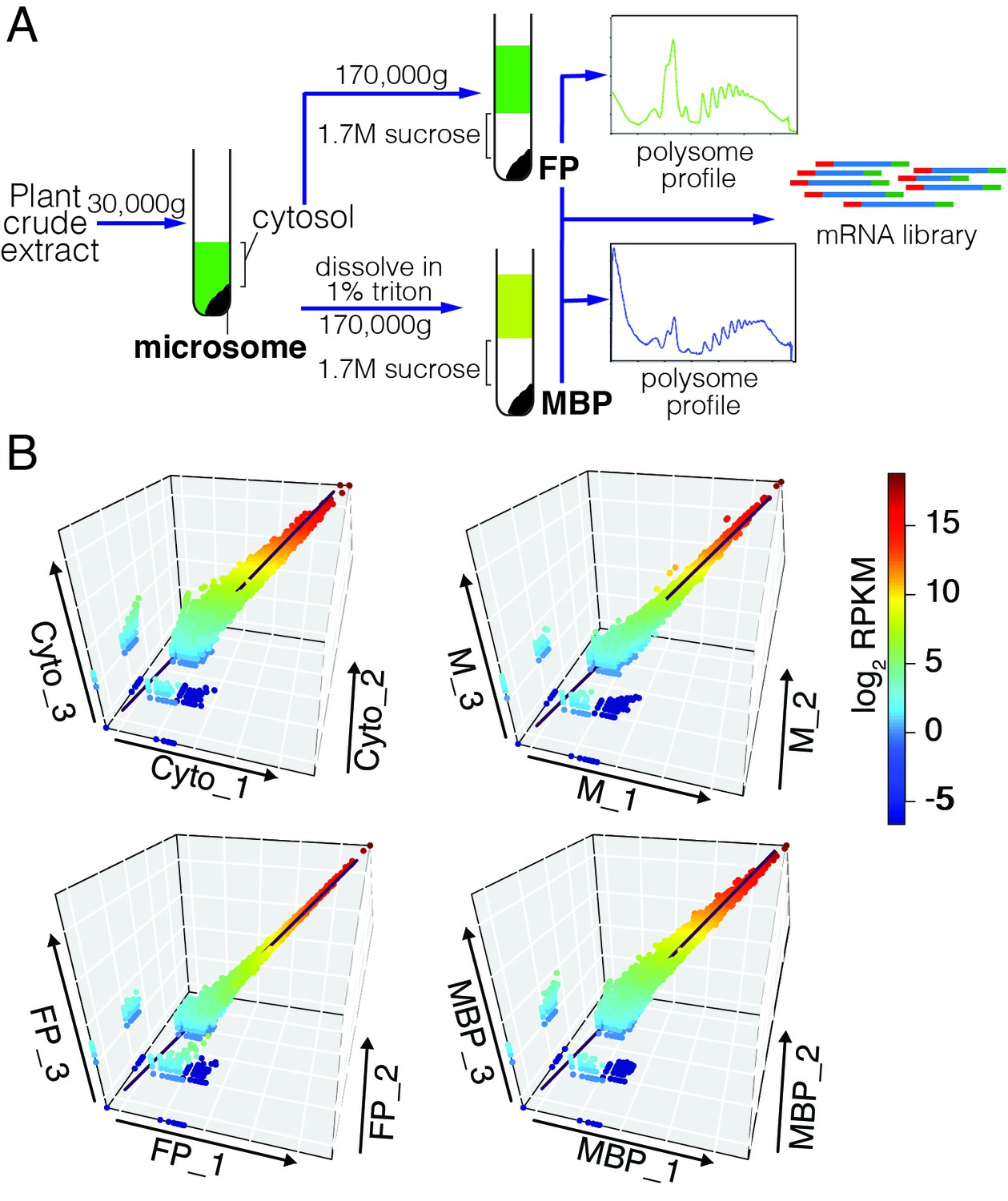
Simultaneous isolation of microsome, cytosol, free polysome (FP) and MBP followed by RNA-seq.
(A) A simplified isolation scheme. Representative FP and MBP profiles are shown. (B) The three biological replicates of RNA-seq for each sample type were highly reproducible.
