A map of human PRDM9 binding provides evidence for novel behaviors of PRDM9 and other zinc-finger proteins in meiosis
- University of Oxford, United Kingdom
- École Polytechnique Fédérale de Lausanne, Switzerland
Figures
Figure 1 with 4 supplements
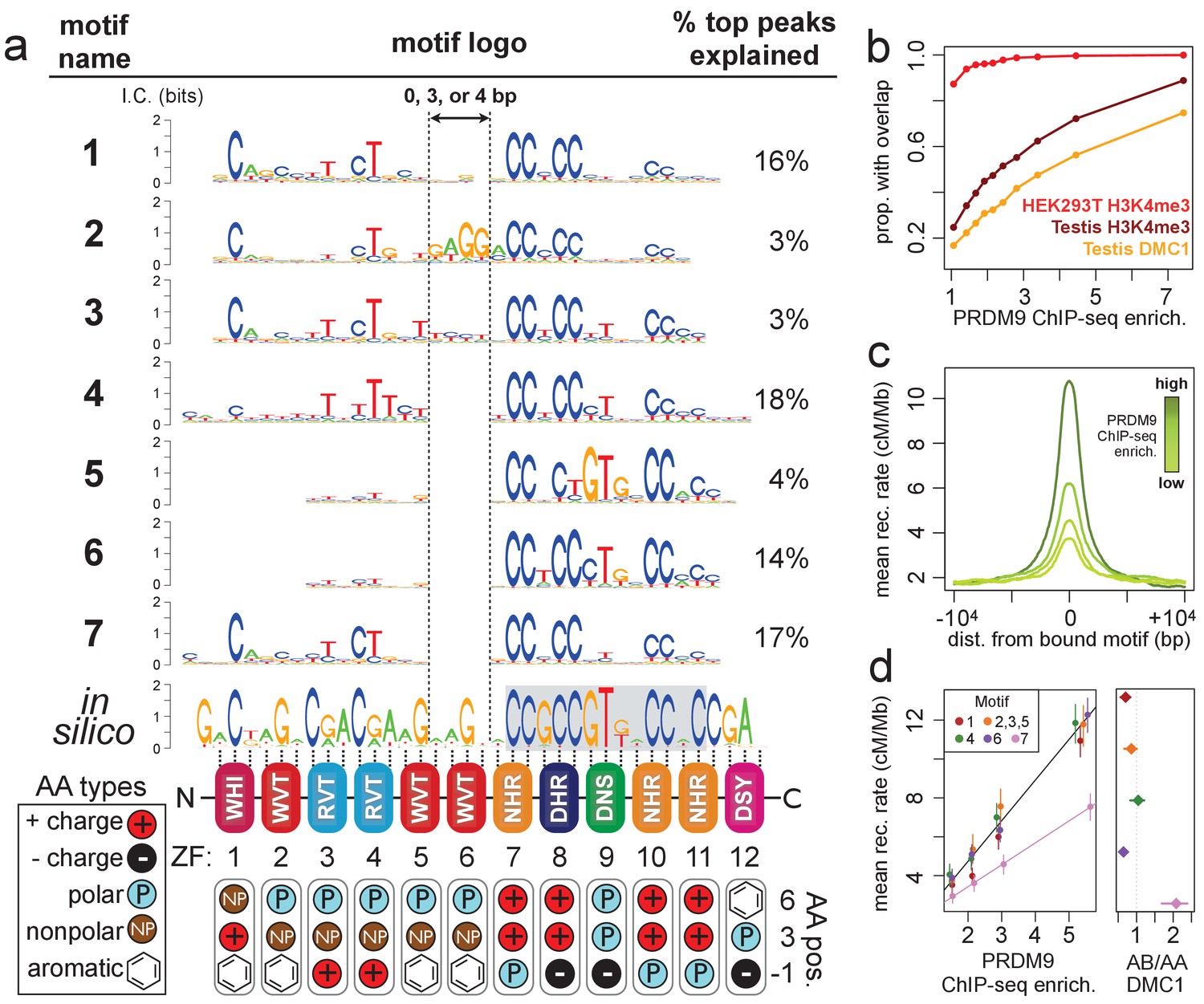
Comparison of seven distinct motifs bound by human PRDM9 (B allele).
(a) Seven motif logos produced by our algorithm (applied to the top 5,000 PRDM9 binding peaks ranked by enrichment, after filtering out repeat-masked sequences) were aligned to each other and to an in silico binding prediction (Myers et al., 2010; Persikov et al., 2009; Persikov and Singh, 2014, maximizing alignment of the most information-rich bases. The position of the published hotspot 13-mer is indicated by the gray box overlapping the in silico motif (Myers et al., 2008). On the right is the percentage of the top 1,000 peaks (ranked by enrichment without further filtering) containing each motif type. Zinc-finger residues at 3 DNA-contacting positions (labeled −1, 3, 6) are illustrated below each ZF position, classified by polarity, charge, and presence of aromatic side chains. ZFs 5 and 6 lack positively charged amino acids and contain aromatic tryptophan residues, and they coincide with a variably spaced motif region (indicated by vertical dotted lines). Motif 4 is truncated here. (b) H3K4me3 ChIP-seq data from PRDM9-transfected HEK293T cells (this study) and H3K4me3/DMC1 data from testes (Pratto et al., 2014) were force-called to provide a p-value for enrichment of each sample in a 1 kb window centered on each PRDM9 peak (filtered to remove coverage outliers and those overlapping H3K4me3 peaks in untransfected cells). PRDM9 enrichment values are unitless (equal to the estimated signal divided by background, minus 1 and set to 0 if negative, at the base with the smallest p-value within each peak). Peaks were split into deciles according to their PRDM9 enrichment values, and the proportion of peaks with a force-called H3K4me3 or DMC1 p-value <0.05 is plotted within each decile. (c) Peaks were stratified into quartiles based on increasing PRDM9 enrichment (light green to dark green) after filtering out promoters. Mean recombination rates (from the HapMap LD-based recombination map, Frazer et al., 2007) at each base in the 20 kb region centered on each bound motif are plotted for each quartile, with smoothing (ksmooth, bandwidth 25). (d) Left plot: Peak enrichment quartiles (filtered to remove promoters as in c) were separated by motif type (Motifs 2, 3, and 5 were combined due to low abundance), and the mean HapMap CEU recombination rate overlapping peak centers was plotted against median PRDM9 enrichment in each quartile, with lines of best fit added for Motif 7 (pink) versus all other motifs. Right plot: Fold enrichment of each motif in AB-only DMC1 peaks versus AA-only DMC1 peaks (Pratto et al., 2014). Error bars indicate two standard errors of the mean (left plot) or 95% bootstrap confidence intervals (right plot).
-
Figure 1—source data 1
List of all ChIP-seq samples.
- https://doi.org/10.7554/eLife.28383.008
-
Figure 1—source data 2
PWMs for all motifs, in MEME format.
- https://doi.org/10.7554/eLife.28383.009
Figure 1—figure supplement 1
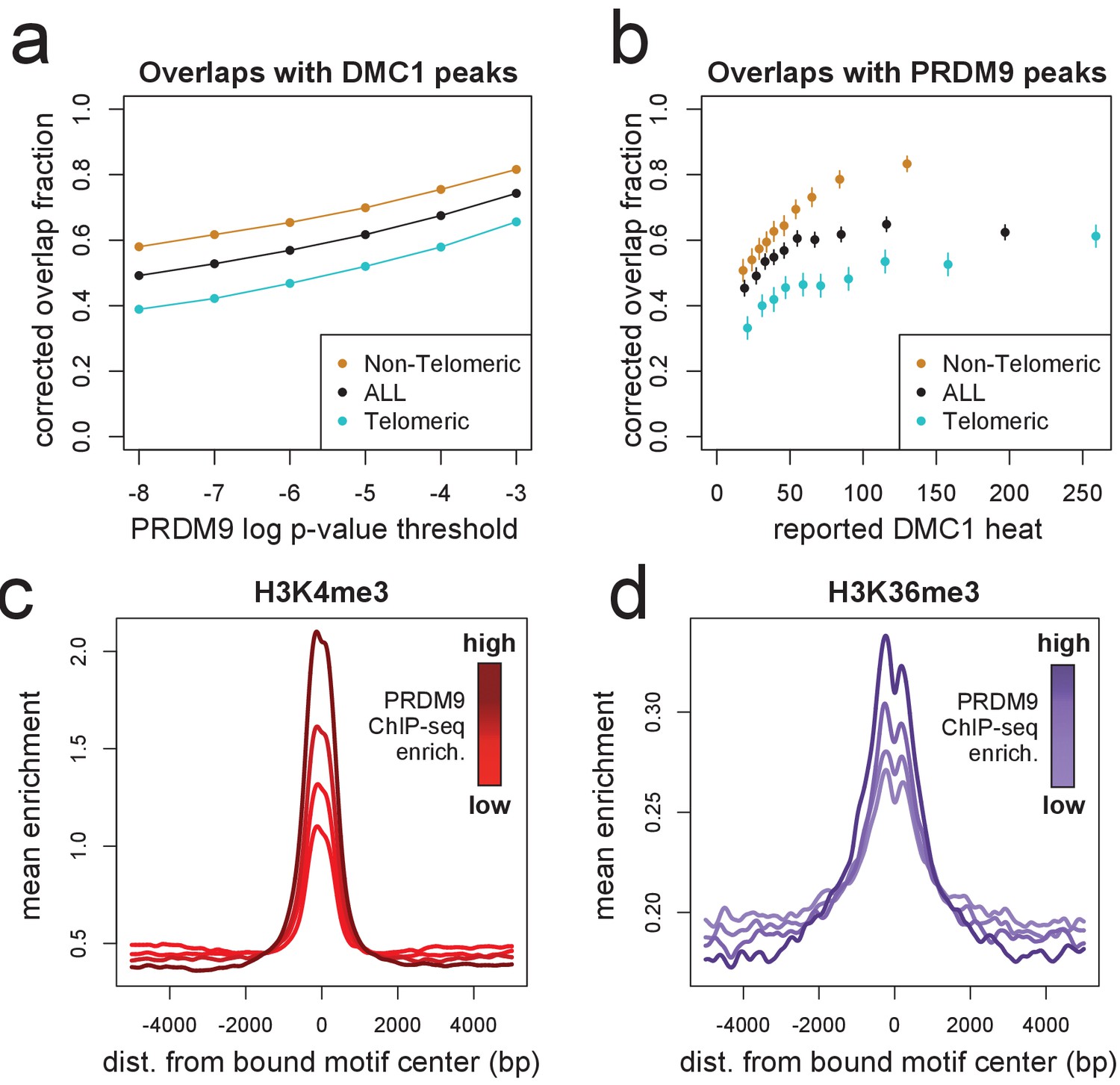
DMC1, H3K4me3, and H3K36me3 signals surrounding human PRDM9 peaks.
(a) A comparison our autosomal PRDM9 peaks, called at various p-value thresholds ranging from 10−8 to 10−3 (minimum peak separation 250 bp), to a set of published DSB hotspots corresponding to the human A allele (from a set of 18,343 ‘Intersect’ DMC1 hotspots found in multiple individuals, filtered to remove hotspots wider than 3 kb; Pratto et al., 2014). Hotspots were further split into subsets occurring within 15 Mb of a telomere (turquoise) or not (orange). ‘Overlap’ requires a PRDM9 peak center to fall within a reported DMC1 hotspot interval, and overlap fractions were corrected downward to account for chance overlaps (see Materials and methods). (b) DMC1 hotspots were split into decile bins by reported DMC1 heat, and the proportion of hotspots in each bin overlapping one or more of our PRDM9 peaks is indicated (error bars represent two standard errors of the proportion). (c) Profile plot showing the mean H3K4me3 enrichment (measured in HEK293T cells transfected with human PRDM9) at bound human motifs conditioned not to have any H3K4me3 enrichment measured in untransfected cells, and split into quartiles of increasing PRDM9 enrichment (smoothing: ksmooth, bandwidth 200) (d) Profile plot showing the mean H3K36me3 enrichment (measured in HEK293T cells transfected with human PRDM9) at bound human motifs conditioned not to have any H3K36me3 enrichment measured in untransfected cells, and split into quartiles of increasing PRDM9 enrichment. NB: absolute enrichment values cannot be compared across samples. (smoothing: ksmooth, bandwidth 25) .
