A lncRNA fine tunes the dynamics of a cell state transition involving Lin28, let-7 and de novo DNA methylation
- University of Cambridge, United Kingdom
- University of California Berkeley, United States
- Cold Spring Harbor Laboratory, United States
- Babraham Institute, United Kingdom
- Wellcome Trust Sanger Institute, United Kingdom
- Integrated DNA Technologies, United States
Abstract
Execution of pluripotency requires progression from the naïve status represented by mouse embryonic stem cells (ESCs) to a state capacitated for lineage specification. This transition is coordinated at multiple levels. Non-coding RNAs may contribute to this regulatory orchestra. We identified a rodent-specific long non-coding RNA (lncRNA) linc1281, hereafter Ephemeron (Eprn), that modulates the dynamics of exit from naïve pluripotency. Eprn deletion delays the extinction of ESC identity, an effect associated with perduring Nanog expression. In the absence of Eprn, Lin28a expression is reduced which results in persistence of let-7 microRNAs, and the up-regulation of de novo methyltransferases Dnmt3a/b is delayed. Dnmt3a/b deletion retards ES cell transition, correlating with delayed Nanog promoter methylation and phenocopying loss of Eprn or Lin28a. The connection from lncRNA to miRNA and DNA methylation facilitates the acute extinction of naïve pluripotency, a pre-requisite for rapid progression from preimplantation epiblast to gastrulation in rodents. Eprn illustrates how lncRNAs may introduce species-specific network modulations.
https://doi.org/10.7554/eLife.23468.001Introduction
Mouse embryonic stem cells (ESCs), in vitro counterparts of the pre-implantation epiblast, exhibit dual properties of self-renewal and differentiation (Boroviak et al., 2015; Bradley et al., 1984; Evans and Kaufman, 1981; Martin, 1981). These properties make them an attractive system for investigating cell fate decision making. In the embryo, spatially and temporally coordinated signals direct the rapid and continuous transition of the epiblast towards lineage specification (Acampora et al., 2016; Smith, 2017). In contrast, ESCs can be suspended in a ground state of pluripotency, where self-renewal is decoupled from lineage specification, using two inhibitors (2i) of glycogen synthase kinase 3 (GSK3) and mitogen-activated protein kinase kinase (MEK1/2), along with the cytokine leukaemia inhibitory factor (LIF) (Ying et al., 2008). Therefore, ESCs provides a unique experimental system to explore the principles and molecular players underlying the developmental progression of pluripotency (Kalkan and Smith, 2014).
While it is increasingly clear that the ESC state is maintained by a core network of transcription factors (Chen et al., 2008; Dunn et al., 2014; Ivanova et al., 2006), less is known about how cells progress from this state to lineage specification (Buecker et al., 2014; Kalkan and Smith, 2014; Smith, 2017). Loss-of-function screens have highlighted a multi-layered machinery that dismantles the naïve state transcription factor network (Betschinger et al., 2013; Leeb et al., 2014). The latency period for transition depends on the clearance kinetics of network components (Dunn et al., 2014). The orchestration of multiple regulators thus ensures rapid and complete dissolution of this core network and consequent timely extinction of ESC identity upon 2i withdrawal (Kalkan and Smith, 2014).
In addition to protein coding genes, accumulating evidence suggests that non-coding RNAs can contribute to the regulation of cell fate transitions. Within this class, long non-coding RNAs (lncRNAs) comprise a large fraction of the transcriptome in diverse cell types and exhibit specific spatio-temporal expression (Carninci et al., 2005; Guttman et al., 2009; Necsulea et al., 2014). The genomic distribution of lncRNAs is non-random (Luo et al., 2016). A subclass of lncRNAs are divergently transcribed from neighbouring genes and thought to regulate proximal gene expression in cis, either due to the process of transcription (Ebisuya et al., 2008; Engreitz et al., 2016; Martens et al., 2004) or through local lncRNA-protein interactions that recruit regulatory complexes (Lai et al., 2013; Lee, 2012; Luo et al., 2016; Nagano et al., 2008). However, the functions and mode of action of the vast majority of lncRNAs remain unknown and require case-by-case experimental determination. In mouse ESCs, knockdowns of a number of lncRNAs have been reported to exert effects on the transcriptome (Bergmann et al., 2015; Dinger et al., 2008; Guttman et al., 2011; Lin et al., 2014; Sheik Mohamed et al., 2010) and in some cases impair self-renewal (Lin et al., 2014; Luo et al., 2016; Savić et al., 2014).
We investigated the potential involvement of lncRNAs in transition from the naïve ESC state and identified a dynamically regulated lncRNA (linc1281) that we named Ephemeron (Eprn). We present functional evaluation of Eprn and delineation of a downstream genetic interaction network, which is an additional component of the regulatory machinery driving the irreversible and rapid progression from naïve pluripotency in rodent.
Results
Identification of lncRNAs associated with transition from naïve pluripotency
Post-implantation epiblast derived stem cells (EpiSCs) represent a primed state of pluripotency developmentally downstream of naïve state ESCs (Brons et al., 2007; Nichols and Smith, 2009; Tesar et al., 2007). To identify lncRNA candidates with a possible role in ESC transition, we analysed in silico the effect of genetic perturbation on expression of ESC and EpiSC states based on published data. We first selected genes that are over ten-fold differentially enriched in ESCs (182 genes) and EpiSCs (131 genes) relative to each other as molecular signatures to represent these two states (Tesar et al., 2007). Using published data, we investigated the impact on these two signature sets when individual lncRNAs (147 in total) and known protein coding regulators (40 in total) were knocked down in ESCs grown in LIF/serum (Guttman et al., 2011) (Figure 1A, Figure 1—source data 1). Serum culture supports a heterogeneous mixture of naïve, primed and intermediate cells (Chambers et al., 2007; Kolodziejczyk et al., 2015; Marks et al., 2012). Therefore, analysis in this condition could potentially reveal regulators of the ESC and EpiSC states. The effect of each gene knockdown was plotted based on the percentage of genes significantly altered within ESC and EpiSC signature sets (FDR < 0.05 and fold change >2 or<0.5 over negative control defined by the original study). We validated the approach by analysing the knockdown effects of known ESC self-renewal regulators. As predicted, depletion of factors that maintain the ESC state, such as Stat3, Esrrb, Sox2 and Klf4, led to a decrease in ESC and increase in EpiSC signature (Figure 1A), while knockdown of Oct4 gave rise to a decrease in both ESC and EpiSC signatures, consistent with its requirement in both states (Niwa et al., 2000; Osorno et al., 2012). With this system, we identified lncRNAs that increased ESC and decreased EpiSC signatures when knocked down, suggestive of a possible role in transition from the ESC state (Figure 1A bottom right quadrant).
Figure 1 with 2 supplements see all
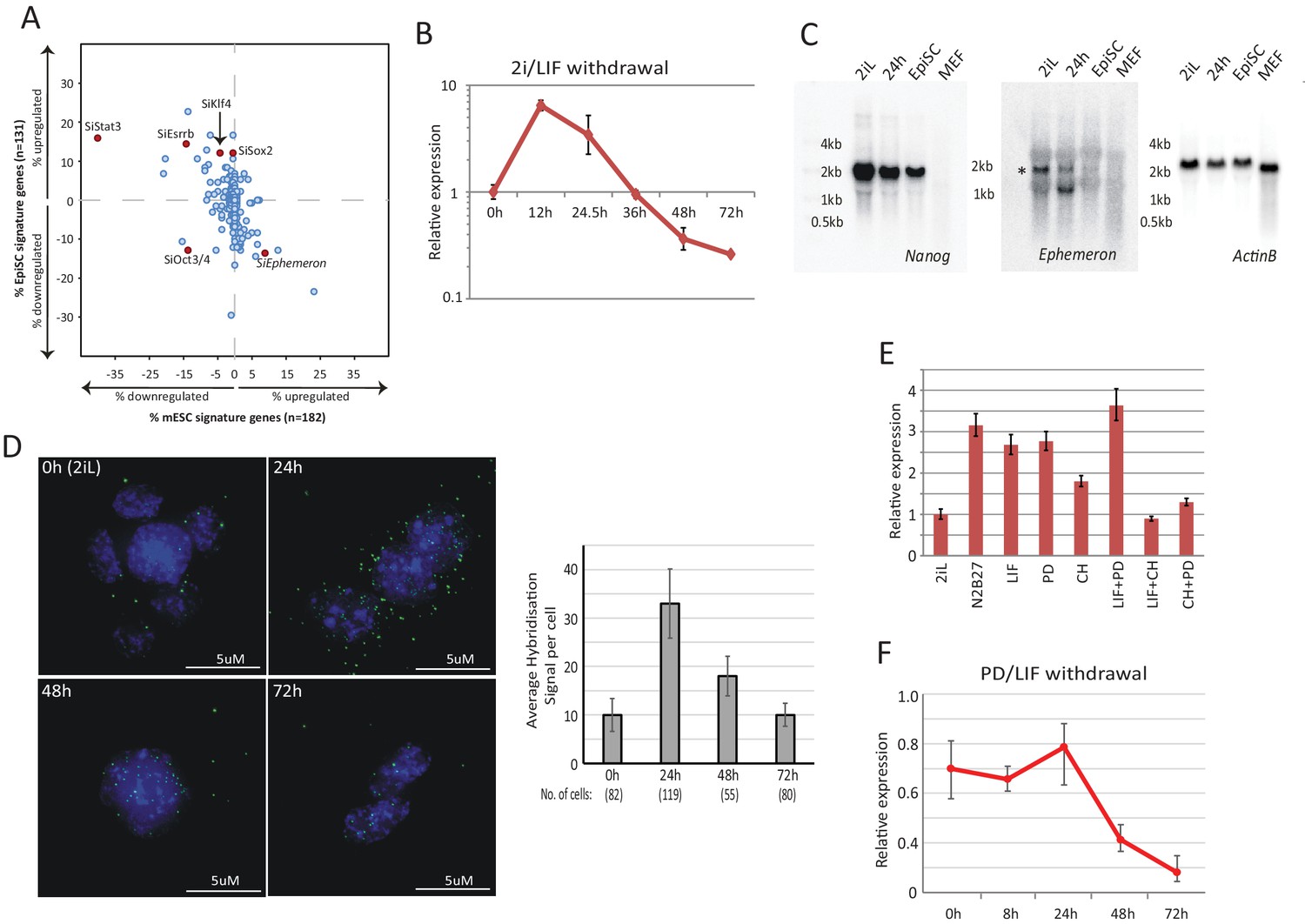
Dynamic expression of lncRNA Ephemeron during exit from naïve pluripotency.