Figure 4 with 1 supplement
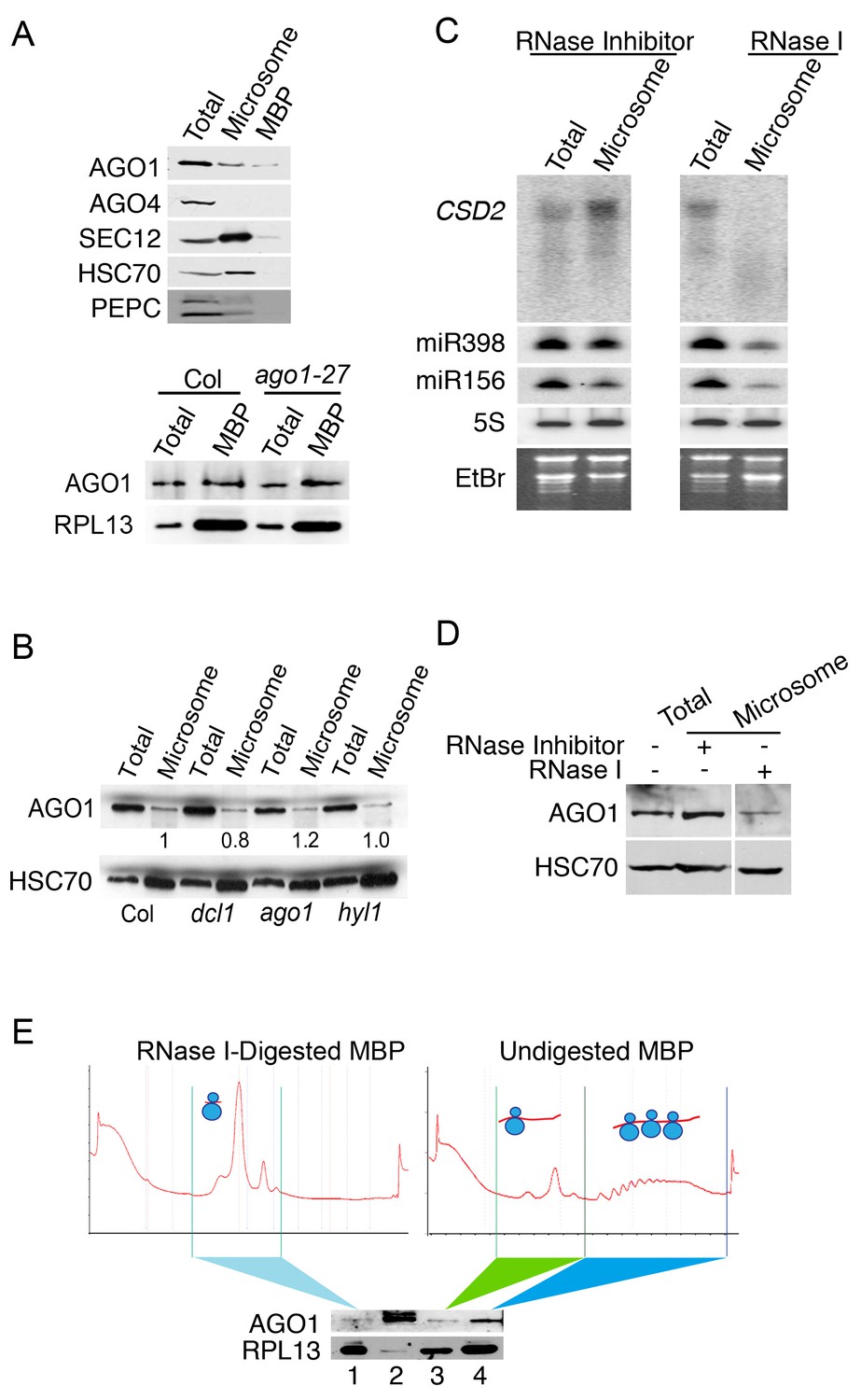
The microsome- and MBP-association of AGO1.
(A) Western blot to detect various proteins in total extract and microsomal and MBP fractions. SEC12 is an ER transmembrane protein; HSC70 is an ER luminal protein; PEPC is a cytosolic protein; RPL13 is a ribosomal protein. AGO1 but not AGO4 was present at detectable levels in microsomal and MBP fractions. The bottom panel shows that the AGO1-27 protein was still associated with MBPs. (B) Microsomal AGO1 and HSC70 levels as determined by western blotting in wild type (Col), dcl1-20 (dcl1), ago1-27 (ago1), and hyl1-2 (hyl1). Note that dcl1-20 is a newly isolated, strong dcl1 allele (see Figure 4—figure supplement 1). (C–D) Microsomal miRNA (C) and AGO1 (D) levels with or without RNase I treatment. Microsomes were isolated from extracts that were treated with RNase inhibitor or RNase I and subjected to northern blotting to detect CSD2 mRNA and miRNAs and western blotting to detect the AGO1 protein. 5S rRNA and the stained gels (EtBr) indicate relative sample loading. (E) The association of AGO1 with monosomes and polysomes. MBPs were treated or not with RNase I and fractionated in a sucrose gradient. The marked fractions were subjected to western blotting to detect AGO1 and the ribosomal protein L13 (RPL13). Lane 2, input (total extract). Note that for ‘undigested MBP’, the polysome fraction is expected to have more ribosomes than the monosome fraction. Thus, the higher RPL13 levels in the polysome fraction do not reflect unequal loading. On the other hand, RPL13 levels in the ‘RNase I-digested MBP’ can be compared to those of the polysomal ‘Undigested MBP’ to reflect relative sample loading.