Figure 1—figure supplement 2
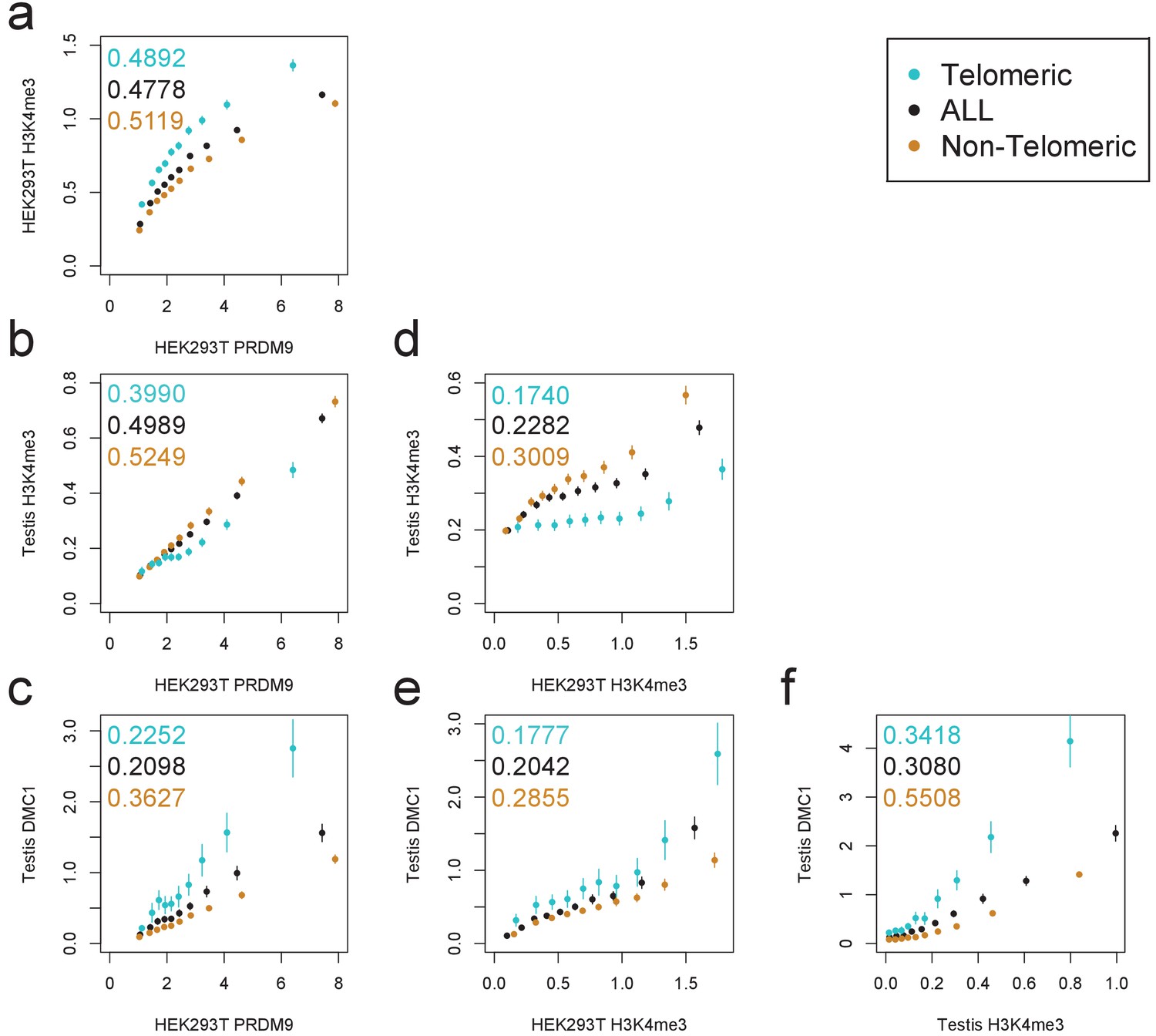
Comparison of PRDM9 and H3K4me3/DMC1 enrichment values.
H3K4me3 ChIP-seq data from transfected HEK293T cells (this study) and H3K4me3/DMC1 data from testes (Pratto et al., 2014) were force-called in a 1 kb window centered on each PRDM9 binding peak center (p<10−6, minimum peak separation 1000 bp) to provide an enrichment value for each H3K4me3/DMC1 sample at each PRDM9 peak. Peaks were further split into subsets occurring within 15 Mb of a telomere (turquoise) or not (orange). Pairwise comparisons plot the mean force-called enrichment value of each sample (y axis) in each enrichment decile bin of each other sample (x axis). Points are positioned at the median value of each decile and error bars represent two standard errors of the mean. Raw Pearson correlation values are printed on each plot. All comparisons show a significant positive correlation (p<2×10−16). Peak windows with fewer than five input reads from cells or testes were filtered out, to improve enrichment estimates, and windows with excessive genomic coverage (in the top 0.1%ile) or IP coverage (>500 combined fragments) were removed to avoid outliers due to mapping errors. PRDM9 peaks overlapping H3K4me3 peaks from untransfected cells were removed, leaving 37,188 peaks passing all filters. Interestingly, we observe an enrichment of H3K4me3 in telomeric peaks in our HEK293T cells but not in testes. .
Figure 1—figure supplement 3
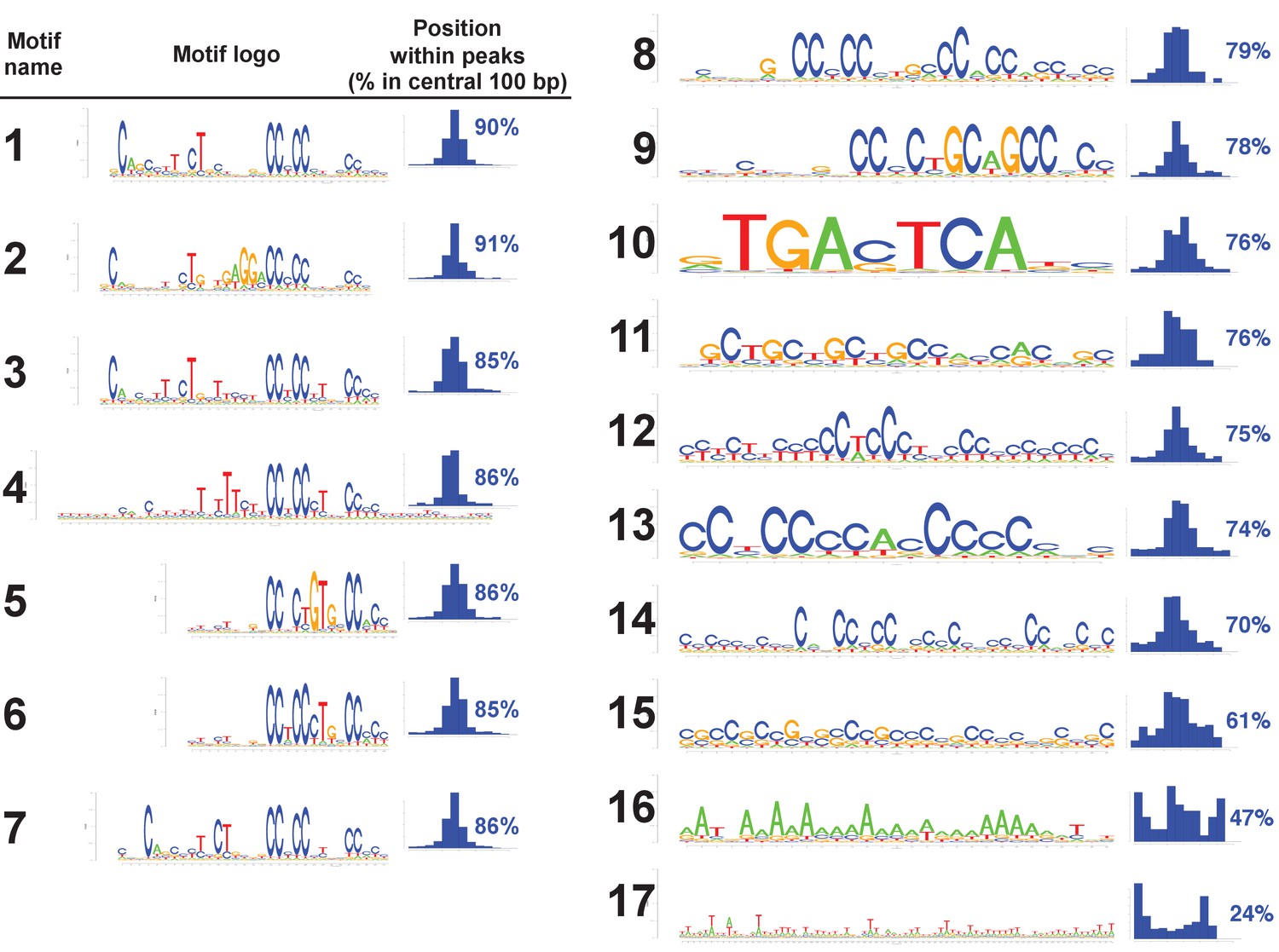
All motifs found in human PRDM9 peaks.
All 17 motif logos returned by our motif-finding algorithm are listed, along with histograms indicating their positions within the central 300 bp of our human PRDM9 peaks, as a measure of how centrally enriched they are (and therefore likely to represent true binding targets). Only the seven motifs for which greater than 85% of occurrences within peaks are within 100 bp of the peak center were retained for downstream analyses. The remaining, less centrally enriched, motifs are either degenerate (as seen in mice containing the human allele: (Davies et al., 2016) or may arise as a consequence of PRDM9 binding to promoter regions (this would explain Motif 10, which is a near identical match to the binding motif for the transcription factor AP1).
Figure 1—figure supplement 4
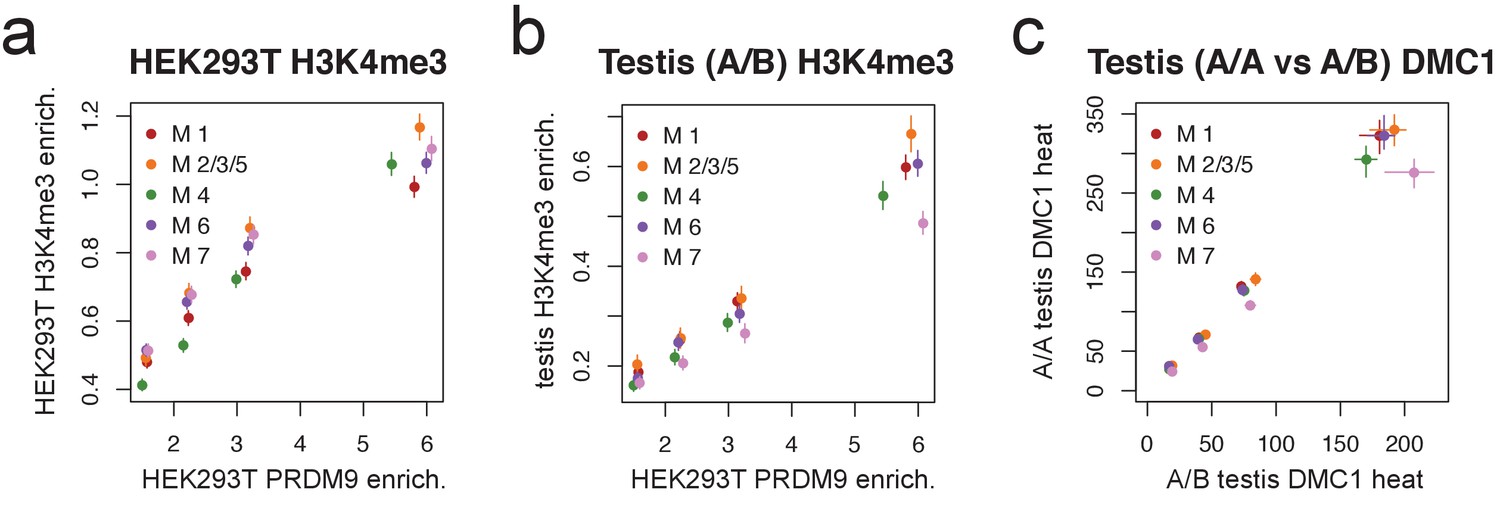
Motif 7 represents a binding mode favored by the B allele.