(A) Bioinformatic analysis of potential lncRNA candidates in naïve state regulation based on published transcriptome data for lncRNA and pluripotency related gene knockdowns. Each dot represents the effect on ESC (x-axis) and EpiSC (y-axis) gene signatures when a given gene is knocked down. (B) RT-qPCR detection of Eprn expression relative to β-actin upon 2i/LIF withdrawal. Mean ±SD, n = 3. (C) Northern blotting of Eprn, Nanog and β-actin in ESCs in 2i/LIF or withdrawn from 2i/LIF for 24 hr, EpiSCs and MEF. * indicates a cross-hybridising RNA species since part of the probe region overlaps with LINE1-L1 and ERVK TEs. (D) RNA-FISH for Eprn upon 2i/LIF withdrawal with quantification of average hybridisation signals per cell. Mean value of total hybridisation signals for all cells ± SD, n = 2. (E) Eprn expression relative to β-actin upon 2i/LIF component withdrawal quantified by RT-qPCR. Cells cultured in 2i/LIF and were transferred to N2B27 containing indicated single or dual factors for 24 hr. Mean ±SD, n = 3. F, Eprn expression relative to β-actin upon PD/LIF withdrawal quantified by RT-qPCR. Mean ± SD, n = 3.
-
Figure 1—source data 1
Bioinformatics analysis of all lncRNAs and protein coding genes plotted in Figure 1A.
- https://doi.org/10.7554/eLife.23468.003
-
Figure 1—source data 2
Expression of potential lncRNA candidates in facilitating naïve state exit.
The genomic coordinates are based on mouse GRCm38/mm10 assembly.
- https://doi.org/10.7554/eLife.23468.004
We examined expression profiles of these candidate lncRNAs during exit from self-renewal in defined conditions, exploiting the Rex1::GFP (RGd2) reporter ESC cell line (Kalkan et al., 2017; Wray et al., 2011) (Figure 1—source data 2). Upon 24 hr of 2i withdrawal, Rex1 expression status can discriminate subpopulations of cells with distinct functional properties, with Rex1-GFP high cells corresponding to undifferentiated ESCs and loss of GFP marking extinction of ESC identity (Kalkan et al., 2017). Amongst the 16 candidates analysed, linc1281 (Refseq entry D630045M09Rik) (Figure 1—figure supplement 1A) was the third highest expressed lncRNA across all time points. Notably this lncRNA showed a distinctive profile during the first 24 hr, with differential expression observed between Rex1-GFP high and low cells (Figure 1B, Figure 1—figure supplement 1B). Due to its dynamic and transient expression profile, we designated linc1281 as Ephemeron (Eprn). Ribosomal profiling analysis indicated that Eprn is indeed a non-coding RNA, with the longest predicted open reading frame (80 amino acids) possessing a ribosome release score typical of a non-coding sequence (Guttman et al., 2013). Eprn is located in a region of high transposable element (TE) content, with its exons comprised of 76.4% annotated TE sequences (including ERV-K, LINE L1, and SINE B2 elements, Figure 1—figure supplement 1A). This genomic region exhibits minimal sequence conservation in mammals (Figure 1—figure supplement 1A) and we failed to identify any human homologue either within the syntenic region or elsewhere in the human genome. However, a positionally conserved spliced transcript (CA504619) that shares 79% sequence identity to exon 3 of mouse Eprn is present within the rat syntenic region (Figure 1—figure supplement 1C). Therefore, it is likely that Eprn is conserved in rodents over 30 million years since the mouse-rat lineage divergence.
We conducted RT-qPCR, Northern blotting and RNA-FISH to evaluate expression, transcription variants and subcellular localisation of Eprn in ESCs. Eprn showed strong induction within 12 hr of 2i/LIF withdrawal, but decreased subsequently (Figure 1B–D). In EpiSCs or mouse embryonic fibroblasts (MEFs), Eprn expression was below the detection limit (Figure 1C). Consistent with the UCSC gene annotation, Northern blotting of total ESC RNA confirmed the expression of a single Eprn transcript over 1 kb in length (Figure 1C, Figure 1—figure supplement 1D). Transcription start and end sites of Eprn mapped by 5’ and 3’ RACE were in agreement with the annotation (Figure 1—figure supplement 1E,F). After 24 hr of 2i/LIF withdrawal, Eprn RNA-FISH hybridisation signals displayed predominantly cytoplasmic localisation, but from 48 hr onwards the remaining signals were mostly in the nucleus (Figure 1D).
To explore the regulation of Eprn, two inhibitors and LIF were withdrawn singly or dually for 24 hr. In conditions lacking Gsk3 inhibitor CHIRON99021 (CH), Eprn was upregulated (Figure 1E). When transferred to non-supplemented N2B27 medium from PD/LIF, Eprn expression was maintained for 24 hr before declining (Figure 1F). The addition of CH to LIF/serum culture reduced Eprn expression within 24 hr irrespective of the presence of MEK inhibitor PD0325901 (PD) (Figure 1—figure supplement 1G,H). Therefore, Eprn is suppressed by CH in self-renewing ESCs.
Through analysis of published data, we found that during early mouse development, Eprn expression peaked at E4.5 and was present in both epiblast and primitive endoderm of the mature blastocyst, but absent or low in E5.5 post-implantation epiblast (Figure 1—figure supplement 2A) and later stages between E7 and E17 (Figure 1—figure supplement 2B). Amongst adult tissues analysed, Eprn was only detected in kidney, and at a much lower level than in ESCs. We also observed that Eprn expression is restored upon naïve state resetting from EpiSCs (Guo et al., 2009; Yang et al., 2010) (Figure 1—figure supplement 2C,D). We conclude that Eprn expression is highly specific to ESCs and the early mouse embryo.
LINE and ERVL-MaLR elements are present within the Eprn proximal promoter region (2 kb upstream of TSS) (Figure 1—figure supplement 1A). Such repetitive elements gain DNA CpG methylation dramatically during pre- to post-implantation transition (Smith et al., 2014). By examining published data from embryos (Seisenberger et al., 2012; Wang et al., 2014) and ESC progression in vitro (Kalkan et al., 2017), we found that CpG methylation gain at the Eprn promoter was more extensive in the primed E6.5 epiblast (3% to 80%) than the average changes across all promoters (9% to 35%) or the genome (24% to 70%) (Figure 1—figure supplement 2E). In contrast, no major CpG methylation gain at Eprn promoter was present 24 hr post 2i withdrawal. These data suggest that promoter methylation does not initiate Eprn repression, but could contribute to maintain silencing in later epiblast.
Loss of Ephemeron delays exit from naïve pluripotency
Initiation of ESC differentiation in defined media upon withdrawal of self-renewal factors recapitulates features of peri-implantation epiblast development (Kalkan et al., 2017). The latency of naïve state exit varies, however, according to the starting self-renewal condition (Dunn et al., 2014; Wray et al., 2011). Higher activity of the core network in PD/LIF compared with 2i results in slower network dissolution, reflected in later onset of RGd2 downregulation (Dunn et al., 2014). These two conditions feature different levels of Eprn due to CH mediated suppression in 2i (Figure 1E). We generated Eprn knockout (KO) ESCs via sequential gene targeting (Figure 2—figure supplement 1) and examined the phenotype in each condition. In steady state self-renewal, Eprn loss did not affect the Rex1-GFP profile in either case (Figure 2A,B). Upon transfer to N2B27, Eprn KO cells displayed delayed downregulation of GFP compared to parental cells, measured at 24 hr from 2i culture and 40 hr from PD/LIF culture (Figure 2B). By 72 hr, however, GFP expression was fully extinguished from either starting condition (Figure 2—figure supplement 2A). A transient delay in GFP downregulation in both culture conditions was also evident upon Eprn knockdown using siRNAs (Figure 2—figure supplement 2B). To assess the effect of Eprn depletion functionally, we conducted colony forming assays. Cells maintained in PD/LIF were subjected to 40 hr culture in N2B27 and then replated at clonal density in 2i/LIF to assay the persistence of ES self-renewal potential (Betschinger et al., 2013). Eprn KO and knockdown cells both gave rise to substantially more undifferentiated colonies than wild type controls (Figure 2C,D, Figure 2—figure supplement 2C). Considered together, these results indicate that Eprn deficiency impairs timely exit from naïve pluripotency.