Figure 4—figure supplement 1
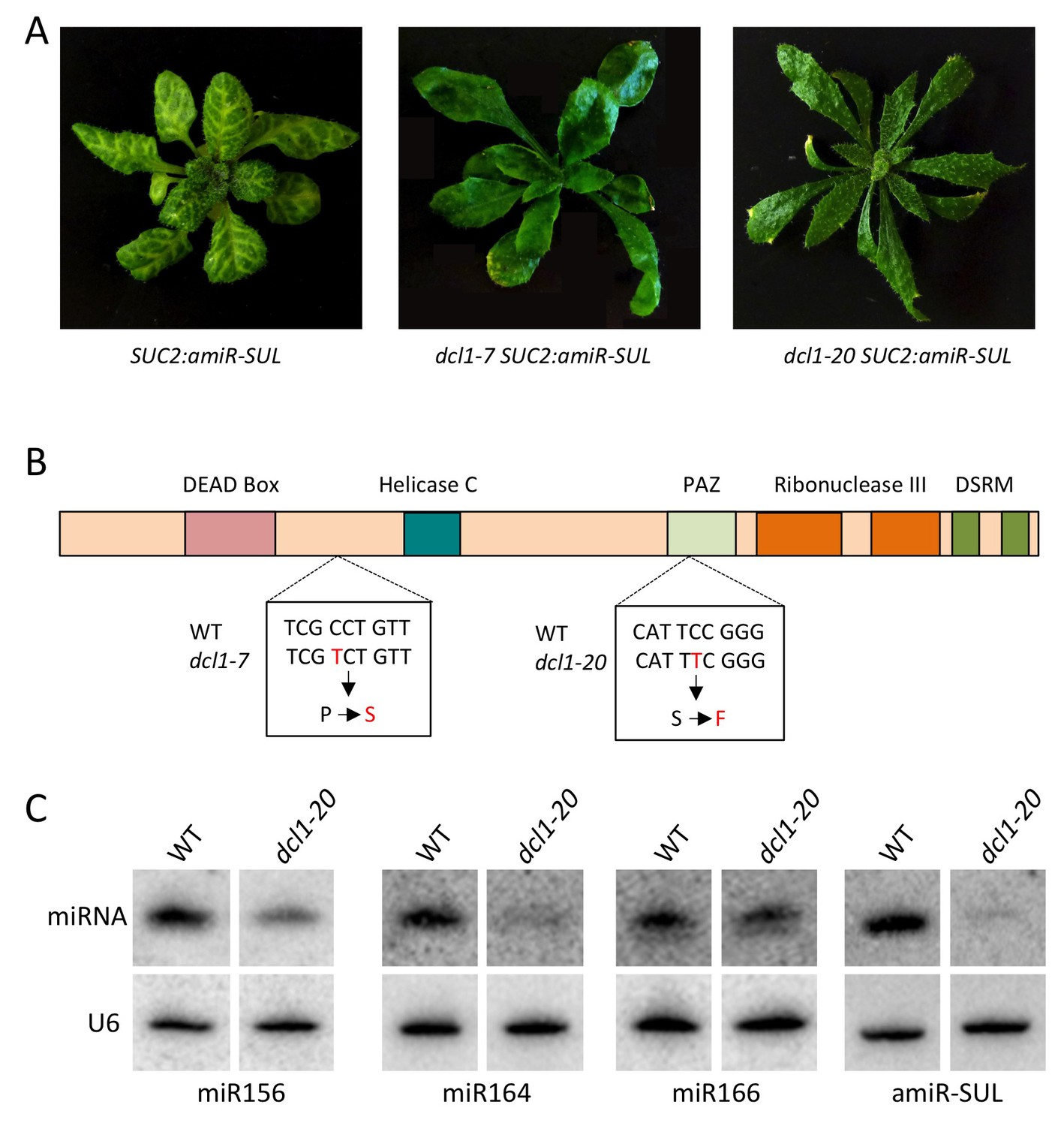
Characterization of a new dcl1 allele.