(a) Peak enrichment quartiles (filtered to remove promoters) were separated by motif type (Motifs 2, 3, and 5 were combined due to low abundance), and mean force-called H3K4me3 enrichment was plotted against median PRDM9 enrichment in each quartile. Error bars indicate two standard errors of the mean. This shows that the lower recombination rates for Motif 7 do not result from lower histone methylation activity of PRDM9 at those sites. (b) Peak enrichment quartiles as in a, but with force-called testis H3K4me3 enrichment values from (Pratto et al., 2014) in an individual with an A/B genotype. Motif 7 shows lower testis H3K4me3 enrichment for each level of PRDM9 binding, consistent with it being bound less efficiently by the A allele. Error bars indicate two standard errors of the mean. (c) At DMC1 hotspots found in both A/A and A/B individuals (from Pratto et al., 2014), a q-q plot of reported DMC1 heats for each motif type (quantiles 0.125,0.375,0.625,0.875). Motif 7 peaks are relatively hotter in the A/B samples than in the A/A samples. Error bars represent 95% bootstrap confidence intervals.
Figure 2 with 4 supplements
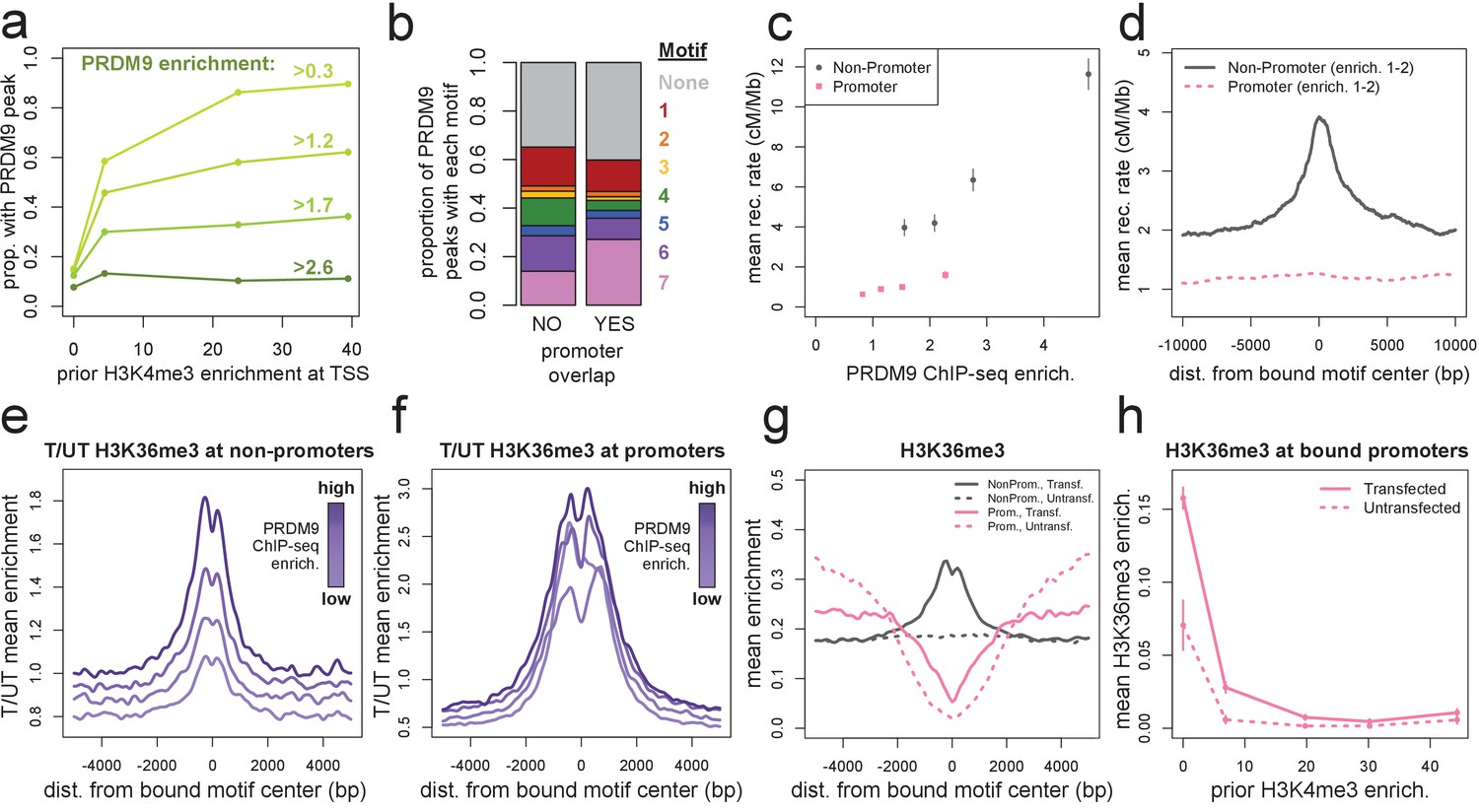
Human PRDM9 can bind promoters, though recombination is suppressed.
(a) The chance-corrected proportion of protein-coding genes that have a PRDM9 peak center occurring within 500 bp of the TSS, stratified by different PRDM9 enrichment value thresholds (shades of green, with thresholds listed), in each quartile of force-called H3K4me3 enrichment surrounding the TSS in untransfected cells. The power to detect weaker binding events increases at more active promoters (as measured by H3K4me3), though strong PRDM9 binding events appear at roughly 10% of all promoters regardless of activity. (b) Barplot illustrating the proportion of promoter or non-promoter PRDM9 peaks assigned to each of the 7 motifs (or no motif, in gray). Motif 7 appears 2-fold enriched in promoter peaks. (c) Mean HapMap CEU recombination rates are reported for promoter (pink squares) and non-promoter (gray circles) human PRDM9 peaks split into quartiles of PRDM9 enrichment (filtered not to overlap repeats or occur within 15 Mb of a telomere; error bars represent two standard errors of the mean). Both median enrichment values and recombination rates are greater for non-promoter peaks, even in overlapping ranges of PRDM9 enrichment. (d) Mean recombination rate in 20 kb windows centered on bound motifs, for promoter (pink) and non-promoter (gray) peaks further filtered only to include peaks with PRDM9 enrichment values between 1 and 2 (smoothing: ksmooth bandwidth 200). (e) Mean H3K36me3 enrichment in transfected cells divided by mean H3K36me3 enrichment in untransfected cells at 36,000 non-promoter PRDM9 binding sites split into quartiles of PRDM9 enrichment (shades of purple). (f) same as e but for 10,000 promoter PRDM9 binding sites split into quartiles of PRDM9 enrichment. (g) The absolute mean enrichment values used to generate plots e and f, split into transfected (solid) and untransfected (dotted) samples at promoter (pink) and non-promoter (gray) PRDM9 binding sites in the top quartile of PRDM9 enrichment. There is a depletion of H3K36me3 coverage surrounding promoters in untransfected cells, but the magnitude of this depletion decreases in transfected cells. (h) At 4,000 protein-coding genes with a strong PRDM9 binding peak within 500 bp of the TSS (PRDM9 enrichment >2 and <10), we show the relationship between force-called H3K4me3 enrichment and force-called H3K36me3 enrichment in the 1 kb surrounding each TSS, for both transfected and untransfected cells (solid and dotted lines). Error bars indicate two standard errors of the mean H3K36me3 enrichment within each quintile of H3K4me3 enrichment. H3K36me3 enrichment increases in transfected cells at all strongly bound promoters, but this effect diminishes almost to 0 as promoter activity increases (which forces H3K36me3 close to 0 in all cells). This effect cannot be accounted for by the modest decrease in PRDM9 enrichment at more active promoters (mean PRDM9 enrichment decreases from 4.3 in the first H3K4me3 quintile to 3.1 in the fifth quintile).
Figure 2—figure supplement 1
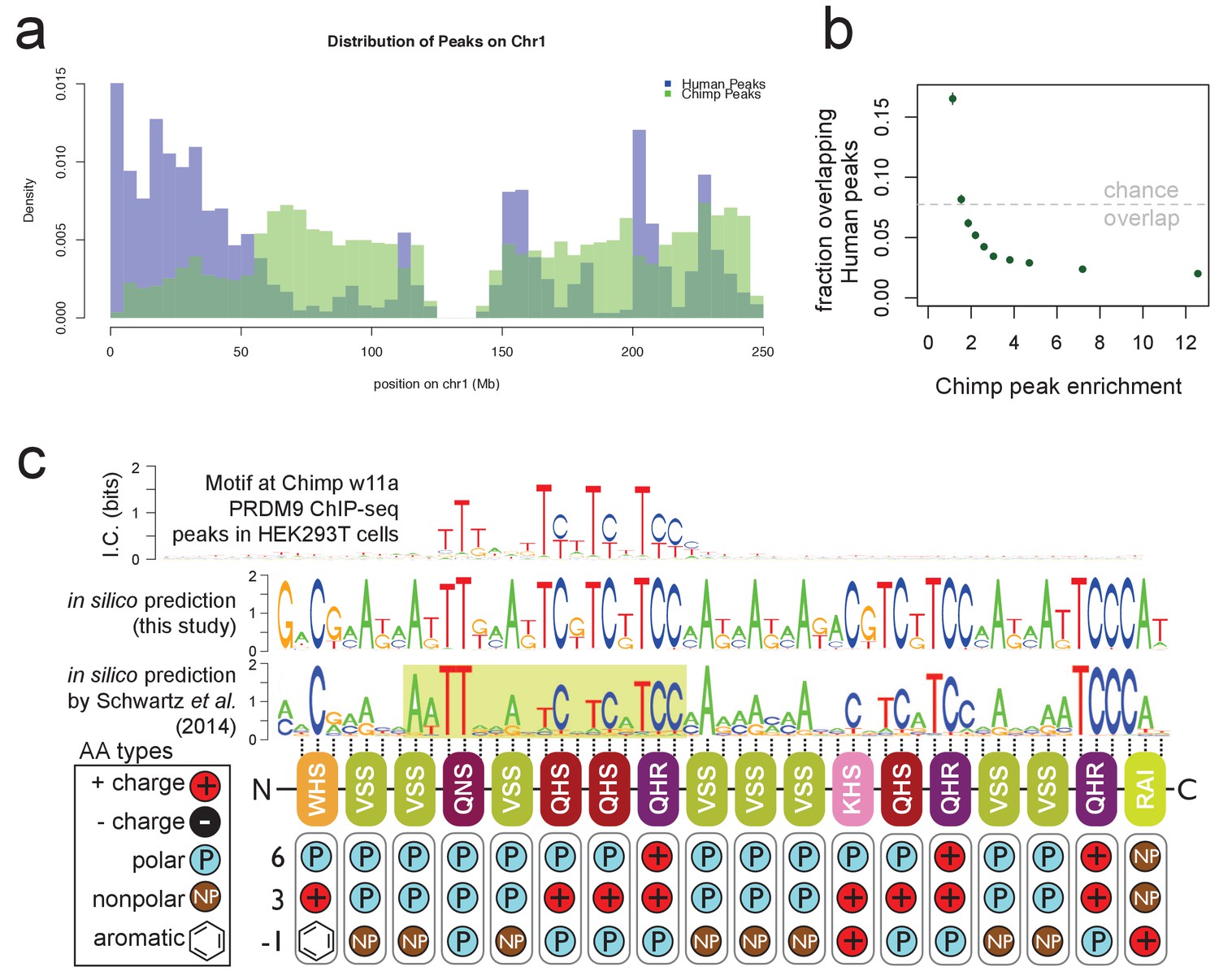
Chimp w11a PRDM9 binds a T-rich motif away from human binding sites.
