A unifying mechanism for the biogenesis of membrane proteins co-operatively integrated by the Sec and Tat pathways
- University of Dundee, United Kingdom
- John Innes Centre, United Kingdom
Figures
Figure 1 with 4 supplements
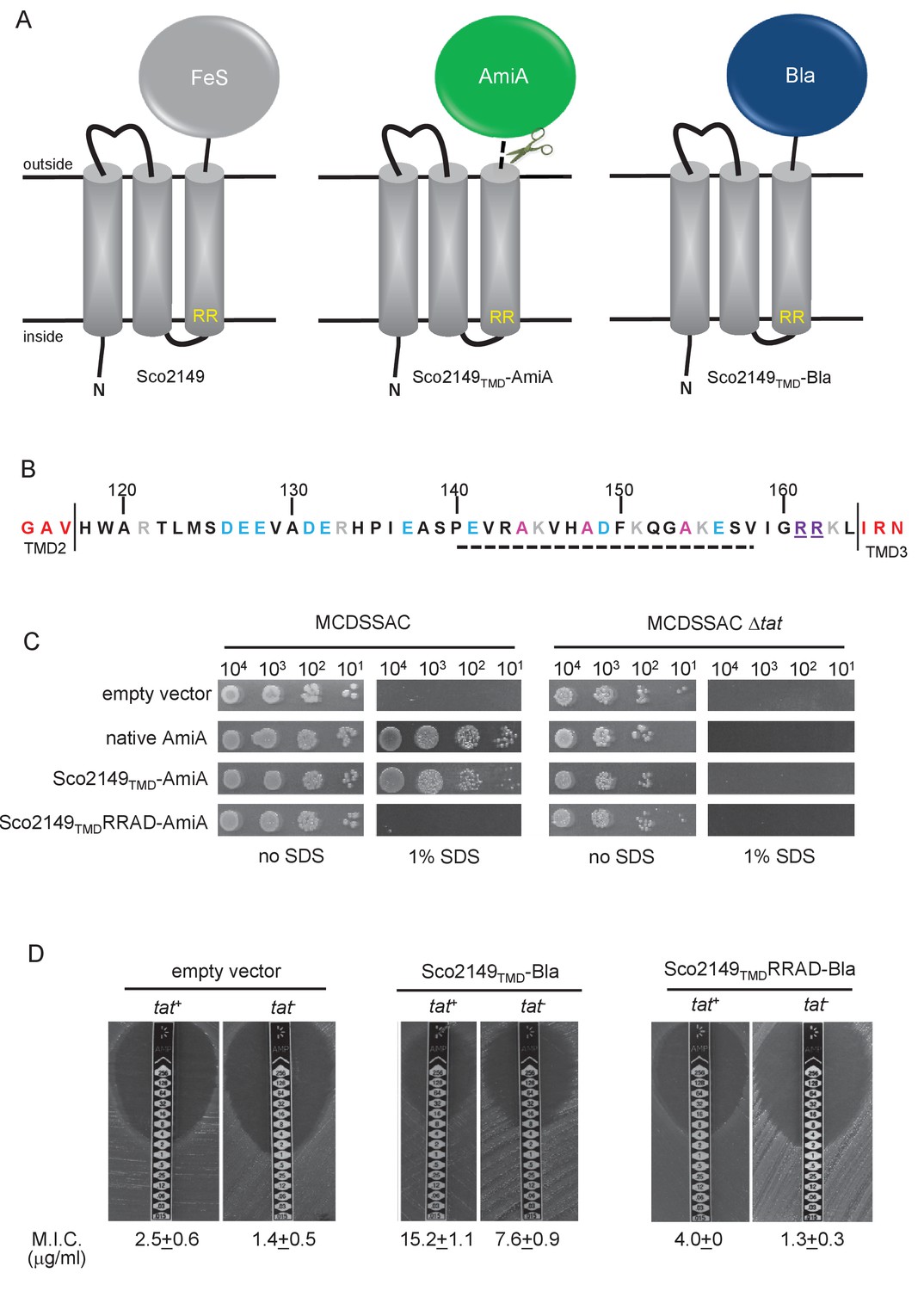
Sco2149TMD-reporter fusions to follow membrane insertion.
(A) Cartoon representations of the S. coelicolor Rieske protein, Sco2149, and the Sco2149TMD-AmiA and Sco2149TMD-Bla fusions. A signal peptidase I cleavage site (indicated by scissors) was introduced between the end of TMD3 and the AmiA sequence to allow release of AmiA from the membrane (Keller et al., 2012). The position of the twin-arginine motif is indicated by RR. (B) Sequence of the Sco2149 cytoplasmic loop region between TMDs 2 and 3. Amino acids predicted to be part of TMDs 2 and 3 are shown in red. The twin arginines of the Tat recognition motif are given in purple underline. Predicted α-helical secondary structure is shown with a dotted line, and alanine residues within this region that were mutated to proline are shown in pink. Negatively charged amino acids in the loop region are shown in blue, positively charged ones in grey. (C) E. coli strain MCDSSAC (which carries chromosomal deletions in the signal peptide coding regions of amiA and amiC) or an isogenic tatABC mutant containing either pSU-PROM (empty vector), or pSU-PROM producing native AmiA, Sco2149TMD-AmiA or a variant where the twin-arginines were substituted to AD (Sco2149TMDRRAD-AmiA), were spotted, after serial dilution, on LB medium in the absence or presence of 1% SDS. The plates were incubated for 20 hr at 37°C. (D) Representative images of M.I.C.Evaluator strip tests of strains MC4100 (tat+) and DADE (tat-) harbouring pSU-PROM (empty vector), pSU-PROM Sco2149TMD-Bla or pSU-PROM Sco2149TMDRRAD-Bla are shown. The mean M.I.C ± s.d. for strains harbouring these constructs is given at the bottom of each test strip (where n = 4 biological replicates for each strain harbouring the empty vector, n = 5 biological replicates for each strain harbouring pSU-PROM Sco2149TMD-Bla and n = 3 biological replicates for each strain harbouring pSU-PROM Sco2149TMDRRAD-Bla).
Figure 1—figure supplement 1
Sequence alignment of selected actinobacterial Rieske proteins.
(A) Sequences of the Rieske (QcrA) proteins from Streptomyces coelicolor (ScoQcrA), Mycobacterium tuberculosis (MtuQcrA) and Corynebacterium glutamicum (CglQcrA) were aligned using ClustalW (http://www.ch.embnet.org/software/ClustalW.html) and Boxshade (http://www.ch.embnet.org/software/BOX_form.html). Predicted positions of the TMDs, using the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016), are shown in blue. Positively charged amino acids immediately downstream of TMD3 are shown in orange. The consensus twin arginine (Tat) motif is boxed in red and conserved boxes I and II that are predicted coordinate the 2Fe-2S cluster are boxed in yellow. The positions after which reporter proteins were fused to the S. coelicolor or M. tuberculosis proteins are indicated. (B) Partial sequence alignment of the same three actinobacterial Rieske proteins with the single TMD Rieske proteins from Paracoccus denitrificans and Rhodobacter sphaeroides.