The dcl1-20 mutation was isolated from a forward genetic screen in the SUC2:amiR-SUL background (de Felippes et al., 2011), in which an artificial miRNA (amiR-SUL) silences the chlorophyll biosynthesis gene SULFUR (SUL) to lead to vein-centered clearing. (A) dcl1-7, a known dcl1 mutation with strong defects in miRNA biogenesis (Kasschau et al., 2007), and dcl1-20 both led to suppression of the SUC2:amiR-SUL silencing effect. Note that the dcl1-20 mutant exhibited stronger leaf shape defects (narrower and more pointy leaves) than dcl1-7. (B) Diagram of the DCL1 protein showing the various domains. The locations and nature of the two mutations are also shown. (C) Northern blotting to detect three endogenous miRNAs and the artificial miRNA (amiR-SUL) in SUC2:amiR-SUL (WT) and SUC2:amiR-SUL dcl1-20. The latter showed reduced levels of the miRNAs examined.
Figure 5

miRNAs and ta-siRNAs are recruited to membranes by AGO1.
(A) Detection of three miRNAs in total extracts and the microsomal fraction in wild type (Col) and ago1-36. The ago1-36 mutant lacks the full-length AGO1 protein as shown by western blotting. HSC70 was a loading control. The three miRNAs were detected by northern blotting. 