(a) Comparison of the number of human (B allele; blue) and chimp (w11a allele; semitransparent green) PRDM9 ChIP-seq peaks in 1 Mb bins across human Chr1. Consistent with their different predicted binding motifs, we observe very different binding landscapes across the genome. (b) The proportion of chimp PRDM9 peaks with a human PRDM9 peak center occurring within 500 bp, for each decile of chimp PRDM9 enrichment. Overall, chimp and human peaks overlap less than expected by chance (gray line), especially for stronger chimp peaks. (c) Top: the only non-degenerate motif returned by our motif-finding algorithm when trained on the top 5,000 chimp PRDM9 peaks ranked by enrichment, representing the first empirically determined direct binding motif for any chimp PRDM9 allele. Although the motif extends 53 bp (the expected span of the zinc fingers), only a 17 bp core region shows high specificity, and this region overlaps and matches in-silico binding predictions Persikov et al., 2009; Persikov and Singh, 2014; Schwartz et al., 2014; Auton et al., 2012), in particular a submotif (highlighted in yellow) shown to be common to many western chimp PRDM9 alleles (reproduced from Schwartz et al., 2014 Supplementary Material, Supplemental Figure 2, available under the terms of the https://creativecommons.org/licenses/by/4.0/ Creative Commons Attribution License). Zinc-finger residues at DNA-contacting positions (labeled −1, 3, 6) are illustrated below each zinc-finger position, classified by polarity, charge, and presence of aromatic side chains. In contrast to the human B allele, this chimp allele has 18 instead of 12 canonical zinc fingers, and they differ in amino acid types at the DNA-contacting positions.
Figure 2—figure supplement 2
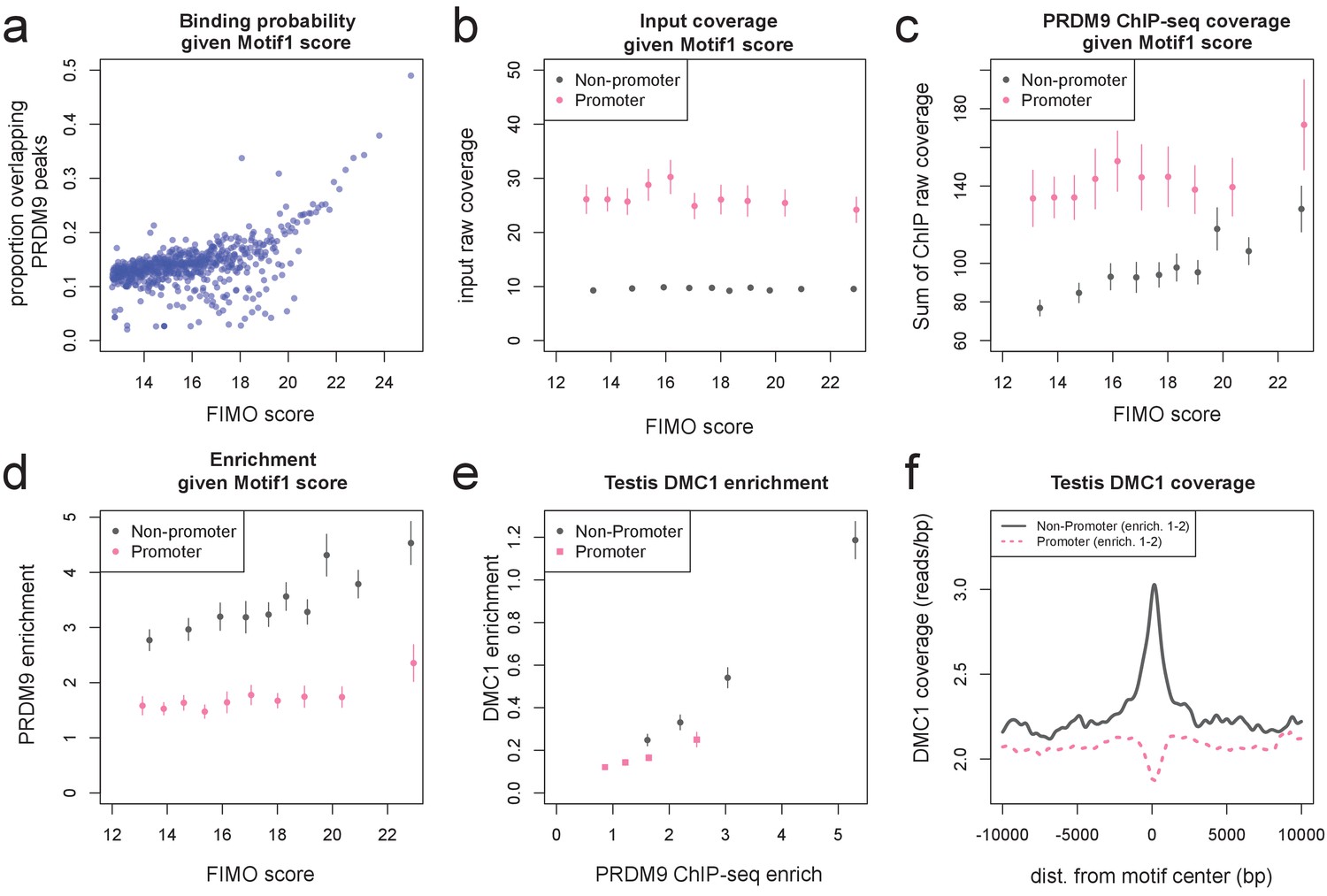
Human PRDM9 can bind promoters, though DSBs do not occur.
(a) FIMO was used to identify the top 1 million matches for Motif 1 in hg19 (Bailey et al., 2015. For 0.1 percentile bins of increasing FIMO score, the proportion of motif matches occurring within 150 bp of a PRDM9 peak center is plotted (p<10−6, minsep 250). Even the strongest 0.1% of motif matches are only bound 50% of the time. (b) PRDM9 peaks overlapping Motif 1 (and having more than 5 input reads overlapping the peak center) were divided into those overlapping promoters (stringently, those within 1 kb of a TSS, overlapping an H3K4me3 peak in untransfected cells, and overlapping a DNase HS site; gray), and non-promoters (failing those criteria and further not overlapping an H3K4me3 peak reported by any ENCODE data; see Materials and methods; pink). Mean raw input coverage values are plotted in decile bins of FIMO score, with error bars representing ±2 s.e.m. (c,d) Same as b, but with mean sum of raw ChIP fragment coverage values in each bin (c) or mean computed enrichment values in each bin (d). Overall, promoters show greater input sequencing coverage and thus we have greater power to detect weak binding in these regions. When corrected for this sequencing bias, we see that promoter binding sites tend to have weaker binding enrichment for a given FIMO score. (e) Mean force-called testis DMC1 enrichment values (Pratto et al., 2014) are reported for promoter (pink squares) and non-promoter (gray circles) human PRDM9 peaks split into quartiles of PRDM9 enrichment (filtered to not overlap repeats or occur within 15 Mb of a telomere; error bars represent two standard errors of the mean). Both median PRDM9 enrichment values and DMC1 enrichment values are greater for non-promoter peaks, even in overlapping ranges of PRDM9 enrichment, as observed for LD-based recombination rate estimates (Figure 2c). (f) Mean raw testis DMC1 coverage in 20 kb windows centered on bound motifs, for promoter (pink) and non-promoter (gray) peaks further filtered only to include peaks with PRDM9 enrichment values between 1 and 2 (smoothing: ksmooth bandwidth 200). There is a local spike in DMC1 coverage around non-promoter binding sites, similar to the peak observed in LD-based recombination rate (Figure 2d), and an apparent depletion of DMC1 coverage at promoter binding sites. .
Figure 2—figure supplement 3
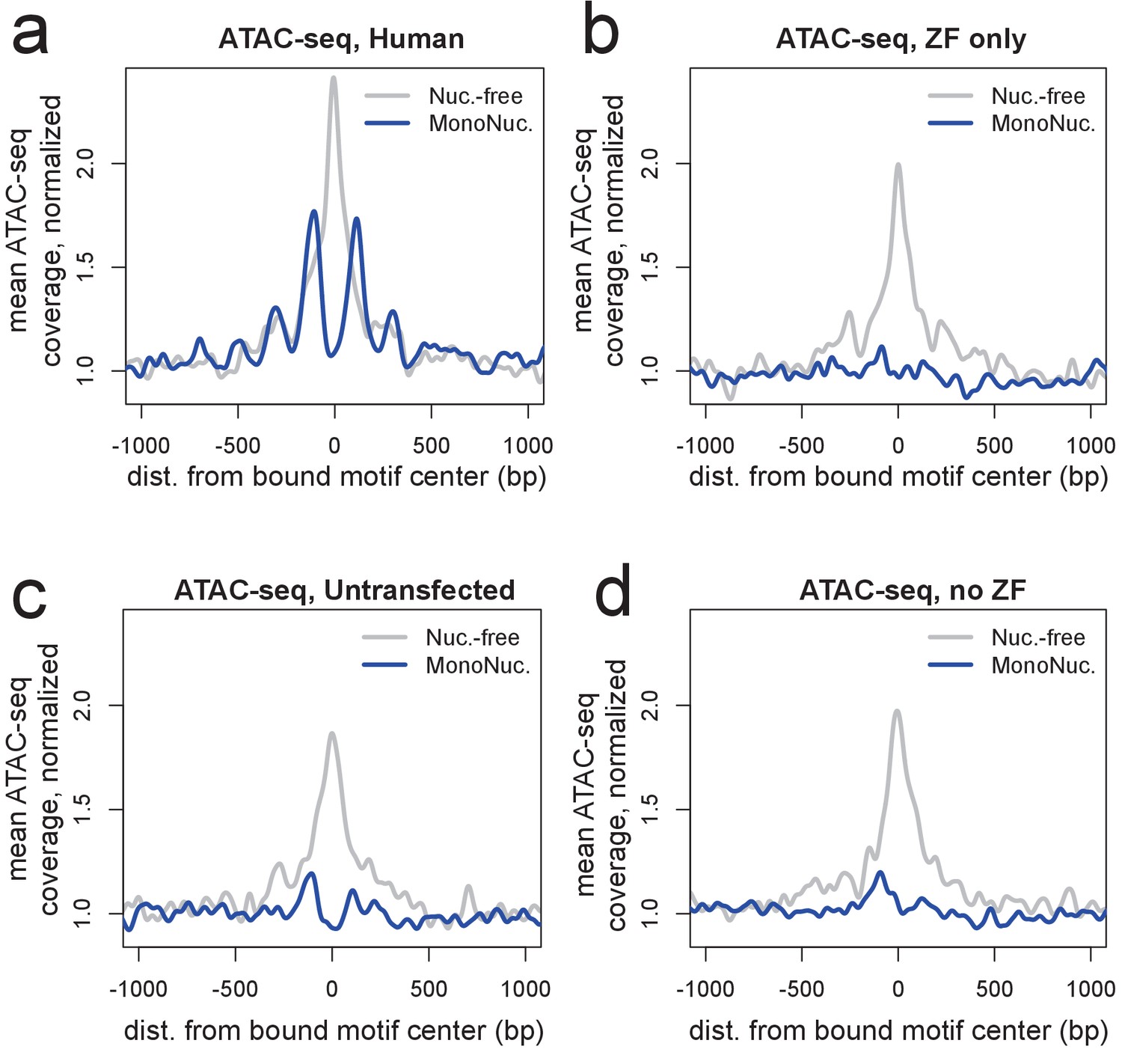
ATAC-seq profiles showing nucleosome phasing around PRDM9 binding sites.
(a) ATAC-seq profile plots surrounding a set of the ~15,000 strongest human PRDM9 ChIP-seq peaks (filtered to require a motif match and to not overlap an annotated DNase hypersensitive site) in cells transfected with human PRDM9. ‘Coverage’ here refers to the frequency with which an ATAC-seq fragment center occurs at each position, such that 'Nuc.-free' coverage tracks the centers of nucleosome-depleted regions, and ‘MonoNuc.’ coverage tracks the centers of single nucleosomes. Coverage values are normalized to the mean values observed between 1,500 and 3,000 bases away from each site, as a measure of background, and smoothed (ksmooth bandwidth = 50). The human-transfected cells show strongly phased nucleosomes centered at ~100 bp to either side of the motif and an elevated signature of nucleosome depletion at the center. (b-d) The same plot at the same sites as a but for cells transfected with the zinc-finger domain alone (b), untransfected cells (c), or cells transfected with a truncated construct excluding the ZF domain (d). These plots show that the ZF domain is insufficient to phase nucleosomes and confirm that PRDM9 binding favors nucleosome-depleted regions. They also confirm that PRDM9 creates a nucleosome-depleted region near its binding site (Baker et al., 2014).