Figure 2 with 4 supplements see all
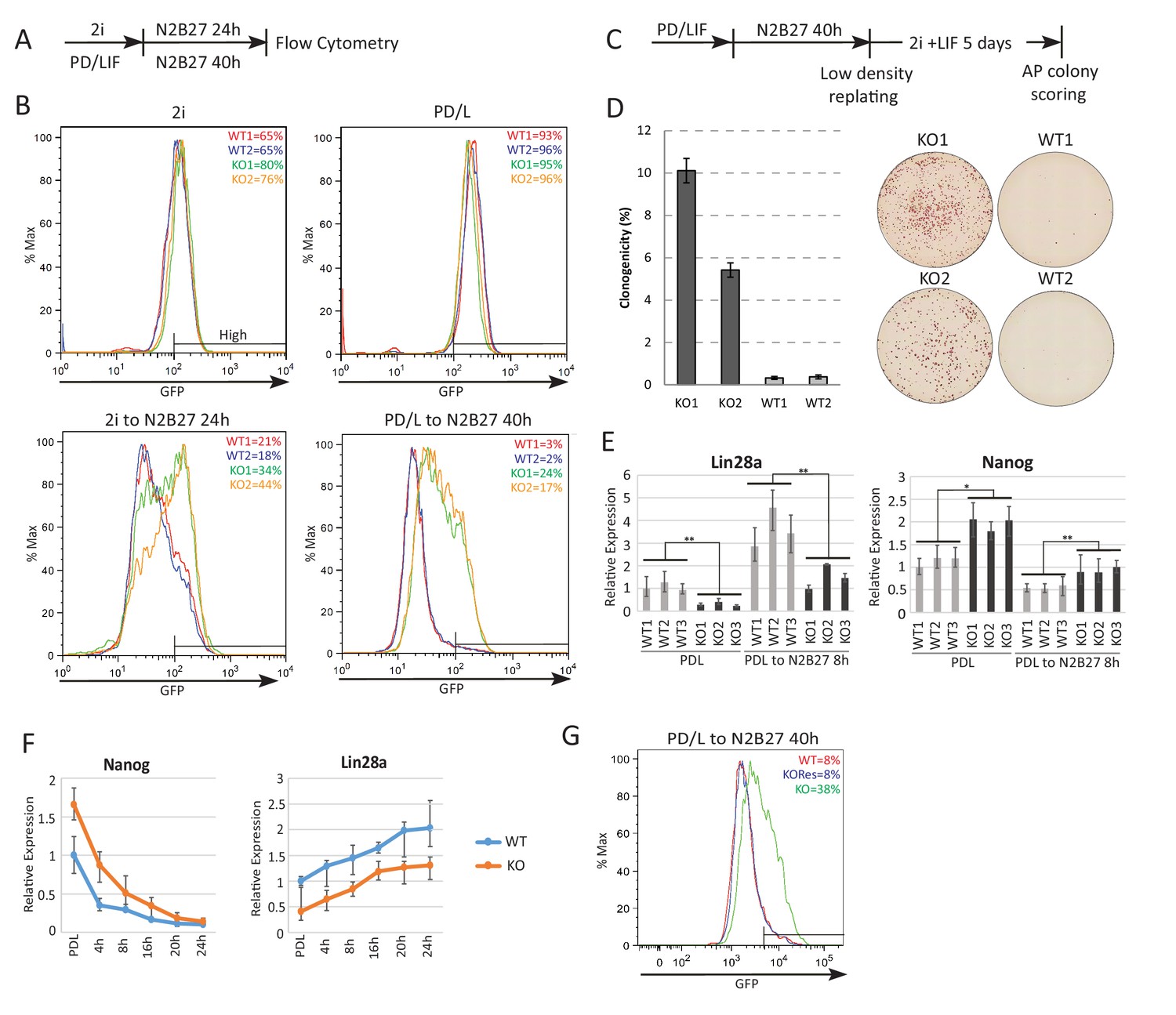
Absence of Ephemeron delays exit from naïve pluripotency.
(A) Experimental scheme for analysing naïve state exit using Rex1GFPd2 reporter cells. (B) Rex1-GFP flow cytometry profiles of wild type and Eprn KO cells in 2i and PD/LIF and during transition from these starting conditions. Two independent clones for wild type and Eprn KO cells were analysed. Percentage of GFP high cells were quantified. (C) Experimental scheme for colony formation assay. (D) Colony formation assay for wild type and Eprn KO cells in 2i/LIF 40 hr post PD/LIF withdrawal. Colonies were stained with alkaline phosphatase (AP), with representative images shown. Percentage clonogenicity was calculated by the number of AP positive colonies divided by the total number of cells plated. Mean ± SD, n = 3. (E) Lin28a and Nanog expression relative to β-actin in three independent wild type and Eprn KO cell lines measured by RT-qPCR. Mean ± SE, n = 3. *p<0.05, **p<0.01, student’s t-test. (F) Nanog and Lin28a expression kinetics upon PD/LIF withdrawal in wild type and Eprn KO cells. Mean ± SD, n = 3. (G) Rex1-GFP flow cytometry profile for wild type, Eprn KO and Eprn rescue cells 40 hr post PD/LIF withdrawal. Percentage of GFP high cells were quantified.
Molecular consequences of Ephemeron loss
We performed RNA-sequencing and compared the transcriptome of wild type and Eprn KO ESCs using three independently targeted KO ESC lines and three subclones of the parental wild type ESCs. Sixteen genes were differentially expressed between wild type and Eprn KO cells both in PD/LIF and after 8 hr withdrawal (Benjamini-Hochberg adjusted p<0.05, fold change >1.5 or<0.7) (Figure 2—figure supplement 2D) (Figure 2—figure supplement 2E). These include Tcf15, which has been associated with transition from the naïve state and has an inverse expression pattern compared to naïve pluripotency factors (Davies et al., 2013). Lin28a was the most differentially expressed gene in the group, with Eprn KO cells displaying a twofold reduction in mean expression level (Figure 2E). Although Lin28a is commonly considered as a core pluripotency factor, its expression is actually increased when cells transition out of the naïve state in vivo and in vitro (Boroviak et al., 2015; Kalkan et al., 2017; Kumar et al., 2014; Marks et al., 2012). Attenuated downregulation of members of the naïve transcription factor network is one explanation for delayed exit from the ESC state (Kalkan and Smith, 2014). We hypothesised that Lin28a could be a negative regulator of the network. We examined expression of naïve pluripotency transcription factors in Eprn KO cells and found a higher level of Nanog mRNA (Figure 2E, Figure 2—figure supplement 2F). To characterise the profile of naïve pluripotency dissolution further in Eprn KO cells, a PD/LIF withdrawal time course was monitored over 24 hr. The two-fold reduction in Lin28a mRNA in Eprn KO cells was constant throughout this time course (Figure 2F). Conversely, Nanog transcript and protein levels remained higher at 16 hr and 24 hr respectively (Figure 2F, see also 3E,F for protein). Mean Klf2 transcript levels appeared higher in Eprn KO cells, but below statistical significance. Other members of the naïve network showed similar expression profiles in wild type and Eprn KO cells (Figure 2—figure supplement 2F). Among peri-implantation epiblast markers, upregulation kinetics for Fgf5 were unchanged in Eprn KO cells, but transcripts for Dnmt3a, Dnmt3b and Oct6 remained lower from 16 to 24 hr (Figure 2—figure supplement 2F). Although not statistically significant, Otx2 transcripts appeared modestly reduced throughout the time course, which could be related to the elevated expression of Nanog (Acampora et al., 2016).
We restored Eprn expression in KO cells by inserting the Eprn genomic region under control of the human EF1α promoter into the deleted locus (Figure 2—figure supplement 3A,B). The rescue cells displayed a wild type exit profile as measured by GFP profile 40 hr post PD/LIF withdrawal (Figure 2G). Lin28a and Nanog expression levels were similar to wild type cells (Figure 2—figure supplement 3C).
To explore the differentiation capacity of Eprn KO cells, we conducted in vitro differentiation assays directing ESCs towards EpiSCs and somatic lineages (Figure 2—figure supplement 4). Both wild type and Eprn KO ESCs could be differentiated into EpiSCs using N2B27 supplemented with ActivinA/Fgf2/XAV939 (Sumi et al., 2013) on fibronectin. Such in vitro differentiated EpiSCs could be stably propagated over multiple passages, and displayed typical morphology and gene expression irrespective of genotype (Figure 2—figure supplement 4A,B). We also applied neuronal, mesendoderm and definitive endoderm differentiation protocols to Eprn KO ESCs and found that lineage markers were induced, with a slight delay for mesendoderm (Figure 2—figure supplement 4C–E). Thus retarded naïve state exit does not notably impair subsequent lineage commitment capacity.
The Ephemeron genetic network includes Lin28a and Nanog
Based on the preceding data, we hypothesised that Lin28a could be a downstream effector of Eprn, acting to reduce expression of Nanog. To characterise further the relationship between Eprn, Lin28a and the naïve transcription factor network, we carried out a series of genetic perturbation experiments and measured both Rex1-GFP reporter dynamics and colony formation upon withdrawal from PD/LIF. In Eprn KO cells, Nanog knockdown partially restored downregulation of Rex1-GFP 40 hr after PD/LIF withdrawal, and colony formation was reduced to the low level observed in wild type cells subjected to Nanog siRNA (Figure 3A). Knockdown of Klf4 had no effect on exit kinetics from PD/LIF in either wild type or Eprn KO cells. Knockdowns of other naïve transcription factors, Esrrb, Tfcp2l1 and Klf2, accelerated exit in wild type cells but in contrast to Nanog depletion this phenotype was attenuated in Eprn KO cells (Figure 3—figure supplement 1B). Resistance of Eprn KO cells to accelerated transition upon Esrrb, Tfcp2l1 and Klf2 knockdown could be attributed to elevated Nanog. We therefore conducted dual knockdown experiments (Figure 3—figure supplement 1C). Simultaneous depletion of Esrrb, Tfcp2l1 or Klf2 together with Nanog largely abolished the effect of Eprn KO on GFP downregulation (Figure 3—figure supplement 1C,D). These data are consistent with Eprn acting, at least in part, via modulation of Nanog expression.