Figure 1—figure supplement 2

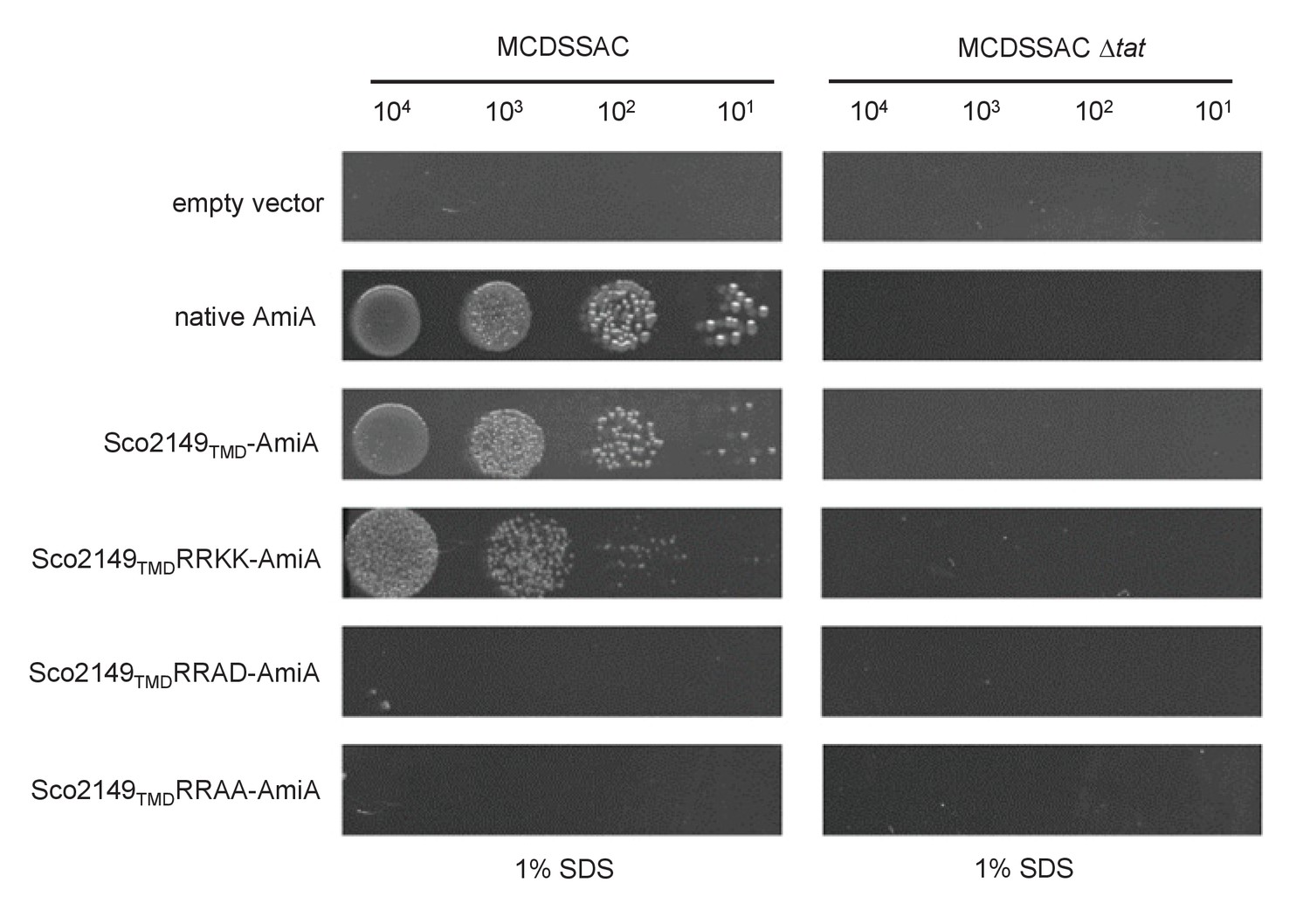
A twin lysine substitution of the twin arginine motif of Sco2149 still retains some interaction with the Tat pathway.
E. coli strains MCDSSAC or MCDSSAC △tatABC, as indicated containing either pSU-PROM (empty vector), or pSU-PROM producing native AmiA, Sco2149TMD-AmiA or variants where the twin-arginines were substituted to KK (Sco2149TMDRRKK-AmiA), AD (Sco2149TMDRRAD-AmiA) or AA (Sco2149TMDRRAA-AmiA) were spotted, after serial dilution, on LB medium in the absence or presence of 1% SDS. The plates were incubated for 20 hr at 37°C.
Figure 1—figure supplement 3
Sequence alignment TMD2/TMD3 loop region for a selection of actinobacterial Rieske proteins.
Sequences were aligned using ClustalW (http://www.ch.embnet.org/software/ClustalW.html) and Jalview (Cole et al., 2008). The approximate end position of TMD2 and start of TMD3 is indicated and the dotted black line indicates the position of predicted α-helical secondary structure determined using Jpred3 (http://www.compbio.dundee.ac.uk/www-jpred/index.html).
Figure 1—figure supplement 4

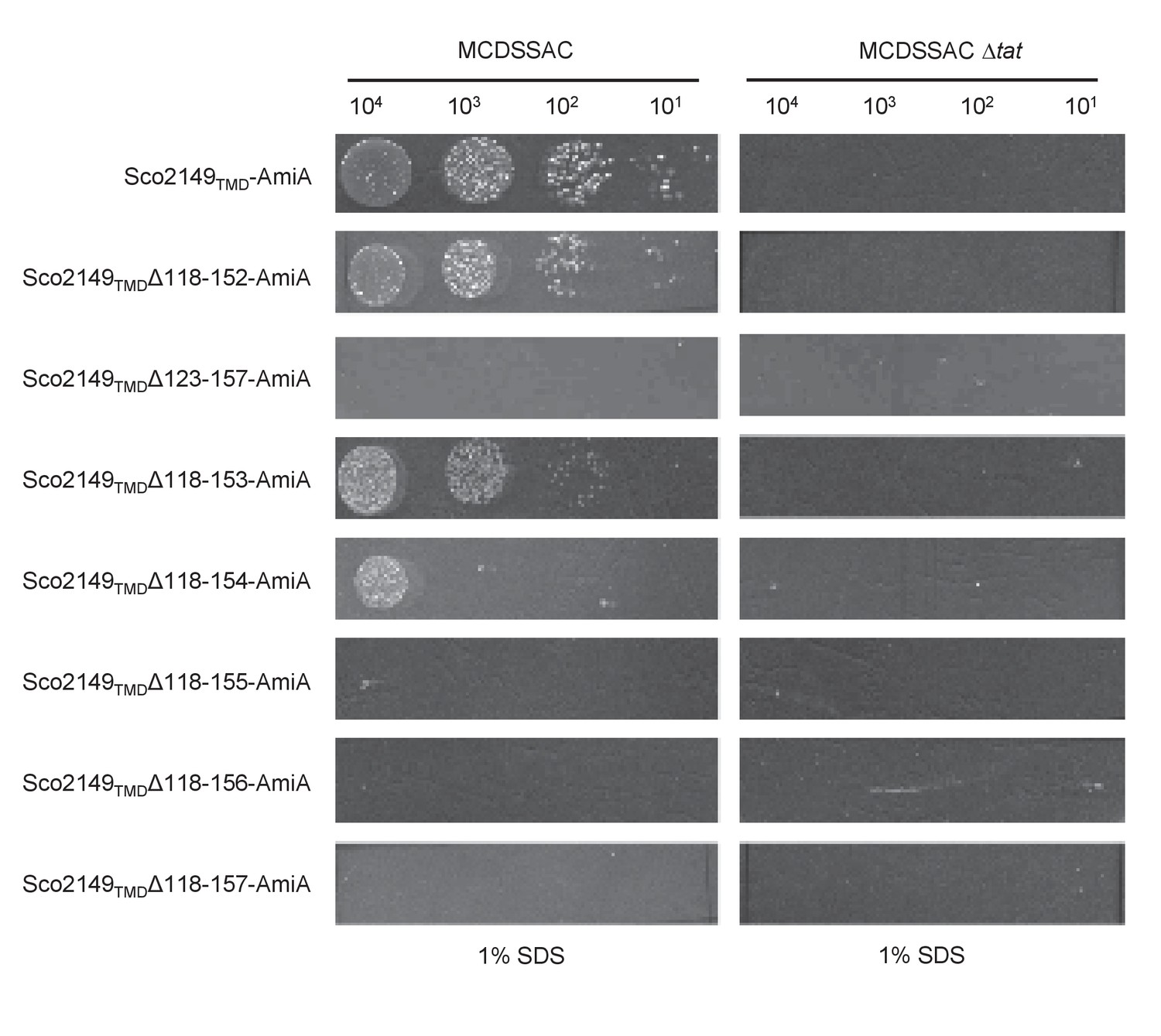
Effect of >35 residue truncations in the Sco2149 cytoplasmic loop region on the ability of Sco2149TMD-AmiA to support growth on SDS.