Figure 2—figure supplement 4
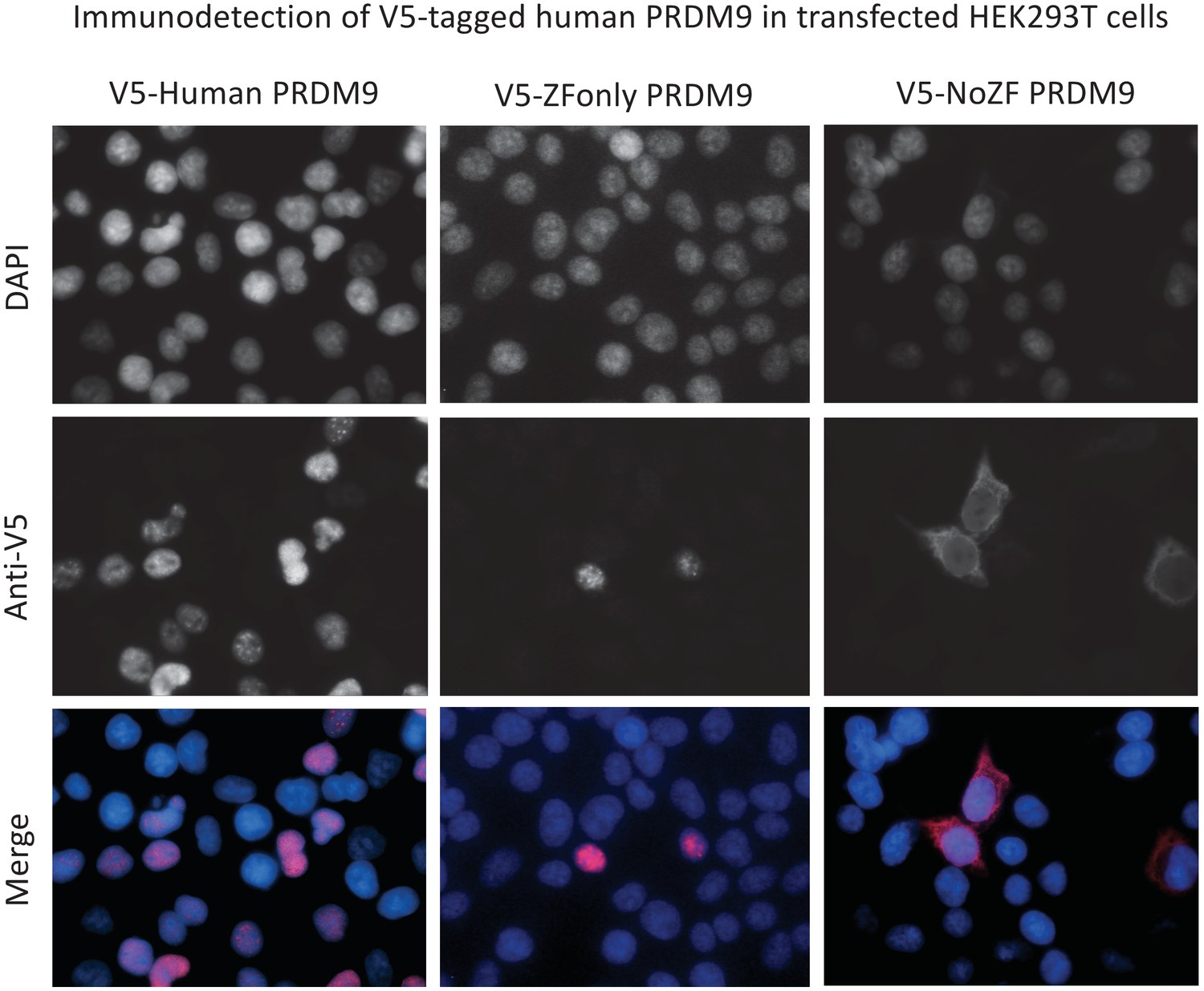
PRDM9s ZF domain is necessary and sufficient for nuclear localization.
Representative results of ImmunoFluorescence detection of V5 tags in HEK293T cells transfected with full-length human PRDM9 (first column), the ZF domain alone (‘ZF-only’, second column), or everything but the ZF domain (‘no-ZF’, third column). Grayscale images show fluorescence intensities of DAPI to mark the nucleus (first row), or of the anti-V5 antibody to mark transfected protein localization (second row). False-colored merged images (DAPI blue, anti-V5 red) show that the full-length and ZF-only proteins are restricted to the nucleus, while the no-ZF construct localizes throughout the cell. Note these images are not representative of the overall transfection efficiencies of these constructs.
Figure 3 with 1 supplement
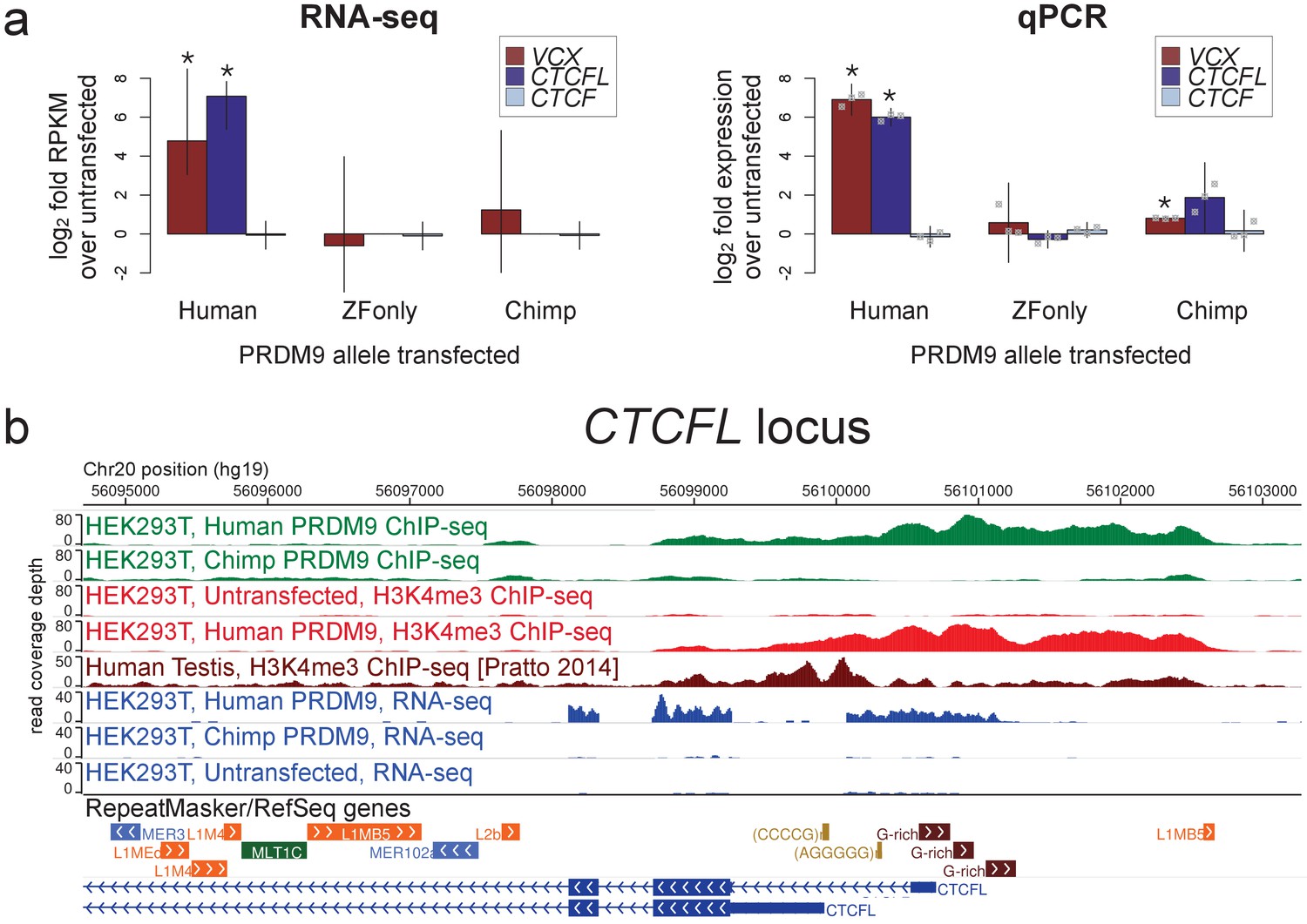
Spermatogenesis-specific genes VCX and CTCFL are activated by human PRDM9 in HEK293T cells.
(a) left: Bar plots showing the log2 fold change relative to untransfected HEK293T cells in computed FPKM values (fragments per kilobase of transcript per million mapped RNA-seq reads) for HEK293T cells transfected with the human allele, the chimp allele, or a construct containing only the human Zinc-Finger domain, for CTCFL and VCX, with CTCF as a negative control. Error bars conservatively represent maximum ranges of the ratios given confidence intervals for FPKM values computed by cufflinks (Trapnell et al., 2012). Asterisks indicate significant differential gene expression, as reported by CuffDiff (p<0.0001). right: qPCR validation results for the same genes from 3 independent biological replicates. Y-axis values are log ratios of C values for each gene relative to the untransfected sample (normalized to the TBP housekeeping gene; see Materials and methods). Error bars represent 95% confidence intervals from 3 biological replicates (t distribution; gray points represent individual replicate values), and asterisks indicate p<0.001 (one-tailed t test). (b) A browser screenshot (Zhou et al., 2011) from Chr20 near the promoter region of CTCFL with custom tracks indicating ChIP-seq and RNA-seq raw read coverage data. Human PRDM9, but not chimp PRDM9, (green) binds a G-rich repeat near the TSS, adding an H3K4me3 mark (light red) where none is present in untransfected cells. RNA-seq coverage (blue) spikes in the coding regions in transfected cells, while it is nearly flat in untransfected cells or chimp-transfected cells. Testis H3K4me3 coverage (dark red, from Pratto et al., 2014) peaks at a slightly different locus, corresponding to an alternative TSS.
-
Figure 3—source data 1
qPCR primers, Ct values, and calculations.
- https://doi.org/10.7554/eLife.28383.017
-
Figure 3—source data 2
PRDM9-bound genes with differential expression.
- https://doi.org/10.7554/eLife.28383.018
Figure 3—figure supplement 1
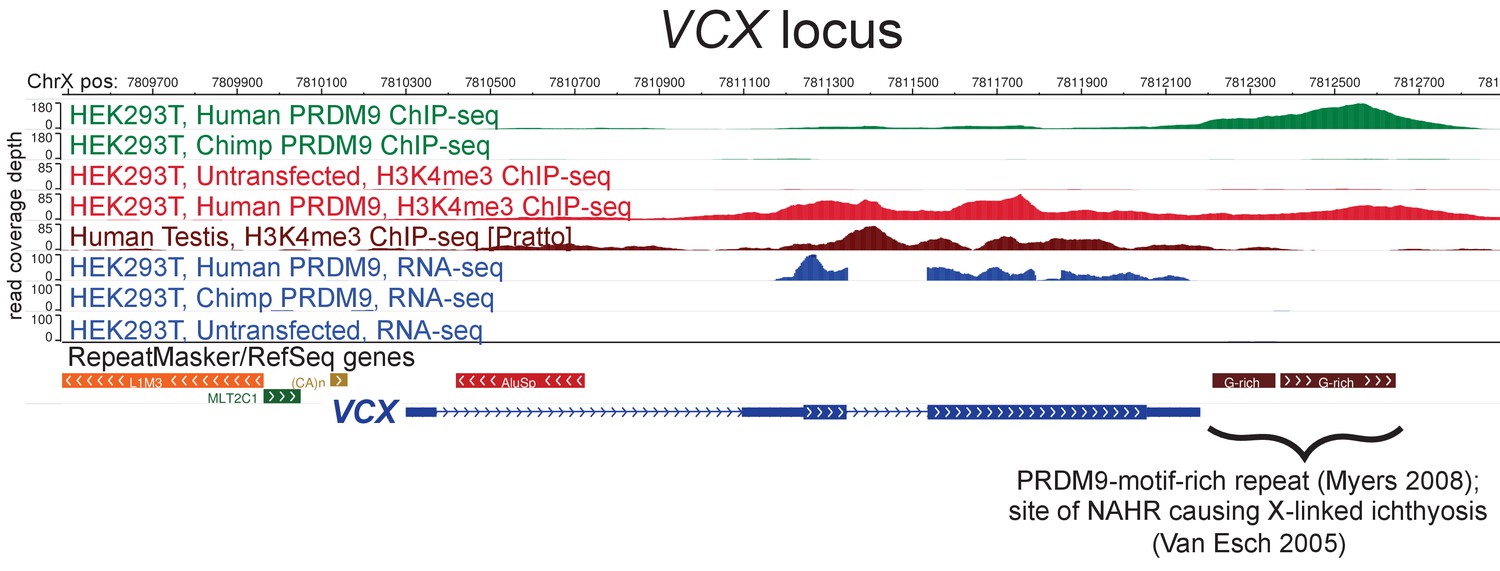
Raw coverage values surrounding the VCX promoter.