Figure 3 with 2 supplements see all
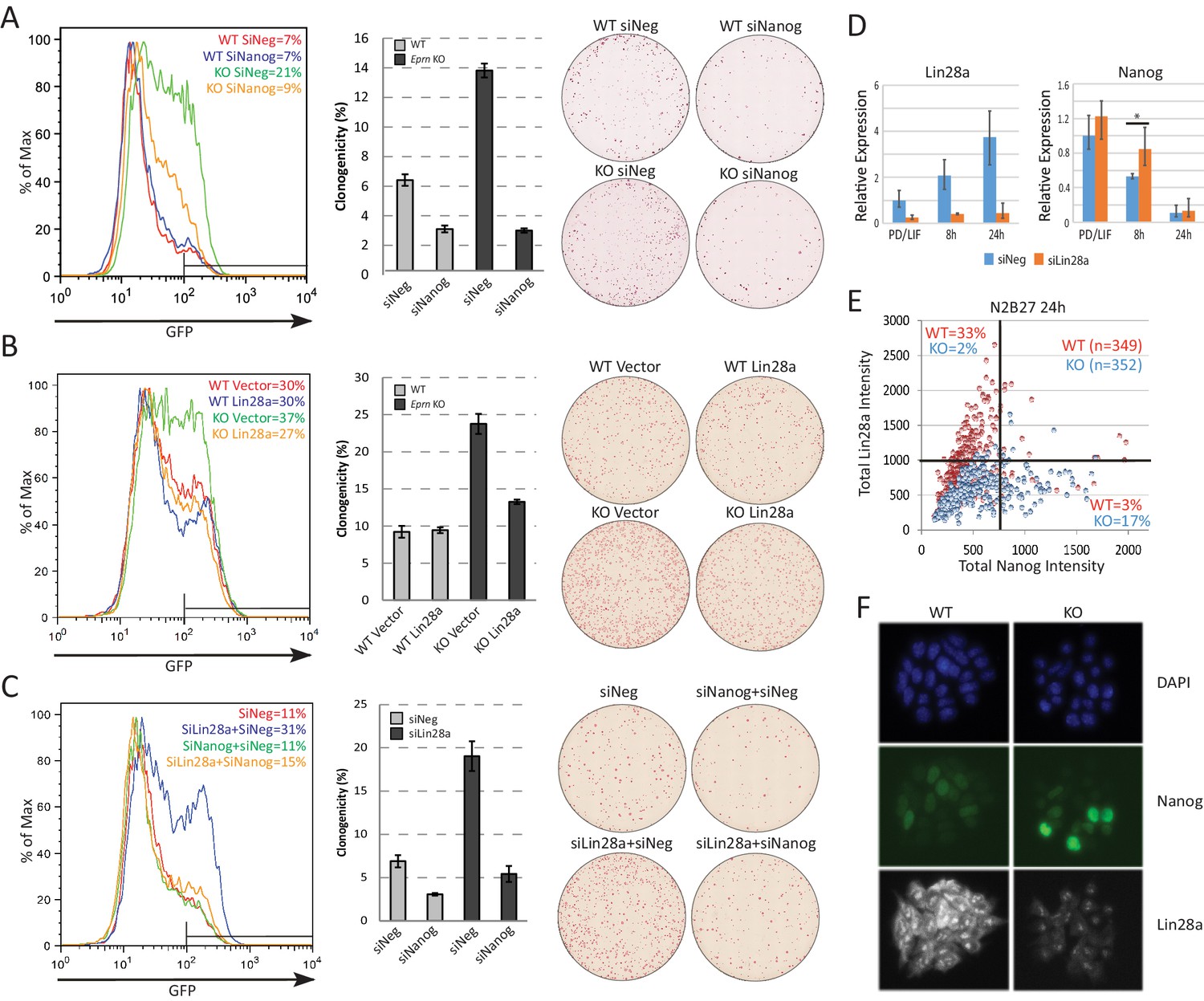
Lin28a is downstream of Ephemeron and regulates Nanog expression.
(A) Rex1-GFP flow cytometry profiles (Left) and colony formation capacity (Right) 40 hr post PD/LIF withdrawal for wild type and Eprn KO cells transfected with indicated siRNAs. (B) Rex1-GFP flow cytometry profiles and colony formation capacity 40 hr post PD/LIF withdrawal for wild type and Eprn KO cells transfected with Lin28a expression vector. (C) Rex1-GFP flow cytometry profile and colony formation capacity 40 hr post PD/LIF withdrawal with Nanog and Lin28a single or dual knockdowns in wild type cells. Quantification of percentage of GFP high cells were shown in (A-C). Percentage clonogenicity in (A-C) is measured by the number of AP positive colonies divided by the total number of cells plated, with representative AP staining images shown. Mean ± SD, n = 3. (D) Lin28a and Nanog expression relative to β-actin upon PD/LIF withdrawal in Lin28a knockdown and control cells. Mean ± SD, n = 3. *p<0.05, Student’s t-test. (E) Correlation of Nanog and Lin28a protein expression immunostaining in wild type and Eprn KO cells 24 hr post PD/LIF withdrawal. (F) Representative images of cells co-immunostained with Nanog and Lin28a and quantified in E.
We investigated whether lowered expression of Lin28a contributes to the slower exit from naïve pluripotency and the increased Nanog expression. We manipulated Lin28a dosage by either overexpression or knockdown in Eprn KO cells. In wild type cells, Lin28a overexpression had no significant effect. In Eprn KO cells, however, it restored normal transition kinetics (Figure 3B). Conversely, Lin28a knockdown phenocopied Eprn loss, delaying exit from naïve pluripotency (Figure 3C). Concomitant knockdown of Nanog and Lin28a abolished this effect (Figure 3C). Lin28a knockdown cells exhibited marginally elevated Nanog mRNA in PD/LIF and persistence at higher levels after 8 hr of PD/LIF withdrawal (Figure 3D). At the protein level, Eprn null cultures displayed more cells with high Nanog and low Lin28a expression at the 24 hr time point as quantified by co-immunostaining (Figure 3E,F, Figure 3—figure supplement 1E). Interestingly, Lin28a was detected as concentrated foci in the nucleus and also in the cytoplasm (observed with two independent antibodies), and both nuclear and cytoplasmic expression were increased after PD/LIF withdrawal (Figure 3—figure supplement 1F). During early embryo development, expression of Lin28a and Eprn are positively correlated, while Lin28a and Nanog are negatively correlated (Figure 3—figure supplement 1G)(Ohnishi et al., 2014). These data are consistent with the proposition that Lin28a is genetically downstream of Eprn and may facilitate exit from naïve pluripotency by accelerating downregulation of Nanog.
To assess whether Eprn could regulate Lin28a or Nanog expression directly, we employed chromatin isolation by RNA purification (ChIRP) (Chu et al., 2011). Using this method, we were able to selectively pull down endogenous Eprn RNA (Figure 3—figure supplement 2A). However, we did not detect chromatin enrichment at the Lin28a or Nanog promoter regions (Figure 3—figure supplement 2B–D). Indeed, no significant enrichment genome-wide was observed in wild type compared to Eprn KO cells (Figure 3—figure supplement 2E). Thus we found no evidence that Eprn functions by chromatin association (Rinn and Guttman, 2014).
The H3K4me3 modification was reduced at the Lin28a promoter in Eprn KO cells, in line with reduced transcription (Figure 3—figure supplement 2F). One explanation for anti-correlated expression could be direct negative regulation of Lin28a by Nanog. We inspected two published Nanog chromatin immunoprecipitation (ChIP) sequencing datasets (Chen et al., 2008; Marson et al., 2008) but observed no localisation of Nanog at the Lin28a locus (Figure 3—figure supplement 2G). Furthermore, we did not observe Lin28a upregulation in Nanog knockdown cells (Figure 3—figure supplement 2H). Therefore, Nanog does not appear to be a direct upstream regulator of Lin28a.
The function of Lin28a in ESC transition may be mediated by suppression of let-7g
Lin28a is an RNA binding protein with a well-established function in suppressing maturation of let-7 family miRNAs (Cho et al., 2012; Viswanathan et al., 2008). We investigated whether the role of Lin28a in naïve state exit is let-7 dependent. We profiled mature miRNA expression of let-7 family members using RT-qPCR. Expression of let-7a, let-7d, let-7e, let-7g and let-7i decreased 24 hr after 2i/LIF withdrawal, coincident with the increase in Lin28a expression (Figure 4A). Mature miRNA let-7c expression was unaffected, suggesting that let-7c expression is independent of Lin28a. This observation is in agreement with a recent finding that let-7c-2, the major let-7c isoform expressed in mouse ESCs, bypasses Lin28a regulation due to lack of a GGAG recognition motif in its loop region (Triboulet et al., 2015). The Lin28a regulated let-7 miRNAs, but not let-7c, are expressed at higher levels in ESCs in 2i/LIF than in LIF/serum (Pandolfini et al., 2016) (Figure 4—figure supplement 1A), consistent with lower Lin28a in 2i/LIF.
Figure 4 with 1 supplement see all

Lin28a function is mediated via members of let-7 miRNAs.
(A) Mature let-7 family microRNA expression quantified by RT-qPCR in 2i/LIF, 24 hr post 2i/LIF withdrawal, and EpiSCs. (B) Rex1-GFP flow cytometry profile upon forced expression of mature let-7g mimic during transition from 2i and PD/LIF. Quantification of percentage of GFP high cells was shown. (C) Colony formation assay in 2i/LIF of cells with forced expression of let-7g mimic and control 40 hr post PD/LIF withdrawal. Colonies were stained with alkaline phosphatase (AP). Percentage clonogenicity was calculated by the number of AP positive colonies divided by the total number of cells plated. Mean ± SD, n = 3. (D) Predicted target sites of let-7g in 3’UTRs of Dnmt3a and Dnmt3b by RNA22. (E) Dual luciferase assay measuring repression by let-7g mediated through 3’UTRs of Dnmt3a and Dnmt3b. Fold repression of Luc/Rluc relative to scramble was plotted. Mean ± SD, n = 3. (F) Relative expression normalised to β-actin of naïve and peri-implantation epiblast associated genes in ESCs with forced expression of let-7g mimics. Mean ± SD, n = 3.