E. coli strains MCDSSAC or MCDSSAC △tatABC, as indicated containing pSU-PROM producing Sco2149TMD-AmiA or variants with 35 or more residues removed (Sco2149TMDΔ118–152-AmiA, Sco2149TMDΔ123–157-AmiA, Sco2149TMDΔ118–153-AmiA, Sco2149TMDΔ118–154-AmiA, Sco2149TMDΔ118–155-AmiA, Sco2149TMDΔ118–156-AmiA and Sco2149TMDΔ118–157-AmiA) were spotted after serial dilution on LB medium in absence or presence of 1% SDS. The plates were incubated for 20 hr at 37°C.
Figure 2
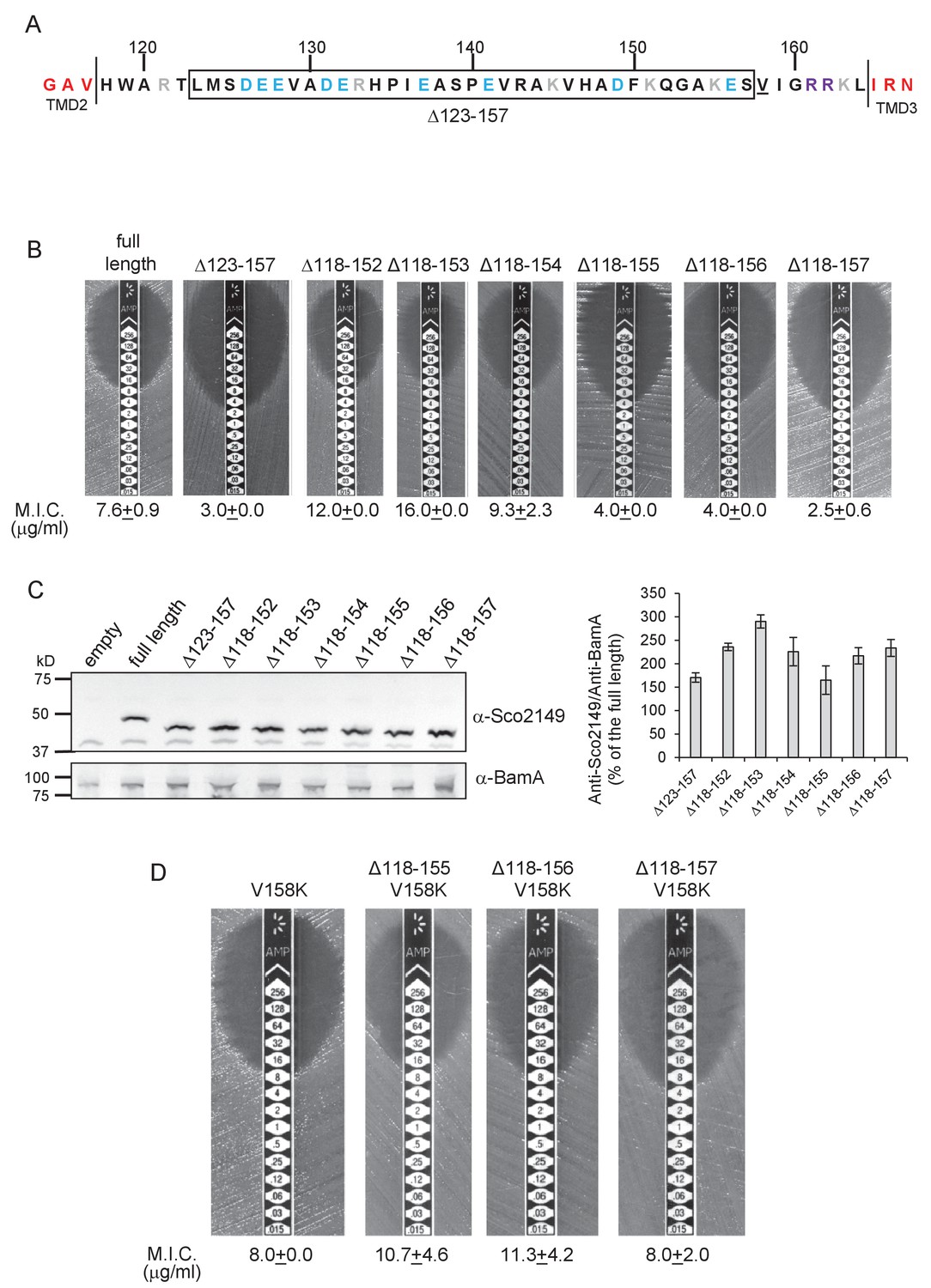
Interaction of Sco2149TMD-Bla with the Sec pathway.
(A) The Sco2149 cytoplasmic loop region between TMDs 2 and 3. Color-coding is as described in Figure 1. The extent of the 123–157 deletion is shown boxed and V158 that was substituted to K in this study is underlined. (B) and (D) Representative images of M.I.C.Evaluator strip tests of strain DADE (tat-) harbouring pSU-PROM producing the indicated variants of Sco2149TMD-Bla. The mean M.I.C ± s.d. is given at the bottom of each test strip (where n = 3 biological replicates for each strain). (C) Membrane extracts prepared from the same strains used in (B) along with DADE harboring the empty plasmid vector as a negative control, were separated by SDS-PAGE (12% acrylamide), transferred to nitrocellulose membrane and probed with anti-Sco2149 or anti-BamA (an unrelated outer membrane protein was used as a loading control). To the right, the Sco2149-associated signal was quantified and normalised against the BamA signal for each sample. The quantification results were expressed as percentage of the normalised signal obtained for the full length fusion (which was set at 100%). The results represent mean ± s.e.m. of three biological replicates, a representative blot is shown.
-
Figure 2—source data 1
Images of Sco2149 and BamA western blots used for quantification in Figure 2C.
- https://doi.org/10.7554/eLife.26577.012
-
Figure 2—source data 2
Quantification of density associated with Sco2149 and BamA signals from western blots from Figure 2—source data 1 used to generate graph in Figure 2C.
- https://doi.org/10.7554/eLife.26577.013
Figure 3
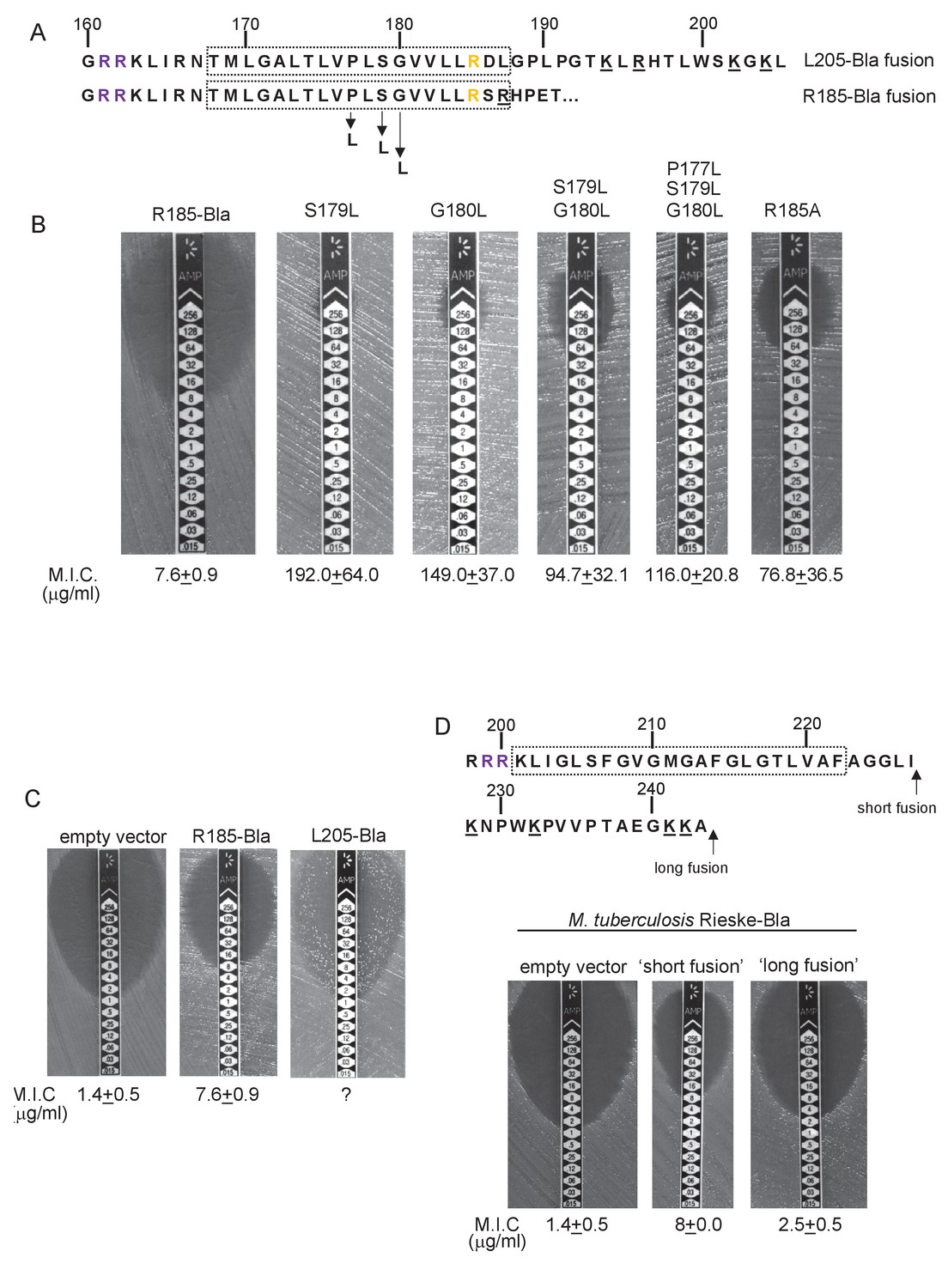
Hydrophobicity of TMD3 and C-terminal positive charges modulate interaction with the Sec pathway.