A browser screenshot (Zhou et al., 2011) from ChrX containing the VCX gene with custom tracks indicating ChIP-seq and RNA-seq raw coverage data. Human PRDM9, but not chimp PRDM9, (green) binds a G-rich repeat near the terminus of VCX as well as two loci in the middle of the gene, adding H3K4me3 marks (light red) where none were present in untransfected cells. RNA-seq coverage (blue) spikes in the coding regions in transfected cells, while it is nearly flat in untransfected cells or chimp-transfected cells. Testis H3K4me3 coverage (dark red, from Pratto et al., 2014) also increases in the gene body, instead of near the annotated TSS.
Figure 4 with 2 supplements
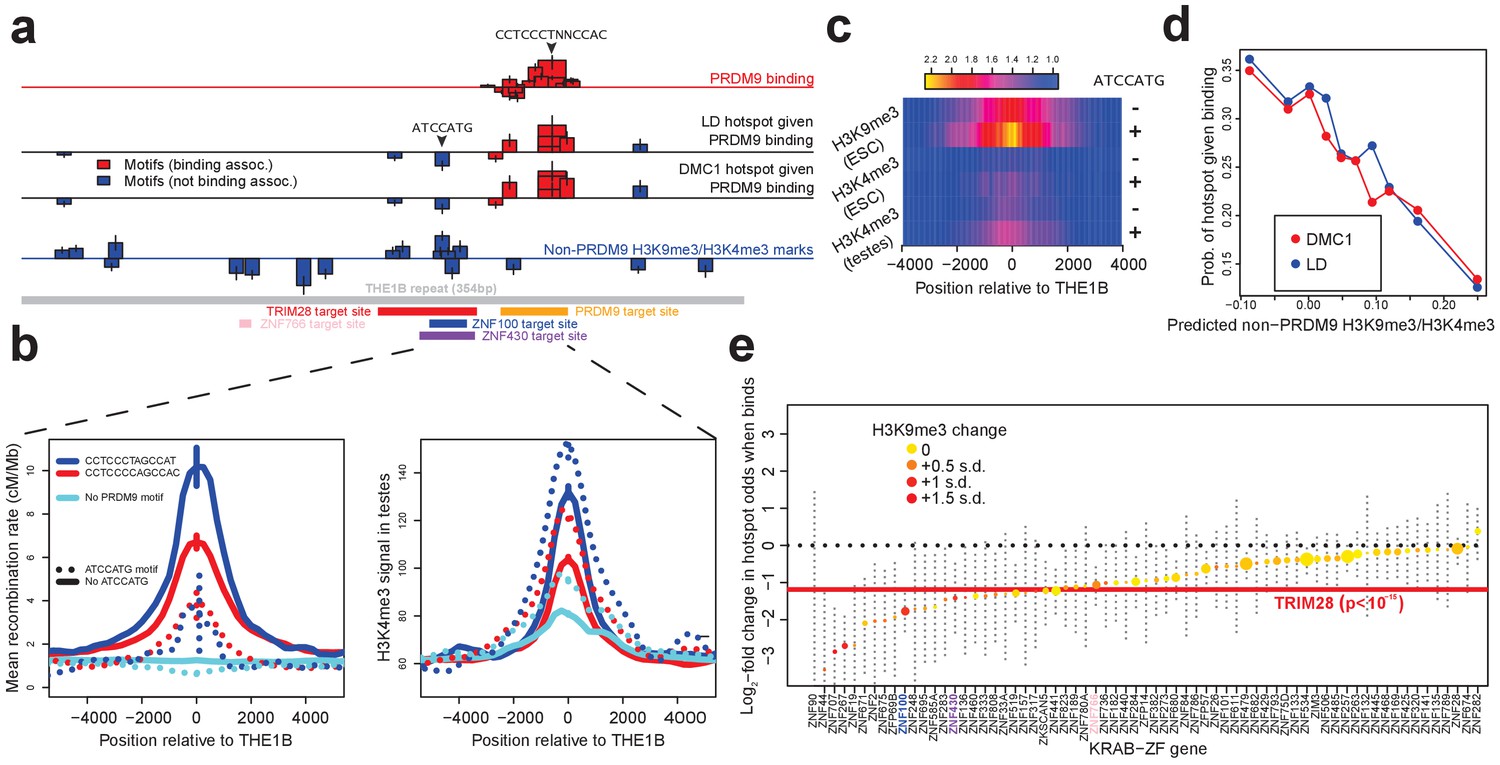
Influences on recombination in cis downstream of PRDM9 binding.
(a) Analysis of THE1B repeats shows the positions along the THE1B consensus (bottom, gray) of motifs influencing PRDM9 binding (top row), motifs influencing recombination hotspot occurrence at bound sites (middle two rows), and motifs influencing H3K4me3/H3K9me3 in testes and somatic cells (bottom row). Rectangle widths show motif size, and heights show log-odds-ratio or effect size (two standard errors delineated). Rectangles below the lines have negative effects. Motifs associated with PRDM9 binding are in red; others in blue. Binding motifs for labeled proteins are at the plot base. (b) Left plot shows LD-based recombination rates around the centers of THE1B repeats containing different approximate matches to the PRDM9 binding motif CCTCCC[CT]AGCCA[CT] (colors) and the motif ATCCATG (lines dotted if present). Right plot is the same but shows mean H3K4me3 in testes (from Pratto et al., 2014). ATCCATG presence reduces recombination and increases H3K4me3. (c) Impact of ATCCATG presence (+) or absence (-) on normalized enrichment values around the centers of THE1B repeats, of H3K4me3 and H3K9me3 in different cells (labeled pairs of color bars, normalized to equal 1 at edges). H3K9me3 shows the strongest signal increase. (d) Predicted non-PRDM9 H3K9me3/H3K4me3 versus probability DMC1-based or LD-based hotspots occur at PRDM9-bound sites. For the x-axis repeats were binned according to an additive DNA-based score, using the bottom row of part A and the combination of motifs they contained. (e) Estimated impact on whether a hotspot occurs of co-binding by individual KRAB-ZNF proteins (labels; Imbeault et al., 2017) near a PRDM9 binding peak (genome-wide, not only within THE1B repeats, after filtering out promoter regions). For each KRAB-ZNF protein, a GLM was used to estimate the impact of KRAB-ZNF binding (binary regressor) on hotspot probability. We show the estimated log-odds, with 95% CIs. Colors indicate H3K9me3 enrichment increase at co-bound sites. Horizontal line shows the results for TRIM28. Features below the horizontal dotted line have a negative estimated impact on downstream recombination.
-
Figure 4—source data 1
Detailed information on all THE1B motifs.
- https://doi.org/10.7554/eLife.28383.022
Figure 4—figure supplement 1
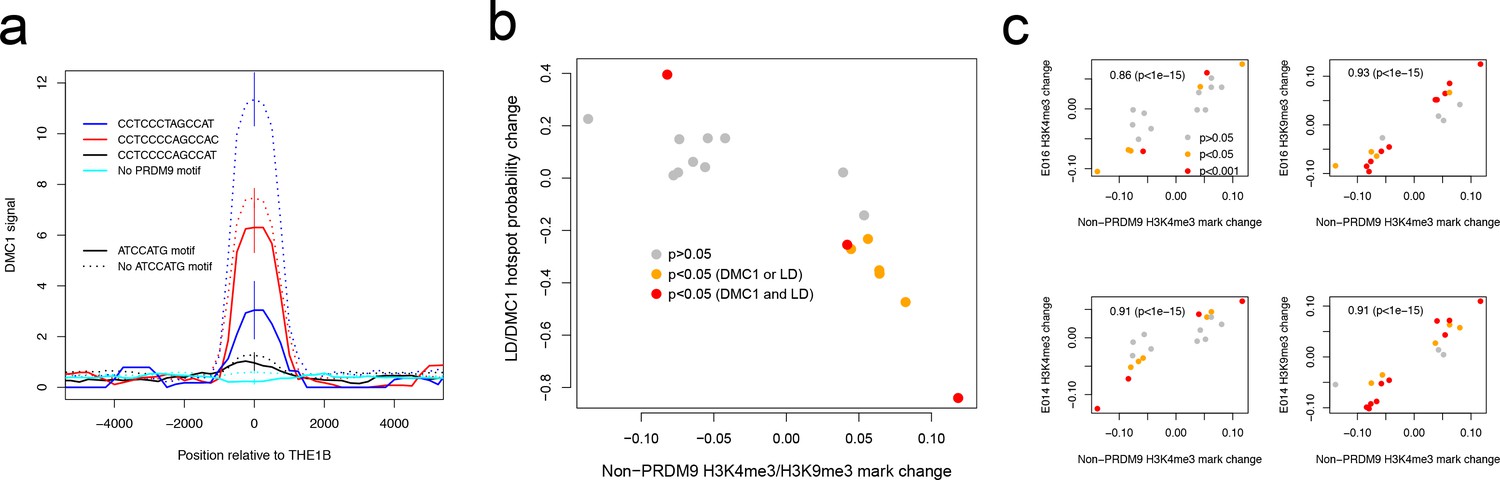
Features associated with recombination outcomes given PRDM9 binding.
(a) DMC1-based recombination rates around the centers of THE1B repeats containing different approximate matches to the PRDM9 binding motif CCTCCC[CT]AGCCA[CT] (colors) and the motif ATCCATG (lines dotted if absent). ATCCATG presence reduces recombination. Vertical lines: 2 s.e. (b) For 18 motifs identified to influence H3K4me3 signal strength at THE1B repeats in testes (and H3K9me3 in other cell types, see d) but not PRDM9 (Appendix 2) we fit a joint generalized linear model of each motifs effect size on H3K4me3 in testes (x-axis). For the same set of motifs, we fit two joint generalized linear models to estimate each motif’s effect size on the probability a THE1B repeat overlaps respectively a DMC1 or LD-based hotspot, and average the estimated effect sizes, corresponding to an odds ratio for each motif (y-axis). Points are colored according to whether coefficients for the second linear models differ significantly from zero (legend). The strong negative correlation on the plot implies that motifs increasing H3K9me3/H3K4me3 associate with decreased recombination, and conversely. (c) Four panels correspond to two different histone modifications H3K9me3 and H3K4me3, in two distinct somatic embryonic stem cell types (E014 and E016) studied by ROADMAP (Kundaje et al., 2015) and labeled accordingly on the y-axis. In each panel the x-axis is as for b. Each y-axis gives estimated coefficients under a generalized linear model fitted in the same way as the x-axis (Appendix 2), predicting enrichment of that particular histone modification in a particular cell type in THE1B repeats by presence/absence of each motif. Points are colored according to whether coefficients for this linear model differ significantly from zero (legend). Note strong positive correlations (each plot is labeled with rank-based correlation and p-value of rank-based correlation test) of 0.86 to 0.93, slightly higher for H3K9me3 than H3K4me3 and showing larger coefficients. The same motifs are then associated with both H3K9me3 and H3K4me3 changes across cell types including the cells lacking PRDM9 expression.