To examine the role of Lin28a regulated let-7 miRNAs in naïve state exit, we first transfected ESCs with mature let-7g mimic. We used let-7g as a representative member since all apart from let-7e share the same seed sequence (Figure 4—figure supplement 1B). Forced expression of let-7g in RGd2 cells resulted in delayed GFP downregulation upon both 2i and PD/LIF withdrawal (Figure 4B). Elevated ESC colony formation capacity post PD/LIF withdrawal was also observed (Figure 4C). To identify downstream targets of let-7g, we curated genes that are upregulated upon 2i withdrawal in our RNA-sequencing dataset and searched for known or predicted let-7g targets using the RNA22 tool (Miranda et al., 2006). DNA methyltransferases Dnmt3a and Dnmt3b emerged as prime candidates, as has previously been proposed (Kumar et al., 2014). Expression of both increases during ESC transition (Kalkan et al., 2017). Dnmt3a/3b transcript levels were lower in Eprn KO cells than wild type control (Figure 2—figure supplement 2F). Multiple let-7g target sites were predicted by RNA22 within the Dnmt3a 3’UTR and one site in the Dnmt3b 3’UTR (Figure 4D, Figure 4—figure supplement 1C). ESCs were co-transfected with mature let-7g mimic and luciferase constructs containing the entire 3’UTRs of Dnmt3a and Dnmt3b. let-7g reduced luciferase expression by more than 60% relative to scrambled control (Figure 4E), suggesting that Dnmt3a/3b transcripts are indeed let7-g targets. To test specificity of this repression, we generated two Dnmt3b 3'UTR luciferase reporter constructs with the let-7 seed recognition site mutated (Figure 4—figure supplement 1D). These mutant reporters escaped repression by the let-7g mimic (Figure 4—figure supplement 1D).
Dnmt3a and Dnmt3b methylate the Nanog promoter during naïve state exit
Epiblast progression is associated with genome-wide de novo methylation during pre-to post-implantation development (Auclair et al., 2014). This phenomenon is recapitulated when naïve ESCs are withdrawn from 2i (Kalkan et al., 2017). Previous studies demonstrated hypomethylation of the Nanog promoter in mouse ESCs compared to lineage committed cells (Farthing et al., 2008; Yu et al., 2007). We speculated that impeded de novo DNA methylation could allow perdurance of Nanog expression at the onset of naïve state exit. To investigate this hypothesis, we carried out bisulfite sequencing analysis across the Nanog proximal promoter region, 1 kb upstream of the TSS, after siRNA knockdown of Dnmt3a/3b singly or together (Figure 5A). We observed a marked reduction of CpG methylation in the −1 kb to −761 bp region (region 1) 40 hr after PD/LIF withdrawal (Figure 5A). Cells transfected with scrambled control siRNA exhibited 40% CpG methylation at scored sites, whereas Dnmt3b depleted cells displayed 18% CpG methylation and Dnmt3a or Dnmt3a/b double knockdown cells showed only around 8%. Effects were less obvious in the −538 bp to +18 bp region (region 2), which was barely methylated at this time point. These data suggest that Dnmt3a and Dnmt3b have overlapping roles in mediating de novo methylation at the Nanog proximal promoter. Eprn KO cells also exhibited reduced methylation at the Nanog promoter, with region one again showing a more prominent reduction (Figure 5—figure supplement 1A).
To explore the role of de novo DNA methylation in ESC transition, we investigated functional consequences of Dnmt3a and Dnmt3b depletion. We created Dnmt3a and Dnmt3b single and compound knockouts in RGd2 ESCs using CRISPR/Cas9. Using two guide RNAs (gRNAs), we generated deletions of highly conserved PC and ENV motifs (motifs IV and V) within the catalytic domain for both Dnmt3a and Dnmt3b, recapitulating the previously characterised Dnmt3b and Dnmt3b mutant gene structures (Okano et al., 1999) (Figure 5—figure supplement 1B). Dnmt3a and Dnmt3b single and double KO cells exhibited delayed Rex1-GFP downregulation (Figure 5B). Colony formation capacity of the single and double KO cells 40 hr post PD/LIF withdrawal confirmed slower extinction of ESC identity (Figure 5C). Interestingly, however, as with Eprn KO, the delay in GFP downregulation did not endure (Figure 5—figure supplement 1C). Absence of Dnmt3a and Dnmt3b singly or together was associated with transient perdurance of Nanog expression (Figure 5D). At 8 hr after PD/LIF withdrawal, Nanog mRNA in Dnmt3a and/or Dnmt3b mutants was equivalent to wild type cells in PD/LIF, whereas wild type cells had downregulated Nanog expression by 50% (Figure 5D). We also observed elevated expression of Tfcp2l1, Klf2, Klf4 and Tbx3 in Dnmt3a/3b single or compound KO cells (Figure 5—figure supplement 1E). The promoters of these genes are methylation refractory in the 2i withdrawal time course (Kalkan et al., 2017). Therefore, the elevated expression should be secondary to some other factor(s) such as increased Nanog. Dnmt3a/3b compound KO also resulted in impeded upregulation of peri-implantation markers Fgf5, Oct6 and Otx2 at 24 hr post PD/LIF withdrawal (Figure 5—figure supplement 1F). These data indicate that de novo DNA methylation facilitates timely progression from the ESC state. Importantly, however, methylation by Dnmt3a/3b is not essential for the exit from naïve pluripotency.
Figure 5 with 1 supplement see all
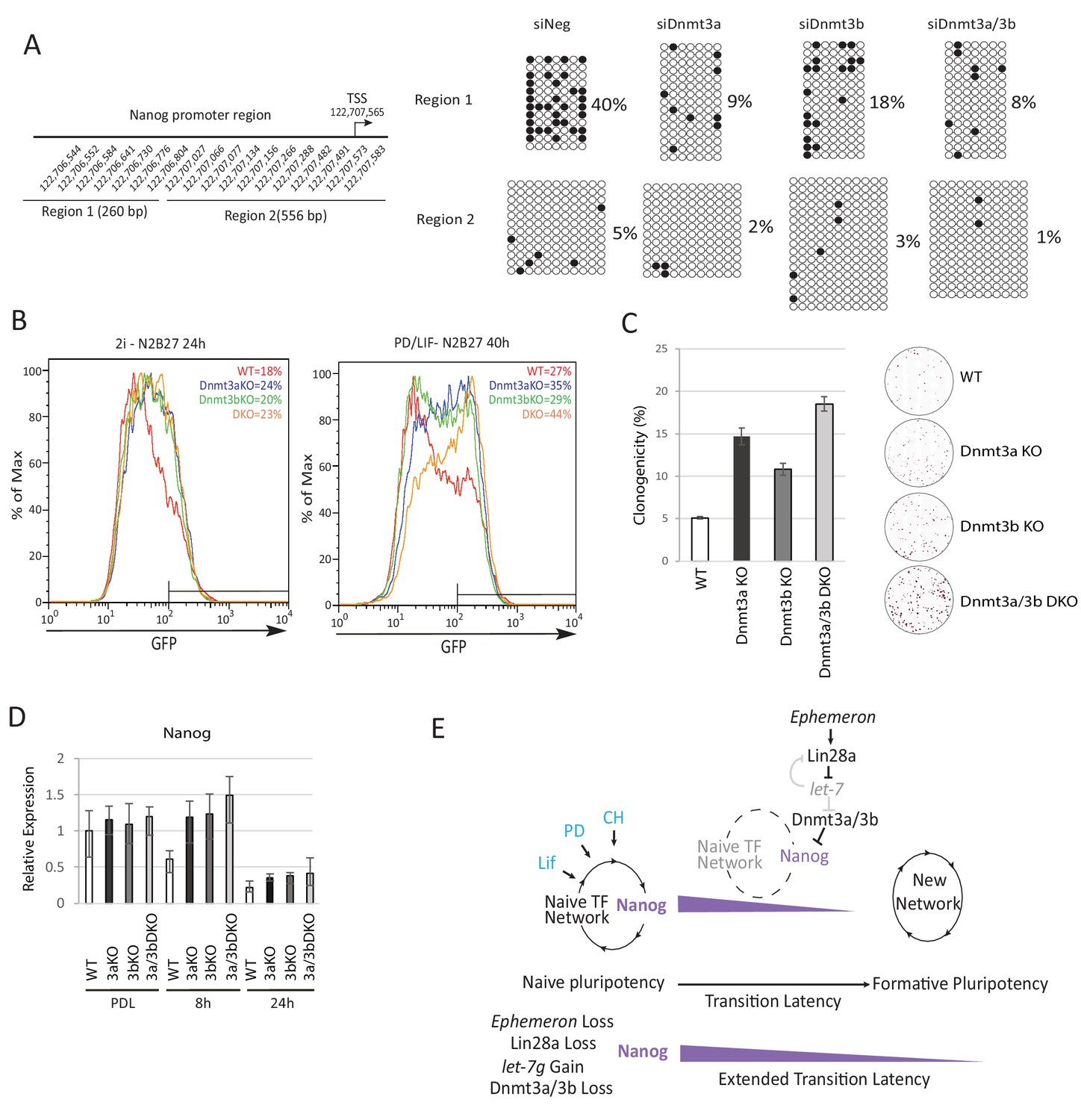
Loss of Dnmt3a and Dnmt3b delays naïve state exit associated with transient persistence of Nanog expression.
(A) Bisulfite sequencing analysis of Nanog proximal promoter CpG island DNA methylation in Dnmt3a and Dnm3b single and dual knockdown cells at 40 hr post PD/LIF withdrawal. The positions of cytosines analysed (mm10) are indicated on the left panel. Black and while circles represent methylated and unmethylated cytosine respectively. (B) Rex1-GFP flow cytometry profiles of Dnmt3a and Dnmt3b single and dual KO cells withdrawn from 2i or PD/LIF for 24 and 40 hr respectively. Percentage of GFP high cells were quantified. (C) colony formation capacity 40 hr post PD/LIF withdrawal for Dnmt3a and Dnmt3b single and compound KO cells. Percentage clonogenicity was measured by the number of AP positive colonies formed divided by the total number of cells plated, with representative AP staining images shown. Mean ± SD, n = 3. (D) Expression of Nanog relative to β-actin in Dnmt3a and Dnmt3b single and compound KO cells quantified by RT-qPCR. Mean ± SD, n = 2. (E) Schematic representation of the inferred Eprn pathway. Legends for Figures and Source Data.