(A) Sco2149 TMD3 and flanking sequences. Top shows the native Sco2149 sequence up to amino acid 205 (position of the L205-Bla fusion) and below the sequence of the R185 Sco2149-Bla fusion. In each case the predicted position of TMD3 was determined using the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016) and is shown boxed. The twin arginines are shown in purple and R185 (the position after which Bla was fused in the R185 construct) is shown in yellow. Positively charged amino acids C-terminal to R185 are underlined. (B–D) Representative images of M.I.C.Evaluator strip tests of strain DADE (tat-) harbouring pSU-PROM producing (B) the indicated variants of the R185 Sco2149TMD-Bla fusion or (C) the R185 or L205 Sco2149TMD-Bla fusions, as indicated, or (D) M. tuberculosis QcrA fused to Bla. In (D) the amino acid sequence around TMD3 of M. tuberculosis QcrA is shown, with TMD3 boxed. ‘short fusion’ refers to a Bla fusion after I227 and ‘long fusion’ to a Bla fusion after A243. In each panel the mean M.I.C ± s.d. is given at the bottom of each test strip (where n = 3 biological replicates for each strain).
-
Figure 3—source data 1
Images of M. I.C.Evaluator strip tests used to generate mean M.I.C. values in Figure 3.
- https://doi.org/10.7554/eLife.26577.016
Figure 4

Further families of Tat-dependent polytopic membrane proteins.
(A) Schematic representation of a polytopic predicted molybdenum cofactor (MoCo) binding protein (Sco3746, left) a polytopic polyferredoxin (PFD, centre) and a polytopic metallophosphoesterase of the YkuE family (right) identified bioinformatically as candidate dual-inserted membrane poteins. The twin arginines of the Tat recognition sequence are highlighted in yellow. Four histidines in the TMDs of Sco3746 and homologues that are predicted to ligate two b hemes are shown in red. Note that three of these histidines are conserved (Figure 5) but the one at the N-terminal end of TMD3 (H?) is not. (B) Fusions of the TMDs of Sco3746 and of Q1NSB0 (PFD) to maltose binding protein (MBP) or AmiA are shown as cartoons. As before, a signal peptidase I cleavage site (indicated by scissors) was introduced at the end of predicted TMD5 to allow release of AmiA from the membrane (Keller et al., 2012). (C) E. coli tat+ strain HS3018-A (that lacks chromosomally-encoded MBP) harboring pSU18 (empty vector), or the same vector producing native MBP, Sco3746TMD-MBP, PFDTMD-MBP or the twin-arginine substituted variants Sco3746TMDRRKK-MBP and PFDTMDRRKK-MBP, as indicated, was cultured overnight, resuspended in minimal medium containing 1% Maltose and 0.002% Bromocresol purple. Cells were diluted either 2.5 fold (left hand panel) or 10 fold (right hand panel) in the same medium and incubated without shaking at 37°C for 24 hr (left hand panel) or 48 hr (right hand panel). (D) E. coli strains MCDSSAC or an isogenic tatABC mutant harboring pSU18 (empty vector), pSU18 producing native AmiA, or pSU18 producing either Sco3746TMD-AmiA or PFDTMD-AmiA fusion proteins, or variants of these where the twin-arginine motif was substituted to twin lysine (Sco3746TMDRRKK-AmiA/PFDTMDRRKK-AmiA) were serially diluted and spotted onto LB or LB containing 2% SDS. The plates were incubated for 20 hr at 37°C.
Figure 5

Sequence alignment of selected polytopic MoCo-binding proteins.
Sequences of polytopic predicted MoCo-binding proteins from the indicated prokaryotes were aligned using ClustalW (http://www.ch.embnet.org/software/ClustalW.html) and Boxshade (http://www.ch.embnet.org/software/BOX_form.html). Predicted positions of the TMDs, using the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016), are shown in blue. Positively charged amino acids immediately downstream of TMD5 are shown in orange. The consensus twin arginine (Tat) motif is boxed in red. Histidine residues that may co-ordinate heme b are shown in red font. The positions after which Bla was fused to the S. coelicolor protein are indicated.
Figure 6
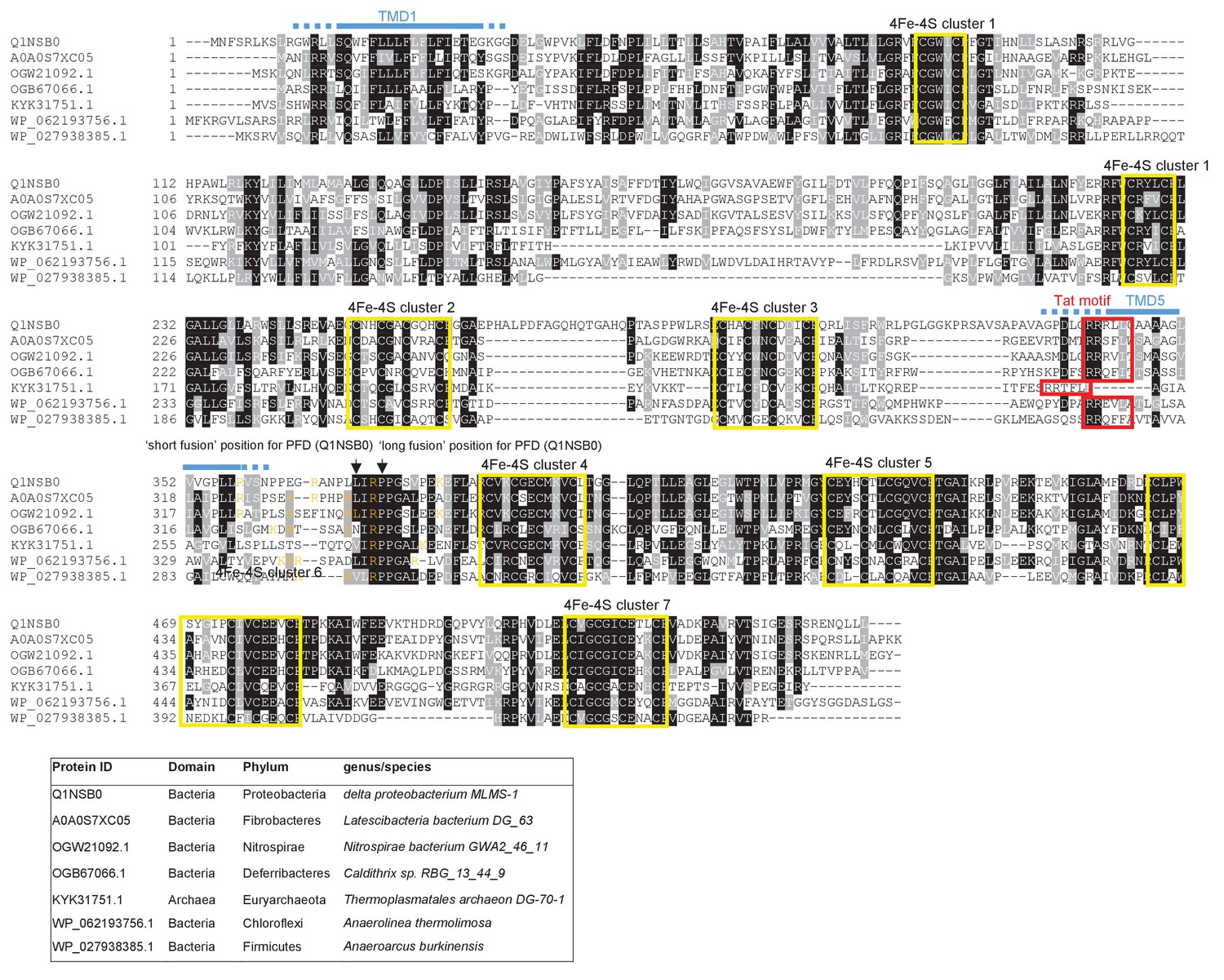
Sequence alignment of selected polytopic polyferredoxin proteins.