Figure 4—figure supplement 2
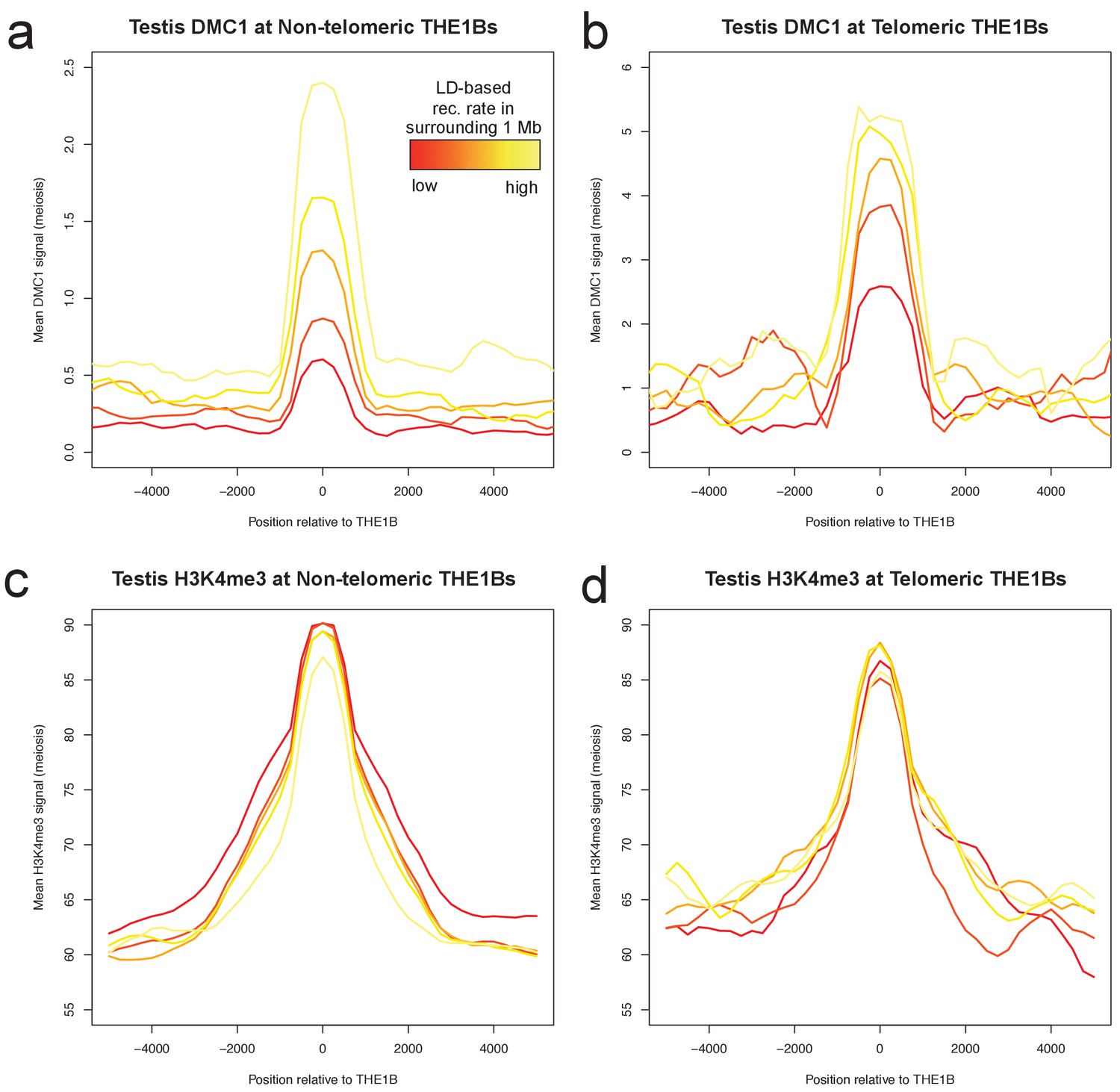
Large-scale recombination rate affects testis DMC1 but not H3K4me3.
Profiles of mean DMC1 and H3K4me3 read coverage from human male testes (with a PRDM9 A/B genotype; Pratto et al., 2014) around all THE1B repeats, stratified into quantiles based on the pedigree-based recombination heat in the surrounding 1 Mb of DNA (Kong et al., 2002, excluding the surrounding 20 kb and the repeat itself, by color (red to yellow are increasing 20% quantiles). H3K4me3 shows no impact whatsoever from surrounding recombination rate, implying PRDM9 binding is completely unaffected (c,d). However DMC1 signal increases dramatically (a,b), implying that broad-scale recombination control at these repeats occurs completely independently of PRDM9 binding or local sequence. Note the y-axes are different for telomere and non-telomere DMC1 (a,b) but not H3K4me3 (c,d). Telomeric sites were defined as those occurring within 10 Mb of a telomere, and H3K4me3 values were capped at 500 to reduce outlier effects. .
Figure 5 with 3 supplements
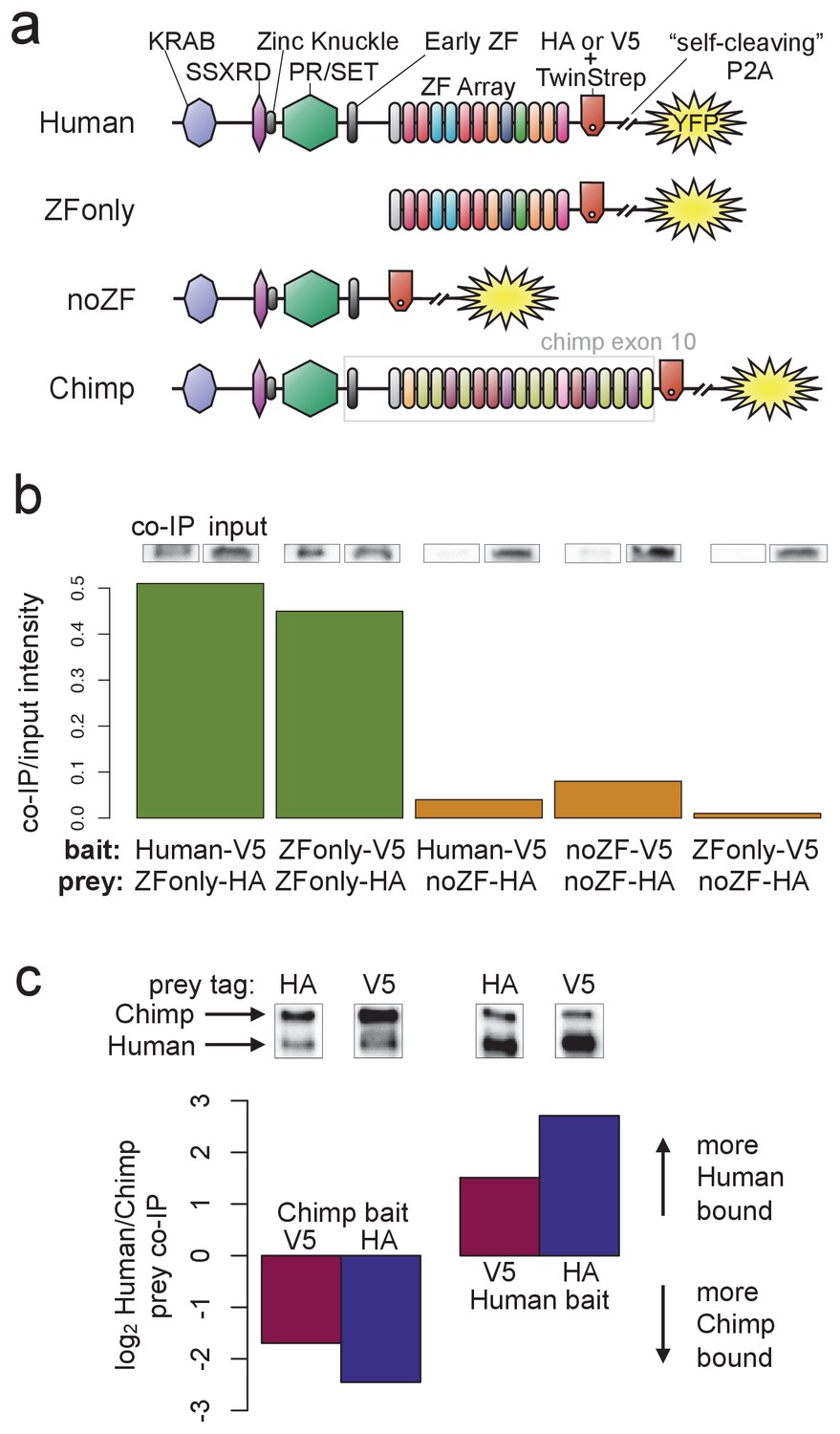
PRDM9 multimer formation is mediated by the ZF domain in an allele-biased manner.
(a) Overview of the different C-terminally tagged PRDM9 constructs used. Both an HA and a V5 version of each construct were generated for co-IP experiments. (b) Barplot showing the relative intensity of western blot co-IP bands normalized to input bands (from 50-μg of total lysate protein) for each combination of bait and prey constructs. Whenever both bait and prey contain the zinc-finger domain (green bars), the co-IP signal is much stronger than when either or both constructs lack a ZF domain (orange bars). See Figure 5—figure supplements 1 and 2 for complete westerns with mock controls. (c) Barplot showing the results of competitive co-IP experiments performed in cells transfected with both Human and Chimp as prey (with the same epitope tag) and either Human or Chimp as bait (with a complementary epitope tag). Bars indicate the relative co-IP band intensity for Human and Chimp prey constructs when pulled down with either Chimp or Human bait. When Human is used as bait, more Human prey is pulled down; when Chimp is used as bait, more Chimp prey is pulled down (and this holds for both directions of HA/V5 tagging).
Figure 5—figure supplement 1
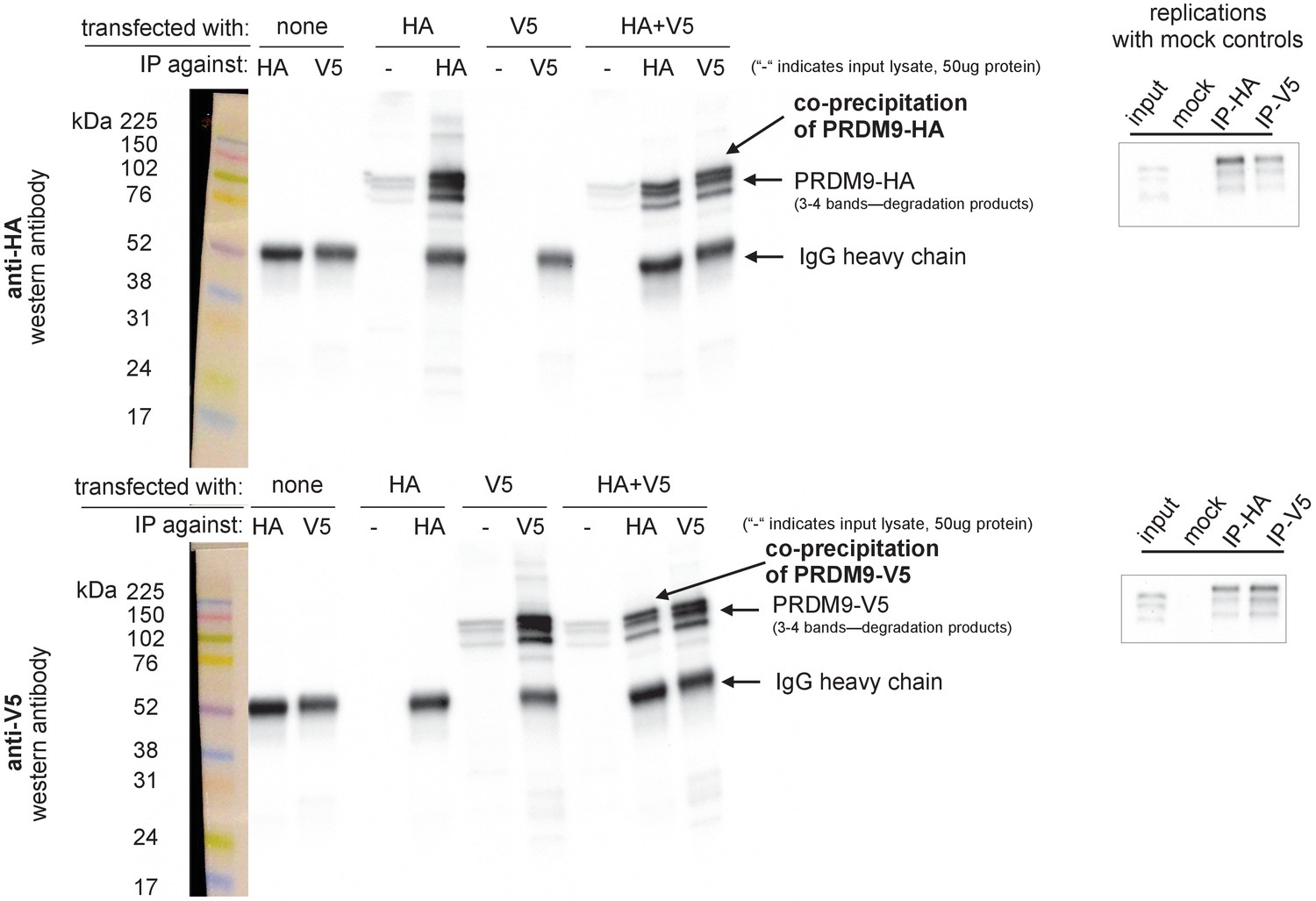
Confirmation of PRDM9 multimer formation.