Discussion
Mouse ES cell self-renewal is robust due to recursive wiring of a core transcription factor network (Dunn et al., 2014; Martello and Smith, 2014; Young, 2011). Rapid developmental progression from such a resilient state is achieved through parallel mechanisms. In this study, we find that a lncRNA, Ephemeron, participates in the timely dissolution of naïve identity. Genetic interactions link Eprn with known players in post-transcriptional and epigenetic regulation (Figure 5E). Eprn lies upstream of Lin28a/let-7g and Dnmt3a/3b, and ultimately contributes to timely downregulation of the pivotal naïve transcription factor Nanog. Eprn depletion reduces Lin28a expression, although the molecular mechanism underlying this effect remains unclear. Lower Lin28a stabilises expression of the let-7 miRNAs whose targets include de novo DNA methyltransferases Dnmt3a and Dnm3b. Resulting decreased Dnmt3a/3b reduces Nanog proximal promoter CpG methylation, correlating with transiently perduring expression. This lncRNA/miRNA/DNA methylation module provides an additional layer in the multi-layered machinery that enforces transition from naïve to formative pluripotency (Acampora et al., 2016; Jang et al., 2017; Kalkan et al., 2017; Kalkan and Smith, 2014; Smith, 2017).
Eprn promotes ESC transition and is suppressed by Gsk3 inhibition during ground state self-renewal in 2i or 2i/LIF. ESCs cultured without Gsk3 inhibition can self-renew in the presence of PD/LIF or LIF/serum. In these conditions they express Eprn and higher levels of Lin28a. The lack of overt consequence is presumably due to the dominant self-renewal environment provided by Stat3 activation and MEK inhibition that sustain expression of Nanog and other naïve factors. Nonetheless, loss of Eprn in PD/LIF resulted in elevated Nanog and delayed transition kinetics.
We observed a Mendelian ratio of homozygous Eprn mutant mice from heterozygous intercrosses (5:18:7, wild type: heterozygous: homozygous offspring). Therefore, in common with Lin28a (Shinoda et al., 2013), Eprn is dispensable for development of laboratory mice. Some protein-coding genes that exhibit demonstrable loss-of-function phenotypes in ESC self-renewal or transition also show no early embryo phenotype (Leeb et al., 2014; Martello and Smith, 2014). Our interpretation is that ESCs provide a sensitised platform for identifying components whose functions may be compensated during in vivo development.
The majority of lncRNAs are not phylogenetically conserved (Necsulea et al., 2014). Due to their rapidly evolving nature, it is thought that lncRNAs are likely to acquire species or lineage-restricted functions and several examples have recently been described (Durruthy-Durruthy et al., 2016; Paralkar et al., 2014; Rani et al., 2016). The presence of Eprn exclusively in rodent may be associated with rapid embryonic progression from pre-implantation epiblast to gastrulation (Rossant and Tam, 2017), which necessitates acute extinction of the naïve pluripotency programme (Smith, 2017). LncRNAs are more tolerant to TE integration than protein coding genes, which could drive more rapid evolution (Kelley and Rinn, 2012; Necsulea et al., 2014). Non-coding transcripts harbouring TE sequences are enriched in ESCs and early embryo development in both mouse and human (Fort et al., 2014; Göke et al., 2015; Kelley and Rinn, 2012) and in several instances have been proposed to regulate pluripotency (Durruthy-Durruthy et al., 2016; Fort et al., 2014; Lu et al., 2014). Eprn is comprised of 76.4% TEs, compared to the average of 41.4% TE composition in the mouse genome and 33% reported for mouse multi-exon lincRNA sequences (Kelley and Rinn, 2012). The aligned sequence between Eprn and the rat transcript from the syntenic region includes ERVK LTR and SINE B2 elements. These sequences have been preserved for over 30 million years since the mouse-rat lineage divergence.
Lin28a is known as a human somatic cell reprogramming factor (Yu et al., 2007). However, Lin28a is expressed at a lower level in ground state mouse ESCs (Marks et al., 2012) and pre-implantation epiblast, than in post-implantation epiblast and EpiSCs (Boroviak et al., 2015). The expression pattern is consistent with our evidence that up-regulation of Lin28a at the onset of mouse ESC differentiation functions to facilitate transition from the naïve state. During human iPSC generation, Lin28a may promote acquisition of primed pluripotency, the endpoint for current human somatic cell reprogramming. Lin28a itself is a target of let-7 miRNAs (Kumar et al., 2014; Melton et al., 2010) and the reciprocal negative feedback loops can act as a bimodal switch. Our findings are consistent with the recent report that loss of Lin28a reduced ESC heterogeneity in serum/LIF, favouring a more naïve state (Kumar et al., 2014). This effect was attributed to let-7g. We note, however, that Lin28a can post-transcriptionally regulate the expression and/or translation of many RNAs independently of let-7 (Cho et al., 2012; Zhang et al., 2016) that could also contribute to ESC transition.
De novo methyltransferases Dnmt3a/3b have previously been proposed as targets of let-7g (Kumar et al., 2014). Our results show that loss of Dnmt3a and Dnmt3b, individually and in combination, delays naïve state exit. Naïve ESCs (Ficz et al., 2011; Habibi et al., 2013; Leitch et al., 2013) and pre-implantation epiblast (Monk et al., 1987; Sanford et al., 1987) have low expression of Dnmt3a/3b and display global DNA hypomethylation. However, the post-implantation epiblast rapidly acquires global DNA methylation and this process is dependent on Dnmt3a/3b (Auclair et al., 2014). A similar acute trend is observed upon naïve ESC withdrawal from 2i (Kalkan et al., 2017). Early de novo methylation may have functional consequences for specific naïve pluripotency associated factors, such as Nanog, enduring rapid downregulation. It is noteworthy, however, that the Dnmt3a/3b KO phenotype is transient and ESC identity still collapses. Therefore, although de novo DNA methylation facilitates ESC transition it is not mandatory for the exit from naïve pluripotency.
Human naïve pluripotency shares molecular and cellular features with mouse, consistent with a conserved core pluripotency programme in mammals (Guo et al., 2017, 2016; Smith, 2017; Takashima et al., 2014; Theunissen et al., 2014). However, species-specific differences are evident. Notably, Gsk3 inhibition has less impact on human naïve state maintenance (Guo et al., 2017; Theunissen et al., 2016). This is partly explained by the lack of ESRRB expression in human pluripotent cells (Blakeley et al., 2015; Martello et al., 2012; Takashima et al., 2014; Theunissen et al., 2014), but absence of Eprn may be an additional factor that reduces requirement for Gsk3 inhibition.
In summary, we have mapped a genetic interaction module consisting of a novel lncRNA, proteins and miRNAs that is integrated into the multi-pronged molecular machinery that propels mouse ESCs towards lineage competence. The fine-tuning effect of Eprn could be representative of lncRNA actions in the regulation of molecular networks and illustrative of their potential contribution to species diversification.
Materials and methods
Targeting, expression and gRNA vector construction
Request a detailed protocolBAC RP24-353A19 (C57BL/6J background) was obtained from Wellcome Trust Sanger Institute for constructing the Eprn knockout targeting vectors by recombineering using bacterial strain EL350 (Lee et al., 2001). Floxed drug resistant cassettes containing hygromycin B phosphotransferase gene (Hygro) or Blasticidin S-resistance gene (Bsd) were PCR amplified using chimeric primers miniU and miniD (Supplementary file 1A) harbouring 80 bp mini-homologies to the genomic region flanking Eprn locus. The purified PCR fragments loxP-PGK-Hygro-bghpA-loxP and loxP-PGK-Bsd-bghpA-loxP were used to replace the entire genomic region of Eprn locus with the drug resistant cassettes. The retrieval homology arms were PCR amplified using primers ReUF and ReUR for upstream and ReDF and ReDF for downstream mini-arms (Supplementary file 1B). The mini arms were subsequently cloned into pBS-MC1-DTA vector by 3-way ligation using restriction enzymes, SpeI, HindIII and XhoI. The mini-arm containing vector was linearised by HindIII and used to retrieve the targeting vectors from the modified BACs, giving rise to the final targeting vectors, HygroTV and BsdTV.
Lin28a overexpression vector was constructed by PCR cloning mouse Lin28a from cDNA using forward primer AATTGTCGACATGGGCTCGGTGTCCAACCAGCAGT and reverse primer AATTGCGGCCGCTCAATTCTGGGCTTCTGGGAGCAG and cloned into pENTR2B vector. It was subsequently cloned into PiggyBac-based expression vector using Gateway LR clonase (Thermo Fisher Scientific, Waltham, MA, USA, 11791020) to generate pCAG-Lin28a-pA:PGK-hygro-pA plasmid.
The gRNA design was conducted using online CRISPR gRNA design tool https://www.atum.bio/eCommerce/cas9/input. The chosen gRNAs were based on minimal off-target scores. Deletions were designed to recapitulate the original Dnmt3a and Dnmt3b KO mutations (Okano et al., 1999), excising the highly conserved PC and ENV motifs (motifs IV and V) within the catalytic domain. The gRNAs were generated by annealing the indicated oligos (Supplementary file 2A), which were subsequently ligated into pX458 vector (Addgene) digested with BbsI. The constructs were sequence validated before transfection.
Cell culture
Request a detailed protocolESCs were cultured on 0.1% gelatin in 2i/LIF medium (homemade N2B27 base medium, supplemented with 1 μM PD0325901, 3 μM CHIR99021, and 20 ng/ml LIF) as described (Ying et al., 2008). For gene targeting, ESC were maintained with serum containing medium supplemented with 2i/LIF as above (KO-DMEM high glucose, 15% FCS, 2 mM L-Glutamine, NEAA, 1 mM Sodium Pyruvate (Thermo Fisher Scientific), 100 mM β-Mercaptoethanol (Sigma Aldrich, St. Louis, MO, USA). Correctly targeted clones were transferred to N2B27 based 2i/LIF medium for expansion and experimentation. The RGd2 reporter wild type subclones and Eprn KO ESC clones are of V6.5 origin (RRID:CVCL_C865). An independent wild type RGd2 reporter line is of E14 origin (RRID:CVCL_C320). All cell lines are mycoplasma negative by PCR screening in house.