Sequences of polytopic predicted polyferredoxin proteins from the indicated prokaryotes were aligned using ClustalW (http://www.ch.embnet.org/software/ClustalW.html) and Boxshade (http://www.ch.embnet.org/software/BOX_form.html). Predicted positions of TMD1 and 5 (using the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016)) are shown in blue. Positively charged amino acids immediately downstream of TMD5 are shown in orange. The consensus twin arginine motif is boxed in red and cysteine-rich regions that are predicted coordinate 4Fe-2S cluster are boxed in yellow. The positions after which Bla was fused to the delta proteobacterium MLMS-1 protein are indicated.
Figure 7

Sequence alignment of selected YkuE-related proteins.
Sequences of polytopic YkuE-like metallophosphoesterase proteins from the indicated prokaryotes were aligned, alongside the shorter homologues from E. coli and B. subtilis using ClustalW (http://www.ch.embnet.org/software/ClustalW.html) and Boxshade (http://www.ch.embnet.org/software/BOX_form.html). Predicted positions of the TMDs (using the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016)) are shown in blue. Note that residues in predicted TMD2 and TMD3 are not well aligned across the homologues and therefore amino acids predicted to be in TMD2 and TMD3 for each protein are individually marked in blue font. Positively charged amino acids immediately downstream of TMD5 are shown in orange. The consensus twin arginine motif is boxed in red and amino acids predicted to coordinate the metal ion cofactor are shown in red font.
Figure 8
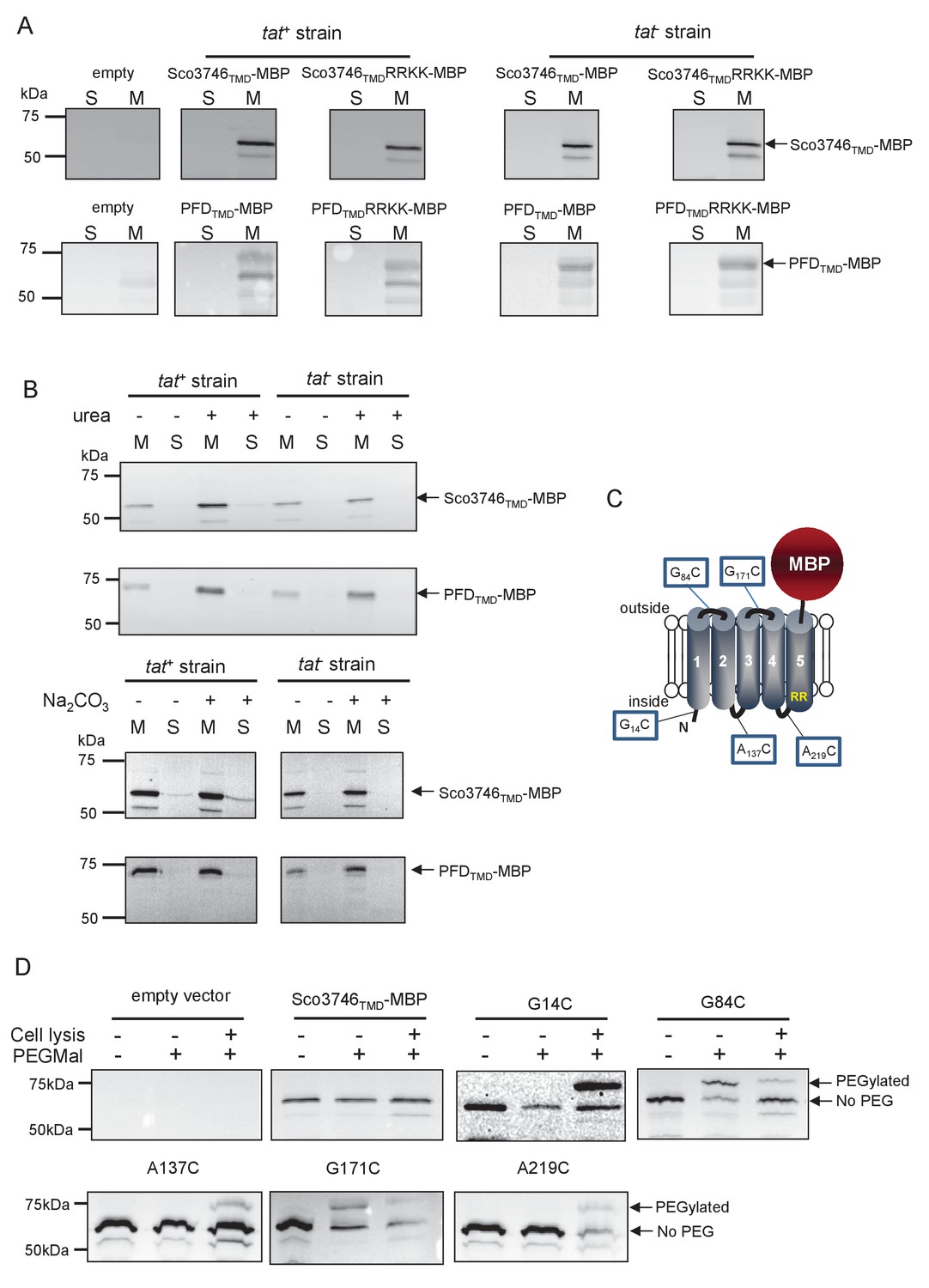
Topological analysis and membrane integration of Sco3746TMD-MBP and PFDTMD-MBP.
(A) Membrane (M; 100 µg protein) and soluble (S; 50 µg protein) fractions of E. coli HS3018-A (△malE, tat+) and HS3018-A△tat strains harboring pSU18 (empty vector), pSU18 encoding Sco3746TMD-MBP or PFDTMD-MBP fusion proteins, or variants of these where the twin-arginine motif was substituted to twin-lysine were separated by SDS-PAGE (12% acrylamide), transferred to nitrocellulose membrane and immunoblotted with an anti-MBP antibody. (B) Crude membranes of the same strains and plasmids were treated with 4M urea or 0.2M carbonate, and the presence of the fusion proteins in the wash supernatant (S) and pelleted membrane (M) was analyzed by immunoblotting as in (A). (C) Predicted locations of cysteine substitutions of Sco3746TMD-MBP used for topology analysis. (D) Cell suspensions of strain HS3018-A harboring pSU18 alone (empty vector), or pSU18 encoding Sco3746TMD-MBP or the indicated single cysteine substitutions of Sco3746TMD-MBP were incubated with buffer alone, with 5 mM MAL-PEG, or were lysed by sonication and incubated with 5 mM MAL-PEG. Subsequently all samples were quenched, lysed and membranes pelleted by ultracentrifugation. Membrane samples (150 µg of protein) were separated by SDS PAGE and immunblotted as in (A).