5S rRNA was an internal control and shown below each miRNA blot. The microsomal levels of the miRNAs were reduced in ago1-36. (B) A scatter plot showing comparable miRNA abundance in wild type (WT) and ago1-27 total extracts. Inset: box plots illustrating the distribution of miRNA RPMR values in WT and ago1-27. (C) A scatter plot showing that miRNAs have reduced microsomal enrichment in ago1-27 as compared with wild type. Microsomal enrichment was measured by M/T (ratio of levels in microsome vs. those in total extract). The degree of microsomal enrichment of miR390 (yellow dots) is largely unchanged, while that of most known ta-siRNA/phasiRNA triggers (red dots) is decreased in ago1-27. miR173’s (green dot) microsomal enrichment was weakly affected. (D) A scatter plot showing that 21-nt and 22-nt ta-siRNAs from TAS1-4 loci have reduced microsomal enrichment in ago1-27 as compared with wild type.
Figure 6
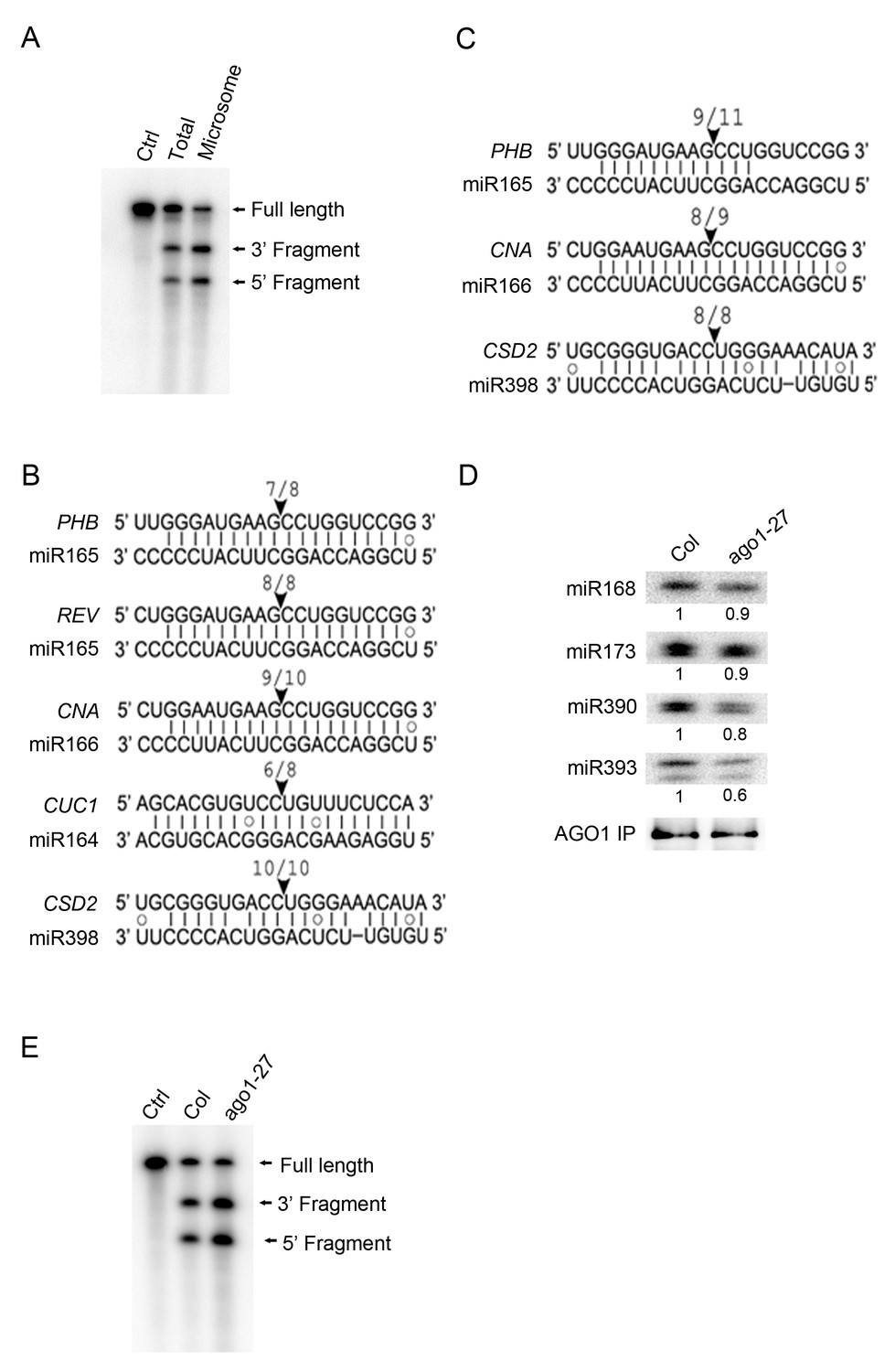
miRNA-guided cleavage occurs in the microsomal and MBP fractions and in ago1-27.
(A) In vitro slicer assay using AGO1 immunoprecipitates (IP) from total extracts or the microsomal fraction. A fragment of PHB RNA containing the miR165/6-binding site was used as the substrate (marked ‘Full Length’). The two cleavage products are indicated. The control (Ctrl) lane was the RNA alone without AGO1 IP. (B–C) Detection of 3’ cleavage fragments from various miRNA target RNAs in vivo using 5’ RACE RT-PCR from the microsomal (B) or MBP (C) fraction. The cloned PCR products were