Naïve pluripotency exit assays
Request a detailed protocolESCs were plated at 1 × 104/cm2 in 2i without LIF or PD/LIF. The next day, cells were carefully washed with PBS before switching to N2B27 medium. Rex1-GFP profile was analysed at indicated time points in at least two independent experiments using a Cyan or Fortessa FACs analyser. Live dead discrimination was performed using TO-PRO-3 (Thermo Fisher Scientific, T3605). For clonal assay, post 24 hr or 40 hr 2i or PD/LIF withdrawal respectively, 300–500 cells were plated per well of a 12 well plate coated with Laminin (1:100 dilution, Sigma Aldrich, L2020) and cultured in 2i/LIF for 6 days. Alkaline Phosphatase staining (Sigma Aldrich, 86R-1KT) was conducted to visualise ES colonies. AP-stained plates were imaged using an Olympus IX51, DP72 camera with CellSens software and subsequent colony counting was conducted manually using ImageJ software.
EpiSC derivation from ESCs and EpiSC resetting
Request a detailed protocolESCs were plated at 1 × 104/cm2 in 2i/LIF on a gelatin coated plate. The next day, cells were washed with PBS before medium switch to N2B27 medium supplemented with 20 ng/ml Activin A and 12 ng/ml Fgf2 together with 2 µM XAV939 (Sigma Aldrich, X3004), A/F/X. Cells were then passaged to fibronectin coated plate in A/F/X medium. EpiSCs were passaged for at least seven times before gene expression analysis and resetting. For EpiSC resetting, EpiSCs were stably transfected with GY118F construct by piggyBac transposition (Yang et al., 2010). 1 × 104 cells were plated in a one well of a 12 well plate in A/F/X, the next day, 2i plus GCSF was supplied to initiate resetting.
Differentiation assays
Request a detailed protocolFor neuronal differentiation, ESCs were plated at 1 × 104/cm2 in N2B27 medium on laminin (1:100 in PBS) for up to 4 days for gene expression analysis. For mesendoderm differentiation, cells were plated at 0.6 × 104/cm2 in N2B27 based medium containing 10 ng/ml ActivinA, 3 µM CHIR99021 on Fibronectin for up to 4 days for gene expression analysis. For definitive endoderm differentiation, cells were plated at 1.5 × 104/cm2 in N2B27 based medium containing 20 ng/ml ActivinA, 3 µM CHIR99021, 10 ng/ml FGF4, 1 µg/ml Heparin, 100 nM PI103. On day 2, the media was switched to SF5 based medium containing 20 ng/ml ActivinA, 3 µM CHIR99021, 10 ng/ml FGF4, 1 µg/ml Heparin, 100 nM PI103 and 20 ng/ml EGF2. Per 100 ml SP5 basal medium, it contains 500 µl N2, 1 ml B27 without VitaminA supplement, 1% BSA, 1 ml L-glutamine and 100 µl β-mercaptoethanol. Detailed protocols can be found in Mulas et al (Mulas et al., 2017).
siRNA, miRNA mimics and plasmid transfection
Request a detailed protocolsiRNAs and miRNA mimics were obtained from Qiagen and the catalogue numbers are listed in Supplementary file 3. Transfection was performed using Dharmafect 1 (Dharmacon, Lafeyette, CO, USA, T-2001–01) in a reverse transfection protocol with the final siRNA or miRNA mimics concentration to be 10 nM. Two siRNA combination were used per transfection for each target gene knockdown.
Plasmid transfection was performed using Lipofectamine 2000 (Thermo Fisher Scientific, 11668027) following the manufacturers protocol. For piggyBac based stable integration, a piggyBac transposon and hyperactive PBase (hyPBase) ratio of 3:1 was used.
Generation of Dnmt3a and Dnmt3b KO ESCs with CRISPR/Cas9
Request a detailed protocolA pair of gRNA containing plasmids based on px458 backbone (Ran et al., 2013) were transfected using Fugene HD (Promega, Madison, WI, USA, E2311). 100 ng of each plasmid were transfected with 0.6 ul Fugene HD (1:3 ratio) to 2 × 105 ESCs in suspension in 2i/LIF medium overnight. The next day, the media was refreshed and 48 hr post transfection, 1,000 GFP high cells were sorted into a well of a six well plate for colony formation. Individual colonies were picked and genotyping was conducted from extracted genomic DNA by triple primer PCR to identify clones with designed deletion (Supplementary file 2B). For Dnmt3a KO, deletion resulted in genotyping PCR product shift from 760 bp representing the wild type allele to 1132 bp. For Dnmt3b KO, deletion resulted in shift from 344 bp representing the wild type allele to 653 bp. Only homozygous mutants were chosen for subsequent experimentation.
Southern blotting
Request a detailed protocolGenomic DNA of individually picked ESC clones was extracted and digested with XmnI, size-fractionated on a 0.8% agarose gel and transferred to Hybond-XL blotting membrane (GE Healthcare, Chicargo, IL, USA, RPN20203) using standard alkaline transfer methods. The 5’ and 3’ external probes were generated by PCR with primer sequences shown in Supplementary file 4A. Southern blot hybridization was conducted as described previously (Li et al., 2011).
Northern blotting
Request a detailed protocol10 µg of purified RNA was resolved by denaturing formaldehyde agarose gel electrophoresis with MOPS buffer. RNA was transferred to Hybond-XL membrane in 1xSSC buffer overnight by capillary transfer. RNA was UV cross-linked to the membrane and pre-hybridised with Expresshyb (CloneTech, Mountainview, CA, USA, 636831) for 2 hr at 65°C. The DNA probe was generated by PCR (primers are shown in Supplementary file 4B) and 25 ng of probe DNA was labelled with [32P]-dCTP using Radprime DNA labeling system (Thermo Fisher Scientific, 18428–011). The free-nucleotide was removed from labelled probe using G-50 column (GE Healthcare, 27-5330-01), and was heat-denatured followed by snap cooling. The probe was added to the pre-hybridised membrane and incubated overnight at 65°C in a rolling incubator. Membrane was washed with wash buffer containing 0.1 x SSC and 0.1% SDS 3 times at 65°C with 10 min intervals. The membrane was placed in a phosphoimager and exposed for at least overnight at −80°C before scanned using Typhoon 9410 phosphoimager system (GE Healthcare).
5’ and 3’ RACE
Request a detailed protocol5’ RACE was conducted using 5'-Full RACE Core Set (Takara, Kusatsu, Japan, #6122) following manufacture’s protocol. The sequences for RT-primer and nested PCR primers A1, A2, S1, and S2 are shown in Supplementary file 5A. 3’ RACE was conducted by using a polyT RT-primer with a unique sequence tag to synthesis cDNA. The 3’ end region was PCR amplified using a primer specific to the RT-primer and a gene specific primer. The primers are shown in Supplementary file 5B. Both 5’ and 3’ RACE PCR products were cloned into plasmids using Zero blunt TOPO PCR cloning kit (Thermo Fisher Scientific, 451245) for subsequent sequencing.
RNA extraction, reverse transcription and Real-time PCR
Request a detailed protocolTotal RNA was isolated using Trizol (Thermo Fisher Scientific, 15596026) or RNeasy kit (Qiagen, Hilden, Germany, 74136) and DNase treatment was conducted either after RNA purification or during column purification. cDNA was transcribed from 0.5 ~ 1 ug RNA using SuperScriptIII (Thermo Fisher Scientific, 18080044) and oligo-dT priming. Real-time PCR was performed using StepOnePlus machine (Applied Biosystems) with Fast Sybrgreen master mix (Thermo Fisher Scientific, 4385612). Target gene primer sequences are shown in Supplementary file 6. Expression level were normalised to Actinβ. Technical replicates for at least two independent experiments were conducted. The results were shown as mean and standard deviation calculated by StepOnePlus software (Applied Biosystems). The cDNA library for E7-E17 embryos and adult tissues were purchased from Clontech (Mouse Total RNA Master Panel, 636644).
RNA-FISH
Request a detailed protocolRNA-FISH was conducted using ViewRNA ISH Cell Assay for Fluorescence RNA In Situ Hybridization system (Thermo Fisher Scientific, QVC0001) with modifications and imaged on a DeltaVision Core system (Applied Precision), as described in Bergmann et al. (2015). The probe set used for Ephemeron was VX1-99999-01.
Mature miRNA expression profiling
Request a detailed protocolTotal RNA was extracted using Trizol. 1 ug RNA was reverse transcribed using Taqman MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific, 366596). Mature miRNA expression was analysed using Taqman Array Rodent MicroRNA A + B Cared Set V3.0 (Thermo Fisher Scientific, 444909).
Luciferase assay
Request a detailed protocolThe Entire 3’UTR of both Dnmt3a and Dnmt3b were PCR cloned downstream of the firefly luciferase coding region into pGL3 vector. For Dnmt3a 3’UTR, forward primer AATTGGCCGGCCGGGACATGGGGGCAAACTGAAGTAG and reverse primer AATTGGATCCGGGAAGCCAAAACATAAAGATGTTTATTGAAGCTC were used for PCR cloning. For Dnmt3b 3’UTR, forward primer AATTGGCCGGCCTTCTACCCAGGACTGGGGAGCTCTC and reverse primer AATTGGATCCTTATAGAGAAATACAACTTTAATCAACCAGAAAGG were used for PCR cloning. To generate mutant Dnmt3b reporter constructs, let-7g binding site was mutated to include BsrGI (mutation V1) and EcoRI (mutation V2) sites by PCR cloning. Each firefly luciferase construct (500 ng) together with Renilla luciferase construct (10 ng) were con-transfected with either let-7g mimic or scrambled control (20 nM). The firefly and Renilla luciferase activity was determined by dual luciferase assay (Promega, E1960) 48 hr post-transfection.