Figure 9

Relative hydrophobicity of TMD5 coupled with C-terminal positive charges modulate interaction of Sco3746 with the Sec pathway.
(A) The sequence flanking TMD5 of Sco3746. The lower sequence extends to the position of the ‘short’ Sco3746-Bla fusion, and the amino acids in TMD5 substituted for leucine in this construct are shown. Top is the sequence fused to Bla in the ‘long fusion’. The predicted position of TMD5 was determined using the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016) and is shown boxed. The twin arginines are shown in purple and positively charged amino acids C-terminal to TMD5 are underlined. (B) Representative images of M.I.C.Evaluator strip tests of strain DADE (tat-) harbouring pSU18 producing the indicated variants of Sco3746TMD-Bla In each panel the mean M.I.C ± s.d. is given at the bottom of each test strip (where n = 3 biological replicates for each strain).
-
Figure 9—source data 1
Images of M. I.C.Evaluator strip tests used to generate mean M.I.C. values in Figure 9B.
- https://doi.org/10.7554/eLife.26577.024
Figure 10
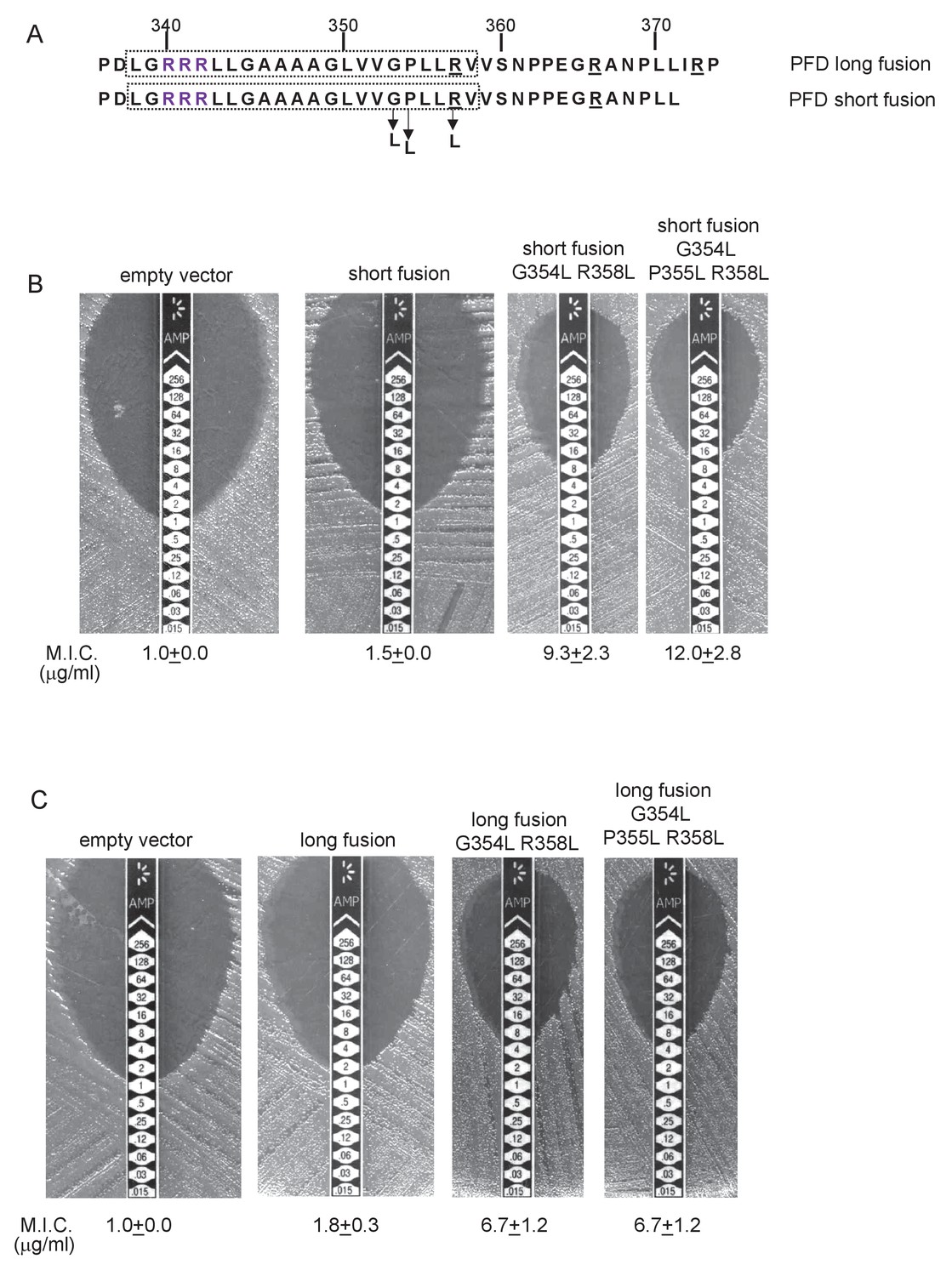
Relative hydrophobicity of MLMS-1 PFD TMD5 coupled with C-terminal positive charges governs interaction with the Sec pathway.
(A) The sequence flanking TMD5 of MLMS-1 PFD. The lower sequence (corresponding to ‘short fusion’ in parts B and C) extends to the position of the shorter PFD-Bla fusion, whereas the top sequence is the sequence fused to Bla in the ‘long fusion’. The predicted position of TMD5 was determined using the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016) and is shown boxed. The twin arginines are shown in purple and positively charged amino acids C-terminal to TMD5 are underlined. Amino acids in TMD5 substituted for leucine in both constructs are shown. (B and C) Representative images of M.I.C.Evaluator strip tests of strain DADE (tat-) harbouring pSU18 producing the indicated variants of PFDTMD-Bla In each panel the mean M.I.C ± s.d. is given at the bottom of each test strip (where n = 3 biological replicates for each strain).
-
Figure 10—source data 1
Images of M. I.C.Evaluator strip tests used to generate mean M.I.C. values in Figure 10B and C.
- https://doi.org/10.7554/eLife.26577.027
Figure 11
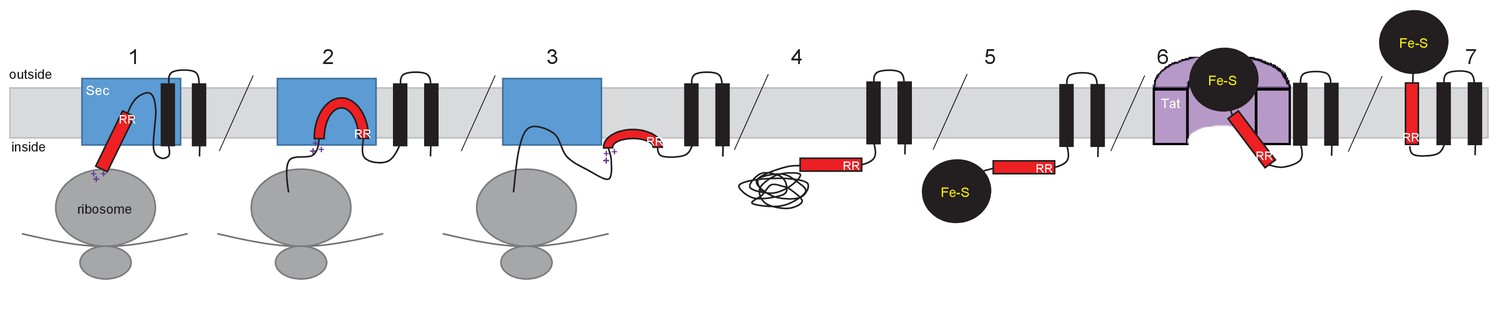
Model for actinobacterial Rieske protein assembly.