sequenced to identify the 5’ ends of the 3’ cleavage fragments. The arrowheads indicate the positions of miRNA-guided cleavage. The numbers above indicate the number of clones with 5’ ends at the predicted cleavage site out of total sequenced clones. (D) The AGO1-27 protein associated with miRNAs in vivo. IP was performed in wild type and ago1-27 and the IP was subjected to western blotting to detect AGO1 and northern blotting to detect several miRNAs. The levels of miRNAs were quantified against the levels of AGO1 in the IP and compared between wild type and ago1-27. (E) In vitro slicer assay using AGO1 IP from wild type and ago1-27 total extracts. The assay was performed as in (A).
Figure 7 with 1 supplement
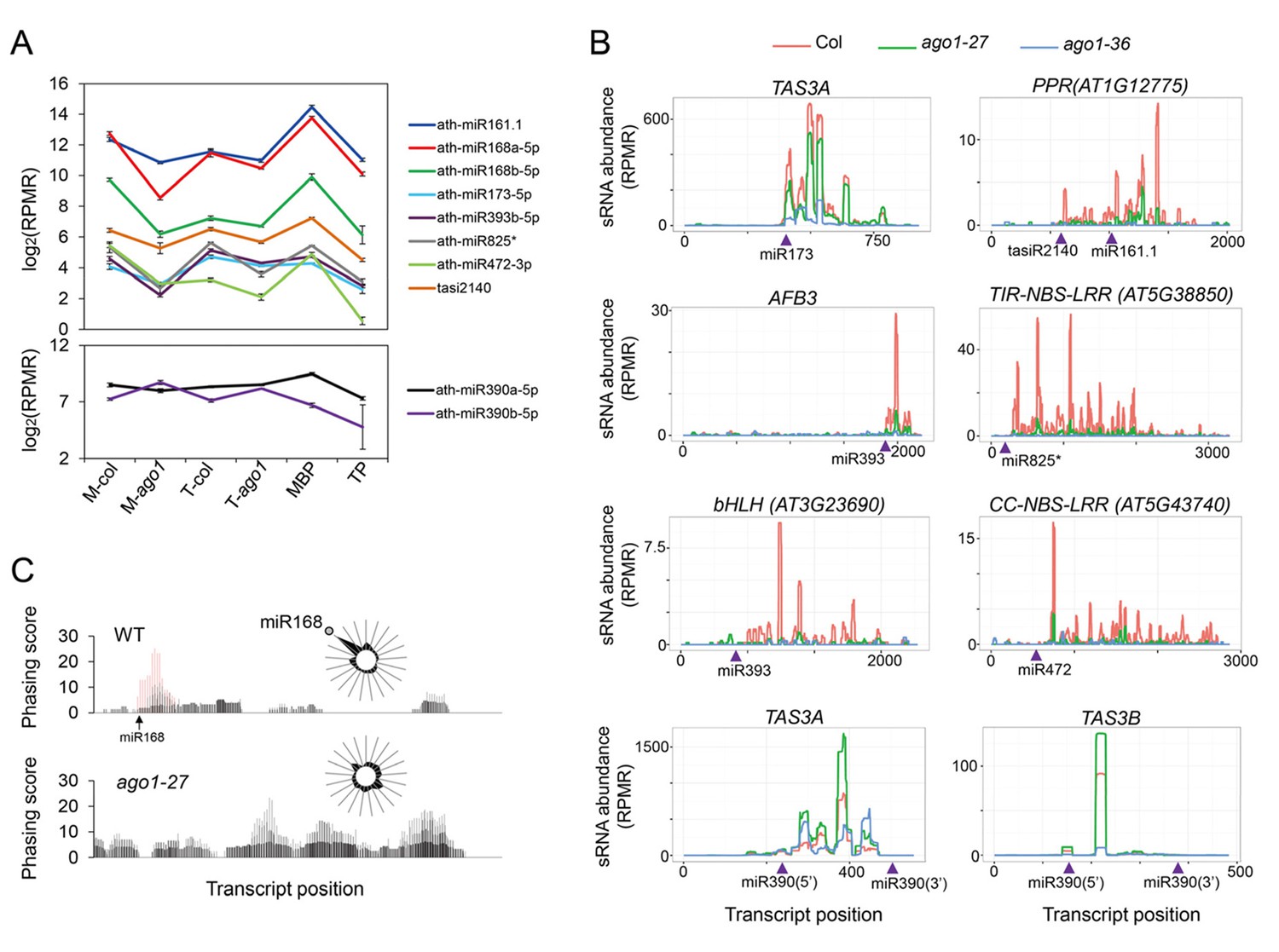
Biogenesis of phasiRNAs is defective in ago1-27.
(A) top panel: abundance of known AGO1-dependent ta-siRNA/phasiRNA triggers in microsome (M), total (T), MBP and TP as determined by sRNA-seq; lower panel: abundance of miR390 in M, T, MBP and TP as determined by sRNA-seq. Wild type: col; ago1-27: ago1. Error bars indicate standard error of the mean (n = 3). (B) sRNAs from known ta-siRNA/phasiRNA loci, except for TAS3 loci, were reduced in ago1-27 (green) as compared with wild type (red), and nearly diminished in ago1-36 (blue). The loci IDs are shown above each plot. The miRNA triggers for these