Left: Western blots illustrating controls and experimental results. Samples were split and run on two blots separately, one imaged using an anti-HA antibody (upper) and one using an anti-V5 antibody (lower). Exposure time was 4 min. Ladder lanes are overlaid on the left, with approximate sizes in kiloDaltons noted. Lanes are labeled according to which full-length Human construct (HA or V5) was used, as well as which antibody was used for immunoprecipitation. IgG heavy chains are visible around 50 kDa, while the Human allele is visible as a band around 100 kDA with two or three smaller bands beneath it, likely representing degradation products (Grey et al., 2011; Cole et al., 2014). ‘-' is a short-hand label for input lanes, for which 50 μg of input chromatin was loaded in each well. The first six lanes demonstrate the specificity of the antibodies and their lack of cross-reactivity. The last two lanes show the co-IP experimental results confirming multimerization. Right: Two independent replicates were performed to confirm the formation of multimers with the full-length human constructs, using IgG mock control lanes to rule out nonspecific co-precipitation. Images were cropped to include only the PRDM9 bands. Input lane bands appear to have run lower than expected due to the use of a higher concentration of loading buffer in the IP lanes, an issue which was avoided in subsequent experiments.
Figure 5—figure supplement 2
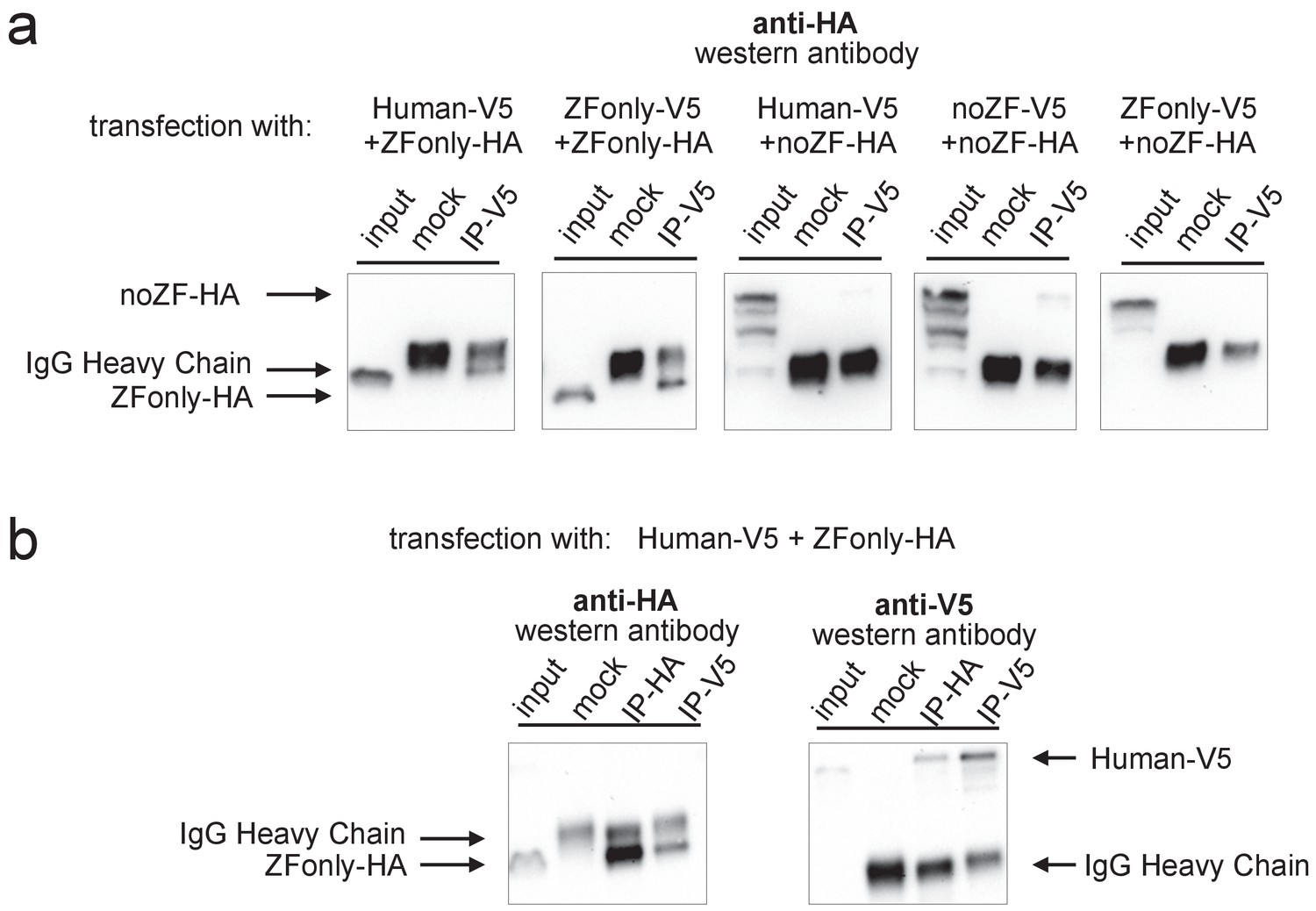
Multimerization is mediated primarily by ZF-ZF binding.
Western blots illustrating co-IP results for various combinations of full-length Human, noZF, and ZFonly constructs. (a) The third and fourth blots show only a very faint co-IP signal despite strong input expression of the noZF construct, indicating that the non-ZF portion of PRDM9 cannot form multimers efficiently with itself or full-length PRDM9. The first and second blots show strong co-IP signals for the ZFonly construct, indicating that the ZF domain binds itself and binds the full-length Human construct. The fifth plot shows that the ZFonly and noZF constructs do not bind each other and confirms that multimerization is not mediated by the C-terminal tags. (b) A replication of the experiment shown in the first blot above, but performing the IPs and western blots with both tag combinations. This confirms that the full-length Human construct can pull down the ZFonly construct, and the ZFonly construct is sufficient to pull down the full-length Human construct.
Figure 5—figure supplement 3
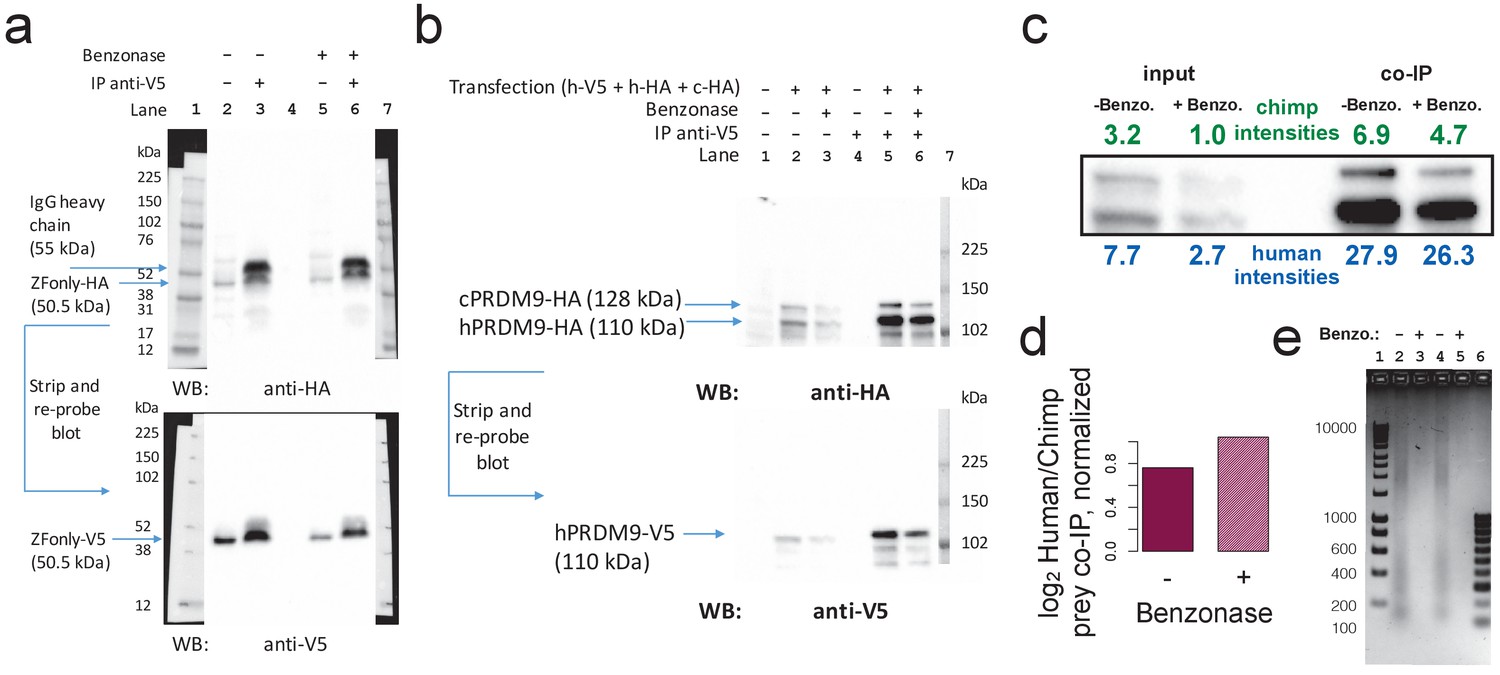
Benzonase treatment does not affect co-IP results.
(a) Replication of ZFonly-ZFonly co-IP western experiment with and without benzonase treatment. Lanes 1 and 7 are protein ladder. Lane 4 is empty. Lanes 2 and 5 are input lysate (50 μg protein measured by BCA). Lanes 3 and 6 are elutions from IP with anti-V5. Upper image shows the blot using a western antibody against the HA tag. A strong band corresponding to the ZFonly-HA construct appears in the IP lanes with and without benzonase treatment, confirming multimerization by this domain. Lower image shows the same blot after stripping and re-probing with anti-V5 antibody. (b) Replication of Chimp-Human competition co-IP western experiment with and without benzonase treatment. Lanes 1–3 are input lysate (50 μg protein measured by BCA) from untransfected cells, and transfected cells without or with benzonase treatment, respectively. Lanes 4–6 are IP elutions from untransfected cells, and transfected cells without or with benzonase treatment, respectively. Lane 7 is a protein ladder. Lower image shows the same blot after stripping and re-probing with anti-V5 antibody. (c) Relative quantification of band intensities for lanes 2–6 from b showing increased pull-down of the human prey relative to the chimp prey by the human bait. (d) co-IP prey band intensity ratios (similar to Figure 5c), though normalized to input band intensity ratios, with and without benzonase treatment. This confirms that the human allele preferentially multimerizes with the human allele over the chimp allele, and that this multimerization is not directly mediated by DNA. (e) 2% agarose DNA gel confirming complete digestion of DNA in transfected cell lysates (2 μg DNA per lane). Lane 1 contains a 1 kb DNA ladder. Lanes 2–3 correspond to cells transfected with ZFonly-ZFonly (used to make blot a) without or with benzonase treatment. Lanes 3–4 correspond to cells transfected with Human-V5 plus Chimp-HA plus Human-HA (used to make blot b) without or with benzonase treatment. Lane 6 contains a 100 bp DNA ladder.
Additional files
-
Transparent reporting form
- https://doi.org/10.7554/eLife.28383.027
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A map of human PRDM9 binding provides evidence for novel behaviors of PRDM9 and other zinc-finger proteins in meiosis
eLife 6:e28383.
https://doi.org/10.7554/eLife.28383




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}