1. TMDs 1 and 2 are inserted into the membrane cotranslationally by the Sec machinery (blue box). The Sec machinery interacts with TMD3 in an N-in, C-out orientation. 2. The positive charges at the C-terminal end of TMD3 force an orientational preference on the helix and it is not inserted by the Sec machinery. 3. The hydrophobic segment of TMD3 is released from the Sec machinery as a re-entrant loop. As there are no further TMDs within the Riekse sequence the Sec machinery releases the polypeptide. 4 and 5. Translation is completed and the iron-sulfur cluster is inserted into the protein. 6. The assembled Tat machinery (pink half-cylinder) interacts with TMD3 to translocate the folded globular domain across the membrane. 7. The fully assembled Rieske protein is released into the membrane to interact with partner proteins.
Tables
Table 1
Effect of amino acid substitutions, small deletions and insertions in the Sco2149 cytoplasmic loop region on the ability of Sco2149TMD-AmiA and Sco2149TMD-Bla to support growth on SDS or ampicillin, respectively. Note that growth on ampicillin was scored using the tat- strain DADE and therefore assesses Sec translocation only. Y indicates growth on 1% SDS, N indicates no growth, nd – not determined. Mean M.I.C for growth on ampicillin is given in µg/ml + one standard deviation, n = at least 3. *Insertion of 3 additional amino acids, DEE between E128 and V129.
| Variant | Growth on 1% SDS (Tat translocation) | Mean M.I.C. for ampicillin (Sec translocation) |
|---|---|---|
| wild type | Y | 7.6 ± 0.9 |
| R161K R162K | Y | 3.8 ± 0.5 |
| R161A R162D | N | 1.3 ± 0.3 |
| R161A R162A | N | 5.5 ± 1.0 |
| R161K R162Q | N | 3.3 ± 0.6 |
| ∆R161 ∆R162 | N | 9.3 ± 2.3 |
| R133H H134R | Y | 7.3 ± 1.2 |
| R133K H134K | Y | 7.0 ± 2.0 |
| M124L | Y | 6.0 ± 2.0 |
| M124A | Y | 6.0 ± 2.0 |
| S125L | Y | 6.7 ± 2.3 |
| S125A | Y | 8.0 ± 0.0 |
| D126L | Y | 7.3 ± 1.2 |
| D126A | Y | 7.3 ± 1.2 |
| E127L | Y | 6.0 ± 2.0 |
| E127A | Y | 8.0 ± 2.8 |
| A144P | Y | 6.7 ± 1.2 |
| A148P | Y | 8.0 ± 2.8 |
| A154P | Y | 8.0 ± 0.0 |
| ∆126–8 | nd | 8.0 ± 0.0 |
| ∆126–127 | nd | 7.5 ± 1.0 |
| ∆127–128 | nd | 8.0 ± 2.8 |
| 131–2 | nd | 7.3 ± 1.2 |
| ∆137∆141 | nd | 4.0 ± 0.0 |
| ∆131–2∆141 | nd | 6.0 ± 2.0 |
| Δ149Δ156 | nd | 8.0 ± 0.0 |
| ∆126–8 ∆137∆141 | nd | 8.0 ± 0.0 |
| ∆126–8∆131–2 | nd | 8.0 ± 2.4 |
| ∆126–8∆131–2 ∆137∆141 | nd | 10.4 ± 2.2 |
| ∆126–8∆131–2 ∆137 ∆141 Δ149 Δ156 | nd | 10.0 ± 2.8 |
| Ins D129 E130 E131* | nd | 6.7 ± 1.2 |
| D131A E132A | nd | 6.0 ± 2.3 |
| E137A E141A | nd | 6.7 ± 2.3 |
| D126A E127A E128A | nd | 7.2 ± 1.1 |
| D131K E132K | nd | 7.3 ± 1.2 |
| E137K E141K | nd | 6.5 ± 1.9 |
| D126K E127K E128K | nd | 9.0 ± 2.0 |
| D126K E127K E128K E137K E141K | nd | 8.0 ± 0.0 |
| D126K E127K E128K D131K E132K E137K E141K | nd | 10.0 ± 2.2 |
-
Table 1—source data 1
Images of SDS growth tests and M.I.C.Evaluator strip tests of all strain and plasmid combinations used in Table 1.
- https://doi.org/10.7554/eLife.26577.008
Table 2
Effect of amino acid truncation in the Sco2149 cytoplasmic loop region on the ability of Sco2149TMD-AmiA and Sco2149TMD-Bla to support growth on SDS or ampicillin, respectively. Note that growth on ampicillin was scored using the tat- strain DADE and therefore assesses Sec translocation only. Y indicates growth on 1% SDS, N indicates no growth. Mean M.I.C for growth on ampicillin is given in µg/ml + one standard deviation, n = at least 3.
| Variant | Growth on 1% SDS (Tat translocation) | Mean M.I.C. for ampicillin (Sec translocation) |
|---|---|---|
| wild type | Y | 7.6 ± 0.9 |
| 5 residue truncations | ||
| ∆118–122 | Y | 10.0 ± 2.3 |
| ∆123–127 | Y | 5.3 ± 2.3 |
| ∆128–132 | Y | 12.0 ± 0.0 |
| ∆133–137 | Y | 6.7 ± 2.3 |
| ∆138–142 | Y | 7.0 ± 2.0 |
| ∆143–147 | Y | 6.7 ± 2.3 |
| ∆148–152 | Y | 10.0 ± 3.3 |
| ∆153–157 | Y | 9.6 ± 3.6 |
| 10 residue truncations | ||
| ∆118–127 | Y | 6.0 ± 0.0 |
| ∆128–137 | Y | 9.5 ± 3.0 |
| ∆138–147 | Y | 8.0 ± 0.0 |
| ∆148–157 | Y | 9.3 ± 2.3 |
| 15 residue truncations | ||
| ∆118–132 | Y | 9.0 ± 3.8 |
| ∆123–137 | Y | 8.0 ± 0.0 |
| ∆128–142 | Y | 10.0 ± 2.3 |
| ∆133–147 | Y | 9.3 ± 2.3 |
| ∆138–152 | Y | 5.0 ± 1.4 |
| ∆143–157 | Y | 4.8 ± 1.5 |
| 20 residue truncations | ||
| ∆118–137 | Y | 7.3 ± 1.2 |
| ∆138–157 | Y | 5.5 ± 1.9 |
| 25 residue truncations | ||
| ∆118–142 | Y | 12.0 ± 0.0 |
| ∆123–147 | Y | 7.6 ± 0.9 |
| ∆128–152 | Y | 8.0 ± 0.0 |
| ∆133–157 | Y | 6.0 ± 0.0 |
| 30 residue truncations | ||
| ∆118–147 | Y | 10.0 ± 2.8 |
| ∆123–152 | Y | 13.6 ± 2.2 |
| ∆128–157 | Y | 7.3 ± 1.2 |
| >35 residue truncations | ||
| ∆118–152 | Y | 12.0 ± 0.0 |
| ∆123–157 | N | 3.0 ± 0.0 |
| ∆118–153 | Y | 16.0 ± 0.0 |
| ∆118–154 | Y/N | 9.3 ± 2.3 |
| ∆118–155 | N | 4.0 ± 0.0 |
| ∆118–156 | N | 4.0 ± 0.0 |
| ∆118–157 | N | 2.5 ± 0.6 |
-
Table 2—source data 1
Images of SDS growth tests and M.I.C.Evaluator strip tests of all strain and plasmid combinations used in Table 2.