ta-siRNAs/phasiRNAs are shown below each plot with the miRNA-binding sites indicated by triangles. (C) The phasing of sRNAs over the AGO1 transcript in wild type and ago1-27. miR168 triggers the production of phasiRNAs (with positions marked by red lines) in wild type, and the phasing caused by this miRNA was nearly lost in ago1-27. The radial graphs show that the miR168 cleavage site fell in the most prominent phasing register of 21-nt sRNAs in wild type but not in ago1-27.
Figure 7—figure supplement 1
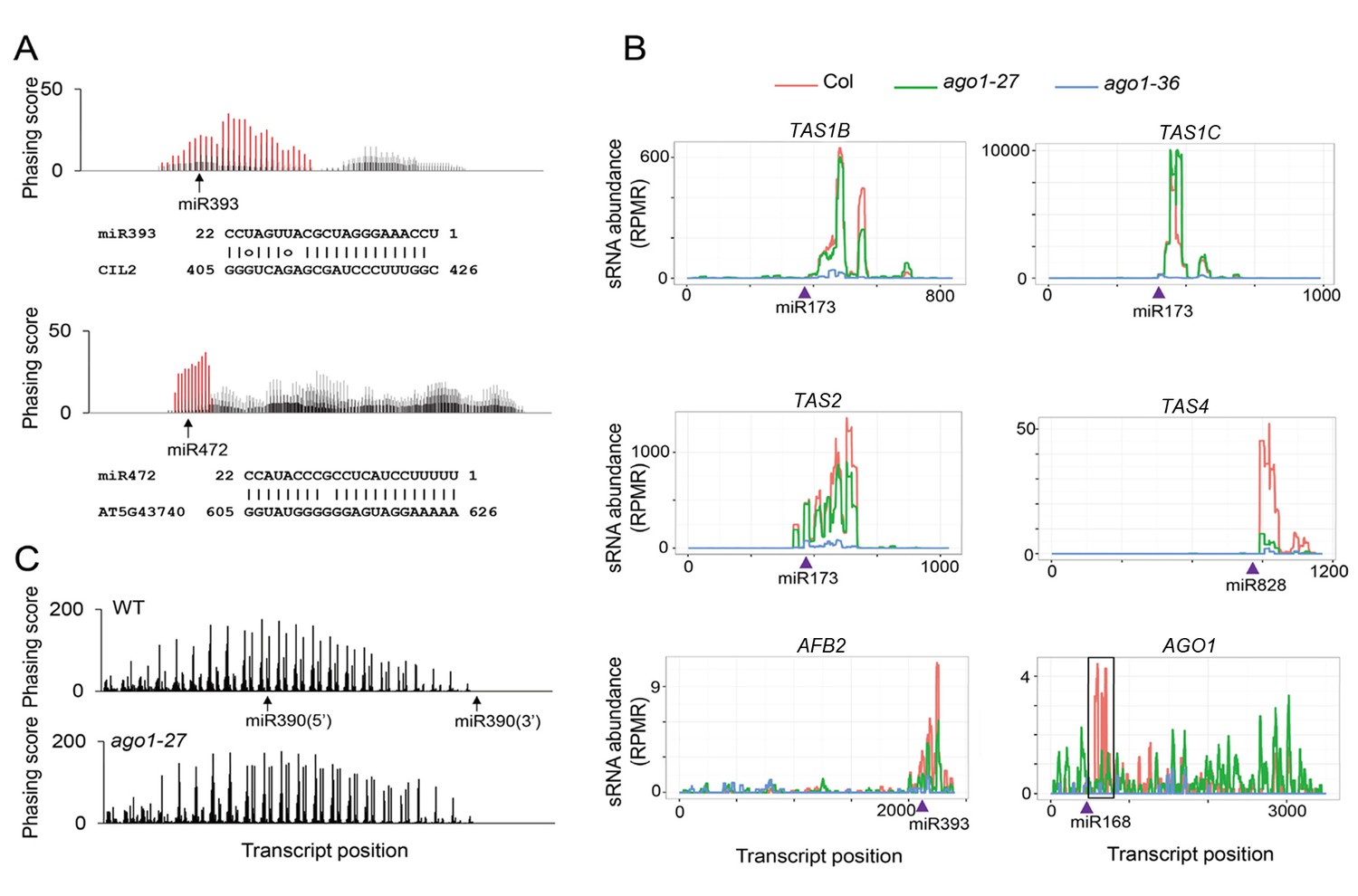
The effect of ago1-27 on ta-siRNAs/phasiRNAs.
(A) Two newly identified phasiRNA loci, CIL2 and an NBS-LRR gene, triggered by miR393 and miR472, respectively. The phasing scores along the two transcripts are shown, with phasiRNA producing positions marked in red. The pairing between the miRNAs and the target sites is presented below the plots. (B) ta-siRNA/phasiRNA levels from loci targeted by various AGO1-bound miRNA triggers in WT (red), ago1-27 (green) and ago1-36 (blue). ta-siRNAs/phasiRNAs were nearly eliminated in ago1-36 at all loci and were reduced in ago1-27 at most loci. TAS1C is the only example showing an increase in ta-siRNAs among 23 non-TAS3 phasiRNA-generating loci in ago1-27 (see Supplementary file 4). (C) The phasing of AGO7-dependent ta-siRNAs from the TAS3A locus was not affected in ago1-27.