Immunostaining
Request a detailed protocolCells were fixed in 4% paraformaldehyde for 10 min at room temperature and were blocked with blocking buffer (5% semi-skimmed milk with 0.1% Triton in PBS) for 2 hr at room temperature. Primary antibodies were diluted in blocking buffer and incubated at 4°C overnight. Primary antibody was carefully washed away with 0.1% Triton in PBS three times with 10 min incubation between each wash. Secondary antibody diluted in blocking buffer (1:1000) was incubated at room temperature for 1 hr followed by 3 washes with 0.1% Triton in PBS. Nuclei were counterstained with DAPI. Primary antibodies used were Nanog (eBioscience, 14–5761, RRID:AB_763613, 1:200) and Lin28a (Cell signalling, 3978, RRID:AB_2297060, 1:800; 8706, RRID:AB_10896850, 1:200). Images from random fields were taken with Leica DMI3000 and the images from different fields at each time point were combined and analysed using CellProfiler software (Broad Institute, RRID:SCR_007358) to conduct nuclear and cytoplasmic compartmentalisation and total fluorescent intensity for each sub-cellular compartments as well as the whole cell for each cell was extracted for correlation analysis.
Chromatin isolation by RNA purification (ChIRP)
Request a detailed protocolThe antisense oligo probes were selected with GC content in the range of 40–50% in regions of the Eprn exons without repetitive sequences (Figure 1—figure supplement 1A). The probes sequences are in shown in Supplementary file 7. CHIRP was conducted following published protocol (Chu et al., 2011). The data is available at the NCBI Gene Expression Omnibus (accession number: GSE90574). The link to the data is as follows: http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE90574.
ChIP
Request a detailed protocolThe experimental procedure was conducted as described previously (Betschinger et al., 2013). 2 ug of H3K4me3 antibody (Diagenode, Ougrée, Belgium pAb-003–050) and IgG control (Santa Cruz, Dallas, TX, USA, sc-2345) was used for 4 × 106 cells per ChIP. qPCR was performed with primers shown in Supplementary file 8.
Nanog promoter DNA methylation analysis
Request a detailed protocolGenomic DNA was extracted using GenElute Mammalian Genomic DNA miniprep kit (Sigma Aldrich, G1N70-1KT). 500 ng purified genomic DNA was treated with sodium bisulfite to convert all unmethylated cytosine residues into uracil residues using Imprint DNA modification Kit (Sigma Aldrich, MOD50-1KT) according to the manufacturer's protocol. Nanog proximal promoter regions (Region 1 and 2 as indicated in Figure 5a) were amplified using a nested PCR approach with KAPA HiFi Uracil + Readymix (KapaBiosystems/Roche, Basel, Switzerland, KK2801). The PCR condition for both nested rounds of PCR is as follows: denaturation at 98°C for 5 min followed by 10 cycles of gradient PCR, 98°C for 15 s, 62°C (starting annealing temperature) for 15 s with annealing temperature reduced by 1°C per cycle and 72°C for 1.5 min. Followed by this, a 35 cycles of 98°C for 15 s, 58°C for 15 s and 72°C for 1.5 min were conducted. 2 µl first round PCR product was used as template for the nested PCR. All primer sequences are shown in Supplementary file 9. The PCR products were verified and purified by gel electrophoresis and subsequently subcloned by TOPO cloning. Reconstructed plasmids were purified and individual clones were sequenced (Eurofins).
Transcriptome sequencing and analysis
Request a detailed protocolTotal RNA was isolated with RNeasy RNA purification. Ribo-zero rRNA depleted RNA was used to generate sequencing libraries for wild type and Ephemeron knockout cells in PD/LIF and 8 hr withdrawal from PDL from three independent cell lines. Single end sequencing was performed and the reads were mapped using NCBI38/mm10 with Ensembl version 75 annotations. RNA-seq reads were aligned to the reference genome using tophat2. Only uniquely mapped reads were used for further analysis. Gene counts from SAM files were obtained using htseq-count with mode intersection non-empty, -s reverse. Differential gene expression analysis was conducted using Bioconductor R package DESeq2 version 1.4.5. DESeq2 provides two P-values, a raw P-value and a Benjamini-Hochberg P-value (adjusted p value). An adjusted p-Value threshold of 0.05 was used to determine differential gene expression (95% of the results are not false discoveries, error rate 0.05 = 5%). The data is available at the NCBI Gene Expression Omnibus (accession number: GSE90574, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE90574).
Eprn promoter CpG methylation analysis
Request a detailed protocolUsing published genome-wide bisulpite sequencing data (Kalkan et al., 2017; Seisenberger et al., 2012; Wang et al., 2014), Eprn promoter region was defined as the 2 kb region upstream of the TSS and the percentage of CpG methylation within the region was quantified. For promoter average, percentage of CpG methylation around the 2 kb promoter region of each annotated gene was quantified and averaged for all values. For genome average, percentage of CpG methylation of all 50 kb tiling windows was quantified and averaged all values.
Data availability
-
A lncRNA/Lin28/let7 axis coupled to DNA methylation fines tunes the dynamics of a cell state transitionPublicly available at the NCBI Gene Expression Omnibus (accession no: GSE90574).
References
-
Regulation of the ESC transcriptome by nuclear long noncoding RNAsGenome Research 25:1336–1346.https://doi.org/10.1101/gr.189027.114
-
Ripples from neighbouring transcriptionNature Cell Biology 10:1106–1113.https://doi.org/10.1038/ncb1771
-
Epigenetic resetting of human pluripotencyDevelopment 144:2748–2763.https://doi.org/10.1242/dev.146811
-
Mapping the route from naive pluripotency to lineage specificationPhilosophical Transactions of the Royal Society B: Biological Sciences 369:20130540.https://doi.org/10.1098/rstb.2013.0540
-
Epigenetic regulation by long noncoding RNAsScience 338:1435–1439.https://doi.org/10.1126/science.1231776
-
Naive pluripotency is associated with global DNA hypomethylationNature Structural & Molecular Biology 20:311–316.https://doi.org/10.1038/nsmb.2510
-
Mobilization of giant piggyBac transposons in the mouse genomeNucleic Acids Research 39:e148.https://doi.org/10.1093/nar/gkr764
-
The retrovirus HERVH is a long noncoding RNA required for human embryonic stem cell identityNature Structural & Molecular Biology 21:423–425.https://doi.org/10.1038/nsmb.2799
-
The nature of embryonic stem cellsAnnual Review of Cell and Developmental Biology 30:647–675.https://doi.org/10.1146/annurev-cellbio-100913-013116
-
Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo developmentDevelopment 99:371–382.
-
Naive and primed pluripotent statesCell Stem Cell 4:487–492.https://doi.org/10.1016/j.stem.2009.05.015
-
Genome engineering using the CRISPR-Cas9 systemNature Protocols 8:2281–2308.https://doi.org/10.1038/nprot.2013.143
Article and author information
Author details
Funding
Wellcome (Sir Henry Wellcome Postdoctoral Fellowship)
- Meng Amy Li
California Institute for Regenerative Medicine (Research Grant)
- Lin He
Wellcome (Research Grant)
- Austin Smith
Medical Research Council (Research Grant)
- Austin Smith
National Institute of General Medical Sciences (Research Grant)
- David L Spector
National Cancer Institute (Research Grant)
- David L Spector
- Lin He
Cancer Research UK (Research Grant)
- Tony Kouzarides
European Research Council (Research Grant)
- Tony Kouzarides
Biotechnology and Biological Sciences Research Council (Research Grant)
- Austin Smith
European Commission (Research Grant)
- Austin Smith
American Cancer Society (Research Scholar Award)
- Lin He
Tobacco-Related Disease Research Program (Research Grant)
- Lin He
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Kosuke Yusa and Graziano Martello for comments on the manuscript. We are grateful to Carla Mulas for assisting the miRNA expression plot, Yiping Zhang for lncRNA candidate prediction analysis and Rosalind Drummond for technical support. We thank Heather Lee for providing Dnmt3a and Dnmt3b siRNAs and Wolf Reik for support. We thank Nicholas Ingolia for useful discussion on lncRNA ribosomal footprinting. We also thank Peter Humphreys and Andy Riddell for technical support for imaging analysis and flow cytometry respectively. AS is supported by Medical Research Council (G1100526/1), Biotechnology and Biological Sciences Research Council (BB/M004023/1), European Commission (HEALTH-F4-2007-200720 EUROSYSTEM), and Wellcome Trust (091484/Z/10/Z). LH is supported by National Cancer Institute (R01 CA139067, 1R21CA175560-01) and California Institute of Regenerative Medicine (RN2-00923-1), American Cancer Society (123339-RSG-12-265-01-RMC), Tobacco-related Disease Research Program (21RT-0133). DLS is supported by NIGMS 42694 and NCI 5PO1CA013106-Project 3. TK is supported by programme grants from Cancer Research UK (C6/A18796) and European Research Council CRIPTON Grant (268569) and core grants from the Wellcome Trust (092096) and Cancer Research UK (C6946/A14492). The Cambridge Stem Cell Institute receives core funding from the Wellcome Trust and the Medical Research Council. MAL was a Siebel postdoctoral fellow at the University of California, Berkeley and a Sir Henry Wellcome postdoctoral fellow (096125/Z/11/Z). AS is a Medical Research Council Professor.
Copyright
© 2017, Li et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,829
- views
-
- 555
- downloads
-
- 43
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 43
- citations for umbrella DOI https://doi.org/10.7554/eLife.23468
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A lncRNA fine tunes the dynamics of a cell state transition involving Lin28, let-7 and de novo DNA methylation
eLife 6:e23468.
https://doi.org/10.7554/eLife.23468




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}