- https://doi.org/10.7554/eLife.26577.010
Table 3
Predicted △Gapp values (in kcal mol−1) for membrane insertion of each of the three TMDs of the indicated Rieske proteins. Sequences were analysed using the △Gapp prediction server (http://dgpred.cbr.su.se/) that are based on hydrophobicity scales generated from (Hessa et al., 2005, 2007). This server uses the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016) to predict the positions of the TMDs and for S. coelicolor Rieske predicts TMD1 to span aa 58–80, TMD two to span aa 96–117 and TMD3 to span aa 168–187.
| Predicted △Gapp | |||||
|---|---|---|---|---|---|
| Family/Species | Uniprot ID | TM1 | TM2 | TM3 | TM3/Bla fusion* |
| Mycobacterium tuberculosis | P9WH23 | −4.252 | −0.715 | 0.248 | |
| Corynebacterium glutamicum | Q79VE8 | −2.873 | −0.761 | 0.564 | |
| Gordonia malaquae | M3VAA9 | −2.569 | −0.214 | 1.291 | |
| Corynebacterium diphtheriae | Q6NGA2 | −2.402 | −1.089 | 0.619 | |
| Dietzia cinnamea | E6JC04 | −2.997 | −0.391 | 0.409 | |
| Salinispora tropica | A4 × 9Y7 | −2.315 | −1.205 | 1.162 | |
| Streptomyces sp. | D9VGG2 | −1.291 | −1.203 | 0.967 | |
| Verrucosispora maris | F4F1U1 | −2.248 | −1.946 | 1.162 | |
| Stackebrandtia nassauensis | D3Q1119 | −1.876 | −1.289 | 0.556 | |
| Rhodococcus erythropolis | C3JJ95 | −2.692 | −0.208 | 0.912 | |
| Streptomyces coelicolor | Q9 × 807 | −2.374 | −0.117 | 0.614 | 0.714 |
| S. coelicolor S179L | −0.389 | −0.205 | |||
| S. coelicolor G180L | −0.037 | 0.044 | |||
| S. coelicolor S179L, G180L | −1.117 | −0.987 | |||
| S. coelicolor P177L, S179L, G180L | −2.510 | −2.409 | |||
| S. coelicolor R185A | 0.517 | 0.620 | |||
-
*Bla is fused to S. coelicolor Rieske after amino acid 185 (full sequence of all of the fusion proteins used in this study can be found in Supplementary file 1D).
Table 4
Predicted △Gapp values (in kcal mol−1) for membrane insertion of each of the five TMDs of the indicated predicted MoCo-binding proteins. Sequences were analysed using the △Gapp prediction server (http://dgpred.cbr.su.se/) that are based on hydrophobicity scales generated from (Hessa et al., 2005, 2007). This server uses the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016) to predict the positions of the TMDs and for S. coelicolor Q9L0V6 (Sco3746) predicts TMD1 to span aa 39–61, TMD2 to span aa 99–121, TMD3 to span aa 139–161, TMD four to span aa 173–194 and TMD5 to span aa 223–242.
| Predicted △Gapp | ||||||
|---|---|---|---|---|---|---|
| Family/Species | Protein ID | TM1 | TM2 | Tm3† | Tm4† | TM5* |
| Phycicoccus sp. | WP_056916643.1 | −1.666 | −0.199 | −1.593 | 1.595 | 0.964 |
| Solirubrobacter soli | WP_053225688.1 | −1.778 | 1.394 | −2.239 | 1.681 | 1.135 |
| Alicyclobacillus acidocaldarius | C8WRE4 | −0.540 | −1.802 | −1.128 | 0.143 | 0.231 |
| Ktedonobacter sp. | OLB54713.1 | −0.479 | −2.647 | −2.375 | −1.257 | 1.762 |
| Haloprofundus marisrubri | WP_058581003.1 | 1.067 | −1.424 | −0.421 | −2.107 | 1.299 |
| Streptomyces coelicolor | Q9L0V6 | −2.268 | −0.011 | 0.328 | 0.512 | 1.376 |
| S. coelicolor G234L, S235L | −0.757 | |||||
| S. coelicolor G234L, S235L, M239L, F2440L | −1.240 | |||||
-
*Bla is fused to S. coelicolor Sco3746 (Q9L0V6) after amino acid 247 after amino acid 252 (full sequence of all of the fusion proteins used in this study can be found in Supplementary file 1D).
-
†Positive values for ΔGapp values noted for some internal TMDs. These marginally hydrophobic TMDs are, however, still likely to be integrated by the Sec pathway. Many individual TMD in multi-spanning membrane proteins have an unfavourable free energy of membrane insertion and are unable to stably integrate by themselves, requiring TMD sequence-extrinsic features for membrane insertion. It is, however, usual for the first and last TMD to be more hydrophobic as they lack these sequence-extrinsic features (Hedin et al., 2010; Virkki et al., 2014; Elofsson and von Heijne, 2007; White and von Heijne, 2008b).
Table 5
Predicted △Gapp values (in kcal mol−1) for membrane insertion of the first and last TMDs of the indicated predicted polyferredoxin proteins. Sequences were analysed using the △Gapp prediction server (http://dgpred.cbr.su.se/) that are based on hydrophobicity scales generated from (Hessa et al., 2005, 2007). This server uses the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016) to predict the positions of the TMDs and for delta proteobacterium MLMS-1 Q1NSB0 (PFD) predicts TMD1 to span aa 9–31 and TMD5 to span aa 338–359.
| Predicted △Gapp | |||
|---|---|---|---|
| Family/Species | Protein ID | TM1 | TM5* |
| Latescibacteria bacterium DG_63 | A0A0S7XC05 | −1.792 | 0.371 |
| Nitrospirae bacterium GWA2_46_11 | OGW21092.1 | −1.062 | 1.222 |
| Caldithrix sp. RBG_13_44_9 | OGB67066.1 | −3.560 | 1.438 |
| Thermoplasmatales archaeon DG-70–1 | KYK31751.1 | −1.474 | 1.715 |
| Anaerolinea thermolimosa | WP_062193756.1 | −2.317 | 1.107 |
| Anaeroarcus burkinensis | WP_027938385.1 | −0.975 | 0.836 |
| delta proteobacterium MLMS-1 | Q1NSB0 | −2.382 | 0.297 |
| delta proteobacterium MLMS-1 G354L, R358L | −0.701 | ||
| delta proteobacterium MLMS-1 G354L, P355L, R358L | −1.741 | ||
-
*Bla is fused to delta proteobacterium MLMS-1 PFD (Q1NSB0) after amino acid 364 (full sequence of all of the fusion proteins used in this study can be found in Supplementary file 1D).
Table 6
Predicted △Gapp values (in kcal mol−1) for membrane insertion of each of the four TMDs of the indicated metallophosphoesterase (YkuE) proteins. Sequences were analysed using the △Gapp prediction server (http://dgpred.cbr.su.se/) that are based on hydrophobicity scales generated from (Hessa et al., 2005, 2007). This server uses the SCAMPI2/TOPCONS servers (Tsirigos et al., 2015, 2016) to predict the positions of the TMDs.
| Predicted △Gapp | |||||
|---|---|---|---|---|---|
| Family/Species | Protein ID | TM1 | TM2 | Tm3† | TM4 |
| Capnocytophaga canimorsus | F9YSF9 | −1.595 | −1.327 | −1.266 | 0.957 |
| Desulfitibacter alkalitolerans | WP_028307559.1 | −1.409 | −2.217 | −2.075 | 1.087 |
| Nitrospirae bacterium RBG_16_64_22 | A0A1G1HRI1 | −1.326 | −0.017 | 0.628 | 1.579 |
| Gemmatimonadetes bacterium 13_2_20 CM_69_27 | A0A1Q6W294 | −1.397 | −0.016 | −0.881 | 1.282 |
-
†Positive values for ΔGapp value noted internal TMD. This marginally hydrophobic TMD is, however, still likely to be integrated by the Sec pathway. Many individual TMD in multi-spanning membrane proteins have an unfavourable free energy of membrane insertion and are unable to stably integrate by themselves, requiring TMD sequence-extrinsic features for membrane insertion. It is, however, usual for the first and last TMD to be more hydrophobic as they lack these sequence-extrinsic features (Hedin et al., 2010; Virkki et al., 2014; Elofsson and von Heijne, 2007; White and von Heijne, 2008b).
Additional files
-
Supplementary file 1
Bacterial strains, plasmids and oligonucleotides used in this study, and full amino acid sequences of the fusion proteins.
- https://doi.org/10.7554/eLife.26577.030
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A unifying mechanism for the biogenesis of membrane proteins co-operatively integrated by the Sec and Tat pathways
eLife 6:e26577.
https://doi.org/10.7554/eLife.26577




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}