Figure 8
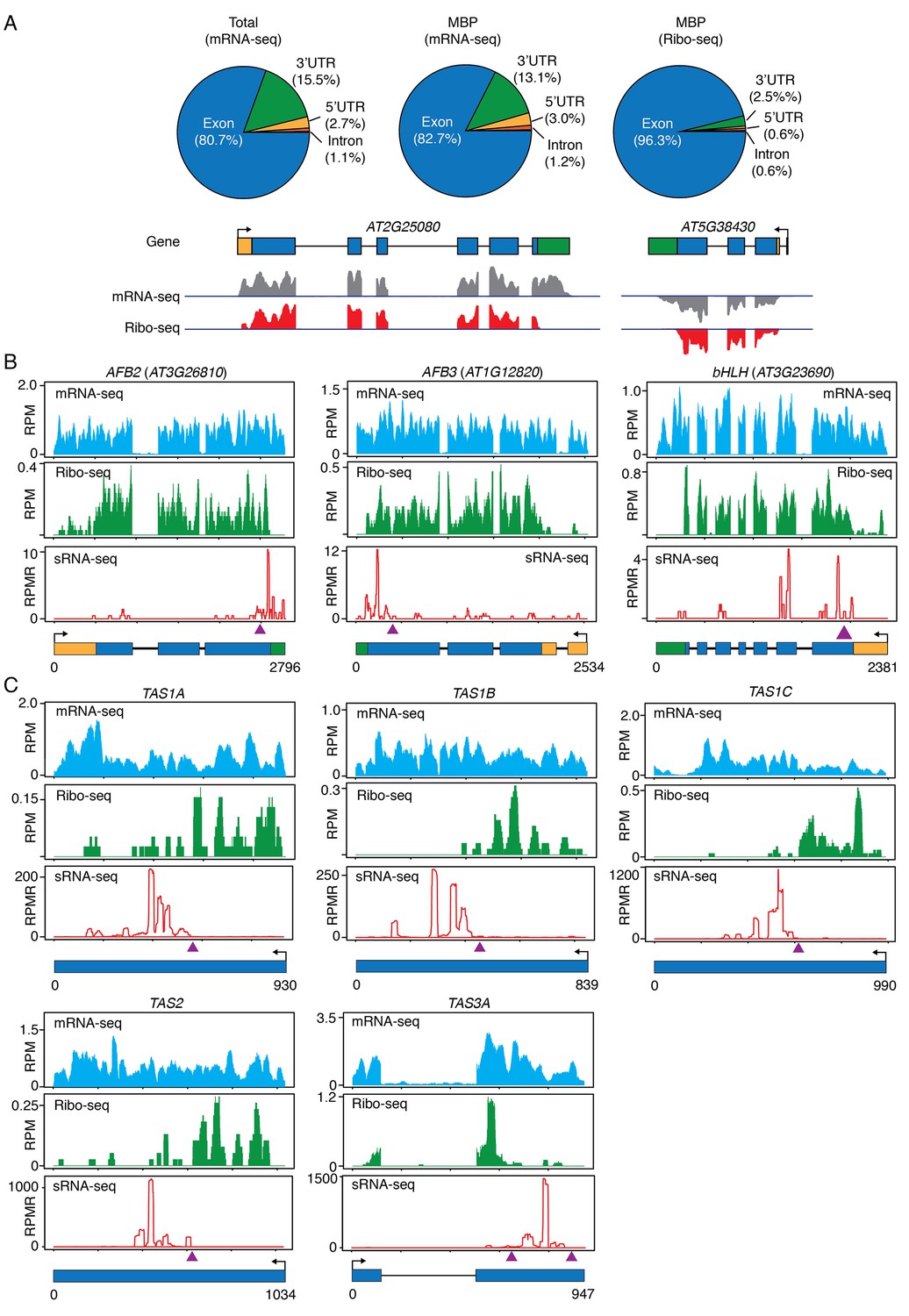
Ribo-seq to examine ribosome occupancy of transcripts known to spawn phasiRNAs.
(A) Composition of the genomic features represented by reads from mRNA-seq from total extract (Total), mRNA-seq from MBP, and ribo-seq from MBP. The overwhelming representation of exons by ribo-seq shows that ribo-seq successfully captured the expected sequences. Two genes are shown as examples. In the gene models, rectangles and lines represent exons and introns, respectively. 5’ and 3’ UTRs are in yellow and green, respectively. Arrows mark the direction of transcription. The lack of most 3’ UTR reads is consistent with the fact that ribosomes only protect a small portion of the 3’ UTR. (B–C) Browser views of MBP mRNA-seq (top panels), ribo-seq (middle panels), and sRNA-seq (lower panels) for three protein-coding genes (B) and five TAS loci (C). The gene IDs are shown above the plots. The gene models are shown below the plots. The same scheme is used to represent exons, introns, UTRs, and transcription start sites as in (A). The triangles represent the positions of the binding sites of the triggering miRNAs.
Additional files
-
Supplementary file 1
Lists of differential small RNA regions (DSR) in the MBP versus TP comparison.
- https://doi.org/10.7554/eLife.22750.015
-
Supplementary file 2
A list of 178 miRNAs in 12 sRNA-seq libraries.
- https://doi.org/10.7554/eLife.22750.016
-
Supplementary file 3
Transcript levels in microsome, cytosol, free polysome and membrane-bound polysome fractions as determined by RNA-seq.
- https://doi.org/10.7554/eLife.22750.017
-
Supplementary file 4
Changes in the abundance of phasiRNAs from known phasiRNA-generating loci caused by ago1-27 and ago1-36 mutations.
- https://doi.org/10.7554/eLife.22750.018
-
Supplementary file 5
Oligonucleotides used in this study.
- https://doi.org/10.7554/eLife.22750.019
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Biogenesis of phased siRNAs on membrane-bound polysomes in Arabidopsis
eLife 5:e22750.
https://doi.org/10.7554/eLife.22750




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}